

Abstract
Human Mesenchymal stem cells (hMSCs) secrete products (supernatants) that are anti-inflammatory and antimicrobial. We have previously shown that hMSCs decrease inflammation and Pseudomonas aeruginosa infection in the in vivo murine model of Cystic Fibrosis (CF). Cystic Fibrosis (CF) is a genetic disease in which pulmonary infection and inflammation becomes the major cause of morbidity and mortality. Our studies focus on determining how MSCs contribute to improved outcomes in the CF mouse model centering on how the MSCs impact the inflammatory response to pathogenic organisms. We hypothesize that MSCs secrete products that are anti-inflammatory in scenarios of chronic pulmonary infections using the murine model of infection and inflammation with a specific interest in Pseudomonas aeruginosa (gram negative). Further, our studies will identify whether the MSCs are impacting this inflammatory response through the regulation of peroxisome proliferator activator receptor gamma (PPARγ) which aides in decreasing inflammation.
Keywords: Mesenchymal stem cells, Chemokines, Cytokines, Anti-Inflammatory Cytokines, Anti-Inflammation, chemotaxis, PPARγ, cystic fibrosis
Introduction
Mesenchymal stem cells (hMSCs) have been shown to be anti-inflammatory and anti-microbial in vitro as well as in vivo using the murine model of CF lung infection and inflammation1,2,3. Cystic fibrosis (CF) is a genetically inherited fatal disease in which the cystic fibrosis transmembrane receptor (CFTR) gene is mutated resulting in defective protein causing patients to have failure to thrive, lung congestion and infection with a variety of bacteria4. Defective CFTR results in impeded mucocilliary clearance, which is associated with a bacterial infection requiring treatment with antibiotics and mucus thinning reagents5. Pseudomonas aeruginosa is the most prevalent bacteria in CF infections, but is not the only bacteria involved in creating serious infection and damage in the CF lung. Organisms such as Streptococcus pnuemoniae and Staphylococcus aureus create a complex pulmonary niche and microbiome6,7. In the end, despite new developments in small molecular correctors and CFTR expression enhancers, the ensuing inflammatory response and pulmonary failure continue to be a serious issue leading to significant morbidity and eventual mortality in CF8. This is mostly due to the immune response in CF individuals which is too aggressive, and ultimately contributing to lung damage. In healthy lungs, bacterial infiltration results in acute neutrophil influx and eradication of bacteria followed by inflammation resolution. However, in CF patients, the neutrophil influx is excessive with increased release of neutrophils containing neutrophil elastase (NE), which is extremely damaging to the lung milieu. NE induces an increased gland secretion, and an increase in bacterial binding to the airway surface9. Increased levels of neutrophils also lead to an influx in oxygen radical production, perpetuating interleukin 8 (IL-8) which recruits more neutrophils. hMSCs secrete products that are anti-inflammatory, anti-microbial, angiogenic, chemotactic, anti-apoptotic, and anti-scarring10,11. hMSCs have been investigated in several lung disease models suggesting their therapeutic potential in redirecting the excessive CF and inflammatory based pathophysiology12,13. We have studied hMSC efficacy in the infection/inflammation using the murine model of cystic fibrosis (CF)14. Wild Type (WT) and cystic fibrosis transmembrane conductance regulator (Cftr) deficient mice (CF) mice were chronically infected with Pseudomonas aeruginosa (PA) and followed for several days for clinical score, survival and weight loss kinetics. At euthanasia, bronchoalveolar lavage (BAL) showed that hMSCs shifted the pulmonary differential away from neutrophils towards macrophages which was statistically significant for CF animals since they traditionally have an augmented neutrophilic response. In the studies reported here, we have begun to pursue the mechanisms associated with the anti-inflammatory function of hMSCs in the Cftr deficient murine models. We report that the anti-inflammatory potency of the hMSCs can affect cell recruitment and aide in attenuating the robust response inherent in CF pathophysiology. Our data also suggest that the mechanism of anti-inflammatory action is through regulating the inflammatory transcription regulator PPARγ as well as the production of chemokines ultimately impacting the phenotype of the inflammatory response. We further provide data to suggest that deficient CFTR function alters hMSC’s activity implicating the need for allogeneic sources of hMSCs for clinical trials. The novelty of the results in this manuscript demonstrate the unique niche which defines the function of hMSCs and their powerful therapeutic potential for diseases associated with chronic inflammation, in the context of unrelenting infection. Further we have begun to define the mechanistic impact of the hMSCs in the context of inflammation and how it might be impacted in scenarios of deficient CFTR activity.
Methods
Human Cell Preparations
hMSCs
hMSC preparations were obtained from the National Center for Regenerative Medicine, Case Comprehensive Cancer Center, and SCC Cellular Therapy Laboratory. The hMSCs were isolated and grown following the standard operating procedures associated with hMSCs in clinical trials15,16 .hMSCs were expanded and grown in hMSC condition media treated with 10% fetal calf serum. hMSCs were taken from first or second passage.
PBMCs
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy control individuals as previously described using Institutional IRB approval17. We experimented with the protocol to optimize the yield of monocytes in anticipation of obtaining limited blood volumes from CF patients. The isolated PBMCs were either cultured with control media (RPMI 1641) or hMSC preparations (1:1). Control conditions of these cultures were left unstimulated and experimental conditions were stimulated with lipopolysaccharide (LPS) at 10ug/ml. There was a 24-hour incubation period at 37°C.
Animal Studies
All Animal Studies were done with approval by Case Western Reserve IACUC committee and approval of application 2011–0146. Cystic fibrosis transmembrane receptor deficient animals (Cftrtm1Kth, CF) and congenic background controls (C57BL/6J, WT) were anesthetized and inoculated with agarose beads embedded with 106 Pseudomonas aeruginosa (PA-M5715) or Staphylococcus aureus (ATCC# SA 29523). Animals were followed with daily weights and clinical scores out to 10 days. Animals were euthanized and evaluated for lung inflammation by bronchoalveolar lavage (BAL) for cytokines, chemokines, cellular recruitment and differentials as described previously3,18.
