

Abstract
Atherosclerosis is an arterial disease process characterized by the focal subendothelial accumulation of apolipoprotein B-lipoproteins, immune and vascular wall cells, and extracellular matrix. The lipoproteins acquire features of damage-associated molecular patterns and trigger first an innate immune response, dominated by monocyte-macrophages, and then an adaptive immune response. These inflammatory responses often become chronic and non-resolving, leading to arterial damage and thrombosis-induced organ infarction. The innate immune response is regulated at various stages, from hematopoiesis through monocyte changes and macrophage activation. The adaptive immune response is regulated primarily by mechanisms that affect the balance of regulatory vs. effector T cells. Mechanisms related to cellular cholesterol, phenotypic plasticity, metabolism, and aging play key roles in regulating these responses. Herein we review select topics that shed light on these processes and suggest new treatment strategies.
A Brief Overview of Atherogenesis and Atherosclerotic Plaque Progression
Atherogenesis is initiated by the entry and retention of apolipoprotein B-containing lipoproteins (apoB LPs) into the subendothelial space, or “intima,” at regions of disturbed blood flow in medium-sized arteries (Williams and Tabas, 1995; Fogelstrand and Boren, 2012). The amount of apoB LP retention is determined by their concentration in the blood, age of the individual, metabolic state, and genetic and environmental factors. These considerations affect arterial wall biology, including variations in subendothelial proteoglycans that retain apoB LPs and factors that alter endothelial permeability. Initially, some of the LP lipid is internalized by resident CD11c+ myeloid cells, and experimental depletion of these cells suppresses the accumulation of foam cells and intracellular lipids within 5 days after cellular depletion (Paulson et al., 2010). Then, certain lipid and protein components of subendothelial apoB LPs, particularly after oxidative modification, take on properties of damage-associated molecular patterns (DAMPs) and thereby trigger an inflammatory response (Glass and Witztum, 2001; Lusis, 2000). The response activates endothelial cells, which, together with flow-mediated changes in these cells (Jongstra-Bilen et al., 2006; Gimbrone, Jr. and Garcia-Cardena, 2013), promotes the entry into the intima of bone marrow-derived monocytes (Tacke et al., 2007; Swirski et al., 2016). The Ly6Chi subpopulation of monocytes in the intima differentiate into macrophages, which, in progressing lesions, take on an inflammatory phenotype (Tacke et al., 2007; Swirski et al., 2007). In part as a result of the accumulation of inflammatory macrophages and dendritic cell activation, an inflammatory adaptive immune response develops involving primarily T helper-1 (Th1) T cells, but also Th17 and Th2 T cells and B cells, and there is a progressive decrease in regulatory T cells (Treg) (Witztum and Lichtman, 2014). Other immune cells, including neutrophils and platelet-neutrophil aggregates, innate immune cells, natural killer cells, mast cells, and eosinophils are present in human atheroma and have been shown to promote atherosclerosis via additional mechanisms in mouse models (Witztum and Lichtman, 2014). Accompanying this immune cell reaction is the accumulation of myofibroblasts in the intima that arise from medial smooth muscle cells and other sources and are referred to as vascular smooth muscle cells (VSMC) (Bennett et al., 2016). These cells are rich sources of extracellular matrix (ECM), which likely represents a “scar” response to inflammation and the ongoing vascular injury.
In a physiologic post-inflammatory response, macrophages and other inflammatory cells secrete molecules and carry out functions that dampen the inflammatory response and promote tissue repair (Serhan et al., 2007; Nathan and Ding, 2010). However, as will be explained later in this review, this so-called resolution response can go awry in the setting of atherosclerosis. Impaired resolution in atherosclerotic lesions leads to sustained, non-resolving, and maladaptive inflammation that promotes plaque progression and, in humans, triggers acute thrombo-occlusive cardiovascular events (Merched et al., 2008; Tabas, 2010; Viola and Soehnlein, 2015) (below). The pathological features of clinically dangerous plaques include large areas of necrosis and thinning of an overlying collagenous, or fibrous, cap. When a breach forms in the fibrous cap, the blood is exposed to thrombogenic material in the lesion, and acute occlusive thrombosis with tissue infarction can ensue (Virmani et al., 2002; Libby, 2013). However, acute thrombotic vascular events can also occur in the vicinity of more fibrous, non-necrotic plaques that are characterized by endothelial erosion (Libby, 2017). Studies in mice have suggested that this latter process is promoted by flow disturbance and neutrophil-mediated effects on endothelial cells (Franck et al., 2017).
In the sections that follow, we will review a selective subset of innate and adaptive immune processes that have recently come to light as affecting atherogenesis and/or plaque progression. The reader is referred to the reviews and original references cited above for the many important immune processes in atherosclerosis that are not included herein.
Changes in Monocyte Dynamics Contribute to Atherogenesis
The abundance of monocytes in the circulation, particularly those of the CD14++ subpopulation in humans and Ly6Chi subpopulation in mice, is strongly correlated with atherosclerotic vascular disease in humans and with atherosclerotic lesion development in mice (Olivares et al., 1993; Murphy and Tall, 2016). In this context, recent studies have provided fascinating new insight into the regulatory mechanisms of monocytosis relevant to atherosclerosis (Figure 1). The role of the sympathetic nervous system (SNS) has come to light as researchers sought to explain why atherosclerosis accelerates after myocardial infarction (MI). In the setting of an inflammatory response, Ly6Chi monocytes initially give rise to macrophages on the inflammatory end of the inflammatory-resolution spectrum, and, as will be discussed below, these macrophages promote atherosclerosis progression (Swirski et al., 2007). A substantial portion of Ly6Chi monocytes that contribute to atherosclerosis originate from the spleen, which becomes populated with bone marrow-derived hematopoietic stem and progenitor cells (HSPCs) and carries out extramedullary hematopoiesis (Robbins et al., 2012). In this context, mouse studies suggest that a key mechanism of post-MI atherosclerosis is SNS-mediated release of HSPCs from the bone marrow, leading to seeding of the spleen, elevated extramedullary hematopoiesis, and increased release of Ly6Chi monocytes, which are the subset of monocytes that drive atherogenesis (Dutta et al., 2012). Further evidence suggests that other stress-related events that are known to be risk factors for atherosclerotic disease, such as psychosocial stress, may work through a similar mechanism (Heidt et al., 2014). How these concepts apply to human atherothrombotic vascular disease remains an important area for future study, particularly in view of uncertainties related to the functions of CD14++ monocytes in humans (Hilgendorf and Swirski, 2012).
Figure 1. Regulation of Innate Immune Processes Related to Monocyte-Macrophages in Atherosclerosis.
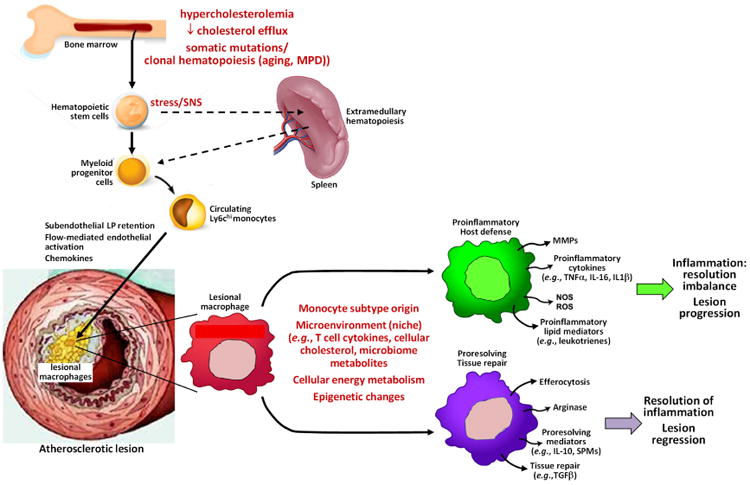
Lesional macrophages originate from bone marrow-derived hematopoietic stem cells, which give rise to circulating monocytes. In certain instances, these stem cells first populate the spleen and then undergo extramedullary hematopoiesis. Proliferation and release of hematopoietic stem cells can be exacerbated by elevation of cellular cholesterol and by somatic mutations leading to clonal hematopoiesis, such as those that occur in aging and myeloproliferative disease (MPD). This process can also be stimulated by stress-induced sympathetic nervous system (SNS) activation. The major subpopulation of monocytes that contribute to atherosclerosis progress are Ly6chi monocytes, which enter lesions in response to subendothelially retained apoB-lipoproteins (LP) and subsequent chemokine release by activated endothelial cells. After differentiation into macrophages, they undergo a variety of phenotypic changes under the influence of the factors listed in the figure. Those macrophages on the inflammatory end of the spectrum secrete proteins and carry out processes that promote atherosclerosis progression, while those on the resolution end of the spectrum promote lesion regression. See text for details.
Hypercholesterolemia also promotes monocytosis in mice, particularly Ly6Chi monocytosis (Swirski et al., 2007), and genetically engineered defective cholesterol efflux in HSPCs can also exacerbate Ly6Chi monocytosis (Murphy and Tall, 2016) (Figure 2). The mechanism responsible for cholesterol-induced monocytosis involves expansion of Lin− cKit+ Sca1+ (LSK) HSPCs in the marrow compartment (Murphy and Tall, 2016). Mechanistic studies revealed that cholesterol-mediated changes in the plasma membrane of HSPCs leads to elevated expression of the common β-subunit of the interleukin-3 (IL-3) receptor and granulocyte-monocyte colony stimulating factor (GM-CSF) receptor on the cell-surface and increased sensing of two key HSPCs growth factors, IL-3 and GM-CSF (Yvan-Charvet et al., 2010). There is also evidence that hypercholesterolemia, presumably by increasing the cellular content of cholesterol, decreases the expression of Rb, a tumor suppressor that limits HSPC proliferation, and increases the expression cyclins B1, D1, and E1 in HSPCs (Seijkens et al., 2014). Interestingly, when normocholesterolemic recipient mice are transplanted with bone marrow from either normocholesterolemic or hypercholesterolemic donor mice, the HSPCs of hypercholesterolemic donor origin showed increased proliferation 10 weeks later (Seijkens et al., 2014). These data suggest a long-lived, cell-intrinsic effect of hypercholesterolemia on HSPCs, perhaps due to epigenetic changes in the donor HSPCs. Moreover, when Ldlr-/- mice are transplanted with bone marrow cells from hypercholesterolemic versus normocholesterolemic mice and then fed a Western-type diet rich in cholesterol and saturated fats, the mice that had received that hypercholesterolemic bone marrow developed larger and more advanced lesions. This increase in atherosclerosis is accompanied by a higher number of lesional leukocytes derived from the hypercholesterolemic mouse bone marrow cells and overall increases in plaque macrophages, granulocytes, and T cells.
