

Abstract
We previously reported that alcohol drinkers with and without cirrhosis showed a significant increase in fecal bile acid secretion compared to nondrinkers. We hypothesized this may be due to activation by alcohol of hepatic cyclic adenosine monophosphate responsive element‐binding protein 3‐like protein 3 (CREBH), which induces cholesterol 7α‐hydroxylase (Cyp7a1). Alternatively, the gut microbiota composition in the absence of alcohol might increase bile acid synthesis by up‐regulating Cyp7a1. To test this hypothesis, we humanized germ‐free (GF) mice with stool from healthy human subjects (Ctrl‐Hum), human subjects with cirrhosis (Cirr‐Hum), and human subjects with cirrhosis and active alcoholism (Alc‐Hum). All animals were fed a normal chow diet, and none demonstrated cirrhosis. Both hepatic Cyp7a1 and sterol 12α‐hydroxylase (Cyp8b1) messenger RNA (mRNA) levels were significantly induced in the Alc‐Hum and Ctrl‐Hum mice but not in the Cirr‐Hum mice or GF mice. Liver bile acid concentration was correspondingly increased in the Alc‐Hum mice despite fibroblast growth factor 15, fibroblast growth receptor 4, and small heterodimer partner mRNA levels being significantly induced in the large bowel and liver of the Ctrl‐Hum mice and Alc‐Hum mice but not in the Cirr‐Hum mice or GF mice. This suggests that the normal pathways of Cyp7a1 repression were activated in the Alc‐Hum mice and Ctrl‐Hum mice. CREBH mRNA was significantly induced only in the Ctrl‐Hum mice and Alc‐Hum mice, possibly indicating that the gut microbiota up‐regulate CREBH and induce bile acid synthesis genes. Analysis of stool bile acids showed that the microbiota of the Cirr‐Hum and Alc‐Hum mice had a greater ability to deconjugate and 7α‐dehydroxylate primary bile acids compared to the microbiota of the Cirr‐Hum mice. 16S ribosomal RNA gene sequencing of the gut microbiota showed that the relative abundance of taxa that 7‐α dehydroxylate primary bile acids was higher in the Ctrl‐Hum and Alc‐Hum groups. Conclusion: The composition of gut microbiota influences the regulation of the rate‐limiting enzymes in bile acid synthesis in the liver. (Hepatology Communications 2017;1:61–70)
Abbreviations
- Alc‐Hum
humanized with stool from an actively drinking patient with alcoholism and cirrhosis
- BA
bile acid
- CB1R
cannabinoid receptor type 1
- Cirr‐Hum
humanized with stool from an abstinent patient with cirrhosis
- CREBH
cyclic adenosine monophosphate responsive element‐binding protein 3‐like protein 3
- Ctrl‐Hum
humanized with stool from a healthy control
- Cyp7a1
cholesterol 7α‐hydroxylase
- Cyp8b1
cholesterol 12α‐hydroxylase
- DCA
deoxycholic acid
- FGF‐15
fibroblast growth factor
- FGFR4
FGF receptor 4
- FXR
farnesoid X receptor
- GF
germ free
- IL‐6
interleukin‐6
- LEFSe
linear discriminant analysis size effect
- MCP1
monocyte chemoattractant protein 1
- MLN
mesenteric lymph nodes
- mRNA
messenger RNA
- NGRRC
National Gnotobiotic Rodent Resource Center
- PCR
polymerase chain reaction
- SHP
small heterodimeric protein
- UNC
University of North Carolina
- UniFrac
unique fraction metric
Introduction
Our previous studies showed that patients with cirrhosis who actively drank alcohol had significantly higher fecal total and secondary bile acids (BAs) compared to nondrinking patients with cirrhosis.1 Moreover, alcohol‐consuming control individuals also had significantly higher levels of total fecal and secondary BAs compared to abstinent individuals. Serum BA concentrations were higher in drinkers without cirrhosis and in individuals with alcoholism and cirrhosis compared to healthy controls and individuals with cirrhosis but without alcoholism, respectively.1 Our initial interpretation of these data was that alcohol might be activating hepatic cannabinoid receptor type 1, which has been reported to activate endoplasmic reticulum‐bound cyclic adenosine monophosphate responsive element‐binding protein 3‐like protein 3 (CREBH), a transcription factor that up‐regulates cholesterol 7α‐hydroxylase (Cyp7a1), the rate‐limiting enzyme in the neutral pathway of BA synthesis.2 In this manner, alcohol may overcome the repression of Cyp7a1 by hepatic small heterodimer partner (SHP) and intestinal fibroblast growth factor 15/19 (FGF‐15/19) by BAs.3, 4 Alternatively, the gut microbiota composition could be a regulator of hepatic BA synthesis.