Gene Expression
hMSCs that were used both in the in vivo murine CF model and the in vitro bactericidal and anti-inflammatory assays were processed for messenger ribonucleic acids (mRNA) followed by synthesis of complementary deoxyribonucleic acid (cDNA) for chemokine gene expression by real-time-PCR19. hMSCs were grown in the presence and absence of CFTR inhibitor (I-172, 1ug/ml) to mimic CF hMSCs3. Both baseline and CFTR inhibited hMSCs were stimulated with and without lipopolysaccharide (LPS, 10ug/ml) to mimic exposure to gram negative bacteria. The quality of mRNA and synthesized cDNA was assessed using Nanodrop spectrophotometry technology (optimal threshold 260–280nm) as well as validated using peptidyl prolyl isomerase A (PPIA) as the human ubiquitous housekeeping gene. We evaluated the expression of interleukin −8 (IL-8), interleukin 6 (IL-6), chemokine ligand 2(CCL-2), chemokine ligand 20 (CCL20) and peroxisome proliferator activated receptor gamma (PPARγ) as a comparison of PPIA ubiquitous gene expression. In the animal studies, gene expression was compared to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). All PCR samples were compared to the PPIA control for the fold change for each target genes threshold cycle (ΔCt) value followed by the normalization to the baseline control to measure the overall response with and without I-172 exposure for the ΔΔCt response as previously described20. All data is expressed as ddCT response (ΔΔCT value; n = 10 Mean ± SEM).
Luminex Studies
hMSC supernatants or PBMC supernatants were evaluated for cytokines and chemokines using Luminex multi-bead array technology (R&D Systems, Minneapolis, MN) as previously described21,22 using a multi-array purchased which focused on IL-8, IL-6, macrophage inflammatory protein 1a(MIP-1α), CCL-2, and stem cell factor 1 (SCF-1). Briefly, supernatants from samples were interfaced with luminescence beads with ratios of proprietary fluorophores. Each bead is specific for a cytokine or chemokine. As the sample interacts with the beads, the capture antibodies conjugated to the beads binds to the cytokine/chemokine which is followed by a second antibodies labeled with phycoerythrin (PE). The detection of the cytokine/chemokine is based upon detection of the unique bead idea and the detection of the PA using a dual laser system with a lower end sensitivity of 3.2 pg/ml. Each cytokine/chemokine is compared to its specific standard curve, also done in multiplex. Data is expressed as mean (pg/ml ±SEM, n=10).
Aushon Bioscience Ciraplex Studies
The peripheral blood mononuclear cells (PBMC) studies were evaluated using Aushon Bioscience Ciraplex® Technology in which capture antibodies were spotted as a solid array chip, with the read-out being exquisitely sensitive chemi-luminescence. The advantage of this system was the small volume of sample required, the lower end sensitivity and the ease of the assay system. The sensitivity of the study was 1.0 pg/ml, n=10; MSC preparations and n=4 PBMC preparations (see below). For the PBMC gene expression data, cells were cultured in vitro with and without LPS and hMSC supernatants. After 24 hours, cells were removed and supernatants were evaluated for cytokine and chemokines using the Aushon Ciraplex® multi-array technology using Trizol® RNA extraction and TaqMan® real-time PCR assays.
Chemotaxis
PBMCs at 1×106 cells/ml were put into the top chamber of a 0.4μl transwell tissue culture plate. In the bottom well, 0.5mls of fresh hMSC supernatant utilized as previously described20. After 24 hours, the PBMCs in the top and the cells recruited into the bottom were counted for total cell count and made into cytospins to determine the cell differential. Cytospins were dried followed by staining with Wright Giemsa, which stains lymphocytes, monocytes, neutrophils, eosinophils and basophiles with a specific pattern.
PPARγ Studies
The in vitro assay for measuring the overall impact of hMSCs on PPARγ was done using RAW-267 cells, a macrophage cell line with known parameters for expressing PPARγ. Gene expression was done using real-time PCR as previously described23.
Data Evaluation and Statistics
ΔCTs were calculated by subtracting the relative cycling number of the probed sample (i.e. samples probed with IL-8, IL-6, CCL20 and CCL2) from the cycling number of the same sample probed with the background or housekeeping gene PPIA. In each group there were 10 different donor MSC preparations. ΔΔ CTs were calculated by subtracting the ΔCT of stimulated samples (i.e. LPS, I-172, I-172 LPS) from the unstimulated samples ΔCT for each preparation. Statistics were performed using GraphPad Prism 3.0 technology using non-parametric ANOVA analysis and standard T-cells when appropriate. Data is expressed as a Mean±Standard Error of the Mean (SEM) through non-parametric Mann-Whitney tests. P < 0.05 denotes significance.
Results
In Vivo Potency and Efficacy
We have studied hMSC therapeutic potential in the infection/inflammation murine model of inflammation and infection focusing on chronic Pseudomonas aeruginosa and Staphylococcus aureus pneumonia in WT and CF mice. Animals were infected followed 24 hours later by 106 hMSCs infused through the retro-orbital sinus and then monitored for 10 days prior to euthanasia for bronchoalveolar lavage (BAL). The CF BAL fluid had significantly more colony forming units (CFUs) that the WT BAL fluid regardless of whether it was a gram negative (Pseudomonas aeruginosa) or gram positive (Staphylococcus aureus) infection (p≤0.05, n=4 experiments, 10 mice per condition)3. When evaluating the inflammatory infiltrate of each of the models, both Pseudomonas aeruginosa and Staphylococcus aureus infections increased the total cell counts (Figure 1A and 1B respectively, which was decreased with hMSC treatment (p≤0.05, n=4 experiments, 10 mice per condition). When looking at the phenotype of the cells in the infection models, as previously described neutrophil recruitment was significantly higher in the CF model as compared to the WT controls (Figure 1A and 1B, p<0.05 for each, n=4 experiments, 10 mice per condition). Treatment of each of the infection modeling systems with hMSCs resulted in a shift in the BAL differentials changing the lung milieu to preferentially attract macrophages (Figure 1C p≤0.05, n=4 experiments, 10 mice per condition)) while decreasing the number of neutrophils (Figure 1D, p≤0.05, n=4 experiments, 10 mice per condition). Neutrophils are known to participate in the pathophysiologic process associated with CF lung disease. Macrophages are known to be important in infection resolution and attenuation of the inflammatory response.
Figure 1. In Vivo Potency and Efficacy.

Cftrtm1Kth (CF) and C57BL/6 (WT) mice were infected with either 106 CFUs of Pseudomonas aeruginosa or Staphylococcus aureus and monitored for 10 days. At day 10 the total cell count (1A and 1B) were determined by bronchoalveolar lavage (BAL) using cytospin and H&E staining. These studies were done 4 times with n=10 animals in each group using 4 different hMSC donors. Figure 1A and 1B show the total cell count obtained by BAL, Figure 1C and 1D focus on the Pseudomonas aeruginosa model since it is prototypic of CF lung infection and inflammation. These data demonstrate that part of the impact of hMSCs is on on redirecting the inflammatory infiltrate away from neutrophils toward having more macrophages.
hMSCs are Active at Promoting Cellular Recruitment of a Specific Phenotype
hMSCs enhance cellular recruitment overall, which could ultimately be beneficial or detrimental, depending on the cell type. In these studies, we monitored the ability of the hMSCs to recruit cells from peripheral blood mononuclear cells obtained from healthy volunteers using transwell plates of 0.45μM. The hMSC supernatants significantly recruited cells from the upper chamber (Figure 2A, p≤0.05, n=4). Lymphocytes were most heavily recruited (Figure 2B, F variance of p=0.05), followed by monocytes and lastly neutrophils. The preferential recruitment of lymphocytes may aide in lowering neutrophil elastase, translating to less lung damage.