Figure 2. Intracellular Effects of Excess Cholesterol on Myeloid Cells and T Cells That Influence Atherosclerosis.
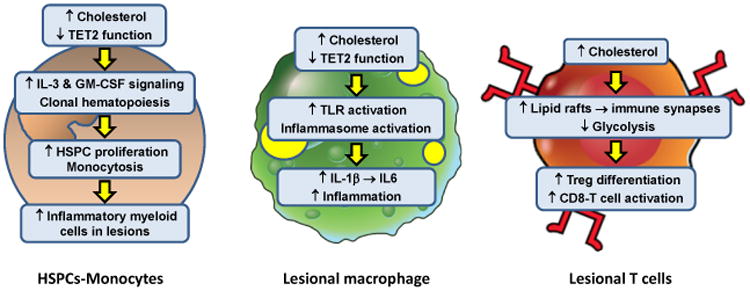
In the setting of hypercholesterolemia or defects in cholesterol efflux, hematopoietic stem and progenitor cells (HSPCs) accumulate excess cholesterol. The consequence is enhanced IL-3 and GM-CSF growth factor signaling, leading to HSPC proliferation and monocytosis. With aging, clonal hematopoiesis can occur owing to loss-of-function mutations in a number of genes, including TET2. This process also contributes to monocytosis. Monocytosis is associated with increased accumulation of inflammatory monocyte-derived macrophages in atherosclerotic lesions and higher risk of atherosclerotic vascular disease in humans. These lesional macrophages are also subject to intracellular cholesterol accumulation owing to their internalization of subendothelial apoB LPs. Excess cholesterol in macrophages has multiple effects that enhance lesion inflammation, including toll-like receptor (TLR) and inflammasome activation. The result is increased production of inflammatory chemokines and cytokines, including inflammasome-derived IL-1β and IL-1β-induced IL-6 production. Moreover, changes associated with clonal hematopoiesis, e.g., loss of TET2 function, can also activate the inflammasome in macrophages, further fueling lesional inflammation. T cells do not have the capacity to accumulate large amounts of excess cholesterol, but several studies have shown that perturbations of T cell cholesterol metabolism can affect T cell differentiation and activation. Impaired ABCG1 cholesterol efflux from T cells results in enhanced Treg differentiation which reduces atherosclerotic lesion development and inflammation. In contrast, impaired esterification of cholesterol by deficiency or inhibition of acyl-coenzyme A:cholesterol acyltransferase (ACAT) increases CD8 effector T cell lipid raft formation and thereby enhances immune synapse formation and killing functions of these cells. The net effect of increases in T cell cholesterol on lesion development and inflammation are likely to reflect changes in the Teff:Treg balance and the influence of Teff cells on lesional macrophages.
Other recent studies have taken advantage of the fact that leukocytosis in myeloproliferative diseases (MPD) is associated with atherothrombotic vascular disease (Murphy and Tall, 2016). For example, a loss-of-function polymorphism in the gene encoding a signaling adaptor protein called LNK (SH2B3) is associated with both MPD and atherosclerosis (McMullin et al., 2011; Deloukas et al., 2013). Moreover, somatic gain-of-function mutations in the JAK2 kinase are also associated with MPD and atherosclerotic disease (Viny and Levine, 2014). While processes related to neutrophils and platelets likely contribute to the mechanism of this association, there is also a link to monocytosis. In this context, genetic targeting of Lnk in Western diet-fed Lnk-/-Ldlr-/- has been found to cause hypercholesterolemia-dependent monocytosis, which is associated with increased amounts of the pro-atherogenic chemokine monocyte chemotactic protein-1 (MCP-1; CCL2) (Wang et al., 2016). Most importantly, these mice demonstrate increases in atherosclerotic lesion area, lesional macrophages, Ly6Chi monocyte entry into lesions, and atherogenic platelet-monocyte aggregates (Wang et al., 2016). There is synergy between hypercholesterolemia and LNK deficiency, leading to further increases in IL-3-GM-CSF receptor signaling in bone marrow HSPCs. Further insight into some of these processes emerge from a study showing that Glut1-mediated glucose uptake by inflammatory myeloid cells promotes myeloproliferation in mice with either defective cholesterol efflux or myeloproliferative disorders (Gautier et al., 2013). In view of the link between glycolysis and inflammatory myeloid cell function (Van den Bossche et al., 2017) (below), the authors propose that Glut1-mediated glucose uptake provides the energy necessary for inflammatory myeloid cell proliferation.
Ly6Clo monocytes derive from Ly6Chi monocytes and serve an endothelial maintenance “patrolling” function in the circulation, but their roles in tissues has become increasingly unclear, and it is not even certain whether they differentiate into macrophages (Jakubzick et al., 2017). As such, the role of Ly6Clo monocytes in atherosclerosis is poorly understood. In one study, investigators have tested the effect of targeting the nuclear receptor Nr4a1 (Nur77), which is required for the differentiation and survival of Ly6Clo monocytes (Hanna et al., 2011). Two models of mouse atherosclerosis with genetic targeting of Nr4a1—Western diet-fed Ldlr-/- mice lacking hematopoietic Nr4a1 and Apoe-/- mice with germline targeting of Nr4a1—demonstrate increased atherosclerosis, and this is associated with an increase in the proportion of lesional macrophages that have an inflammatory vs. resolving phenotype (Hanna et al., 2011). While one interpretation of this finding is that Ly6Clo monocytes directly give rise to resolving macrophages that regulate the inflammatory response, another possible interpretation needs to be considered in view of the uncertainty as to whether Ly6Clo monocytes differentiate into macrophages and based on the results of a study examining post-myocardial infarction (MI) repair (Hilgendorf et al., 2014). Ly6Chi monocytes infiltrate the heart immediately post-MI and then, during the repair phase, give rise to Ly6Clo macrophages with resolving properties. However, in mice lacking hematopoietic Nr4a1, infiltrating Ly6Chi monocytes differentiate into highly inflammatory macrophages that are unable to carry out tissue repair. If applicable to atherosclerosis, the data from the Nr4a1-deficient mouse study above may suggest that Ly6hi monocytes give rise to reparative macrophages in atherosclerotic lesions in a Nr4a1-dependent manner. Consistent with this idea, a recent study shows that Ly6Chi monocytes are the source of resolving macrophages during atherosclerotic plaque regression (Rahman et al., 2017).
Inflammatory Macrophages Drive Atherosclerosis Progression
Macrophage functions can vary widely depending on a number of interacting variables and factors, including local environment (“tissue niche”) (Gosselin et al., 2014; Lavin et al., 2014); intracellular energy metabolism (Van den Bossche et al., 2017); gut microbiota metabolites (Wang et al., 2011); and genetic and epigenetic factors, including non-coding RNAs (Erbilgin et al., 2013; Chen et al., 2015; Wu et al., 2016; Amit et al., 2016; Phan et al., 2017; Aryal et al., 2014). These factors program macrophages for functions on a spectrum from inflammatory and host defense to resolution and repair. In general, inflammatory macrophages carry out processes that promote atherosclerosis progression, whereas resolving macrophages carry out functions that can suppress plaque progression and promote plaque regression (Peled and Fisher, 2014) (Figure 1). By secreting cytokines, proteases, and other factors, inflammatory macrophages increase the cellular expansion of lesions and cause changes in plaque morphology that can trigger plaque rupture and acute lumenal thrombosis. Two key changes in plaque morphology promoted by inflammatory macrophages are plaque necrosis and thinning of a protective collagenous scar (fibrous cap). Conversely, resolving macrophages carry out functions that are associated with plaque stabilization. These functions include clearing dead cells (efferocytosis), which stabilize plaques by preventing post-apoptotic cellular necrosis (below); secreting collagen that can form a protective scar over the lesion; and producing proteins and lipids that quell inflammation and promote tissue repair.
Molecular profiling of lesional macrophages at various stages of lesion progression and regression has demonstrated heterogeneity suggestive of these different functions (Peled and Fisher, 2014). One such study using immunohistochemistry and RNA profiling shows that CD68+ macrophages at both ends of the inflammation-resolution spectrum accumulate as atherosclerotic lesions develop (Stoger et al., 2012). The relative proportion of macrophages with different phenotypic markers vary depending on plaque region. For example, macrophages on the inflammatory end of the spectrum are enriched in regions of the plaque that are prone to rupture. In this regard, inflammatory macrophages can secrete matrix metalloproteinases, e.g., MMP2 and MMP9, which may contribute to plaque rupture, and another study shows that MMPs co-localize with inflammatory macrophages in advanced plaques (Huang et al., 2012). Conversely, plaques that appear more stable based on thicker fibrous caps and smaller areas of plaque necrosis are found to be enriched in macrophages towards the resolution end of the spectrum., which is consistent with the aforementioned plaque-stabilizing functions of resolving macrophages. In a similar vein, regressing plaques, which can be modeled in mouse atherosclerosis by abruptly and markedly lowering plasma cholesterol, show a shift toward the resolution phenotype (Trogan et al., 2006).