In patients with cirrhosis, BA synthesis significantly shifts to the alternative pathway that uses oxysterol 7α‐hydroxylase (Cyp7b1) to form primary BAs.5 Prior studies6, 7 have shown that in patients with cirrhosis the gut microbiota markedly shifts to a more toxic or dysbiotic microbiota, which is correlated with a decrease in BA synthesis and an increase in inflammatory cytokines.1 Bile acids are known to be a major regulator of the structure of the gut microbiome.8, 9 In this regard, feeding cholic acid to rats markedly shifts the gut microbiome to mostly members of the gram‐positive Firmicutes.6, 8 Therefore, we hypothesize that crosstalk between the liver and the gut microbiota could regulate BA synthesis and that BAs serve as interkingdom‐signaling molecules. The current study was undertaken to determine if different human gut microbiota can differentially regulate BA synthesis in mice in the absence of alcohol or cirrhosis.
Materials and Methods
We studied 10‐15‐week‐old, age‐matched, germ‐free (GF) C57BL/6 male mice from the National Gnotobiotic Rodent Resource Center (NGRRC) at the University of North Carolina (UNC)‐Chapel Hill. Sterility was documented by fecal gram stains and aerobic and anaerobic fecal culture every 2 weeks. All mouse groups remained separated in different Trexler isolators throughout the study period to prevent cross‐contamination.
Humane protocols were followed under the UNC Chapel Hill Institutional Animal Care and Use Committee guidelines. The Institutional Animal Care and Use Committee at UNC and Richmond McGuire Veterans Administration Hospital approved all animal activities. The human study was reviewed and approved by the institutional review boards at the Virginia Commonwealth University Medical Center and Richmond McGuire Veterans Administration Hospital. All participants gave written informed consent for this study.
The mice were divided into five groups, and all investigators apart from the NGRRC staff were blinded to the assignment of these groups (experimental design in Supporting Fig. S1): remained germ‐free (GF); were humanized with the stool of a 62‐year‐old healthy man without chronic diseases who was not drinking alcohol (Ctrl‐Hum); were humanized with the stool of a 60‐year‐old man with compensated cirrhosis due to alcohol, who had quit drinking more than 2 years previously (Cirr‐Hum); were humanized with the stool of an actively drinking 60‐year‐old man with compensated alcoholic cirrhosis (Alcohol Use Disorders Identification Test score 14 at the day of collection) (Alc‐Hum); GF mice given 0.04% deoxycholic acid (DCA) in their chow for 30 days (DCA).
None of the human donors were on antibiotics or probiotics during the previous 3 months, and none had a comparable dietary intake during the prior 3 days. The donor with alcoholism had continued drinking until the donation and had a bacterial 16S composition comparable to 42 other individuals with alcoholism and cirrhosis; the nondrinking donor with cirrhosis had a composition comparable to 75 other abstinent patients with cirrhosis; and the control had a composition similar to 24 other controls, indicating that these samples represented changes in those groups.7 The donor batches were collected from 20 g of stool that was immediately spun down with phosphate‐buffered saline. Several batches of feces from donors with alcoholism and from healthy donors were created and frozen; there was no significant variation in bacterial composition between the different batches (unique fraction metric [UniFrac], P = 1, Bonferroni correction).10 Human fecal transfer was performed by gavage, rectal swabbing, and saturating the bedding of the mice over 3 consecutive days, with different Trexler isolators used for each group.
At day 30, all mice were sacrificed humanely and the following organs and tissues were collected: small bowel, cecal and colonic mucosa, mesenteric lymph nodes (MLN), stool, cardiac blood, and liver.
Microbiota and Bile Acid Analysis
We performed multitag sequencing of the microbiota from the stool, MLN, and large bowel mucosa11 to evaluate microbial composition. We also studied serum endotoxin using Limulus amebocyte lysate techniques.7 We used liquid chromatography–mass spectrometry techniques to gauge the fecal and hepatic BA profile.12 Colonic and small bowel expression of farnesoid X receptor (FXR), FGF‐15, and SHP and the hepatic expression of Cyp7a1, Cyp7b1, cholesterol 12α‐hydroxylase (Cyp8b1), FXR, FGF‐15, FGF receptor 4 (FGFR4), CREBH, and SHP were performed with quantitative polymerase chain reaction (PCR). Western blot analysis of hepatic SHP and FXR protein was performed.
