

Summary
Oncogenic mutations in BRAF are believed to initiate serrated colorectal cancers, however the mechanisms of BRAF-driven colon cancer are unclear. We find that oncogenic BRAF paradoxically suppresses stem cell renewal and instead promotes differentiation. Correspondingly, tumor formation is inefficient in BRAF-driven mouse models of colon cancer. By reducing levels of differentiation via genetic manipulation of either of two distinct differentiation-promoting factors (Smad4 or Cdx2), stem cell activity is restored in BRAFV600E intestines, and the oncogenic capacity of BRAFV600E is amplified. In human patients, we observe that reduced levels of differentiation in normal tissue is associated with increased susceptibility to serrated colon tumors. Together, these findings help resolve the conditions necessary for BRAF-driven colon cancer initiation. Additionally, our results predict that genetic and/or environmental factors which reduce tissue differentiation will increase susceptibility to serrated colon cancer. These findings offer an opportunity to identify susceptible individuals by assessing their tissue-differentiation status.
Graphical abstract
Despite high frequency in serrated colon tumors, BRAFV600E inefficiently drives tumorigenesis in mouse models, and paradoxically, BRAFV600E triggers stem cell loss. BRAF-driven tumorigenesis increases in genetic models that reduce differentiation and restore stem cell activity. These findings provide insights into the mechanisms of BRAFV600E driven colon cancer initiation.

Introduction
Colorectal cancer (CRC) arises from diverse mutations of oncogenes and tumor suppressors that ultimately traverse a limited set of defined routes to tumorigenesis (Fearon and Vogelstein, 1990; Rad et al., 2013). The majority of colorectal tumors follow a conventional pathway that is initiated by activating mutations of the WNT pathway (Powell et al., 1992). However, at least 5-10% of CRCs are believed to initiate via activating mutations in the BRAF oncogene, which amplifies MAPK signaling and drives the Serrated Neoplastic Pathway to CRC. Serrated tumors are characterized by a sawtooth pattern of epithelial infolding, with additional histological features according to three distinct tumor categories: hyperplastic polyps (HPs), sessile serrated adenomas (SSAs), and traditional serrated adenomas (TSAs) (Rex et al., 2012). Serrated CRC, though less frequent than WNT-driven CRC, carries a worse prognosis (De Sousa et al., 2013), and is less understood.
The intestinal mucosa is a continuously self-renewing tissue that is organized into defined crypt-villus units: Crypts of Lieberkühn are epithelial pockets tucked into the intestinal wall and contain proliferating stem cells, which feed differentiated progeny onto villi. Villi are luminal projections composed of post-mitotic, differentiated epithelium (Clevers, 2013). Intestinal stem cells are thought to be a key contributor to tumor initiation. Recent mouse studies using genetic techniques for cell-specific manipulation and cell lineage tracking have identified a population of intestinal stem cells called crypt base columnar cells (Barker et al., 2007) that can serve as the cell of origin of CRC (Barker et al., 2009). While intestinal stem cells are not the only cell capable of serving as the cell of origin (Visvader, 2011), they are by far the most efficient, requiring only a single activating mutation of the WNT/β-catenin pathway - a frequent event associated with the progression of the majority of CRCs, such as APC mutations in adenocarcinomas (Clevers and Nusse, 2012; Sekine et al., 2016). Paradoxically, despite a high prevalence among colorectal tumors, ectopic expression of an oncogenic BRAF mutant transgene promotes either senescence or differentiation of intestinal stem cells (Carragher et al., 2010; Riemer et al., 2015). Thus, while oncogenic activation of BRAF can eventually lead to tumor formation in mouse models, these tumors are slow to arise (Rad et al., 2013; Sakamoto et al., 2017). Thus in stark contrast to activating mutations of the WNT pathway, oncogenic BRAF mutations inefficiently trigger tumorigenesis.
Since stem cells are likely the cell of origin of most colon cancers, factors influencing stem cell numbers should influence tumor incidence. Genetic or environmental factors that influence the homeostatic balance between stem and differentiated cells may therefore be expected to alter susceptibility to CRC. Loss of a differentiation-promoting transcription factor, CDX2, has been associated with BRAF-driven serrated tumor development (Bae et al., 2015; Dalerba et al., 2016; Landau et al., 2014; Sakamoto et al., 2017), although it is still unclear whether CDX2 may function in tumor initiation. SMAD4, a differentiation-promoting transcriptional effector of the TGFβ/BMP signaling pathway, is inactivated in 10-15% of CRC (Fearon, 2011), though its role in initiation of serrated tumors has not been functionally explored. Interestingly, both of these transcription factors have been ascribed roles antagonistic to the WNT-signaling pathway (Guo et al., 2010; Liu et al., 2012; Tian et al., 2009). Whether these transcription factors, or the state of tissue differentiation, influences BRAF-driven tumor initiation is still unclear.
In this study, we show that BRAF activation promotes differentiation of the intestinal epithelium, intestinal stem cell loss, and inefficient oncogenesis. However, in genetic backgrounds with reduced levels of the differentiation-promoting transcription factors Cdx2 or Smad4, homeostasis is restored, stem cell activity returns, and oncogenesis ensues at an accelerated pace. Additionally, gene expression analysis of normal human colon tissue indicates an inverse correlation between the differentiation status of the colon mucosa and a patient's susceptibility to serrated tumors. By highlighting oncogenic mechanisms through which BRAF-driven CRC initiates, these observations may directly impact the development of alternative diagnostic and preventative approaches for Serrated CRC by directly targeting the homeostatic balance between stem and differentiated cells.