Figure 2. hMSCs and Cell Recruitment Studies.

hMSCs supernatants (n=4) were cultured in a transwell with peripheral blood monocytes from healthy volunteers. After 24 hours the cells were counted in both the bottom and top chambers to monitor the cell recruitment process. Data is the mean number of cells recruited in response to the MSC supernatants.
hMSC Soluble Mediators
To determine how the hMSCs are contributing to the specific recruitment of cells, hMSCs were evaluated for cytokine secretion. The same supernatants that were utilized in the chemotaxis studies were evaluated in Luminex multiplex assays to determine their chemokine/cytokine profile (Figure 3). The MSCs secreted a variety of cytokines/chemokines that define the potential of the cells to contribute to their environment including IFNγ, IL-1α, IL-1β, IL-2, IL-6, Il-8, IL-10, Il-12p70, TNFα where we all significantly elevated relative to the control medium (p≤0.05 for all n=4). The cytokines that were generated in the largest amount even without stimulation were IL-6 and IL-8 followed by IL-1β and IL-10 which were significantly higher than the medium control that is used to grow the hMSCs. The next series of studies focus on the expression of the chemokines that might predict how the hMSCs contribute to their environment and how the chemokine expression profile of the hMSCs may be impacted by the functionality of CFTR.
Figure 3. hMSC Cytokine Array.
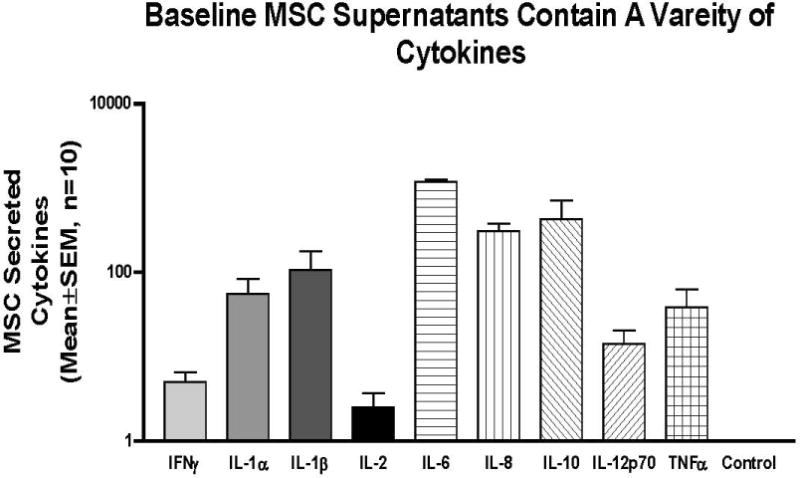
The same hMSC supernatants that were used for the chemotaxis studies were evaluated in a Luminex multiplex assay. Data are Mean±SEM pg/ml, for n=4 different MSC donors.
hMSC IL-8 Production and Impact of CFTR
IL-8 induces chemotaxis in target cells, shifting toward acute neutrophil influx at the sie of infection24. IL-8 has been shown to be significanly expressed in the absence of CFTR activity in a variety of other cells including epithelial cells25. hMSCs also secrete excessive levels of IL-8 when CFTR is inhibited as compared to control CFTR sufficient hMSCs (Figure 4A, −6.58 ± 2.57 versus 6.21± 0.29 respectively; p = 0.01519, n = 10). Stimulation with LPS causes an increase in IL-8 expression in comparison to control mRNA, with CFTR inhibition causing a further increase (LPS : 0.29±1.35 and LPS I-172: 0.40 ±−1.31; p <0.01 and p = 0.003 respectively; n = 10). Similar findings were determined through Luminex technology on secreted products, with differences between I-172 and LPS with I-172 also showing statistical significance (Figure 4B p< 0.01, n = 10).
Figure 4. hMSCs IL-8 response defined by gene expression, secretion with and without CFTR activity and LPS exposure.
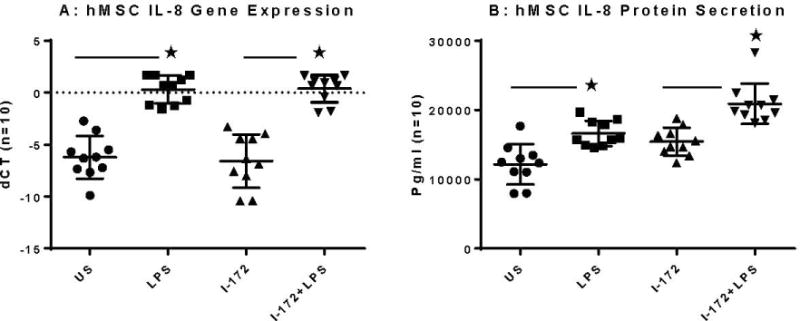
hMSCs (n=10) were cultured in the presence and absence of the CFTR inhibitor I-172 for 48 hours followed by stimulation with LPS. Cells were harvested and processed for gene expression and supernatants were evaluated for IL-8 protein.
hMSC IL-6 Production and Impact of CFTR
IL-6 is an acute phase protein, meaning that it induces other chemokines, such as IL-10. IL-6 has extensive anti-inflammatory functions, many of which could be beneficial in scenarios of chronic infection and inflammation such as CF26. IL-6 is downregulated in CFTR deficient cells in comparison to unstimulated mRNA, a difference of 1.04 cycles in ABI Taqman (Figure 5A, p = 0.01, n=10). Similarily, in comparing LPS stimualted hMSC mRNA, CFTR deficient cells expressed significantly less IL-6 as opposed to LPS stimulated mRNA (1.84 cycles difference; p = 0.05, n=10). Similar results were obtained in secreted product data, with IL-6 downregulated in I-172 and LPS I-172 conditions in comparison to unstimulated controls (Figure 5B, 11225 ± 13, 11834± 25 and 12705± 12.893 respectively; p <0.05, n=10).
Figure 5. hMSCs IL-6 response defined by gene expression, secretion with and without CFTR activity and LPS exposure.