INFLAMMATION
We assessed systemic inflammation in the liver and small and large intestine between groups.
Liver Inflammation
Immediately following sacrifice, a portion of the mouse livers was cut longitudinally into 2‐3‐mm slices and fixed in 10% neutral‐buffered formalin. Tissues were processed on a Sakura Tissue‐Tek Vacuum Infiltration Processor. Sections 3‐µm thick were stained with hematoxylin and eosin, Masson Trichrome, and periodic acid–Schiff for histologic evidence of inflammation and fibrosis. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured in all groups.
Intestinal Inflammatory Markers
We performed quantitative PCR of the intestinal inflammatory markers (interleukin‐6 [IL‐6], monocyte chemoattractant protein 1 [MCP1]) in the large and small intestine.
Systemic Inflammatory Markers
Systematic inflammatory markers were studied using IL‐1β in the serum.
STATISTICAL ANALYSIS
Based on multiple prior studies of mono‐association and polymicrobial inoculation carried out at the NGRRC in which six mice showed changes relative to baseline, at least six mice were considered adequate for the analysis.13 Comparisons were performed overall between GF, Ctrl‐Hum, Cirr‐Hum, Alc‐Hum, and DCA, using analysis of variance and Kruskal‐Wallis tests for messenger RNA (mRNA) expression, BA profile, and endotoxemia.
Microbiome Analysis
Abundances of the bacterial identifications were normalized, and taxa present at 0.1% of the community were tabulated. Microbiota change between groups was assessed using RDP11 (Ribosomal Database Project) Bayesian analysis, UniFrac analysis, and linear discriminant analysis effect size.10, 14, 15 UniFrac analysis was performed using version 1.3.0 of Quantitative Insights into Microbial Ecology, and weighted P values were calculated using the Bonferroni correction.10
Results
ADEQUATE HUMANIZATION OF MICE WAS ACHIEVED USING THREE SEPARATE DONORS
We included forty‐two 10‐15‐week‐old, age‐matched, GF C57BL/6 mice (Supporting Fig. S1); eight remained GF, six underwent control fecal transplant (Ctrl‐Hum), six underwent cirrhotic human fecal transplant (Cirr‐Hum), 12 were humanized with alcoholic cirrhotic feces (Alc‐Hum), and 10 mice were fed 0.04% DCA in their diet (DCA group). Two Alc‐Hum mice died related to gavage; the remaining 10 were followed for 30 days. There were no behavioral or feeding changes observed in any group, and the remaining 40 mice underwent necropsy at the predetermined postcolonization 30‐day endpoint.
STOOL BILE ACID PATTERNS IN HUMANIZED MICE FOLLOWED THAT OF PRIOR HUMAN STUDIES
Different stool BA patterns were induced in the humanized ex‐GF mice via stool transfer (Fig. 1A‐D). Total liver and conjugated BA were also significantly increased in the Alc‐Hum and Ctrl‐Hum colonized mice but not in the Cirr‐Hum or GF mice (Fig. 1E,F).
Figure 1.