Important gaps remain in our understanding of the mechanisms leading to macrophage heterogeneity in various stages of atherosclerosis progression and regression and at different sites within the plaques themselves. A key question is the relative contribution of changes in the types of infiltrating monocytes (above section) versus changes in pre-existing lesional monocytes or macrophages, such as by proliferation of inflammatory macrophages and phenotypic switching. Macrophage proliferation occurs in atherosclerosis, particularly in advanced plaques (Sakai et al., 2000; Robbins et al., 2013), but the mechanisms and consequences of lesional macrophage proliferation remain largely unknown. In one study, investigators have studied the effect of heterozygous deficiency of a Zfp148, zinc-finger protein that suppresses the checkpoint protein P53, in hematopoietic cells in atheroprone Apoe-/- mice (Sayin et al., 2014). These mice demonstrate a decrease in lesional macrophage proliferation, and this was associated with decreases in markers of inflammation and overall lesion area in a P53-dependent manner. Several mechanisms driving macrophage proliferation in atherosclerosis have been proposed, including activation of scavenger receptor class A signaling, activation of the GTPase Ras, and loss of function of the cyclin-dependent kinase Inhibitor 2A (CDKN2A), which encodes two cell cycle inhibitors and is linked to a susceptibility locus for atherothrombotic disease in humans (Sakai et al., 1996; Senokuchi et al., 2005; Kuo et al., 2011). Regarding the Ras hypothesis, a recent study shows that delivery of an HMG-CoA reductase inhibitor (statin), which blocks Ras activation, to lesional macrophages decreased lesional macrophage proliferation and plaque inflammation (Tang et al., 2015).
Much of the effort in understanding phenotypic modulation of lesional macrophages has focused on environmental cues in lesions that can affect macrophage phenotype. For example, accumulation of cholesterol in these cells can trigger inflammatory signaling pathways, presumably by effects on the plasma membrane that activate inflammatory receptors, e.g., toll-like receptors (TLRs) (Westerterp et al., 2014) (Figure 2). Important new work has suggested that this cholesterol-dependent process in macrophages can be exacerbated in the setting of diabetes by the incorporation of endogenously synthesized fatty acids in cholesterol-rich domains of the plasma membrane (Wei et al., 2016). Moreover, cholesterol accumulation in macrophages activates the inflammasome, leading to increased production of pro-atherogenic interleukin 1β. While cholesterol crystals have been implicated in this process (Duewell et al., 2010; Sheedy et al., 2013), other mechanisms likely contribute as well (Hoseini et al., 2017).
Some of the work in this area has been linked to epigenetic changes at early stages of monocyte-to-macrophage differentiation. For example, exposure of monocytes to oxidized LDL, a modified form of LDL that accumulates in atherosclerotic plaques, “primed” the cells for a subsequent increase in their inflammatory response to TLR2 and TLR4 activators (Bekkering et al., 2014). This response is associated with an epigenetic histone mark of active transcription (trimethylation of H3K4) in the promoters of several of the up-regulated inflammatory genes, and the priming response could be blocked by a methyltransferase inhibitor. A similar finding has been found by comparing monocytes from subjects with symptomatic vs. asymptomatic carotid atherosclerosis (Bekkering et al., 2016). Another study shows that HDAC3 inhibitors increase the expression of ABCA1 and ABCG1 in cultured macrophages and that myeloid deletion of HDAC3 in Western diet-fed Ldlr-/- mice suppresses atherosclerosis and promotes a more resolving phenotype in lesional macrophages (Hoeksema et al., 2014). The inhibitors used in these studies cause broad changes in histone marks throughout the genome, and thus future studies will be needed to define specific epigenetic changes that contribute to pro-atherogenic responses in lesional macrophages.
An Increase in the Ratio of Effector T Cells to Regulatory T Cells Promotes Atherosclerosis
As discussed above for macrophages, T cell functional phenotypes can change in response to environmental clues, thereby altering their relative ability to function as regulatory vs. inflammatory cells. The concept that Treg cells can respond to inflammatory mediators in local tissue environments and take on some of the phenotypic properties of effector T cells (Teff) in those sites is well established. In some cases, Teff-like Treg cells retain their suppressive functions, but in other cases such as in autoimmune disease models, Treg cells assume the inflammatory and pathogenic roles of their Teff cell cousins (Sawant and Vignali, 2014). There is a selective decrease in Treg cells compared with Teff cells during atherosclerotic lesion development (Maganto-Garcia et al., 2011), and this could be attributed to phenotypic plasticity, as well as Treg vs. Teff cell differences in migration into lesions or susceptibility to death (Figure 3). FoxP3 and T-bet are the lineage-defining transcription factors required differentiation and function of most CD4+ Treg or Th1 cells, respectively, and plasticity of these subsets is tied to changes in their expression. Recent studies in mouse models provide evidence of Treg cell plasticity driven by the atherosclerotic and hypercholesterolemic environments, resulting in cells with impaired regulatory or enhanced inflammatory functions. In one study, the accumulation of CD4+CCR5+IFNγ+FoxP3+T-bet+ cells (referred to as Th1-Treg cells) were shown to accumulate in atherosclerotic lesions of Apoe−/− mice. Adoptive transfer studies of CD4+ T cells carrying FoxP3 fate mapping reporter genes into Apoe−/− mice indicate the Th1-Treg cells are derived from former Treg but not naïve T cells (Butcher et al., 2016). Furthermore, the conversion to Th1-Treg cells does not occur in non-atherosclerotic C57BL/6 mice. The Th1-Treg cells show impaired regulatory functions in vitro and reduced expression of several genes linked to Treg cell immunosuppressive function, and they fail to attenuate plaque burden in vivo. Data from another independent study shows that up to 40% of the CD4+ T cells in atherosclerotic aorta of Apoe−/− mice have a CCR5+ FoxP3+T-bet+ phenotype and produce abundant IFN-γ and TNFα (Li et al., 2016). These cells, named FoxP3+CCR5+CD25- Teff cells by the investigators, do not suppress Teff cell proliferation and promote atherosclerosis in adoptive transfer experiments. Whether FoxP3+CCR5+ Teff cells identified in that study are derived from plastic conventional Treg cells remains unknown. However, because they are only found in aorta and peri-aortic lymph nodes and not in spleen, it is possible that they are derived from plaque Treg cells in response to factors in the lesional environment. Interestingly, studies in other lineage-specific T-bet deletion and FOXP3-reporter models, without hypercholesterolemia or atherosclerosis have demonstrated that T-bet expressing Treg cells are immunosuppressive and required to prevent autoimmunity (Levine et al., 2017; Yu et al., 2015). This apparent contradiction in the observed functions of T-bet expressing Treg cells may indicate that the development of IFN-γ-expressing inflammatory T-bet+ Treg cells is linked to the unique conditions of systemic hypercholesterolemia and/or the plaque micro-environment.
Figure 3. Regulation and Impact of Adaptive Immune Processes Related to T cells in Atherosclerosis.
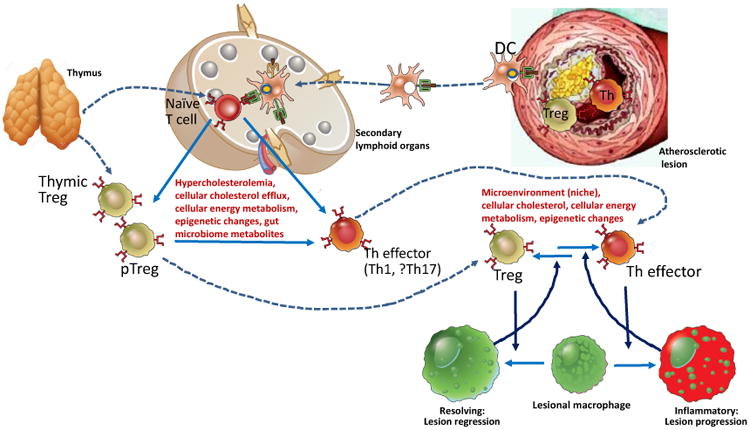
T lymphocyte responses that affect atherosclerosis include a balance between inflammatory effector T cells, mainly interferon γ-producing T helper (Th) cells, and anti-inflammatory regulatory T cells (Treg). Pro-atherogenic Th effector cells differentiate from thymic-derived naïve T cell precursors in secondary lymphoid organs (SLOs) such as lymph nodes, in responses to antigen presentation of LDL-derived peptides by dendritic cells (DC), some of which may have migrated from the arterial wall. Treg develop in the thymus, and peripheral Treg (pTreg) can also be differentiated from peripheral naive T cells in SLOs. The direction of differentiation of naïve T cells into different Th subsets and Treg in SLOs can be influenced by systemic and local metabolic conditions. Treg and Th cells migrate into developing atherosclerotic lesions and modulate the local inflammatory microenvironment, in large part by influencing macrophage phenotypes. Conversely, resolving or inflammatory macrophage phenotypes can shift the plaque T cell balance toward Treg and Th phenotypes, respectively. Change in Treg-Th cell balance may reflect phenotypic plasticity by permitting re-differentiation between regulatory and inflammatory phenotypes, which is also influenced by systemic and plaque metabolic conditions. See text for details.