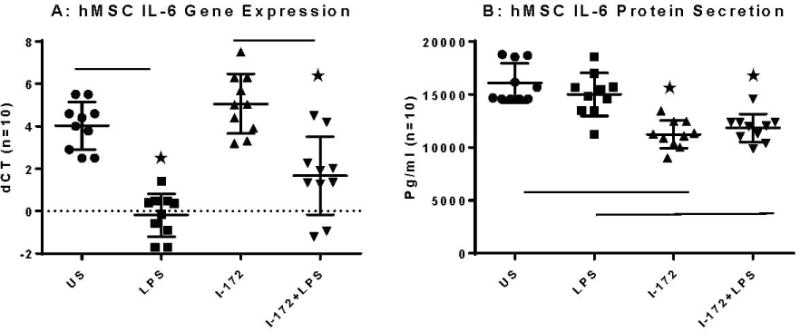
hMSCs (n=10) were cultured in the presence and absence of the CFTR inhibitor I-172 for 48 hours followed by stimulation with LPS. Cells were harvested and processed for gene expression and supernatants were evaluated for IL-6 protein.
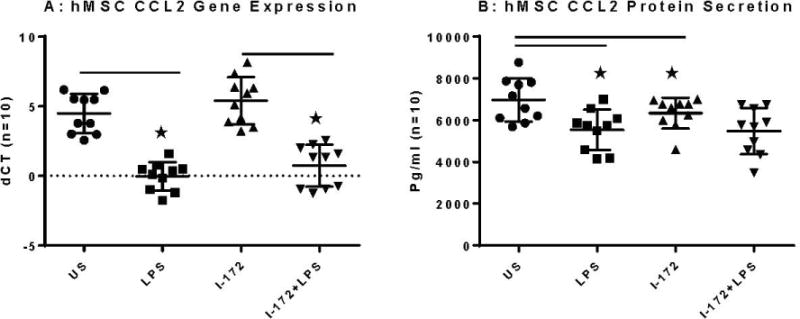
hMSCs CCL2 Gene Expression and Secretion and the Impact of CFTR Activity
CCL2 is a chemokine involved in the recruitment of monocytes and helper-T-cells, both of which are beneficial in scenarios of chronic inflammation27. CCL2 expression was significantly lowered with the addition of LPS, both in the case of otherwise unstimulated cells and in cells with I-172 inhibitor (Figure 6A, −0.03±1.0 and 0.73± 1.5 respectively as compared to US: 4.49±0.98 (p <0.01, n=10). CCL2 expression was significantly lowered with the addition of LPS, both in the case of otherwise unstimulated supernatant and in supernatant with I-172 inhibitor (5402 ± 22., 5480±15. respectively as compared to US: 6683 ±27, p <0.045, n=10). Though not statistically significant, I-172 stimulation of hMSCs alone decreased CCL2 expression in Luminex (Figure 6B, 6337 ± 14 versus US: 6683. ± 27; p = 0.54, n=10).
Figure 6. hMSCs CCL2 response defined by gene expression, secretion with and without CFTR activity and LPS exposure.

hMSCs (n=10) were cultured in the presence and absence of the CFTR inhibitor I-172 for 48 hours followed by stimulation with LPS. Cells were harvested and processed for gene expression and supernatants were evaluated for CCL2 protein.
hMSCs: CCL20 (MIP-3α) Gene Expression and Secretion and the Impact of CFTR Activity
CCL20 is chemotactic for lymphocytes and dendritic cells, while weakly recruiting neutrophils. CCL20 also has antimicrobial properties that would be beneficial in scenarios of acute infection. CCL20 is dependent on activation of amplified IL-6. There was a large decrease in expression between LPS stimulated cells and control cells, both with and without I-172 stimulations (Figure 7A, 9.9±1.7, 4.9± 0.98, 5.2 ± 1.3 respectively; p <0.01, n= 10). There was no significant difference in CCL20 expression in hMSCs between unstimulated and I-172 cells (p = 0.87, n =10). Similar data was determined in secreted products, with LPS stimulation providing significant decreases in expression of CCL20 chemokine as compared to control supernatant (Figure 7B, p < 0.01; n =10).
Figure 7. hMSCs CCL20 response defined by gene expression, secretion with and without CFTR activity and LPS exposure.
hMSCs (n=10) were cultured in the presence and absence of the CFTR inhibitor I-172 for 48 hours followed by stimulation with LPS. Cells were harvested and processed for gene expression and supernatants were evaluated for CCL20 protein.
hMSCs Impact on LPS Stimulated PBMC Cytokine Profile
PBMCs stimulated with LPS produced significant levels of IFNγ, IL-1α, IL-1β, IL-2, IL-6, IL-8, IL-10 IL-12p70 and TNFα (p≤0.05 for all) relative to control unstimulated PBMCs (data not shown). Pro-inflammatory and lymphocyte activating cytokines IL-1α, IL-1β, IL-2 IL-6, TNF-α, IFN-γ were decreased when the PBMCs were cultured in the presence of hMSCs (Figure 8). Cytokine TNFa was decreased from 216. ± 4.3 to 140 ±3.4 pg/ML with hMSC stimulation post LPS stimulation. Cytokine INFγ decreased with hMSC stimulation post LPS stimulation from 98 ± 2 to 83 ± 3.3. Cytokines were minimal in the non –bacterial stimulated samples (no LPS), with levels similarly minimal in both unstimulated hMSC and control conditions. The change in the concentrations of chemokines/cytokine were calculated in GraphPad Prism (C). hMSCs down-regulate the inflammatory response of monocytes exposed to bacterial toxin such as LPS which is summarized in Table I.
Figure 8. PBMCs and hMSCs-Suppression of Response to LPS Exposure Ex Vivo.

Peripheral blood mononuclear cells were obtained from healthy volunteers (n=4) and cultured with and without hMSCs after being stimulated for 24 hours with LPS. Data is the change in cytokine expression post-hMSC treatment as compared to PBMCs without hMSCs.