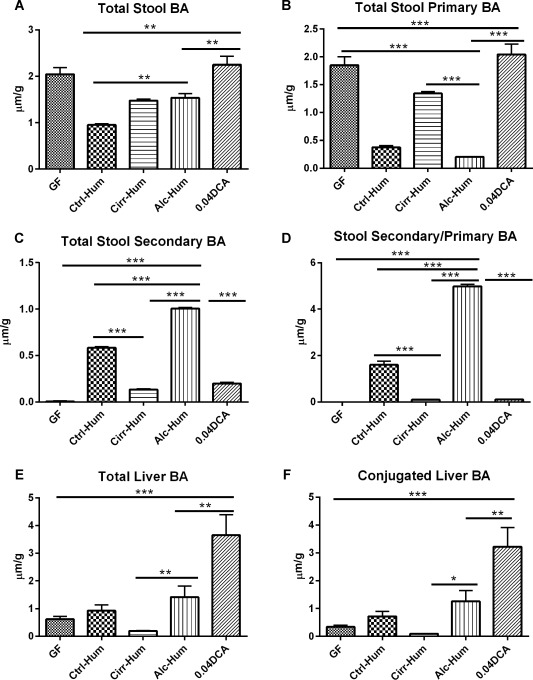
Bile acid content in µM/g of tissue. *P < 0.05, **P < 0.01, ***P < 0.001 using the Kruskall‐Wallis test or unpaired t test based on the number of samples compared. All values are in mean ± SEM. (A) Stool BA content was highest in both GF groups (GF and 0.04 DCA) and shows a significant increase in total BA content in the 0.04 DCA group. This was higher than that in the Alc‐Hum and Cirr‐Hum groups, which in turn were higher than in the Ctrl‐Hum group. (B) Stool primary BA content was highest in the GF groups as expected. Among the humanized groups, it was lowest in the Alc‐Hum and Ctrl‐Hum groups but highest in the Cirr‐Hum group. (C) Stool secondary BA content was absent in the GF group and was intermediate in the DCA and Cirr‐Hum groups. The highest secondary BA content was in the Alc‐Hum group, which was greater than in the Cirr‐Hum and Ctrl‐Hum groups, using unpaired t tests. (D) Stool secondary/primary BA ratio was absent in the GF group and was lowest in the DCA and Cirr‐Hum groups. A significantly higher ratio was seen in the Alc‐Hum group, which was significantly higher compared to the Cirr‐Hum and Ctrl‐Hum groups, using unpaired t tests. (E) Liver BA content was highest in the DCA group followed by the Alc‐Hum group; it was lowest in the Cirr‐Hum group. Alc‐Hum mice had a higher total liver BA content compared to Cirr‐Hum mice, using unpaired t tests. (F) Conjugated liver BA profile was similar to the total liver BA content in that the DCA group had the highest levels followed by the Alc‐Hum group. Levels in the Alc‐Hum group were higher than in the Cirr‐Hum group.
Mice colonized with different human gut microbiota had different levels of serum and intestinal inflammatory markers and liver function enzymes. Mice colonized by Alc‐Hum microbiota had significantly higher levels of serum IL‐1β and endotoxin (Supporting Fig. S2A,B) and increased mRNA levels of small and large intestine IL‐6 and MCP‐1 (Supporting Fig. S3A,B). Mice colonized with Alc‐Hum also had somewhat higher serum levels of AST and ALT (Supporting Fig. S4A,B). However, liver histology showed no apparent differences between animal groups (Supporting Fig. S5).
BACTERIAL TRANSLOCATION AND MICROBIAL CHANGES FOLLOWED THE DONOR AND AFFECTED THE BA PROFILE
There was statistical similarity on UniFrac between respective donors and the microbial distributions in the various mucosal tissues (P > 1.0 in all tissues), indicating excellent transfer of human bacterial profiles using the donor stool samples. Bacterial translocation was defined as the presence of bacterial DNA in the MLN. As expected, no bacteria were isolated from the GF or DCA groups. There was a significant increase in the proportion of bacterial translocation with viable bacterial DNA in the Alc‐Hum group (7 of 10) compared to the Ctrl‐Hum (1 of 6) and Cirr‐Hum groups (0 of 6, P < 0.02) (Supporting Fig. S6A). The bacterial profile in the MLN in Alc‐Hum mice were statistically similar on UniFrac to those found in the mucosa of the Alc‐Hum large intestine (P = 0.13), small intestine (P = 1.0), and stool (P = 0.1). When UniFrac changes were analyzed, there were significant changes between large intestinal mucosally associated bacteria between the Alc‐Hum and Ctrl‐Hum groups and between the Alc‐Hum and Cirr‐Hum groups (both P ≤ 0.01) but not between the Ctrl‐Hum and Cirr‐Hum groups (P = 0.19) (Supporting Fig. S6B,C). The relative abundance of members of the genus Clostridium was significantly higher in the Alc‐Hum and Ctrl‐Hum mice compared to the Cirr‐Hum mice. Clostridium species have been reported to contain genes encoding enzymes in the BA 7α‐dehydroxylation pathway.9 These data are consistent with the BA stool analyses that show much higher amounts of secondary BAs in the Alc‐Hum and Ctrl‐Hum mice (Fig. 1).