Results
BRAFV600E inefficiently drives tumorigenesis and promotes differentiation in a murine model of serrated colon cancer
Studies of melanoma and colon tumorigenesis have shown that the BRAFV600E oncogene, while a prevalent driver of human cancers, is inefficient in promoting tumorigenesis in model systems (Dhomen et al., 2009; Rad et al., 2013). Mutant BRAF activation triggers cellular senescence in some models (Carragher et al., 2010), and intestinal stem cell differentiation in others (Riemer et al., 2015). A third model only observes p21 expression at later stages of tumor progression (Rad et al., 2013). Differences among these models may be attributed to embryonic versus adult-onset of activation, or varying expression levels of mutant BRAF between models. Using a mouse model not previously employed for studies of BRAFV600E-driven intestinal tumorigenesis, we conditionally activated either one or two alleles of the BRAF oncogene from its endogenous locus (Dankort et al., 2007), specifically within the adult intestinal epithelium using the conditional Villin-CreERT2 driver (el Marjou et al., 2004). 5 days after tamoxifen-induced activation, expression of MAPK target genes (Rad et al., 2013) is increased in BRAFV600E/+ mice compared to controls (Figure 1A), confirming activation of downstream BRAF signaling. The BRAFV600E/+ epithelium also took on a more pronounced serrated morphology, a hallmark of serrated colon tumors. To more closely investigate how activation of BRAFV600E immediately impacts the epithelium, we probed tissue markers at 5 days after the onset of tamoxifen treatment (Figure 1B). Interestingly, increased stain intensity of the differentiation marker Alkaline Phosphatase was observed in the BRAFV600E/+ mutants compared to controls, suggesting a more differentiated epithelium. Conversely, levels of the stem cell marker OLFM4 decreased (Figure 1B). These phenotypic changes were further exacerbated in the BRAFV600E/V600E homozygous mutant (from here on indicated as BRAFV600E), and suggest that activation of BRAF shifts the stem-differentiation homeostatic balance towards a differentiated cell state (Figure 1B). Transcriptomic changes in the BRAFV600E/+ epithelium corroborated an increase in differentiation and decrease in stemness in the mutant crypt epithelium. Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) revealed that expression levels of stem cell specific genes (Munoz et al., 2012) are significantly reduced in BRAFV600E/+ mice compared to controls (Figure 1C), consistent with the reduction in OLFM4 staining (Figure 1B). Conversely, expression of differentiation genes (Chong et al., 2009) was increased compared to controls (Figure 1D). We also probed for senescence upon induction of mutant BRAF. Consistent with some previous models of mutant BRAF activation (Riemer et al., 2015), the senescence marker p21 was not expressed upon tamoxifen treatment of BRAFV600E/+ mice. However, p21 was robustly induced upon activation of two BRAF alleles (Figure 1B). These findings suggest that induction of senescence is a function of BRAF activation levels, and may resolve differences between previous studies regarding BRAF-mediated induction of senescence markers in the intestine.
Figure 1. BRAFV600E activation alters intestinal homeostasis, promoting differentiation.

(A) GSEA analysis reveals that MAPK target genes (Rad et al., 2013), were elevated in the BRAFV600E/+ mutants, as expected (K-S test). (B) Assessment of morphological changes (H&E), proliferation (Ki67), differentiation (AP), stem cells (OLFM4), and senescent cells (p21) in BRAFV600E/+ and BRAFV600E/600E mutants. Note increasing levels of differentiation markers and decreasing levels of the stem cell marker with the increased number of activated BRAF alleles. Representative images from 3 biological replicates. Scale bars = 50 μm (C) intestinal stem cell gene expression levels are significantly reduced in BRAFV600E/+ mutants whereas (D) intestinal differentiation gene signatures are significantly increased compared to controls.
Consistent with the promotion of senescence and cellular differentiation, no tumors were observed in 13 BRAFV600E/+ mice between 6 and 16 months after tamoxifen treatment, and dysplastic lesions were only observed in 30% of the mice, with no more than 1 lesion observed per affected animal. Taken together, the Villin-CreERT2; BRAFV600E models recapitulate aspects of BRAF-driven human tumorigenesis (including elevated MAPK signaling and serrated histopathology), but inefficiently induce tumors, potentially owing to an imbalance of the stem-differentiation homeostasis in the gut epithelium.