Table I.
| Cytokine | Concentration change |
|---|---|
| Pro-Inflammatory | |
| IL-1alpha | −11.5% |
| IL-1beta | −8.15% |
| IL-6 | −6.85% |
| TNF-alpha | −39.2% |
| TH1 activators | |
| IL-2 | −16.4% |
| IFN-gamma | −45.85% |
| Anti-Inflammatory | |
| IL-10 | +31.0% |
The Absence of CFTR Decreases PPARγ Gene Expression which can be repaired by hMSCs
PPARγ is constitutively expressed in alveolar macrophages to attenuate the immune response to constant bombardment by environmental antigens28,29. In scenarios of chronic inflammation, PPARγ becomes deficient30. PPARγ has been shown to be deficient in macrophages and epithelial cells31,32,33,34,35. We generated a model of this observation using the CFTR blocker I-172, which decreased PPARγ in WT macrophages (Figure 9A). When CFTR activity is blocked PPARγ levels decrease as shown in the comparison of treated (I-172 inhibited) versus non-treated mRNA PPARγ expression at 24 and 48 hours post treatment with inhibitor. In Figure 9, the comparison of the non–treated ddCT value of 0.925± 0.3, 24 hours and 48-hour treatment to the treated groups, there was significantly less PPARγ (−2.12 ±−0.3 and −3.52± 0.2 respectively for 24 and 48 hours, P≤0.05). We have further established that hMSCs have the capacity to rescue deficient PPARγ expression whether the deficient is due to endotoxin induced down-regulation (Figure 9B) or deficient CFTR activity. Bone marrow derive macrophages were treated with LPS which resulted in decreased PPARγ, TLR-4 expression while TNFα expression is increased as predicted. When hMSCs are added, PPARγ expression was increased while turning down the expression of both TNFα and TLR-4, decreasing the pro-inflammatory phenotype of the macrophages. The increase in TNFα specifically correlated with the decrease in PPARγ ddCT value (normalized to initial start time CT value) from 6 to 24 hours (−1.87±0.74 and 3.35±0.46 respectively; Mean ± SEM, p,0.05).
Figure 9. CFTR Activity and Expression Impact PPARγ Expression which can be reversed by hMSCs Therapeutics.

Bone marrow derived macrophages from CF and WT mice were processed for cDNA and monitored for PPARγ gene expression as compared to GAPDH (A, n=4). Bone marrow derived macrophages treated with the CFTR inhibitor I-172 become deficient in PPARγ by 24 hours (p<0.05) which suggest CFTR function is associated with PPARγ expression (A, n=4). The deficiency in PPARγ correlates with increased TNFα (n, p<0.05) hMSCs cultured in scenarios of deficient PPARγ express increased TNFα, but less TLR-4. The deficient PPARγ can be rescued with hMSCs (p<0.05, B, n=4) contributing to less TNFα. There was also a trend towards decreased TLR-4 with the hMSCs (p=0.06, n=4).
Discussion
We and others have previously demonstrated that hMSCs secrete products that are anti-inflammatory and antimicrobial, making them a suitable candidate to treat diseases associated with severe chronic infection and inflammation such as CF. Defects in the CFTR gene in CF result in increased susceptibility to pulmonary infections from Pseudomonas aeruginosa, Staphylococcus aureus and Streptococcus pneumonia (24). The inability to resolve these infections and the ensuing over-zealous inflammatory response is the major cause of morbidity and mortality in CF. Our previous data has shown that hMSCs decrease inflammation in the in vivo murine model of CF chronic Pseudomonas aeruginosa infection and inflammation. The studies outlined in this manuscript focus on understanding the diversity in the hMSCs anti-inflammatory effectiveness. We have previously shown that growth conditions impact the overall anti-microbial effectiveness of hMSCs and that deficient CFTR function can also decrease antimicrobial potency (24). Furthering these studies on hMSC phenotype, we have demonstrated that hMSCs are potent inflammatory mediators, and have contributed to defining the complexity of these cells. Our studies demonstrated that: 1) hMSCs have the capacity to alter the inflammatory response in both a gram negative (Pseudomonas aeruginosa) and gram positive (Staphylococcus aureus) murine modeling system of CF; 2) hMSCs have the capacity to alter their environment by directly recruiting cells of a specific type recruiting more lymphocytes than monocytes or neutrophils; 3) hMSCs produce a wide variety of factors that have the capacity to define the host response; 4) hMSCs are anti-inflammatory, producing anti-inflammatory chemokines, increasing IL-6 and CCL2 expression, while decreasing IL-8 expression; 5) hMSCs also produce the chemokine CCL20 (MIP-α) which has not only chemotactic properties but also has been shown to be antimicrobial; 6) Deficient CFTR activity alters the expression of all of these cytokines as well as the capacity of the hMSCs to respond to LPS and gram negative bacteria; 7) Deficient CFTR results in the deficiency of the anti-inflammatory transcriptional regulator PPARγ contributing to inflammation by producing factors such as TNFα; 8) h MSCs have the capacity to rescue PPARγ expression as a potential mechanism associated with the MSC anti-inflammatory effectiveness in CF and other chronic inflammatory diseases.
Previous studies have implicated the anti-inflammatory and antimicrobial activity of the hMSCs in both animal models and in preliminary clinical trials36,37,3. In our own studies, we have been able to demonstrate that not only do hMSCs impact inflammation and infection they also alter the direction of inflammatory cell recruitment. In WT and CF mice infected with Pseudomonas aeruginosa, hMSCs decreased both bacterial burden and inflammation14. Mice given the hMSCs had significantly less bacterial burden than mice treated with saline along with attenuated inflammation. In the studies presented in this manuscript, we have demonstrated that hMSCs are active contributors to the host environment by producing cytokines and chemokines that participate, in the host response to pathogen exposure. We believe that the contribution of the hMSCs will be disease specific since in our hands using the same hMSC preparations we have been able to identify very different end-points of hMSC effectiveness whether it is in the context of infection (as in the CF studies) or in acute inflammation12 or chronic asthma38. Certainly our studies demonstrate the unique capacity of the hMSCs to alter inflammation regardless of the inciting source of the pathophysiology.
There are several aspects that need to be considered when thinking about hMSC-based therapeutics including how the cells are cultured and administered36,39 but also important to consider the source of the hMSC preparation. Several reviews have outlined the unique attributes of MSCs and their specific source and processing and the outcome on clinical potency and efficacy40,37. In our studies, we have demonstrated previously that hMSCs deficient in CFTR activity have decreased anti-microbial potency and efficacy3 which may have important implications into whether the autologous therapeutics can be utilized for CF hMSC clinical trials. This may also relate to other diseases in which there is a genetic association with the pathogenicity and may be the reasoning behind the overall variability in hMSC clinical efficacy in clinical trials. The results outlined in this manuscript explore the issue of CFTR expression on hMSC anti-inflammatory potency. Our data indicate that CF hMSCs do not behave similarly to healthy hMSCs in response to bacterial stimulation with healthy and CF hMSCs express different amounts of IL-6, CCL2, CCL20 and IL-841,42. With bacterial stimulation, CF cells express a greater amount of IL-8 than healthy hMSCs when both are exposed to bacteria. The CF hMSCs express less IL-6, CCL2 and CCL20 than healthy hMSCs with bacterial stimulation. Although CF hMSCs and healthy hMSCs express different amounts of IL-6 CCL2, and IL-8, both types of MSCs express roughly equal amounts of CCL20 which may be important in the chemotactic and antimicrobial functions of these chemokine/cytokines27,43. The production of chemokines and cytokine mediators will have a contributory role in defining the tissue niche as a response to the inflammatory stimulus. The contributory fingerprints of the hMSCs secreted products will ultimate change how the host will manage the inflammatory process.