REGULATION OF KEY GENES IN HEPATIC BA SYNTHESIS BY DIFFERENT HUMAN GUT MICROBIOTA
The mRNA levels of Cyp7a1, Cyp8bb1, and Cyp7b1 were measured by quantitative real‐time PCR at day 30 posttransplant of stool. The mRNA levels of Cyp7a1 and Cyp8b1 was significantly up‐regulated in Alc‐Hum and Ctrl‐Hum mice but not in Cirr‐Hum, GF, or GF mice fed 0.04% DCA (Fig. 2A,B). Cyp7a1 and Cyp8b1 are rate limiting for the synthesis of cholic acid. In contrast, Cyp7b1, which is the key enzyme in the alternative pathway and chenodeoxycholic acid synthesis, was significantly induced in all three colonized animal groups. However, there was significantly less induction in the Alc‐Hum mice (Fig. 2C). This indicates that the Alc‐Hum and Ctrl‐Hum mice are primarily using the neutral pathway of BA synthesis and to a lesser extent the alternative pathway. In contrast, the Cirr‐Hum mice appear to be primarily using the alternative pathway of BA synthesis.3, 5
Figure 2.
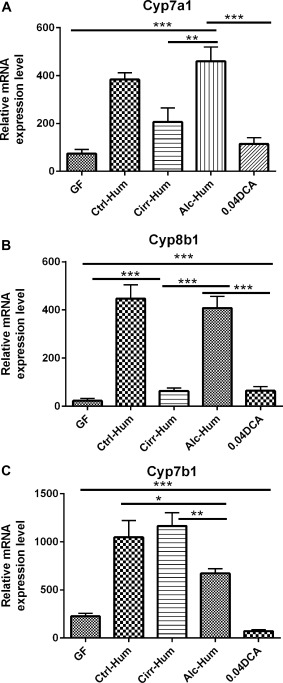
Cholesterol 7α‐hydroxylase (CYP) expression in the liver (relative mRNA expression level). *P < 0.05, **P < 0.01, ***P < 0.001 using the Kruskall‐Wallis test or unpaired t test based on the number of samples compared. All values are in mean ± SEM. (A,B) Cyp7a1 and Cyp8b1 expression was highest in the Alc‐Hum and Ctrl‐Hum groups compared to GF, DCA, and Cirr‐Hum mice. DCA supplementation resulted in appropriate feedback inhibition of Cyp7a1 and Cyp8b1. (C) The alternative pathway enzyme expression Cyp7bB1 was highest in the Cirr‐Hum and Ctrl‐Hum groups but was suppressed in Alc‐Hum, DCA, and GF mice.
LACK OF REPRESSION OF HEPATIC CYP7A1 AND CYP8B1 BY FGF‐15 AND SHP
Because Cyp7A1 and Cyp8B1 were highly up‐regulated, we wanted to determine if intestinal or liver FGF‐15 mRNA or hepatic SHP mRNA was induced. The data showed that both intestinal and hepatic FGF‐15 mRNA was induced in the intestines and liver of the Alc‐Hum and Ctrl‐Hum groups (Fig. 3A‐C). FGF‐15 expression was confirmed by PCR of the product in the sequences (Supporting Fig. S7). The hepatic receptor for FGF‐15 is FGFR4, which was highly induced in the liver of the Ctrl‐Hum and Alc‐Hum mice but not in the Cirr‐Hum mice (Fig. 3D). FGF‐15 and FGFR4 were barely detectable in the Cirr‐Hum colonized animals. Moreover, SHP, a nuclear receptor without a DNA‐binding domain, has been reported to repress hepatic CYP7A1 by interacting with transcription factors (liver receptor homolog 1, hepatocyte nuclear factor 4α) that activate the Cyp7a1 promoter.3, 4 SHP is induced by BAs that activate FXR.3 Therefore, we measured the levels of FXR mRNA and SHP mRNA in the liver and intestines. Liver FXR mRNA and SHP mRNA were both significantly increased in Ctrl‐Hum and Alc‐Hum humanized mice (Fig. 4A,B). In addition, western blot analysis showed that hepatic FXR and SHP protein were also highly expressed in the liver (see Supporting Fig. S7). Moreover, FXR mRNA and SHP mRNA were highly expressed in the small and large intestines in Ctrl‐Hum and Alc‐Hum mice but were expressed much less in Cirr‐Hum mice (Fig. 4C‐F).
Figure 3.