Loss of CDX2 promotes BRAF-driven tumorigenesis by re-balancing stem/differentiation homeostasis
The imbalance of stem–to-differentiation homeostasis triggered by BRAFV600E may underlie the inefficiency of the BRAF mouse model to drive oncogenesis. Therefore, conditions reducing differentiation may restore homeostatic balance and permit BRAF-driven tumor initiation. CDX2 is a homeodomain transcription factor which promotes differentiation in the intestinal epithelium (Mutoh et al., 2002; Silberg et al., 2002; Suh and Traber, 1996). CDX2 directly binds and activates hundreds of enterocyte target genes, promoting differentiation-related cell functions (Verzi et al., 2010), while loss of CDX2 results in mice that lack complete differentiation of the epithelium (Stringer et al., 2012). Furthermore, reduced expression of CDX2 is associated with BRAF-mutant colon cancers (Bae et al., 2015; Dalerba et al., 2016; Landau et al., 2014; Sakamoto et al., 2017). To explore the mechanisms by which CDX2 may modify BRAFV600E-driven tumorigenesis, we generated an allelic series of mutants lacking either one or both copies of Cdx2 (Verzi et al., 2010), in combination with one or two alleles of activated BRAFV600E. As noted before, activation of BRAFV600E alleles diminished expression of stem cell markers (Figure 2A). However, simultaneous loss of Cdx2 alleles in the BRAFV600E models restored OLFM4 expression, indicating an increased population of stem cells in the compound mutant relative to the BRAF mutant (Figure 2A). We next assayed for stem cell activity using the organoid formation assay (Sato et al., 2009). Consistent with reduced levels of OLFM4 staining, BRAFV600E/+ crypts were incapable of giving rise to organoids (Riemer et al., 2015) (Figure 2B). However, organoids could be derived from the Cdx2f/+; BRAFV600E/+ animals, indicating that reduction of CDX2 restores stem cell activity that is lost upon activation of BRAFV600E. We saw a similar pattern of stem cell reporter activity in vivo, using the Lgr5-GFP-CreERT2 reporter line (Barker et al., 2007) to mark Lgr5+ stem cells, and activating mutations with the Villin-CreERT2 driver. GFP+ cells were observed at the crypt base in control and CDX2; BRAFV600E mutant crypts, but were completely depleted in BRAFV600E mutant crypts (Figure 2C). Similarly, using the stem cell specific Lgr5-GFP-CreERT2 driver (Barker et al., 2007), we assayed for stem cell renewal in vivo by monitoring Cre-mediated recombination of loxP elements in the engineered BRAFV600E locus (Dankort et al., 2007). BRAFV600E recombination was not preserved in Lgr5-Cre; BRAFV600E/+mice (Figure 2D), indicating that mutant stem cells could not renew and were depleted over time, presumably replaced by neighboring cells that had not undergone Cre-mediated recombination. By contrast, sustained recombination of Cdx2f/+; BRAFV600E/+ alleles in Lgr5-Cre expressing stem cells was compatible with stem cell renewal, as recombined cells and their progeny were still present as far as 10 months after treatment, indicating that CDX2 loss restores the ability of Lgr5+ stem cells to renew in the presence of BRAFV600E (Figure 2D). It is possible that the loss of stem cells upon BRAFV600E activation is due to a promotion of apoptosis. However, the immunohistochemical analysis of the apoptosis marker, Cleaved-Caspase 3 (CC3), revealed no changes (Figure S1A). We also examined cellular senescence as a possible contributor of stem cell loss, using the marker p21. Interestingly, CDX2 loss reversed the upregulation of p21 induced by BRAFV600E. This was observed at the histological level, the transcript level, and in organoid cultures (Figure S1B-S1D). Thus, while BRAFV600E/+ and BRAFV600E appears to be incompatible with stem cell self-renewal and retention, the reduction of CDX2 is able to prevent the loss of stem cells in a BRAFV600E background.
Figure 2. Ablation of CDX2 reverses BRAFV600E-driven changes in stem/differentiation homeostasis.

(A) Immunohistochemistry of OLFM4 in uninjected control, BRAFV600E/+, Cdx2f/f; BRAFV600E/+, BRAFV600E, and Cdx2f/f; BRAFV600E mice, 5 days post-tamoxifen treatment. (B) Primary organoid cultures isolated from control, BRAFV600E/+, and Cdx2f/+; BRAFV600E/+ mice. BRAFV600E/+ mutant cells do not give rise to viable organoids, but reduction of CDX2 levels permits their growth. Representative of 3 biological replicates. Scale bars=50μm. (C) Lgr5-GFP expression. Scale bars=50μm. (D) Recombination at the BRAFV600E locus, induced by tamoxifen treatment of the stem cell specific, conditional Cre (Lgr5-CreERT2-IRES-eGFP), is not sustained unless Cdx2 is simultaneously inactivated in the stem cells. Samples from crypt epithelium were harvested at 2 days or 10 months after tamoxifen treatment. (E) AP staining reveals increased expression in BRAFV600E/+ and BRAFV600E, which is reduced when Cdx2 is lost. (F) GSEA shows intestinal stem cell genes are elevated while differentiation genes are reduced upon loss of CDX2, reversing gene expression changes induced by BRAFV600E activation. (G) Examples of the types of differentiation-associated genes that are elevated upon BRAF activation, and suppressed upon CDX2-loss.