The difference in expression of cytokines between healthy and CF hMSCs in response to bacterial stimulation indicates that autologous hMSCs are unlikely to be a successful form of therapy for CF patients. The data also suggests the impact of CFTR deficient activity throughout CF patient’s body, not only at the level of the immune system but also the functionality of the autologous hMSCs. This implies an additional contribution of deficient CFTR on CF pathophysiology, encompassing not only the primary major consequence of the defect on the epithelium, but also the additive impact on the CF immune system and the multipotent cell source of hMSCs.
In the pursuit of the mechanistic action of the hMSCs as an anti-inflammatory adjuvant therapeutic, we have begun to identify the potential of hMSCs to alter PPARγ expression in macrophages which is an important regulator of inflammation in chronic inflammation44. In the lung, PPARγ is constitutively expressed in alveolar macrophages in order to attenuate the host response to the continuous exposure to environmental insult23,33. PPARγ quiets inflammation through transcriptional repression of NFκB preventing the ability of NFκB to initiating transcription from its promoter regions which are involved with pro-inflammatory gene expression45. In scenarios of chronic inflammation, PPARγ becomes deficient in macrophages which contributes to the uncontrolled expression of pro-inflammatory genes by NFκB35 a pathophysiological mechanism described in a variety of chronic inflammatory diseases46,47. Like our studies it has been shown that CF macrophages are deficient in PPARγ35. We have been able to demonstrated that hMSCs can up-regulate the expression of PPARγ, in macrophages which in turn has the capacity to down-regulate inflammation regardless of the routes associated with the down-regulation of this inflammation rheostat. Theoretically, the hMSCs likely produce soluble mediators which alter the macrophage phenotype, enhancing the PPARγ expression, allowing for efficient modification of pro-inflammatory cytokine production through the prevention of NFkB promoter activation. In either case of LPS stimulated mouse bone marrow derived macrophages or human peripheral blood mononuclear cells, hMSCs decreased pro-inflammatory cytokine production. The direct mechanism of these effects are the focus of on-going work in our laboratory.
hMSCs have anti-inflammatory potential which has been documented in several different in vivo and in vitro modeling systems48. We propose that one of the mechanisms associated with this hMSC function is the capacity to up-regulate the expression of PPARγ. How CFTR might impact this effect of hMSCs will be pursued in the context of our investigator initiated clinical trial of hMSCs in CF. Further, these studies are consistent with previous benefits of hMSCs in scenarios of acute lung injury (ALI) which is a disease of significant morbidity and mortality in intensive care patients37. ALI is defined by the acute onset of pulmonary infiltrates with severe hypoxemia. In a mouse model of ALI, lipopolysaccharide (LPS) derived from the bacterium Escherichia coli endotoxin was put into the lung followed by administration of murine hMSCs several hours later. Mice treated with murine MSCs had decreased inflammation49,50. The pro-inflammatory response to the LPS endotoxin was decreased, while the resolution response and anti-inflammatory cytokines were enhanced18. These studies suggest the innovative use of adult human MSCs in the treatment of lung diseases associated with infection and inflammation.
Conclusions
hMSCs are active in their environment secreting products that define the host milieu and capacity to regulate immunity and cellular response. The capacity to down-regulate inflammation is reproducible regardless of the modeling system using mouse bone marrow derived macrophages or human peripheral blood mononuclear cells going across species with hMSCs. The capacity of the hMSCs to secrete cytokines and respond to pathogen endotoxin like LPS is impacted by CFTR activity, suggesting that CF hMSCs may not be appropriate for autologous therapy. The inefficiency of the CF hMSCs in these systems along with the established antimicrobial activity, implies that innate hMSC function may altered in CF, potentially contributing to disease. hMSCs are environmentally sensitive and environmentally pro-active emphasizing the complexity of their function and potential therapeutic impact. Future studies will pursue the impact of CFTR on hMSC activity and how hMSC regulates the combined anti-inflammatory and antimicrobial milieu in vivo.
Figure 10. Theoretical Schematic of hMSC Down Regulating Inflammation and Managing Infection.
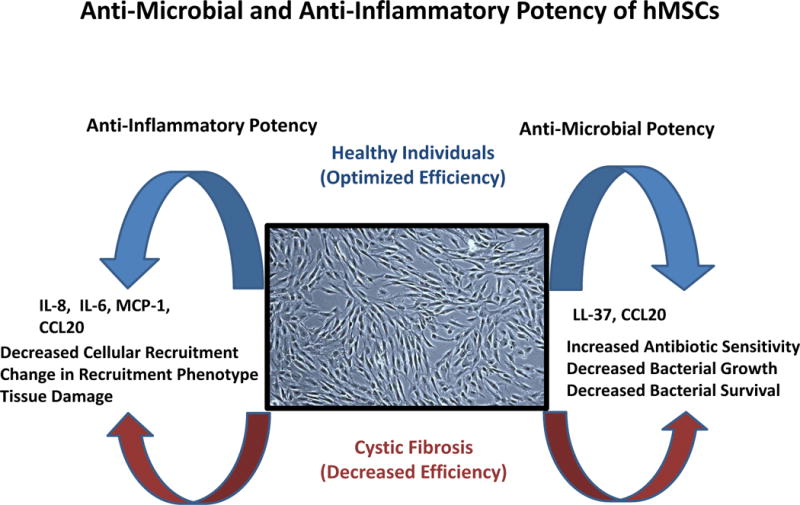
Infection with or without inflammation in CF is poorly regulated and contributes to disease pathogenesis. hMSCs produce soluble mediators which redefine the tissue niche, enhancing macrophage recruitment which may can participate in host immunity. In CF, hMSCs have altered levels of soluble mediators relative to the control suggesting that the immune function of hMSCs in CF maybe alters again re-inforcing the notion of allogeneic therapeutics in the context of genetic diseases.
Acknowledgments
We appreciate the expertise and the services provided by the CTSC Bioanalyte Core and the CF Animal Core. The funding for this research was graciously provided by the David and Virginia Baldwin Fund, The National Center for Stem Cell and Regenerative Medicine, The Case Western Reserve Vision Fund, Rainbow Babies and Children’s Hospital Summer Programs in Undergraduate Research, Rainbow Babies and Children’s Hospitals Strategic Innovator Award and National Institutes of Health HL: R21:104362.
Footnotes
Author Contributions
Morgan T. Sutton: Data Generation, Conception and Design, Collection and Assembly of Data, Data Analysis and Interpretation, Manuscript Writing
David Fletcher: Data Generation Collection and/or Assembly of Data, Data Analysis and Interpretation.