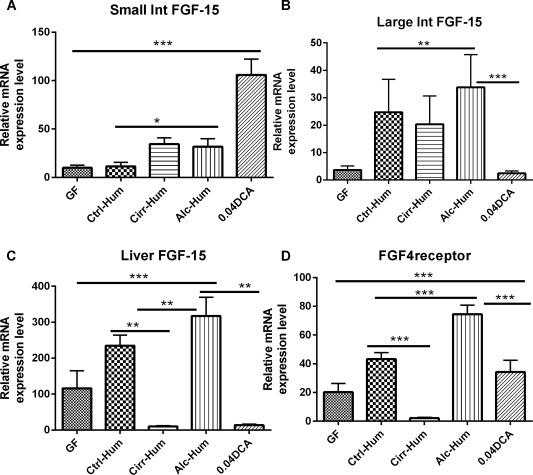
FGF‐15 and FGFR4 expression (relative mRNA expression level). *P < 0.05, **P < 0.01, ***P < 0.001 using the Kruskall‐Wallis test or unpaired t test based on the number of samples compared. All values are in mean ± SEM. (A) Small intestinal FGF‐15 expression was highest in the DCA group. Within the humanized group, this was lowest in the Ctrl‐Hum mice compared to the Alc‐Hum and Cirr‐Hum mice. (B) Large intestinal FGF‐15 expression was high in all three humanized groups but lowest in the Cirr‐Hum group. DCA expression of FGF‐15 was comparable to the GF group. (C) Hepatic FGF‐15 expression was highest in the Alc‐Hum group followed by the Ctrl‐Hum groups. This was lowest in the Cirr‐Hum and DCA groups. (D) FGFR4 receptor expression followed the hepatic FGF‐15 expression patterns. Abbreviation: Int, intestine.
Figure 4.
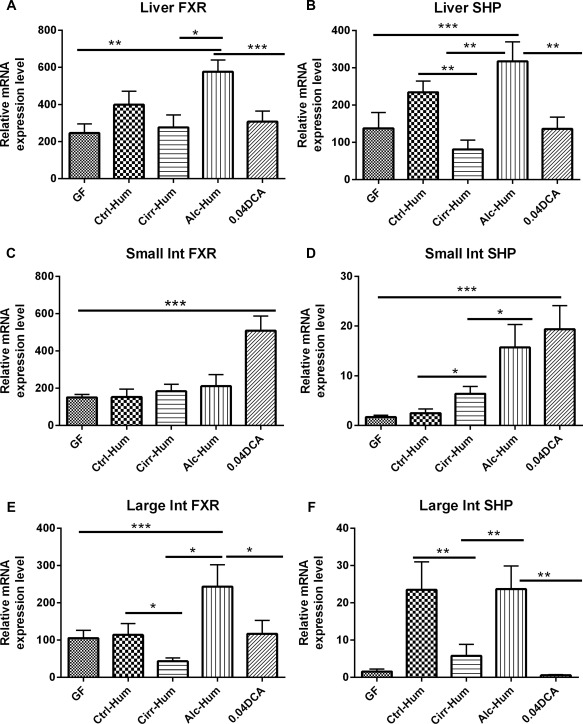
SHP and FXR expression (relative mRNA expression level). *P < 0.05, **P < 0.01, ***P < 0.001 using the Kruskall‐Wallis test or unpaired t test based on the number of samples compared. All values are in mean ± SEM. (A,B) Liver SHP and FXR expression was highest in the Alc‐Hum mice followed by the Ctrl‐Hum mice. Cirr‐Hum mice had the lowest expression of mRNA among the humanized mice. mRNA expression in the Alc‐Hum mice was higher than in the DCA mice. (C,D) Small intestinal FXR and SHP expression was highest in the DCA group compared to the rest. Within the humanized mice groups, Alc‐Hum mice had the highest SHP expression while FXR expression remained statistically similar. (E,F) Large intestinal FXR and SHP expression demonstrated significantly higher levels in the Alc‐Hum mice compared to the Cirr‐Hum and Ctrl‐Hum mice. DCA mice had similar large intestinal FXR or SHP expression to GF mice. Abbreviation: Int, intestine.
CREBH WAS HIGHLY INDUCED BY SPECIFIC GUT MICROBIOTA
CREBH is a liver‐specific transcription factor that induces the gene encoding Cyp7a1 and Cyp8b1.2 Measured mRNA levels of CREBH showed significant induction of this gene only in the Ctrl‐Hum and Alc‐Hum mice (Fig. 5). Unfortunately, we were not able to quantitate CREBH protein as the specific antibody for CREBH is no longer available.2 The Alc‐Hum and Ctrl‐Hum microbiota‐colonized animals appeared to be capable of inducing and activating hepatic CREBH expression to a much greater extent than Cirr‐Hum microbiota or GF mice. Alternatively, the Alc‐Hum and Ctrl‐Hum colonized animals may activate an unknown mechanism that induces Cyp7a1 and Cyp8b1. The results suggest that the microbiota alone can drive the extent and character of the BA pool in the mouse by overriding the classical FXR–SHP–FGF‐15 feedback mechanism. Given the symbiotic relationship that exists, we suggest that the gut microbiota may be able to manipulate BA synthesis to create a better environment for their survival.