If stem cell populations were restored upon CDX2 loss, the differentiation-promoting effects of BRAFV600E may have also reverted in the Cdx2f/f; BRAFV600E compound mutant mice. To assess the differentiation status of the Cdx2f/f; BRAFV600E mice, alkaline phosphatase activity was assayed. Consistent with a restoration of the stem-differentiation balance in the compound mutant epithelium, reduced levels of alkaline phosphatase were observed in the compound Cdx2f/f; BRAFV600E mutants relative to single mutant animals (Figure 2E). These observations were mirrored in transcriptome analyses, which revealed a significant elevation of stem cell and reduction of differentiation gene expression levels in the Cdx2f/f; BRAFV600E/+ epithelium compared to the BRAFV600E/+ single mutant (Figure 2F). A wide array of transcripts associated with differentiation were elevated in the BRAFV600E/+ epithelium and restored to normal levels upon inactivation of Cdx2, including transcripts associated with metabolism, cell adhesion, and digestive/absorptive functions (Figure 2G). Paneth cells, which function in part by supporting the stem cell niche, are diminished in the BRAFV600E/+ epithelium. However, lysozyme staining reveals that CDX2f/f;BRAFV600E mice show similar loss of lysozyme staining at the crypt base, suggesting that stem cell regulation is not dependent on Paneth cells in this model, as has been established in other models of Paneth cell loss (Durand et al., 2012; Kabiri et al., 2014; Kim et al., 2012) (Figure S2).
Since reduced levels of differentiation-promoting CDX2 allowed retention of stem cells in the BRAFV600E condition, reduced CDX2 levels might also lead to increased tumorigenesis in the Cdx2f/+; BRAFV600E/+ model. When compared to controls, the histology of the intestinal epithelium is dramatically altered in age-matched BRAFV600E/+ and Cdx2f/+; BRAFV600E/+ compound mutants (Figure 3A and Figure S3). Both the BRAFV600E/+ and Cdx2f/+; BRAFV600E/+ mice exhibited jagged, undulating epithelium, a hallmark of serrated tumors (Leggett and Whitehall, 2010) that is not observed in control or Cdx2f/f; Vil-CreERT2 mutant mice (Verzi et al., 2010). Dysplastic lesions in Cdx2f/+; BrafV600E/+ mice were broad with a low profile (Figure 3B). While dysplastic characteristics of SSA were found in both mutant strains of mice, serrated lesions were found more frequently in Cdx2f/+; BrafV600E/+ mice than in the BrafV600E/+ mice (Figure 3C), and macroscopic tumors were found exclusively in Cdx2f/+; BrafV600E/+ mutants. These flat tumors exhibited a cloud-like surface without discernible borders, similar to endoscopic features of human precursor SSAs (Hazewinkel et al., 2013). Histopathology of Cdx2f/+; BrafV600E/+ carcinomas revealed significant muscle infiltration, epithelial serrations, eosinophilic cytoplasm, vesicular nuclei and in some cases, excessive mucin production (Figure 3A, S3). Serrated polyps detected in Cdx2f/+; BrafV600E/+ compound mutant mice mainly resembled human hyperplastic polyps and sessile serrated adenomas (Figure 3A, S3). The “hyperplastic polyps” were either of the goblet cell type or the microvesicular type and had a saw-tooth configuration with pleomorphic nuclei, while “sessile serrated polyps” showed significant dilated crypts with mature mucinous cells (Figure S3). These findings indicate that reduced levels of Cdx2 restore the distrupted stem-differentiation balance caused by activation of BRAFV600E and create a permissive environment for the oncogenic BRAFV600E mutation. Importantly, aside from increased susceptibility to BRAF-driven tumors, Cdx2f/+ mutants are otherwise healthy and indistinguishable from littermate controls. This raises the possibility that reduced levels of differentiation in otherwise healthy individuals could underlie susceptibility to serrated neoplasias.
Figure 3. Loss of CDX2 accelerates BRAF-driven tumorigenesis.
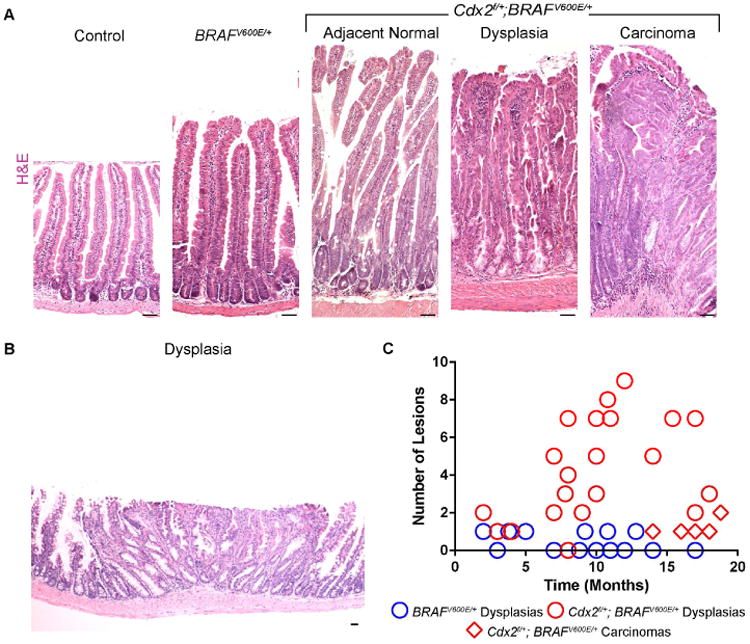
(A) H&E stains of age-matched control, BRAFV600E/+, and Cdx2f/+; BRAFV600E/+ mice reveal characteristics of serrated cancer morphologies. Dysplasias and carcinomas had characteristics of human hyperplastic polyps (HPPs), SSAs, and serrated carcinomas (Also see Figure S3) (B) More serrated lesions were found in Cdx2f/+; BRAFV600E/+ compound mice than age-matched BRAFV600E/+ counterparts. Counts were based upon complete swiss roll sections. Scale bars=50μm.