Many studies of helper T cell subsets in human autoimmune conditions and mouse models of these diseases reinforce the concept that Teff cells are plastic and can differentiate within inflamed issues toward more pathogenic phenotypes during disease progression. For example, under the influence of innate inflammatory cytokines, Th17 cells can be changed from barrier protective effector T cells that secrete IL-17A and IL-10 into pathogenic effectors that secrete IL-17A, IL-22, and IFN-γ (Burkett et al., 2015)(Burkett et al., 2015). This Th17 cell conversion into pathogenic effector cells is promoted by IL-1, IL-6, and IL-23, all of which are known to be expressed in atherosclerotic lesions. The contribution of IL-17 or Th17 cells to atherosclerotic lesion development remains uncertain based on mouse studies, but dual IL-17- and IFN-γ-producing T cells are present in atherosclerotic human coronary arteries at a higher frequency than in the blood (Eid et al., 2009). IL-23 receptor expression on Th17 cells plays a non-redundant role in their pathogenic conversion, and Notch signaling via the DNA binding protein RBPJ is required for induction of IL-23R expression by Th17 cells (Meyer zu et al., 2016). In this context, a recent report shows that abrogation of RBPJ-dependent Notch signaling reduces atherosclerosis in Apoe-/- mice, although the importance of T cell IL-23R expression in that study was not addressed (Nus et al., 2016).
Although T cells do not accumulate large amounts of cholesterol in the setting of hypercholesterolemia as do macrophages, exposure to oxidized LDL and perturbations of T cell intrinsic cholesterol metabolism have profound effects on T cell functions and are implicated in T cell phenotypic modulation (Figure 2). The liver has been long recognized to be an important tissue site for the induction of peripheral T cell tolerance. Given the central role of the liver in handling dietary cholesterol and synthesis of lipoproteins, it is interesting to consider how hepatic T cells respond to changes in cholesterol homeostasis. Hypercholesterolemia in Western diet-fed Ldlr-/- is reported to increase the numbers of intrahepatic Treg cells and expression of TGF-β, a cytokine that both drives Treg cell differentiation and is a principal immunosuppressive mediator secreted by Treg cells(Mailer et al., 2017). Adoptive transfer experiments show that these hepatic Treg cells home to atherosclerotic aortas. Furthermore, hypercholesterolemia induces either Th1 or Th17 cell differentiation of hepatic T cells depending on the presence or absence of LDL receptors. These data suggest that systemic changes in lipoprotein metabolism and dietary cholesterol may change the ratios of Treg and Th cell subsets generated in the liver and then enter plaques.
The mechanisms by which hypercholesterolemia may influence Treg or Teff cell differentiation likely involve effects on the antigen presenting DCs, which are found in atherosclerosis prone regions of arteries before lesion development and readily take up oxidized-LDL (Paulson et al., 2010). DCs maintain their antigen-presenting functions after becoming foam cells (Packard et al., 2008), and respond to cholesterol accumulation by increasing production of cytokines that drive Th cell subset differentiation (Westerterp et al., 2017). However, there also appear to be direct effects of cholesterol on T cells. For example, T cells from mice deficient in the cholesterol transporter ABCG1 show increased proliferative responses to TCR stimulation, and T cell-selective deletion of the Abcg1 gene results in an increase in Treg cell differentiation from naïve T cells (Armstrong et al., 2010). T cell- or Treg-cell-restricted ABCG1 deficiency in Ldlr//- mice results in more Treg, less Teff cells, and less atherosclerosis compared to control mice (Cheng et al., 2016). Interestingly, the ABCG1-deficient T cells accumulate more cholesterol in lysosomes than control T cells, which lead to inhibition of mTOR-mediated STAT5 signaling. These changes in energy metabolism favor Treg over Teff cell phenotypes, as discussed in the next section. T cell activation requires the assembly of antigen receptor and other signaling molecules in cholesterol-rich lipid rafts, which is a prerequisite to the formation of immune synapses with antigen presenting cells. Therefore, alterations in cellular cholesterol may influence T cell activation because of changes in lipid raft formation. This was shown to be the case for CD8+ effector T cells in mice in which cellular unesterified cholesterol was elevated by interventions that block cholesterol ester formation (Yang et al., 2016). CD8+ T cell plasma membrane cholesterol was increased in these mice, and anti-tumor effector functions of the CD8+ T cells were increased. Thus, the net immunologic impact of changes in T cell cholesterol appear to vary depending on the intracellular compartment affected and on the T cell subset.
In two of the previously discussed studies, exposure of T cells to excess cholesterol, either through systemic hypercholesterolemia (Mailer et al., 2017) or by impairing cholesterol efflux by deleting ABCG1 (Cheng et al., 2016), was shown to favor Treg over Teff cell differentiation. This may seem surprising, given the atheroprotective effect of Treg cells and the pro-atherogenic and pro-inflammatory effects of excess cholesterol and Teff cells. Perhaps the direct proinflammatory effects of hypercholesterolemia on macrophages and DCs overwhelms those on Treg cell development. Another possibility is that Treg cells generated in response to the cholesterol stimuli differentiate into Th1-like cells under hypercholesteremic conditions, as discussed earlier. In general, induction of regulatory mechanisms concurrently with inflammatory responses is commonplace in the immune system, which must strike a fine balance between anti-microbial defense and protection of host tissues. This balance and the timing of the counteracting mechanism achieves defense against pathogens followed by resolution and return to homeostasis, thereby limiting collateral damage to healthy tissues and avoiding autoimmunity. In atherosclerosis, an emerging theme is that the balance is pathologically tipped toward the inflammatory side, and the regulatory mechanisms fail to move the scales to achieve resolution.
Energy Metabolism Affects Immune Cell Function in Atherosclerosis
An area of increasing interest is how changes in macrophage metabolism influence their phenotype. Studies using cultured macrophages treated with either lipopolysaccharide (LPS) and interferon γ (IFNγ) to model inflammatory macrophages or with IL-4 to model of so-called alternatively activated macrophages have revealed certain preferences for metabolic pathways, but these associations are more complex than generally portrayed (Van den Bossche et al., 2017). For example, fatty acid oxidation (FAO) and oxidative phosphorylation are needed to support the phenotype of alternatively activated macrophages, but FAO is also involved in inflammasome activation in inflammatory macrophages. Conversely, while glycolytic metabolism has been associated with energy metabolism in inflammatory macrophages, glycolysis is also used by alternatively activated macrophages to fuel the tricarboxylic acid cycle for mitochondrial respiration. The role of metabolic shifts in determining the phenotype of macrophages in atherosclerotic lesions is not yet well-characterized, but a few recent studies have shed light on this interesting topic. As one example, inflammatory macrophages are found to have increased expression of a particular microRNA, miR-33, and when miR-33 is silenced in cultured macrophages, the cells show markers of alternative activation associated with increased expression of AMP kinase and FAO genes, elevated mitochondrial respiration, and decreased glycolysis (Ouimet et al., 2015). In vivo, treatment of Western diet-fed Ldlr-/- mice with an anti-miR-33 leads to a reduction in both plaque size and complexity and a change in lesional macrophage gene expression profile suggestive of alternative activation. Moreover, these changes are accompanied by an increase in athero-protective regulatory T cells in lesions, suggesting a link between immunometabolism in macrophages and innate-adaptive immunity crosstalk in the setting of atherosclerosis. As another example, investigators reported elevated glucose uptake and glycolytic flux in monocytes and macrophages from patients with coronary artery disease (CAD) compared with control subjects (Shirai et al., 2016). In vitro experiments show that this metabolic phenotype leads to activation of a pathway involving mitochondrial oxidative stress (mitoOS) and pyruvate kinase M2-mediated STAT3 activation, resulting in increased production of pro-atherogenic IL-6 and IL-1β. Other studies have suggested additional links between mitoOS and human CAD (Paneni et al., 2017), and mitoOS in macrophages increases plaque progression, activates macrophage NF-κB, and promotes the formation of potentially atherogenic neutrophil extracellular traps (NETs) in mouse models of atherosclerosis (Wang et al., 2014; Wang et al., 2017).
In a follow-up to the glycolytic flux study above, the investigators found that the elevated glycolytic flux in macrophages from CAD subjects leads to excess mitochondrial pyruvate, which drives BMP4- and IRF1-dependent expression of the T cell inhibitory protein PD-L1 (Watanabe et al., 2017). The high PD-L1 expression on these macrophages was shown to block T cell activation, and the investigators correlated this with impaired anti-varicella zoster virus immunity in the CAD patients. Interestingly, one mechanism of the de-activating effect of PD-1 on T cell activation may involve PD-1-mediated suppression of glycolysis in the T cells themselves, as has been shown recently for CD8+ T cells (Bengsch et al., 2016). As with the miR-33 mentioned above, glycolysis-induced PD-L1 in macrophages represents another example of metabolic changes in macrophages linked to regulation of T cells, except in this case glycolysis-driven macrophage inflammation down-regulates T cell activation. This T cell-suppressive process represents another example of a counter-regulatory pathway that is induced concurrently with induction of pro-inflammatory pathways. PD-L1 induction does not prevent lesion development, but likely provides a beneficial effect by limiting lesion growth and inflammation. Consistent with this concept is the finding that deficiency of PD-1 or PD-L1 in high fat-diet-fed Ldlr-/- mice enhances lesion development and plaque inflammation compared with baseline lesion development in wild-type mice that occurs despite the presence of PD-L1 (Lichtman, 2012). Similarly, a recent study shows that hypercholesterolemia induces PD-L1 expression on marginal zone B cells, which inhibits pro-atherogenic T follicular helper cell responses (Nus et al., 2017).
Another environmental factor in advanced atherosclerotic lesions that can affect macrophage metabolism is hypoxia. Hypoxic conditions exist in atherosclerotic plaques, leading to increased expression of hypoxia-inducible factor-1α (HIF-1α) (Marsch et al., 2013). A recent study finds that hypoxia markedly increases glycolysis in cultured macrophages exposed to inflammatory cytokines or oxidized LDL. This effect is associated with a HIF-1α-dependent increase in mRNAs encoding glycolysis-promoting enzymes, notably hexokinase II (Hk2) and the 6-phosphofructo-2-kinase (Pfkfb3), and with an amplified production of inflammatory cytokines (Tawakol et al., 2015). In Western diet-fed Apoe-/- mice, inhibition or silencing of either Pfkfb3 or Hif1a attenuates lesional glycolysis and decreases the expression of inflammatory cytokines, although the net effect on lesion development was not reported. While this study suggests that HIF-1α can be pro-inflammatory, HIF-1α in CD11c-expressing cells may have the opposite effect, as CD11c-cre-mediated deletion of Hif1a in Ldlr-/- mice results in increased lesion development and more T cells in the plaques (Chaudhari et al., 2015). The HIF-1α-deficient APCs express less STAT3 and more IL-12, which is associated with increased Th1 cell differentiation, and lentivirus-induced overexpression of STAT3 in bone marrow cells reverses the effects of the Hif1a deletion Lyz2-cre-mediated Hif1a deletion does not have the same effects on lesion development or T cells, indicating that HIF-1α deficiency in dendritic cells is the likely cause of the pro-atherogenic phenotype.