Figure 5.
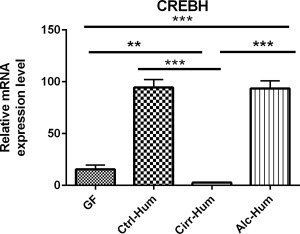
Liver CREBH (relative mRNA expression level). *P < 0.05, **P < 0.01, ***P < 0.001 using the Kruskall‐Wallis test or unpaired t test based on the number of samples compared. All values are in mean ± SEM. Alc‐Hum and Ctrl‐Hum mice had similarly high expressions of CREBH, while Cirr‐Hum and GF mice had significantly lower expressions.
Discussion
Studies of the human microbiome over the past decade have changed our perception of human physiology and microecology. Most biomedical researchers view the human body as a complex ecosystem of interacting prokaryotic and eukaryotic cells. This new concept is particularly relevant to the gastrointestinal system where the circulating BA pool composition is determined not only by primary BA synthesis in the liver but also by synthesis of secondary BAs by specific Clostridium species in the gut.9 Secondary BAs are excellent agonists for specific nuclear receptors (vitamin D, pregnane X receptor) and G protein‐coupled receptors (TGR5) in host cells.16 Bile acids also are important regulators of the structure of the gut microbiome.8 In this regard, a Western diet or providing cholic acids in the diet shifts the gut microbiome strongly toward members of the Firmicutes.8, 17 Moreover, feeding cholic acid to mice has been reported to increase the number of BA 7α‐dehydroxylating bacteria 1,000‐fold.6 Primary BAs induce genes in Clostridium scindens encoding enzymes involved in BA 7α‐dehydroxylation of primary BAs, indicating these bacteria have specific BA‐sensing mechanisms.9 Therefore, BAs could be viewed as important interkingdom‐signaling molecules linking the regulation of metabolic activities in the liver with the regulation of gene expression in specific gut bacteria that metabolize BAs. This liver–gut–BA axis is particularly relevant in cirrhosis and alcohol use where altered intestinal permeability is a major factor behind disease progression.18, 19 Secondary BAs are reported to negatively affect this barrier in conventional and high‐fat‐fed mice.20, 21 We extended these observations to the GF condition where DCA‐fed mice showed significantly higher small intestinal inflammatory cytokine expression. This demonstrates that secondary BAs alone may be responsible for part of the intestinal barrier function that accompanies other insults, such as alcohol abuse. This was reiterated by the relative increase in secondary BAs in the Alc‐Hum mice that demonstrated both large and small intestinal inflammatory cytokine expression and bacterial translocation without significant liver injury. This replicates the human experience without alcohol intake or attendant liver inflammation and extends a prior study into microbiota in alcohol without the setting of alcoholic hepatitis.1, 22
In contrast, transfer of a stool from a patient with cirrhosis (Cirr‐Hum) that had decreased BA synthesis into the GF mice resulted in significantly lower hepatic levels of mRNA encoding key rate‐limiting enzymes in BA synthesis and a higher alternative pathway gene expression, consistent with the human findings.1 These mice also had significantly lower amounts of BAs in the liver and intestines compared to transfers created from samples of patients with active alcoholism and cirrhosis and healthy control samples. This Cirr‐Hum group serves as a comparator to the Alc‐Hum group to differentiate the impact of alcohol compared to cirrhosis on the microbiota and their differential ability to regulate hepatic BA synthesis.