The differentiation-promoting factor SMAD4 also antagonizes BRAF-driven tumorigenesis and alters the stem-differentiation homeostatic balance
To further explore whether an imbalance in stem-to-differentiation homeostasis can increase susceptibility to serrated tumors, we tested whether another differentiation-promoting factor could modify tumor initiation of the BRAFV600E/+ model. The critical role of SMAD4 in the TGFβ/BMP signaling pathway prompted us to consider it a prime candidate, as TGFβ/BMP signaling is a known promoter of intestinal differentiation. We bred Smad4 conditional alleles (Yang et al., 2002) into the BRAFV600E/+ mutant and found suppression of alkaline phosphatase activity (Figure 4A), and a concomitant increase in the stem cell markers OLFM4 and Lgr5-GFP (Figures 4B-C). Stem cell activity was also restored in the context of the organoid forming assay (Figure 5D) when SMAD4 loss was combined with BRAFV600E/+. Again, transcriptomic analysis of BRAFV600E/+ versus Smad4f/f; BRAFV600E/+ compound mutants revealed a shift in gene expression, with an increase in expression of stem cell signature genes and a decrease of differentiated cell gene expression (Figure 4E). Recapitulating the histological analysis, the expression of differentiation genes, such as Alpi and Krt20, were significantly higher upon BRAFV600E activation, but were restored to near normal levels upon simultaneous loss of SMAD4 (Figure S4, ANOVA, p <0.05). Taken together, reduction of SMAD4 restores the stem-to-differentiation balance of the intestinal epithelium that is shifted towards differentiation upon activation of BRAFV600E. Thus, two independent genetic models show that reduced levels of differentiation-promoting factors can restore the homeostatic balance between stem and differentiated cells that is disrupted upon BRAFV600E activation.
Figure 4. Loss of SMAD4 restores the stem-differentiation balance that is altered upon BRAFV600E/+ activation.

(A) AP staining reveals that Smad4 loss reverses the increase in differentiation induced by BRAFV600E/+. (B) OLFM4 staining suggests that loss of SMAD4 in BRAFV600E/+ tissue reverses stem cell loss. Representative of 3 biological replicates. Scale bars=50μm. (C) Lgr5-GFP expression is lost upon BRAFV600E expression, but restored upon SMAD4-loss. (D) Organoid cultures isolated from control, BRAFV600E/+, and Smad4f/f; BRAFV600E/+ mice reveal a rescue in viability in Smad4f/f; BRAFV600E/+ organoids. (E) GSEA shows intestinal differentiation genes are reduced, while stem cell gene expression is elevated upon loss of SMAD4, reversing gene expression changes induced by BRAFV600E/+ activation.
Figure 5. Tumorigenesis in BRAFV600E/+ mice is accelerated upon the loss Smad4.

(A) H&E stain shows overall morphology of adjacent normal and tumor regions of the intestine of control, BRAFV600E/+, and Smad4f/f; BRAFV600E/+ mice. Similar to Cdx2f/+; BRAFV600E/+ mice, dysplasias and carcinomas featured serrated morphologies (See also Figure S5). Counts of macroscopic (B) and microscopic (C) lesions observed in brafv6ooe/+mice are lower than Smad4f/f; BRAFV600E/+ mutants. Scale bars=50um.
Akin to our findings from the Cdx2 study described above, the Smad4f/f; BRAFV600E/+ compound mutant exhibited increased susceptibility to serrated tumors - including serrated dysplastic regions and invasive carcinomas (Figure 5A-B, Figure S5). Large visible tumors were seen within the intestinal tract as early as 2-3 months post-tamoxifen injection (Figure 5C). Furthermore, as early as 1 month post-tamoxifen treatment, dysplastic lesions were observed in the compound mutants, while carcinomas were found as early as 3 months post-treatment. Thus, loss of a differentiation promoting transcription factor SMAD4, like CDX2, results in an alteration of the stem/differentiation balance in the intestinal epithelium and corresponds to increased BRAF-driven tumor susceptibility.
Elevated WNT signaling is common to both genetic models and can rescue stem cell loss in BRAF mutant mice
To elucidate a mechanism through which reduced levels of differentiation factors can predispose to serrated tumorigenesis, we explored common transcriptomic signatures of epithelia expressing activated BRAFV600E/+ and inactivating mutations of either Cdx2 or Smad4. In both cases, WNT signature genes (Van der Flier et al., 2007) were significantly elevated in these models' transcriptomes (Figure 6A). A panel of commonly studied WNT-target genes exemplify these findings (Figure 6B). Notably, just as SMAD4 or CDX2 loss can restore organoid-forming ability of the BRAFV600E epithelium, addition of WNT ligand (WNT3A), a WNT-pathway agonist (CHIR-98014), or genetic activation of the WNT pathway (Harada et al., 1999) rescues the organoid forming potential of the BRAFV600E/+ mutants (Figure 6C). These findings indicate that elevation of the WNT signaling pathway, which functions to promote stem/proliferation in the intestinal epithelium, restores stem cell activity that is normally antagonized by the BRAFV600E oncogene. Consistent with these observations, immunohistochemical staining for the WNT target gene products SOX9 and CD44 were elevated upon loss of either Cdx2 or Smad4 (Figure 6D). Finally, to test whether the loss of Cdx2 or Smad4 rescues BRAFV600E-organoid forming ability via a WNT-dependent mechanism, we inhibited the WNT pathway using the chemical inhibitors IWP-2 or XAV-939, which block WNT secretion or β-catenin stabilization, respectively. These WNT-pathway inhibitors each reverse the effects of SMAD4 and CDX2 loss, indicating that the rescue of stem cell activity in the organoid forming assay is dependent upon WNT signaling (Figure 6E).