As discussed for macrophages (above), changes in energy metabolism in T cells are also linked to functional phenotypes, and these metabolic-functional T cell states may synergize with proatherogenic or resolving functions of monocytes and macrophages within atherosclerotic lesions or systemically. In this regard, two differences between T cells and macrophages that should be kept in mind are (a) the requirement for rapid proliferative expansion to generate effector T cells from naïve or resting memory cells; and (b) the greater diversity of specialized functional phenotypes of T cells. These features are reflected in many different metabolic states in T cells, depending on the stage of the T cell response and the functional subset of the T cell (MacIver et al., 2013). Resting naïve T cells use the TCA cycle to oxidize glucose-derived pyruvate, lipids and amino acids, and efficiently generate ATP. Antigen-induced clonal proliferation requires a shift to aerobic glycolysis with reduced lipid oxidation and increased lipid synthesis. The resting memory T cells that survive at the end of a T cell response go back to efficient energy generation, but mainly by lipid oxidation. Furthermore, there are distinct metabolic phenotypes in different T cell subsets. Effector helper T cells, including Th1, Th2, and Th17 cells require mTOR and aerobic glycolysis for differentiation. However, whereas Th1 and Th17 cells depend on active mTORC1 activity, Th2 cells depend on mTORC2. In contrast, Treg cell generation is regulated by AMP-activated protein kinase (AMPK) and relies on lipid oxidation through the TCA cycle. Given the opposing roles of Th effector cells and Treg cells on atherosclerosis, summarized earlier, and the possible different influences of Th cell subsets, the impact of systemic modulators of cellular metabolism associated with atherosclerotic disease and the local metabolic environment of the plaque are likely to affect how T cells contribute to lesion progression.
Of relevance to how the local plaque environment may influence T cell metabolism and function, a recently published study has shown that effector memory T cells (Tem), the functional phenotype of most T cells found in plaques, responds to hypoxia by increased HIF-1α expression, which in turn increases proliferation, viability, and cytotoxicity (Xu et al., 2016). This is not the case for naïve or central memory T cells, and, as discussed earlier, is in contrast to the finding of increased T cell inflammation in plaques in Western diet-fed Ldlr-/- mice with DC-restricted deletion of Hif1a (Chaudhari et al., 2015). GAPDH has been shown to block HIF-1α translation, and the increased glycolytic activity in Tem reduced GAPDH, resulting in elevated HIF1α expression. A recent study has shown that FoxP3, the defining Treg cell transcription factor, is responsible for maintaining a metabolic state in which glycolysis is repressed and oxidative phosphorylation and nicotinamide adenine dinucleotide oxidation are increased. This state allows Treg cells to function in low-glucose, lactate-rich environments that impair effector T cell function and proliferation (Angelin et al., 2017). The likely influence of the plaque environment on T cell metabolic states and function have not yet been experimentally established.
Aging Affects Immune Cell Function in Atherosclerosis
Aging is major risk factor for atherosclerotic disease (Lakatta and Levy, 2003). One of the consequences of aging is an accumulation of somatic mutations in hematopoietic stem cells (HSC), which has been linked to increased clonal hematopoiesis and cardiovascular disease (Jaiswal et al., 2014; Jaiswal et al., 2017) (Figure 2). One of the genes affected by age-related HSC somatic mutations is TET2, which encodes an epigenetic modifier enzyme that can also repress inflammatory gene expression in human and mouse macrophages and DCs by recruiting histone deacetylases (HDAC) (Zhang et al., 2015). Among the genes repressed by TET2 include IL-6 and several chemokines. In Western diet-fed Ldlr-/- mice, partial bone marrow reconstitution with TET2-deficient hematopoietic cells leads to clonal expansion of these cells, with a myeloid bias, and increased atherosclerosis with an enrichment of Tet2-/- lesional macrophages (Fuster et al., 2017; Jaiswal et al., 2017). The pro-atherogenic effect TET2 deficiency in mice has been ascribed to inflammasome activation (Fuster et al., 2017). Mechanistic studies have shown that the ability of TET2 to suppress the inflammasome does not depend on its canonical activity of oxidizing 5-methylcytosine but rather on HDAC-mediated suppression of Il1b and inflammasome component genes. Because the in vivo causation study in this report used a chemical inhibitor of the inflammasome, the relative contribution of the inflammasome to the pro-atherogenic effect of TET2 deficiency will need to be confirmed using genetic approaches. Other areas for future studies include relevance to human lesions in subjects with somatic TET2 mutations and the impact of somatic mutations on DC function and T cell responses related to atherosclerosis. Importantly, one of the studies cited above has shown strong associations between coronary artery disease and age-related somatic mutations in a number of genes that cause clonal hematopoiesis of indeterminate potential (CHIP), including TET2 but also DNMT3A, ASXL1, and JAK2 (Jaiswal et al., 2017). These findings have potentially important implications related to risk stratification and possibly new and/or more tailored treatment strategies.
Aging is also associated with the accumulation of so-called senescent cells that, in chronic diseases of aging, can contribute to a maladaptive inflammatory response via secretion of inflammation-inducing factors. Advanced atherosclerotic lesions contain cells with markers of senescence, including senescence-associated β-galactosidase, p16Ink4a, p53, p21, and shortened telomeres (Minamino and Komuro, 2007). While indirect causation studies in mice originally suggested a pro-atherogenic role of senescent lesional cells, genetic association studies in mice and humans have called into question this conclusion (Schierwagen et al., 2017). However, a more definitive causation study has been recently conducted using Western diet-fed Ldlr-/- mice in which lesional macrophages, VSMCs, and endothelial cells expressing p16Ink were genetically eliminated (Childs et al., 2016). Compared with the control cohort, the engineered mice had smaller lesion size and decreased lesion complexity without changes in pro-atherogenic systemic factors. In particular, the mice have a marked decrease in cholesterol ester-laden macrophages (foam cells) with markers of senescence, and this is accompanied by decreases in inflammatory cytokines and matrix metalloproteinases and an increase in fibrous cap thickness. In addition to the role of senescent cells in advanced plaque progression suggested by this study, an interesting observation is that senescent-appearing foam cell macrophages also accumulate in very early lesions and initiated processes that promote early atherogenesis.
Resolution of Inflammation is Impaired in Atherosclerosis and Contributes to Plaque Progression
In normal physiology, the inflammatory response is directly linked to a resolution phase that repairs collateral damage and restores tissue homeostasis (Serhan et al., 2007; Nathan and Ding, 2010). Resolution is mediated by (a) endogenous lipids that are generated during inflammation, including lipoxins, resolvins, protectins, and maresins, called specialized pro-resolving mediators (SPMs) (Serhan, 2010); and (b) proteins, such as IL-10, TGFβ, and annexin A1 (Perretti and D'Acquisto, 2009). By activating specific cell-surface receptors, these mediators block inflammatory cell influx and promote their egress; clear pathogens, cellular debris, inflammatory cytokines, and apoptotic cells (efferocytosis); and repair tissue damage (Serhan, 2010; Perretti and D'Acquisto, 2009). In chronic diseases in which the inciting pathologic process is persistent, resolution becomes defective owing to impaired synthesis or increased degradation of resolution mediators and to decreased resolution mediator action on cells that effect the repair process (Serhan, 2010). This defect leads to an amplification cycle of continual tissue injury and DAMP-mediated inflammation and, in the case of atherosclerosis, plays a key role in the clinical progression of plaques (Merched et al., 2008; Tabas, 2010; Viola and Soehnlein, 2015). In this context, several key features of clinically dangerous advanced plaques are characteristic of impaired resolution, including defective efferocytosis, plaque necrosis, and DAMP-mediated inflammation; thinning of the fibrous cap; and oxidative stress. Moreover, the ratio of resolving:inflammatory lipid mediators is markedly lower in advanced murine and human plaques compared with earlier lesions (Fredman et al., 2016; Viola et al., 2016). The therapeutic potential of enhancing resolution in chronic diseases like atherosclerosis could be substantial in that SPMs, unlike anti-inflammatory drugs, suppress inflammation and promote tissue repair in a manner that is less likely to compromise host defense than drugs directly targeting the inflammatory response (Serhan et al., 2007; Tabas and Glass, 2013) (below).
A lesional macrophage resolution process that has received particular attention in the field of atherosclerosis is efferocytosis (Tabas, 2005; Schrijvers et al., 2007; Linton et al., 2016; Van Vre et al., 2012). Efferocytosis is mediated through phagocyte receptors, apoptotic cell ligands, bridging proteins, and chemoattractants. It is normally a high-capacity and efficient process, but when it goes awry, tissue necrosis and subsequent DAMP-mediated inflammation occur (Henson et al., 2001; Camenisch et al., 1999). A critically important example of this principle is advanced atherosclerosis (Geng and Libby, 1995; Schrijvers et al., 2005; Otsuka et al., 2015). Clinically dangerous human coronary plaques show evidence of defective efferocytosis, i.e., abundant uncleared dead cells, and this defect correlates with two key features of these plaques—necrosis and inflammation. Causation is suggested by studies using genetically altered mice. For example, when efferocytosis is compromised through gene targeting of effector molecules, there is an increase in uncleared apoptotic cells, inflammation, and plaque necrosis (Yancey et al., 2010; Tao et al., 2015; Cai et al., 2017).