The Alc‐Hum and Ctrl‐Hum mice had normal or increased levels of stool BAs compared to the Cirr‐Hum mice, similar to the human experience.1 Interestingly, the normal pathways of BA feedback repression by FXR–FGF‐15–SHP pathways were induced in the Ctrl‐Hum and Alc‐Hum mice, suggesting that an overriding mechanism for BA synthesis is in play in these mice. It has been reported that probiotics can promote BA deconjugation and fecal secretion of BA, inducing new BA synthesis in the liver.23 In those studies, there was decreased BA absorption with repression of the FXR–FGF‐15 axis in the intestines allowing increased hepatic BA synthesis. It has also been reported that the gut microbiota can regulate gut levels of the FXR antagonist tauro‐β‐muricholic acid, which decreases FGF‐15 synthesis in the intestines.24 Each of these models relies on manipulation of the intestinal FXR–FGF‐15 axis. However, in the current study, mRNA of both FXR and FGF‐15 was up‐regulated in the intestines and SHP mRNA was up‐regulated in the liver. In both the Ctrl‐Hum and Alc‐Hum mice but not the Cirr‐Hum mice, hepatic CREBH mRNA was significantly up‐regulated, suggesting this may be responsible for the up‐regulation of Cyp7a1 and Cyp8b1.2 Cannabinoid receptor type 1 (CB1R) has been reported to activate CREBH by up‐regulating Cyp7a1, Cyp8b1, and BA synthesis.2 CB1R is normally activated by endocannabinoids (anandamide and 2‐arachidonoyl glycerol), but recent studies report it can be activated by ethanol.2, 25 Therefore, these data suggest that the gut microbiota from Ctrl‐Hum and Alc‐Hum mice may be producing a compound(s) that activates hepatic CB1R. Alternatively, an unknown mechanism may be activated that up‐regulates genes involved in BA synthesis.
In summary, the current study reports novel data indicating that the composition of the gut microbiota can regulate BA synthesis in the liver, and this can be associated with intestinal barrier dysfunction. One might speculate that members of the genus Clostridium may be the most likely component of the gut microbiota that regulates hepatic BA synthesis. Nevertheless, the current studies point to a new direction for investigating the regulation of hepatic BA synthesis and how the interactions between cells in the liver and gut microbiota regulate BA homeostasis in the body.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1020/suppinfo.
Supporting Information
Supporting Information
Potential conflict of interest: Nothing to report.
Supported by the National Institute of Diabetes and Digestive and Kidney Diseases (RO1DK089713) and the U.S. Department of Veterans Affairs (CX10076 VA Merit Review) to J.S.B. The National Gnotobiotic Rodent Resource Center is supported by grants P40OD010995 and P30DK034987 from the National Institutes of Health to R.B.S.
REFERENCES
- 1. Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol 2014;306:G929‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chanda D, Kim YH, Li T, Misra J, Kim DK, Kim JR, et al. Hepatic cannabinoid receptor type 1 mediates alcohol‐induced regulation of bile acid enzyme genes expression via CREBH. PLoS One 2013;8:e68845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 2014;66:948‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell 2000;6:517‐526. [DOI] [PubMed] [Google Scholar]
- 5. Axelson M, Sjovall J. Potential bile acid precursors in plasma‐‐possible indicators of biosynthetic pathways to cholic and chenodeoxycholic acids in man. J Steroid Biochem 1990;36:631‐640. [DOI] [PubMed] [Google Scholar]
- 6. Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes 2013;4:382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol 2014;60:940‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011;141:1773‐1781. [DOI] [PubMed] [Google Scholar]
- 9. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016;7:22‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamady M, Knight R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res 2009;19:1141‐1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Naqvi A, Rangwala H, Keshavarzian A, Gillevet P. Network‐based modeling of the human gut microbiome. Chem Biodivers 2010;7:1040‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kakiyama G, Muto A, Takei H, Nittono H, Murai T, Kurosawa T, et al. A simple and accurate HPLC method for fecal bile acid profile in healthy and cirrhotic subjects: validation by GC‐MS and LC‐MS. J Lipid Res 2014;55:978‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10‐deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005;128:891‐906. [DOI] [PubMed] [Google Scholar]
- 14. White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 2009;5:e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11:55‐67. [DOI] [PubMed] [Google Scholar]
- 17. O'Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun 2015;6:6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tandon P, Garcia‐Tsao G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin Liver Dis 2008;28:26‐42. [DOI] [PubMed] [Google Scholar]
- 19. Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, et al. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol 2012;302:G966‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol 2013;304:G227‐234. [DOI] [PubMed] [Google Scholar]
- 21. Stenman LK, Holma R, Korpela R. High‐fat‐induced intestinal permeability dysfunction associated with altered fecal bile acids. World J Gastroenterol 2012;18:923‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016;65:830‐839. [DOI] [PubMed] [Google Scholar]
- 23. Degirolamo C, Rainaldi S, Bovenga F, Murzilli S, Moschetta A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr‐Fgf15 axis in mice. Cell Rep 2014;7:12‐18. [DOI] [PubMed] [Google Scholar]
- 24. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐beta‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225‐235. [DOI] [PubMed] [Google Scholar]
- 25. Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 2006;58:389‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1020/suppinfo.
Supporting Information
Supporting Information