Figure 6. Elevated WNT signaling may restore oncogenic potential in Cdx2f/f; BRAFV600E/+and Smad4f/f; BRAFV600E/+ double mutant mice.

(A) Transcriptome analysis of WNT signature genes (Van der Flier et al., 2007) indicates increased WNT target gene expression in the double mutant mice when compared to BRAFV600E/+ mice (K-S test). (B) Common intestinal WNT target genes are highlighted. (C) Organoid cultures derived from BRAFV600E/+ crypts survive when treated with either WNT3A or the WNT-pathway agonist CHIR-98014. (D) Immunohistochemistry of WNT targets CD44 and SOX9 show elevated levels in Cdx2f/f; BRAFV600E/+an6 Smad4f/f;BRAFV600E/+ mutants compared to BRAFV600E/+ mutants. (E) Organoid cultures derived from compound mutant mice show sensitivity to WNT inhibitors. Table describes viability in days of mature organoids after treatment with XAV-939 or IWP-2. Representative of 2 biological replicates and 3 technical replicates each. Bars=50 μ.
This work suggests a diminished state of epithelial differentiation is required for oncogenic mutations of BRAF to be sustained and promote serrated tumorigenesis. We observe that 3 different regulators of the stem-to-differentiated transition of the intestinal epithelium can critically influence the potential of BRAF-driven oncogenesis to sustain stemness in the organoid-forming assay.
Expression of differentiation genes in the human epithelium correlates with susceptibility to serrated tumorigenesis
Given that an epithelium with reduced capacity for differentiation (Smad4f/f or Cdx2f/+) predisposed the murine epithelium to BRAFV600E-oncogenesis, we investigated whether differentiation status corresponds to serrated tumor susceptibility. Normal tissue from serrated tumor patients is not abundant in the literature, as most studies focus on tumor tissues. Fortunately, a recent study performed transcriptional profiling from mucosal biopsies of non-tumor tissues from the right colons of serrated tumor patients and control individuals (Delker et al., 2014; Kanth et al., 2016). We queried these data to determine whether the differentiation genes that were reduced in pre-neoplastic tissue of our murine models (Figures 2F & 4F) are also reduced in the normal epithelium of patients exhibiting one (sporadic) or multiple (syndromic) serrated tumors. Interestingly, serrated cancer patients exhibited significantly reduced expression of intestinal differentiation genes in their normal tissues when compared to control patients (Figure 7A). We next applied a k-clustering approach to determine whether patients could be accurately classified into tumor and normal subject groups based upon changes in their stem and differentiation gene expression levels. K-means clustering on 1000 random gene panels of equal size to our gene lists provides a baseline distribution of clustering accuracies by chance alone (measured by Rand Index (RI), median RI = 0.64) (Rand, 1971). Gene expression levels of stem and differentiation genes were indeed a robust classifier for stratifying patients into tumor and non-tumor groups when compared against the clustering accuracy of random gene panels of equal size (RI = 0.8, scoring in the top 2.5%, Figure 7B, Table S2). Notably, clustering was more accurate using the combined gene list than either gene set alone (Figure S6). Thus, it appears that the normal epithelium of serrated tumor patients has a shift in the stem-differentiation homeostasis, as was observed in our mouse models. Analysis of individual patient samples revealed that normal patients had relatively higher expression levels of differentiation genes in the right colon tissue when compared with normal tissue from tumor patients (with patients bearing multiple serrated lesions exhibiting the lowest levels of differentiation gene expression) – further suggesting a correlation between tissue differentiation and tumor susceptibility in humans (Figure 7C-D, Kruskal-Wallis Test, p < 0.05). To corroborate these findings, we scored a separate set of patients bearing hyperplastic polyps and sessile serrated adenomas for the protein expression level of KRT20 in adjacent, non-tumor tissue (Figure 7D). Based on blind assessment of KRT20 stain intensity, tumor-bearing patients exhibited a trend towards lower levels of this differentiation marker (Figure 4E, Mann-WhitneyU, p = 0.13362). Reduced levels of transcripts for KRT20 were also observed in the normal tissues of patients bearing serrated tumors compared to controls (Figure 7F, ANOVA, p<0.05). Taken together, this study indicates that the human population exhibits a range of differentiation, and an inverse correlation exists between the degree of differentiation and the propensity to develop serrated tumors, echoing the findings in mice described above.
Figure 7. Differentiation gene expression levels in normal tissues corresponds to serrated tumor susceptibility in humans.