The unresolved mechanism of defective efferocytosis in advanced plaques represents a major gap in this field. In theory, defects could occur in processes occurring during apoptosis that normally ensure proper recognition or uptake by lesional phagocytes or in the phagocytes themselves. As an example of the former scenario, a recent study has presented evidence that lesional apoptotic macrophages, under the influence of inflammatory signaling, continue to express a “don’t-eat-me” cell-surface protein called CD47 that is normally extinguished when cells die (Kojima et al., 2016). CD47, by interacting with a molecule on phagocytes called SIRPα, suppresses cell internalization by the phagocyte, and thus its persistence on lesional dead cells contributes to defective efferocytosis. With regard to phagocyte defects, there is evidence that lesional hypoxia may lead to decreased expression of a key efferocytosis receptor on macrophages called MerTK and thereby compromise the ability of lesional macrophages to recognize apoptotic cells (Marsch et al., 2014). Another mechanism of defective efferocytosis related to MerTK is ADAM17-mediated MerTK cleavage. There is evidence that MerTK cleavage occurs in advanced, necrotic human coronary plaques (Garbin et al., 2013; Cai et al., 2017), and Western diet-fed Ldlr-/- mice expressing an engineered non-cleavable mutant of MerTK show improved lesional efferocytosis and decreased plaque necrosis compared with mice expressing wild-type MerTK (Cai et al., 2017). Interestingly, these lesions also have additional signs of improved resolution, such as thicker fibrous caps, and they have an increase in the ratio of resolving:inflammatory lipid mediators, which is consistent with a prior examining holo-MerTK deficiency on plaque inflammation (Ait-Oufella et al., 2008). Mechanistic studies reveal a MerTK signaling pathway in macrophages that promotes the biosynthesis of resolving lipid mediators at the expense of inflammatory lipid mediators (Cai et al., 2016). These studies suggest an amplification loop in which resolving mediators promote efferocytosis, and then efferocytosis promotes the synthesis of resolving mediators. Thus, therapeutic strategies that could convert the pro-atherogenic cycle of defective efferocytosis, i.e., impaired efferocytosis exacerbating DAMP-mediated inflammation, to this beneficial feedback cycle might be particularly effective at preventing advanced plaque progression.
Considering the intricate relationships between innate and adaptive effectors in chronic inflammatory processes, the macrophage contributions to defective resolution in plaques are likely to enhance and be aggravated by effector T cell responses. Conversely, pro-resolving interventions would be expected to engage regulatory T cells. There is evidence that Treg cells increase during the resolution phase of innate inflammatory processes in mouse models and help shape the resolution process. For example, after bacterial and worm infections in fate mapping reporter mice, Th17 cells have been shown to transdifferentiate into FoxP3 Treg cells as inflammation resolved (Gagliani et al., 2015). In a lung model of self-limited allergic inflammation, the SPM maresin 1 has been shown to induce Treg cells, which then inhibited type 2 innate lymphoid cells. A recent in vitro study has shown that SPMs can block differentiation of naïve human T cells to Th1 and Th17 cell effectors, block Teff cell inflammatory cytokine production including IFN-γ, and induce Treg cells (Chiurchiu et al., 2016). These in vitro findings are supported by studies in Elovl2−/− mice, which are defective in SPM production and shown to have more Th1 and Th17 cells in lymphoid tissues and less Treg cells than control mice. Moreover, the fraction of aortic T cells that are FoxP3+ is increased in mice with enhanced plaque efferocytosis due to cleavage-resistant MerTK, described above (Cai et al., 2017). These initial reports set the stage for future studies that will further define T cell contributions to the resolution state and determine how these contributions may be leveraged by pro-resolving therapeutics in atherosclerosis.
Summary and Therapeutic Considerations
Our understanding of the ways innate and adaptive immunity influence atherosclerotic disease has continued to evolve along with emerging areas in immunology research. In this review, we have emphasized selected themes, including the dynamics and heterogeneity of macrophage and T cell phenotypes, the impact of cellular and systemic metabolic sates on immune cell phenotypes, and the ways macrophage and T cell functions impact the balance between progression and resolution of plaque inflammation. These emerging areas of investigation have raised new questions, but they have also validated efforts at modifying existing targeted therapeutic approaches and developing new strategies. Viewing the pathogenesis of atherosclerosis as an immunological reaction to retained and modified apoB LPs in the arterial wall emphasizes the fundamental importance and success of plasma apoB LP-lowering therapies, including statins, ezetimibe, and now PCSK9 inhibitors (Boren and Williams, 2016). By lowering the concentration of apoB LPs in the blood, which then lowers their infiltration into developing lesions, these therapies decrease the burden of DAMPs and antigens that drive innate and adaptive immune responses in the artery. Moreover, as discussed above, hypercholesterolemia can promote atherogenic activation of circulating immune cells and their precursors even before they enter developing lesions. In theory, if cholesterol-rich apoB LPs were lowered below a certain threshold concentration, this alone would halt plaque progression and likely induce regression. However, this potential has not been met due to a variety of issues, with the counteracting effects of obesity-induced insulin resistance playing a major role. Insulin resistance is a potent risk factor for atherosclerotic heart disease owing to its links to dyslipidemia and inflammation and to the effects of high insulin and glucose on atherosclerotic lesional cells (Razani et al., 2008; Bornfeldt and Tabas, 2011). Indeed, largely due to the epidemic of obesity and insulin resistance, atherosclerotic vascular disease remains the leading cause of death in the industrialized world, and its prevalence is expected to rise over the next 20 years (Go et al., 2013; Behn and Ur, 2006; American Heart Association and RTI International, 2015). In this regard, a topic of great interest moving forward is how insulin resistance might affect the immune processes discussed in this review and thereby exacerbate atherosclerosis (Han et al., 2006; Kanter et al., 2012; Nagareddy et al., 2013; Willecke et al., 2015). Moreover, as the number of patients being treated with PCSK9 inhibitors increases, we may have an opportunity to learn how lowering cholesterol below the concentrations achieved with statins alters immune parameters of atherosclerosis, even in the face of the obesity epidemic (Dullaart, 2017; Boren and Williams, 2016).
Cytokine-targeted therapies, such as TNF or IL-17 antagonists, have transformed treatment of autoimmune diseases, including rheumatoid arthritis and psoriasis. These diseases carry increased cardiovascular risk, but whether the anti-cytokine therapies used to reduce the autoimmune pathology directly impact plaque inflammation and progression is not clear. The recently completed CANTOS trial of an anti-IL-1β blocking antibody (canakinumab) in subjects with a history of myocardial infarction and elevated serum high-sensitivity C-reactive protein concentration (hsCRP) is the first clinical study of the effectiveness of an anti-cytokine therapy given explicitly for cardiovascular disease (Ridker et al., 2017). Treatment of over 10,000 patients with canakinumab was shown to reduce the risk of secondary nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death by 15% compared with compared to placebo. Canakinumab treatment significantly lowered serum hsCRP and IL-6 concentrations but had no effect on lipids, suggesting that the cardiovascular risk reduction was directly related to anti-inflammatory effects. The currently active Cardiovascular Inflammation Reduction Trial (CIRT) will determine if myocardial infarction or stroke in a high-risk population can be decreased by low-dose methotrexate, which is an immunosuppressive regimen targeting cytokine-producing immune cells that has long been used to treat rheumatoid arthritis (Ridker et al., 2012).
A concern for strategies that attack immune activation for a chronic disease process like atherosclerosis is compromise of host defense. Indeed, the risk of fatal infection was significantly increased in the canakinumab-treated group in the CANTOS trial (Ridker et al., 2017). The realization that physiologic mechanisms underlying resolution of inflammation are impaired in the human plaque environment and that pro-resolution therapy may not compromise host defense as much as anti-inflammatory therapy has led to active work in developing therapies based on delivery of pro-resolution mediators into lesions (Fredman et al., 2015; Hasturk et al., 2015; Viola et al., 2016; Petri et al., 2017). Another challenge to treating chronic autoimmune diseases is the accumulation of autoreactive tissue resident memory lymphocytes whose pathogenic phenotypes are stabilized by epigenetic modifications of various gene loci (Phan et al., 2017). There is also evidence that metabolic conditions can induce epigenetic changes in macrophages, leading to a form of innate immune memory (“trained immunity”) (Netea et al., 2016; Phan et al., 2017) that may promote lesion progression (Bekkering et al., 2016). In this regard, pre-clinical studies have provided support for the principal that targeting physiologic epigenetic modifiers, for example with inhibitors of Bromodomain and Extra-Terminal motif (BET) proteins, can inhibit inflammatory gene expression and reduce atherosclerosis (Brown et al., 2014; Jahagirdar et al., 2014). Many preclinical studies have validated the concept of treating atherosclerosis with therapeutic vaccinations that tolerize against antigens that drive proatherogenic T cell responses. Recent progress in this area has included identification of apoB100 peptide epitopes that bind mouse or human class II MHC glycoproteins, and the demonstration that vaccination of mice expressing MHC molecules that can bind the peptide reduces lesion formation (Gistera et al., 2017; Kimura et al., 2017). Evidence is provided suggesting the mechanism of protection is induction of Treg cells(Kimura et al., 2017) or antibodies that block LDL activation of macrophages (Gistera et al., 2017). Careful assessment of efficacy and adverse effects of these new therapies, as well as future ones that will undoubtedly be conceived as we learn more about the immune mechanisms that drive atherosclerosis, represent important ongoing efforts to complement apoB LP-lowering therapy for the prevention and treatment of atherothrombotic vascular disease.