Nicole Episalla: Data Generation, Collection and Assembly of Data.
Lauren Auster: Provision of Study Materials, Manuscript Review.
Sukhmani Kaur: Collection of Data, Manuscript Review.
Mary Chandler Gwin: Collection of Data; Manuscript Review.
Michael Folz: Manuscript Review Data Analysis and Interpretation.
Dante Velasquez: Data Analysis and Interpretation; Manuscript Review.
Varun Roy: Generation of Peripheral Blood Mononuclear Studies, Data Analysis and Interpretation.
Rolf vanHeeckeren: Provision of Study Materials, Collection and/or Assembly of Data
Donald Lennon: Provision of Study Materials
Arnold Caplan: Financial Support, Provision of Study Materials, Data Analysis and Interpretation, Final Approval of Manuscript.
Tracey L. Bonfield: Conception and Design, Financial Support, Administrative Support, Provision of Study Materials, Data Analysis and Interpretation, Manuscript Writing, Final Approval of Manuscript.
Disclosure of Potential Conflicts of Interest: The author has no conflicts of interest to report.
References
- 1.Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217:318–324. doi: 10.1002/path.2469. 1096–9896(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomic S, Djokic J, Vasilijic S, et al. Immunomodulatory properties of mesenchymal stem cells derived from dental pulp and dental follicle are susceptible to activation by toll-like receptor agonists. Stem Cells Dev. 2011;20:695–708. doi: 10.1089/scd.2010.0145. 1557–8534(Electronic) [DOI] [PubMed] [Google Scholar]
- 3.Sutton MT, Fletcher D, Ghosh SK, et al. Antimicrobial Properties of Mesenchymal Stem Cells: Therapeutic Potential for Cystic Fibrosis Infection, and Treatment. Stem Cells Int. 2016;2016:5303048. doi: 10.1155/2016/5303048.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols D, Chmiel J, Berger M. Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. ClinRevAllergy Immunol. 2008;34:146–162. doi: 10.1007/s12016-007-8039-9. 1080-0549(Print) [DOI] [PubMed] [Google Scholar]
- 5.Drumm ML, Ziady AG, Davis PB. Genetic variation and clinical heterogeneity in cystic fibrosis. AnnuRevPathol. 2012;7:267–282. doi: 10.1146/annurev-pathol-011811-120900. 1553–4014(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dasenbrook EC, Checkley W, Merlo CA, Konstan MW, Lechtzin N, Boyle MP. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA. 2010;303:2386–2392. doi: 10.1001/jama.2010.791. 1538–3598(Electronic) [DOI] [PubMed] [Google Scholar]
- 7.Goss CH, Muhlebach MS. Review: Staphylococcus aureus and MRSA in cystic fibrosis. JCystFibros. 2011;10:298–306. doi: 10.1016/j.jcf.2011.06.002. 1873–5010(Electronic) [DOI] [PubMed] [Google Scholar]
- 8.Kotha K, Clancy JP. Ivacaftor treatment of cystic fibrosis patients with the G551D mutation: a review of the evidence. TherAdvRespirDis. 2013;7:288–296. doi: 10.1177/1753465813502115. 1753–4666(Electronic) [DOI] [PubMed] [Google Scholar]
- 9.Cosgrove S, Chotirmall SH, Greene CM, McElvaney NG. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/Toll-like receptor pathway. J Biol Chem. 2011;286:7692–7704. doi: 10.1074/jbc.M110.183863. 1083-351X(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman MS, Bar-Or A, Atkins HL, et al. The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: consensus report of the International MSCT Study Group. MultScler. 2010;16:503–510. doi: 10.1177/1352458509359727. 1477–0970(Electronic) [DOI] [PubMed] [Google Scholar]
- 11.Roddy GW, Oh JY, Lee RH, et al. Action at a distance: systemically administered adult stem/progenitor cells (MSCs) reduce inflammatory damage to the cornea without engraftment and primarily by secretion of TNF-alpha stimulated gene/protein 6. Stem Cells. 2011;29:1572–1579. doi: 10.1002/stem.708. 1549–4918(Electronic) [DOI] [PubMed] [Google Scholar]
- 12.Bonfield TL, Nolan Koloze MT, Lennon DP, Caplan AI. Defining human mesenchymal stem cell efficacy in vivo. JInflamm(Lond) 2010;7:51. doi: 10.1186/1476-9255-7-51. 1476–9255(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonfield TL, Caplan AI. Adult mesenchymal stem cells: an innovative therapeutic for lung diseases. DiscovMed. 2010;9:337–345. 1944–7930(Electronic) [PubMed] [Google Scholar]
- 14.Bonfield Tracey L, Ghosh Santosh K, Weinberg Aaron, Caplan Arnold I, L D. Cell Based Therapy Aides in Infection and Inflammation Resolution in The Murine Model of Cystic Fibrosis Lung Disease. Stem Cell Discov. 2013 [Google Scholar]
- 15.Lennon DP, Caplan AI. Isolation of human marrow-derived mesenchymal stem cells. Exp Hematol. 2006;34:1604–1605. doi: 10.1016/j.exphem.2006.07.014. 0301-472X(Print) [DOI] [PubMed] [Google Scholar]
- 16.Caimi PF, Reese J, Lee Z, Lazarus HM. Emerging therapeutic approaches for multipotent mesenchymal stromal cells. CurrOpinHematol. 2010;17:505–513. doi: 10.1097/MOH.0b013e32833e5b18. 1531–7048(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonfield TL, Barna BP, John N, et al. Suppression of activin A in autoimmune lung disease associated with anti-GM-CSF. J Autoimmun. 2006;26(1):37–41. doi: 10.1016/j.jaut.2005.10.004. http://www.sciencedirect.com/science/article/B6WHC-4HR75N7-1/2/a02a605bfb4df60b49e576071571fdc1. [DOI] [PubMed] [Google Scholar]
- 18.van Heeckeren AM, Schluchter MD. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim. 2002;36:291–312. doi: 10.1258/002367702320162405. 0023-6772(Print) [DOI] [PubMed] [Google Scholar]
- 19.Bonfield TL, Thomassen MJ, Farver CF, et al. Peroxisome proliferator-activated receptor-gamma regulates the expression of alveolar macrophage macrophage colony-stimulating factor. J Immunol. 2008;181:235–242. doi: 10.4049/jimmunol.181.1.235. 0022-1767(Print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonfield TL, John N, Malur A, et al. Elevated monocyte chemotactic proteins 1,2 and 3 in pulmonary alveolar proteins are associated with chemokine receptor suppression. ClinImmunol. 2004 doi: 10.1016/j.clim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Bonfield TL, John N, Barna BP, Kavuru MS, Thomassen MJ, Yen-Lieberman B. Multiplexed particle-based anti-granulocyte macrophage colony stimulating factor (GM-CSF) assay: A pulmonary diagnostic test. ClinDiagnLab Immunol. 2005 doi: 10.1128/CDLI.12.7.821-824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rieder F, Nonevski I, Ma J, et al. T-helper 2 cytokines, transforming growth factor β1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology. 2014;146(5) doi: 10.1053/j.gastro.2014.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonfield TL, Farver CF, Barna BP, et al. PPAR_g is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol. 2003;29:677–682. doi: 10.1165/rcmb.2003-0148OC. [DOI] [PubMed] [Google Scholar]