(A) Non-tumor, right colon tissue from serrated tumor patients (n=15) exhibit lower levels of differentiation-gene expression when compared to control patient tissues (n=10, K-S test). (B) K-means clustering of patients into tumor and non-tumor classes is improved when combining intestinal stem (Munoz et al., 2012) and differentiation cell (Chong et al., 2009) gene sets that are differentially regulated in mouse models (performing in the top 2.5% of 1000 random gene lists of equivalent size), or by either gene set individually (Figure S6). (C) Patients exhibit a range of differentiation gene expression, with serrated cancer patients exhibiting lower levels of intestinal differentiation genes (K-W Test, p<0.05). (D) Human tissue samples assessed for differentiation marker KRT20 stain intensity. (E) Adjacent normal tissue of serrated tumor patients exhibits a trend towards reducedstain intensity relative to non-tumor patients (Mann-Whitney p=0.13362). (F) KRT20transcript levels in normal tissue is lower in serrated tumor patients when compared tonon-tumor patient samples (ANOVA, p<0.05). Scale bars=50 μm.
Discussion
Despite the ample genomic characterization of BRAF-driven tumors in human CRCs, few animal studies describe the cellular mechanisms driving serrated tumor initiation. An initial BRAFV600E tumorigenesis model recapitulated some characteristics of human serrated tumors, but mice died prematurely due to several tumors that developed outside the intestine (Carragher et al., 2010). In that study, conditional activation of BRAFV600E in the small intestine resulted in induction of senescence, while tumorigenesis required repression of senescence (Carragher et al., 2010; Kriegl et al., 2011). In contrast, other mouse models of BRAF activation have not found evidence of senescence or apoptosis in the intestine (Rad et al., 2013; Riemer et al., 2015). To better understand the magnitude of MAPK signal influence in CRC initiation, we employed both BRAFV600E heterozygous and homozygous mutants, targeted at the endogenous locus, and initiated in the adult epithelium. Our allelic series revealed that a single allele of mutant BRAF expression is tolerated in the epithelium, but higher levels of MAPK signaling are not tolerated, and trigger activation of p21 expression. Together, these results are in agreement with the observed mutual exclusivity of BRAF and KRAS in CRC (Cancer Genome Atlas, 2012; Unni et al., 2015). We propose a model in which only a narrow window of MAPK pathway activity is oncogenic: excessive and early MAPK pathway activation is not tolerated by individual stem cells under normal conditions (such as in BrafV600E; Lgr5-CreERT2), whereas baseline MAPK levels are not sufficient to promote oncogenesis (such as in control mice). Our work shows that altering the pre-existing differentiation status of a tissue is one factor that modulates tissue tolerance to BRAF activity, essentially expanding the “sweet spot” at which BRAF-driven oncogenesis can occur. Thus, individuals with lower levels of tissue differentiation (whether due to genetic or environmental factors) would have stem cells that are more tolerant to MAPK-activity spikes, and are more susceptible to serrated tumorigenesis.
Given that activating mutations of the MAPK pathway drive serrated tumorigenesis in up to 30% of CRC, it is counterintuitive that this pathway would promote differentiation under normal conditions. In our study, stem cells were resistant to retaining oncogenic BRAFV600E, unless differentiation-promoting factors were diminished or deleted. These findings suggest the interesting possibility that tissue-resident stem cells evolved a defense mechanism to differentiate in response to oncogenic levels of signaling. This strategy would be particularly appealing in renewing tissues such as the colon, in which all non-stem cell lineages are immediately fated to death as they transit toward the lumen, where they will ultimately be expelled from the epithelium. The mechanism through which BRAFV600E triggers stem cell differentiation is unclear, and whether differentiation is a cause, consequence, or unrelated to p21 activation is also uncertain. Future studies unraveling these mechanisms could provide new opportunities for chemosuppression.
Finally, we show that a normal tissue biopsy can be screened for gene signatures of stem-to-differentiation homeostasis, and this signature can help classify serrated tumor patients versus unaffected individuals. We therefore suggest the possibility that an otherwise healthy mucosa contains a predictive signature for serrated tumor susceptibility. The observation that differentiation status of the human transcriptome is correlated with serrated tumor susceptibility offers exciting opportunities to determine whether gene expression signatures could serve as diagnostics to direct screening practices, and whether chemoprevention or dietary strategies could be calibrated to promote a more differentiated epithelium in patients at increased susceptibility to SSA.
Experimental Procedures
Animals and Tissue Processing
Animal experiments were conducted in accordance with Rutgers University IACUC. Mice strains are listed in Table S1. Mice 6-weeks of age were treated with intraperitoneal injection of tamoxifen (1mg/20g), for four consecutive days. Both males and females were used in equal consideration and littermates were used when available for matched experimental sets. Day 1 is considered the onset of treatment, and most animals were investigated at day 5, or at the indicated time point. Mouse intestines were collected and fixed in 4% paraformaldehyde solution overnight at 4°C, and paraffin sections (5μm) were generated.
For cryo-embedding, the proximal one third of duodenum was fixed in 4% (vol/vol) paraformaldehyde overnight at 4°C, washed in cold P BS, and equilibrated in 30% (wt/vol) sucrose before freezing in OCT compound (catalog no. 4583; Tissue-Tek).
Immunohistochemistry
Slides were treated with 10mM sodium citrate and a pressure cooker was used for antigen retrieval. Slides were quenched in 0.5% peroxidase for 20 minutes, washed, permeabilized in 5% Triton X-100 (in 1× PBS) for 5 minutes, blocked in 5% Fetal Bovine Serum for 1 hour, and incubated overnight at 4°C with primary antibodies (Table S3). Slides were developed using 0.05% DAB and 0.015% hydrogen peroxide in 0.1 M Tris, after employing secondary antibody and the ABC Vectastain HRP Kit (Vector Labs), and counterstained with Hematoxylin.