Acknowledgments
This work was supported by the NIH (R01 HL132412, HL127464, HL075662 and P01 HL087123 to I.T.; R01 HL087282 and HL131862 to A.H.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, Offenstadt G, Leseche G, Cohen PL, Tedgui A, et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:1429–1431. doi: 10.1161/ATVBAHA.108.169078. [DOI] [PubMed] [Google Scholar]
- American Heart Association & RTI International. Cardiovascular disease: a costly burden for America - projections through 2035 2015 [Google Scholar]
- Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. 2016;17:18–25. doi: 10.1038/ni.3325. [DOI] [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, III, Kopinski PK, Wang L, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25:1282–1293. doi: 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Gebre AK, Parks JS, Hedrick CC. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J Immunol. 2010;184:173–183. doi: 10.4049/jimmunol.0902372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal B, Rotllan N, Fernandez-Hernando C. Noncoding RNAs and atherosclerosis. Curr Atheroscler Rep. 2014;16:407. doi: 10.1007/s11883-014-0407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behn A, Ur E. The obesity epidemic and its cardiovascular consequences. Curr Opin Cardiol. 2006;21:353–360. doi: 10.1097/01.hco.0000231406.84554.96. [DOI] [PubMed] [Google Scholar]
- Bekkering S, Quintin J, Joosten LA, Van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. 2014;34:1731–1738. doi: 10.1161/ATVBAHA.114.303887. [DOI] [PubMed] [Google Scholar]
- Bekkering S, van dMI, Nielen T, Lamfers E, Dinarello C, Rutten J, de GJ, Joosten LA, Netea MG, Gomes ME, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. 2016;254:228–236. doi: 10.1016/j.atherosclerosis.2016.10.019. [DOI] [PubMed] [Google Scholar]
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T Cell exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boren J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–483. doi: 10.1097/MOL.0000000000000330. [DOI] [PubMed] [Google Scholar]
- Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkett PR, Meyer zu HG, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest. 2015;125:2211–2219. doi: 10.1172/JCI78085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N, Boldin M, Lundberg PS, Galkina EV. Atherosclerosis-driven Treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/Tregs. Circ Res. 2016;119:1190–1203. doi: 10.1161/CIRCRESAHA.116.309764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, Spite M, Fredman G, Tabas I. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. 2017;127:564–568. doi: 10.1172/JCI90520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, Spite M, Fredman G, Tabas I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A. 2016;113:6526–6531. doi: 10.1073/pnas.1524292113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol. 1999;162:3498–3503. [PubMed] [Google Scholar]
- Chaudhari SM, Sluimer JC, Koch M, Theelen TL, Manthey HD, Busch M, Caballero-Franco C, Vogel F, Cochain C, Pelisek J, et al. Deficiency of HIF1α in antigen-presenting cells aggravates atherosclerosis and type 1 T-helper cell responses in mice. Arterioscler Thromb Vasc Biol. 2015;35:2316–2325. doi: 10.1161/ATVBAHA.115.306171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HH, Keyhanian K, Zhou X, Vilmundarson RO, Almontashiri NA, Cruz SA, Pandey NR, Lerma YN, Ho T, Stewart CA, et al. IRF2BP2 reduces macrophage inflammation and susceptibility to atherosclerosis. Circ Res. 2015;117:671–683. doi: 10.1161/CIRCRESAHA.114.305777. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Gaddis DE, Wu R, McSkimming C, Haynes LD, Taylor AM, McNamara CA, Sorci-Thomas M, Hedrick CC. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J Clin Invest. 2016;126:3236–3246. doi: 10.1172/JCI83136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurchiu V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, Serhan CN. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med. 2016;8:353r. doi: 10.1126/scitranslmed.aaf7483. a111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dullaart RPF. PCSK9 inhibition to reduce cardiovascular events. N Engl J Med. 2017;376:1790–1791. doi: 10.1056/NEJMe1703138. [DOI] [PubMed] [Google Scholar]
- Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, Sokol SI, Pfau S, Pober JS, Tellides G. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119:1424–1432. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbilgin A, Civelek M, Romanoski CE, Pan C, Hagopian R, Berliner JA, Lusis AJ. Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res. 2013;54:1894–1905. doi: 10.1194/jlr.M037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelstrand P, Boren J. Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutr Metab Cardiovasc Dis. 2012;22:1–7. doi: 10.1016/j.numecd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Franck G, Mawson T, Sausen G, Salinas M, Santos MG, Cole AP, Beltrami MM, Chatzizisis Y, Quillard T, Tesmenitsky Y, et al. Flow perturbation mediates neutrophil recruitment and potentiates endothelial injury via TLR2 in mice - implications for superficial erosion. Circ Res. 2017;121:31–42. doi: 10.1161/CIRCRESAHA.117.310694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Milton J, Perretti M, Farokhzad OC, et al. Targeted nanoparticles containing the pro-resolving peptide Ac2-26 protect against advanced atheosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7:275ra20. doi: 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limon P, Paiva RS, Ching T, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–225. doi: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbin U, Baggio E, Stranieri C, Pasini A, Manfro S, Mozzini C, Vallerio P, Lipari G, Merigo F, Guidi G, et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: a role for oxidized polyunsaturated fatty acids? Cardiovasc Res. 2013;97:125–133. doi: 10.1093/cvr/cvs301. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Westerterp M, Bhagwat N, Cremers S, Shih A, Abdel-Wahab O, Lutjohann D, Randolph GJ, Levine RL, Tall AR, et al. HDL and Glut1 inhibition reverse a hypermetabolic state in mouse models of myeloproliferative disorders. J Exp Med. 2013;210:339–353. doi: 10.1084/jem.20121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng YJ, Libby P. Evidence for apoptosis in advanced human atheroma Colocalization with interleukin-1β-converting enzyme. Am J Pathol. 1995;147:251–266. [PMC free article] [PubMed] [Google Scholar]
- Gimbrone MA, Jr, Garcia-Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013;22:9–15. doi: 10.1016/j.carpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gistera A, Hermansson A, Strodthoff D, Klement ML, Hedin U, Fredrikson GN, Nilsson J, Hansson GK, Ketelhuth DF. Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J Intern Med. 2017;281:383–397. doi: 10.1111/joim.12589. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metabolism. 2006;3:257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol. 2011;12:778–785. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, Van Dyke TE. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35:1123–1133. doi: 10.1161/ATVBAHA.115.305324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur MC, Bode C, Fricchione GL, Denninger J, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754–758. doi: 10.1038/nm.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr Biol. 2001;11:R795–R805. doi: 10.1016/s0960-9822(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–1622. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf I, Swirski FK. Making a difference: monocyte heterogeneity in cardiovascular disease. Curr Atheroscler Rep. 2012;14:450–459. doi: 10.1007/s11883-012-0274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeksema MA, Gijbels MJ, Van den Bossche J, van dV, Sijm A, Neele AE, Seijkens T, Stoger JL, Meiler S, Boshuizen MC, et al. Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6:1124–1132. doi: 10.15252/emmm.201404170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2017 doi: 10.1002/jcp.25930. In press-online. [DOI] [PubMed] [Google Scholar]
- Huang WC, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-kappaB. PLoS ONE. 2012;7:e42507. doi: 10.1371/journal.pone.0042507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahagirdar R, Zhang H, Azhar S, Tobin J, Attwell S, Yu R, Wu J, McLure KG, Hansen HC, Wagner GS, et al. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis. 2014;236:91–100. doi: 10.1016/j.atherosclerosis.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017 doi: 10.1056/NEJMoa1701719. In press-online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–2083. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, Li LO, Becker L, Yuan W, Chait A, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci U S A. 2012;109:E715–E724. doi: 10.1073/pnas.1111600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Tse K, McArdle S, Gerhardt T, Miller J, Mikulski Z, Sidney J, Sette A, Wolf D, Ley K. Atheroprotective vaccination with MHC-II-restricted ApoB peptides induces peritoneal IL-10-producing CD4 T cells. Am J Physiol Heart Circ Physiol. 2017;312:H781–H790. doi: 10.1152/ajpheart.00798.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. doi: 10.1038/nature18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CL, Murphy AJ, Sayers S, Li R, Yvan-Charvet L, Davis JZ, Krishnamurthy J, Liu Y, Puig O, Sharpless NE, et al. Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arterioscler Thromb Vasc Biol. 2011;31:2483–2492. doi: 10.1161/ATVBAHA.111.234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AG, Medoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. 2017;546:421–425. doi: 10.1038/nature22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McArdle S, Gholami A, Kimura T, Wolf D, Gerhardt T, Miller J, Weber C, Ley K. CCR5+T-bet+FoxP3+ effector CD4 T Cells drive atherosclerosis. Circ Res. 2016;118:1540–1552. doi: 10.1161/CIRCRESAHA.116.308648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013;368:2004–2013. doi: 10.1056/NEJMra1216063. [DOI] [PubMed] [Google Scholar]
- Libby P. Superficial erosion and the precision management of acute coronary syndromes: not one-size-fits-all. Eur Heart J. 2017;38:801–803. doi: 10.1093/eurheartj/ehw599. [DOI] [PubMed] [Google Scholar]
- Lichtman AH. T cell costimulatory and coinhibitory pathways in vascular inflammatory diseases. Front Physiol. 2012;3:18. doi: 10.3389/fphys.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J. 2016;80:2259–2268. doi: 10.1253/circj.CJ-16-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maganto-Garcia E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation. 2011;124:185–195. doi: 10.1161/CIRCULATIONAHA.110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailer RKW, Gistera A, Polyzos KA, Ketelhuth DFJ, Hansson GK. Hypercholesterolemia induces differentiation of regulatory T cells in the liver. Circ Res. 2017;120:1740–1753. doi: 10.1161/CIRCRESAHA.116.310054. [DOI] [PubMed] [Google Scholar]