- 24.Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A. Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. AmJ Respir Crit Care Med. 1994;149:655–659. doi: 10.1164/ajrccm.149.3.8118632. 1073-449X. [DOI] [PubMed] [Google Scholar]
- 25.Saadane A, Eastman J, Berger M, Bonfield TL. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. JInflamm(Lond) 2011;8:26. doi: 10.1186/1476-9255-8-26. 1476–9255(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niemand C, Nimmesgern A, Haan S, et al. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol. 2003;170:3263–3272. doi: 10.4049/jimmunol.170.6.3263. 0022–1767. [DOI] [PubMed] [Google Scholar]
- 27.Daly C, Rollins BJ. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: therapeutic opportunities and controversies. Microcirculation. 2003;10:247–257. doi: 10.1038/sj.mn.7800190. 1073–9688. [DOI] [PubMed] [Google Scholar]
- 28.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 29.Berger J, Moller DE. The mechanisms of action of PPARs. AnnRevMed. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Young HA. PPAR and immune system–what do we know? IntImmunopharmacol. 2002;2:1029–1044. doi: 10.1016/s1567-5769(02)00057-7. 1567–5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez A, van Heeckeren AM, Nichols D, Gupta S, Eastman JF, Davis PB. Peroxisome proliferator-activated receptor-{gamma} in cystic fibrosis lung epithelium. AmJPhysiol Lung Cell MolPhysiol. 2008;295:L303–L313. doi: 10.1152/ajplung.90276.2008. 1040-0605(Print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malur A, Abraham S, Culver DA, et al. Interferon -gamma (IFNgamma) represses PPARgamma expression in sarcoidosis. Proc Am Thorac Soc. 2005;2:A843. [Google Scholar]
- 33.Culver DA, Barna BP, Raychaudhuri B, et al. Peroxisome proliferator-activated receptor gamma activity is deficient in alveolar macrophages in pulmonary sarcoidosis. Am J Respir Cell Mol Biol. 2004;30:1–5. doi: 10.1165/rcmb.2003-0304RC. 1044–1549. [DOI] [PubMed] [Google Scholar]
- 34.Bonfield TL, Farver CF, Barna BP, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. AmJ Respir Cell MolBiol. 2003;29:677–682. doi: 10.1165/rcmb.2003-0148OC. 1044–1549. [DOI] [PubMed] [Google Scholar]
- 35.Andersson C, Zaman MM, Jones AB, Freedman SD. Alterations in immune response and PPAR/LXR regulation in cystic fibrosis macrophages. JCystFibros. 2008;7:68–78. doi: 10.1016/j.jcf.2007.05.004. 1569–1993(Print) [DOI] [PubMed] [Google Scholar]
- 36.Kean TJ, Lin P, Caplan AI, Dennis JE. MSCs: Delivery Routes and Engraftment, Cell-Targeting Strategies, and Immune Modulation. Stem Cells Int. 2013;2013:732742. doi: 10.1155/2013/732742. 1687-966X(Print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthay MA, Thompson BT, Read EJ, et al. Therapeutic potential of mesenchymal stem cells for severe acute lung injury. Chest. 2010;138:965–972. doi: 10.1378/chest.10-0518. 1931–3543(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonfield TL, Koloze M, Lennon DP, Zuchowski B, Yang SE, Caplan AI. Human mesenchymal stem cells suppress chronic airway inflammation in the murine ovalbumin asthma model. AmJPhysiol Lung Cell MolPhysiol. 2010;299:L760–L770. doi: 10.1152/ajplung.00182.2009. 1522–1504(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zagoura DS, Trohatou O, Bitsika V, et al. AF-MSCs fate can be regulated by culture conditions. Cell DeathDis. 2013;4:e571. doi: 10.1038/cddis.2013.93. 2041–4889(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caplan AI. Adult mesenchymal stem cells and the NO pathways. ProcNatlAcadSciUSA. 2013;110:2695–2696. doi: 10.1073/pnas.1221406110. 1091–6490(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. 1471–4906. [DOI] [PubMed] [Google Scholar]
- 42.Zeng SL, Wang LH, Li P, Wang W, Yang J. Mesenchymal stem cells abrogate experimental asthma by altering dendritic cell function 5. MolMedRep. 2015;12:2511–2520. doi: 10.3892/mmr.2015.3706. 1791–3004(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vulcano M, Struyf S, Scapini P, et al. Unique regulation of CCL 18 production by maturing dendritic cells. J Immunol. 2003;170:3843–3849. doi: 10.4049/jimmunol.170.7.3843. 0022-1767(Print) [DOI] [PubMed] [Google Scholar]
- 44.Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. MolCell Biol. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. 0270–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. ProcNatlAcadSciUSA. 2003;100:6712–6717. doi: 10.1073/pnas.1031789100. 0027–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belvisi MG, Hele DJ, Birrell MA. Peroxisome proliferator-activated receptor gamma agonists as therapy for chronic airway inflammation. EurJPharmacol. 2006;533:101–109. doi: 10.1016/j.ejphar.2005.12.048. 0014-2999(Print) [DOI] [PubMed] [Google Scholar]
- 47.Clark RB. The role of PPARs in inflammation and immunity. J Leukoc Biol. 2002;71:388–400. [PubMed] [Google Scholar]
- 48.Dimarino AM, Caplan AI, Bonfield TL. Mesenchymal Stem Cells in Tissue Repair. Front Immunol. 2013;4:201. doi: 10.3389/fimmu.2013.00201. 1664–3224(Print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krasnodembskaya A, Song Y, Fang X, et al. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells. 2010;28:2229–2238. doi: 10.1002/stem.544. 1549–4918(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matthay MA, Goolaerts A, Howard JP, Lee JW. Mesenchymal stem cells for acute lung injury: preclinical evidence. Crit Care Med. 2010;38:S569–S573. doi: 10.1097/CCM.0b013e3181f1ff1d. 1530–0293(Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]