- Marsch E, Sluimer JC, Daemen MJ. Hypoxia in atherosclerosis and inflammation. Curr Opin Lipidol. 2013;24:393–400. doi: 10.1097/MOL.0b013e32836484a4. [DOI] [PubMed] [Google Scholar]
- Marsch E, Theelen TL, Demandt JA, Jeurissen M, van GM, Verjans R, Janssen A, Cleutjens JP, Meex SJ, Donners MM, et al. Reversal of hypoxia in murine atherosclerosis prevents necrotic core expansion by enhancing efferocytosis. Arterioscler Thromb Vasc Biol. 2014;34:2545–2553. doi: 10.1161/ATVBAHA.114.304023. [DOI] [PubMed] [Google Scholar]
- McMullin MF, Wu C, Percy MJ, Tong W. A nonsynonymous LNK polymorphism associated with idiopathic erythrocytosis. Am J Hematol. 2011;86:962–964. doi: 10.1002/ajh.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008;22:3595–3606. doi: 10.1096/fj.08-112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer zu HG, Wu C, Wang C, Cong L, Pawlak M, Lee Y, Elyaman W, Xiao S, Regev A, Kuchroo VK. RBPJ controls development of pathogenic Th17 cells by regulating Il-23 receptor expression. Cell Rep. 2016;16:392–404. doi: 10.1016/j.celrep.2016.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37:1113–1121. doi: 10.1093/eurheartj/ehv718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O'Neill LA, Xavier RJ. Trained immunity: A program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nus M, Martinez-Poveda B, MacGrogan D, Chevre R, Amato G, Sbroggio M, Rodriguez C, Martinez-Gonzalez J, Andres V, Hidalgo A, et al. Endothelial Jag1-RBPJ signalling promotes inflammatory leucocyte recruitment and atherosclerosis. Cardiovasc Res. 2016;23:610. doi: 10.1093/cvr/cvw193. [DOI] [PubMed] [Google Scholar]
- Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, Weller S, Tsiantoulas D, Raffort J, Marcus D, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. 2017;23:601–610. doi: 10.1038/nm.4315. [DOI] [PubMed] [Google Scholar]
- Olivares R, Ducimetiere P, Claude JR. Monocyte count: a risk factor for coronary heart disease? Am J Epidemiol. 1993;137:49–53. doi: 10.1093/oxfordjournals.aje.a116601. [DOI] [PubMed] [Google Scholar]
- Otsuka F, Kramer MC, Woudstra P, Yahagi K, Ladich E, Finn AV, de Winter RJ, Kolodgie FD, Wight TN, Davis HR, et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis. 2015;241:772–782. doi: 10.1016/j.atherosclerosis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van SC, Fullerton MD, Cecchini K, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334–4348. doi: 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard RR, Maganto-Garcia E, Gotsman I, Tabas I, Libby P, Lichtman AH. CD11c(+) dendritic cells maintain antigen processing, presentation capabilities, and CD4(+) T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ Res. 2008;103:965–973. doi: 10.1161/CIRCRESAHA.108.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paneni F, Diaz CC, Libby P, Luscher TF, Camici GG. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol. 2017;69:1952–1967. doi: 10.1016/j.jacc.2017.01.064. [DOI] [PubMed] [Google Scholar]
- Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- Peled M, Fisher EA. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front Immunol. 2014;5:579. doi: 10.3389/fimmu.2014.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9:62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- Petri MH, Laguna-Fernandez A, Arnardottir H, Wheelock CE, Perretti M, Hansson GK, Back M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E-/- mice. Br J Pharmacol. 2017 doi: 10.1111/bph.13707. In press-online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan AT, Goldrath AW, Glass CK. Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity. 2017;46:714–729. doi: 10.1016/j.immuni.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. doi: 10.1172/JCI75005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Chakravarthy MV, Semenkovich CF. Insulin resistance and atherosclerosis. Endocrinol Metab Clin North Am. 2008;37:603–621. doi: 10.1016/j.ecl.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017 doi: 10.1056/NEJMoa1707914. In press-online. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, Thuren T. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, Van RN, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M, Kobori S, Miyazaki A, Horiuchi S. Macrophage proliferation in atherosclerosis. Curr Opin Lipidol. 2000;11:503–509. doi: 10.1097/00041433-200010000-00008. [DOI] [PubMed] [Google Scholar]
- Sakai M, Miyazaki A, Hakamata H, Kodama T, Suzuki H, Kobori S, Shichiri M, Horiuchi S. The scavenger receptor serves as a route for internalization of lysophosphatidylcholine in oxidized low density lipoprotein-induced macrophage proliferation. J Biol Chem. 1996;271:27346–27352. doi: 10.1074/jbc.271.44.27346. [DOI] [PubMed] [Google Scholar]
- Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev. 2014;259:173–191. doi: 10.1111/imr.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin VI, Khan OM, Pehlivanoglu LE, Staffas A, Ibrahim MX, Asplund A, Agren P, Nilton A, Bergstrom G, Bergo MO, et al. Loss of one copy of Zfp148 reduces lesional macrophage proliferation and atherosclerosis in mice by activating p53. Circ Res. 2014;115:781–789. doi: 10.1161/CIRCRESAHA.115.304992. [DOI] [PubMed] [Google Scholar]
- Schierwagen R, Uschner FE, Magdaleno F, Klein S, Trebicka J. Rationale for the use of statins in liver disease. Am J Physiol Gastrointest Liver Physiol. 2017;312:G407–G412. doi: 10.1152/ajpgi.00441.2016. [DOI] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73:470–480. doi: 10.1016/j.cardiores.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- Seijkens T, Hoeksema MA, Beckers L, Smeets E, Meiler S, Levels J, Tjwa M, de Winther MP, Lutgens E. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 2014;28:2202–2213. doi: 10.1096/fj.13-243105. [DOI] [PubMed] [Google Scholar]
- Senokuchi T, Matsumura T, Sakai M, Yano M, Taguchi T, Matsuo T, Sonoda K, Kukidome D, Imoto K, Nishikawa T, et al. Statins suppress oxidized low density lipoprotein-induced macrophage proliferation by inactivation of the small G protein-p38 MAPK pathway. J Biol Chem. 2005;280:6627–6633. doi: 10.1074/jbc.M412531200. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177:1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–354. doi: 10.1084/jem.20150900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoger JL, Gijbels MJ, van dV, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Robbins CS, Nahrendorf M. Development and function of arterial and cardiac macrophages. Trends Immunol. 2016;37:32–40. doi: 10.1016/j.it.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, Van RN, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Lobatto ME, Hassing L, van der Staay S, van Rijs SM, Calcagno C, Braza MS, Baxter S, Fay F, Sanchez-Gaytan BL, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1 doi: 10.1126/sciadv.1400223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L, Fazio S, Linton MF. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. 2015:1449–1460. doi: 10.1194/jlr.M056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, Rudd JH, Narula J, Enriquez JA, Traves PG, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc Biol. 2015;35:1463–1471. doi: 10.1161/ATVBAHA.115.305551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J, O'Neill LA, Menon D. Macrophage immunometabolism: Where are we (going)? Trends Immunol. 2017;38:395–406. doi: 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887–893. doi: 10.1161/ATVBAHA.111.224873. [DOI] [PubMed] [Google Scholar]
- Viny AD, Levine RL. Genetics of myeloproliferative neoplasms. Cancer J. 2014;20:61–65. doi: 10.1097/PPO.0000000000000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola J, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Doring Y, Drechsler M, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. 2016;119:1030–1038. doi: 10.1161/CIRCRESAHA.116.309492. [DOI] [PubMed] [Google Scholar]
- Viola J, Soehnlein O. Atherosclerosis - A matter of unresolved inflammation. Semin Immunol. 2015;27:184–193. doi: 10.1016/j.smim.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. 2002;15:439–446. doi: 10.1111/j.1540-8183.2002.tb01087.x. [DOI] [PubMed] [Google Scholar]
- Wang W, Tang Y, Wang Y, Tascau L, Balcerek J, Tong W, Levine RL, Welch C, Tall AR, Wang N. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ Res. 2016;119:e91–e103. doi: 10.1161/CIRCRESAHA.116.308955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol. 2017;37:e99–e107. doi: 10.1161/ATVBAHA.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, Schaefgen B, Harrison DG, Tremmel JA, Giacomini JC, et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J Clin Invest. 2017;127:2725–2738. doi: 10.1172/JCI92167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, Ory DS, Semenkovich CF. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–298. doi: 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014;114:157–170. doi: 10.1161/CIRCRESAHA.114.300738. [DOI] [PubMed] [Google Scholar]
- Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, Wang N, Randolph GJ, D'Agati VD, Yvan-Charvet L, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 2017;25:1294–1304. doi: 10.1016/j.cmet.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke F, Yuan C, Oka K, Chan L, Hu Y, Barnhart S, Bornfeldt KE, Goldberg IJ, Fisher EA. Effects of high fat feeding and diabetes on regression of atherosclerosis induced by low-density lipoprotein receptor gene therapy in LDL receptor-deficient mice. PLoS ONE. 2015;10:e0128996. doi: 10.1371/journal.pone.0128996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014;9:73–102. doi: 10.1146/annurev-pathol-020712-163936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XQ, Dai Y, Yang Y, Huang C, Meng XM, Wu BM, Li J. Emerging role of microRNAs in regulating macrophage activation and polarization in immune response and inflammation. Immunol. 2016;148:237–248. doi: 10.1111/imm.12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Chaudhury A, Zhang M, Savoldo B, Metelitsa LS, Rodgers J, Yustein JT, Neilson JR, Dotti G. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J Clin Invest. 2016;126:2678–2688. doi: 10.1172/JCI85834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, Linton MF, Fazio S. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and AKT activation. Arterioscler Thromb Vasc Biol. 2010;30:787–795. doi: 10.1161/ATVBAHA.109.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531:651–655. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Sharma S, Edwards J, Feigenbaum L, Zhu J. Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat Immunol. 2015;16:197–206. doi: 10.1038/ni.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]