Immunofluorescence
10μm sections of cryo-embedded tissues were examined by direct fluorescence for EGFP expression in Lgr5+ stem cells, and DAPI for nuclei. Zeiss Axiovert 200M was used for fluorescence microscopy using Retiga-SRV CCD (Q-Imaging). Adjustments to constrast and sharpness, when made, were applied to all images using ImageJ (NIH).
Histochemistry and scoring
Alkaline phosphatase activity was carried out using the AP Staining Kit II (Stemgent). To detect mucin, rehydrated slides were immersed in 1% alcian blue solution in 3% aqueous acetic acid (pH 2.5), followed by neutral red counterstain.
Microscopic tumors were quantified based on proliferation markers and morphology, exploring the same tissue space in each mouse, from swiss roll sections containing the entire gut length. Ki67 staining facilitated identification of these lesions. Dysplasias included regions exhibiting proliferation ¼ of the way above the crypts and higher with major morphological changes including atypical branching or hypervesicular mucinous cells. Carcinomas were classified based on an invasive phenotype with a definite morphologic change in the cells as well as a disturbance of the muscle layer in which proliferation spread past the normal epithelial boundary.
Organoid forming assays
Crypt-derived organoids were isolated from duodenum and cultured in Cultrex reduced growth factor matrix R1 (Trevigen) according to established methods (Sato et al., 2009). An average of 100 crypts per biological replicate were seeded in 12.5μl of matrix with Basic Crypt Media (BCM): Advanced DMEM/F12 (Gibco), 1× Penicillin/Streptomycin, 2mM Glutamax, 10mM HEPES (Life Technologies) supplemented with 50 ng ml-1EGF (R&D), 100 ng ml-1 Noggin (Peprotech), A/-acetyl-l-cysteine 1 μM (Sigma-Aldrich), R-Spondin CM 2.5% (v/v), 1× N2, 1× B27 (both from Life Technologies). For rescue experiments of BRAFV600E/+ organoids, growth media was supplemented with 3μM CHIR99021 (Stemgent) or Wnt3a-conditioned media (Willert et al., 2003) at 50% (v/v). For WNT inhibition experiments, duodenal crypts were embedded in matrix and passaged after 7 days with 1× TrypLE (Gibco). Mature organoids were treated with IWP-2, XAV-939 (Tocris), or DMSO vehicle dissolved in complete growth media and changed every two days. Organoids were imaged using an AxioVert 200M inverted microscope (Zeiss) with a Retiga-SRV CCD (Qlmaging).
Cell culture
Wnt3A-conditioned medium was produced using Wnt3A-expressing L cells (ATCC) cultured in Advanced DM EM F/12 supplemented with 10% fetal bovine serum, 2m M GlutaMAX-l and 10mM HEPES (Life Technologies) as previously described (Farin et al., 2016; Willert et al., 2003).
BRAFV6OOE Recombination Assay
PCR was conducted on isolated intestinal crypt DNA using the primers previously described (Dankort et al., 2007) to resolve wild-type, mutant, and recombined alleles.
Human Colon Tissue KRT20 Analysis
The human study was approved by University Medical Center of Princeton IRB. Immunohistochemical studies on the de-identified normal (n=10), HPP (n=10), and SSA (n=10) used human tissues fixed in 10% neutral formalin and cut to 4 μm at a College of American Pathologists-accredited histology lab. Slides were stained for KRT20 as described above. Non-tumor tissue adjacent to the lesions (HPP or SSA) were imaged and assigned a randomly generated number (1-30). 6 coauthors independently scored KRT20 stain intensity (“light” = 1, “medium” = 2, “dark” = 3) in a blinded fashion. The sum of the 6 scores of each sample was used as the sample's composite score for statistical analyses. Mann-Whitney U (Wilcoxon rank-sum) was used to determine the difference in KRT20 stain intensity.
RNA-Seq Data Analysis
Mouse intestinal crypts were isolated and processed for RNA using Trizol and sequenced using Illumina'sTruSeq RNA Library Prep kit v2 RNA reads were aligned using TopHat2 (v2.1.0). All files were run together using CuffNorm (v2.2.1) using default settings (geometric normalization) (Langmead and Salzberg, 2012; Trapnell et al., 2010). For GSEA analysis, genes were pre-ranked based on Signal-to-Noise values, as described (Subramanian et al., 2005).
Human data comprising RNA-Seq from 25 right colon samples (10 control patients, 10 sporadic SSA tumor patients, and 5 syndromic tumor patients) was derived from non-tumor tissue (Delker et al., 2014; Kanth et al., 2016) (GSE76987). FPKM table was normalized by fold change to the average of the 10 control patient FPKM values for each gene. Specific genes associated with intestinal stem cell and differentiation gene signatures were selected and plotted in R (Chong et al., 2009; Munoz et al., 2012). Kruskal-Wallis test was performed to determine significant differences between the control, sporadic, and syndromic patients overall.
Differentiation signature genes (Chong et al., 2009) and stem cell signature genes (Munoz et al., 2012) that were represented on the the human RNA-Seq FPKM table (GSE76987) (Delker et al., 2014; Kanth et al., 2016), were combined (Table S2), and k-means clustering was performed in R. K-means clustering was performed (clustering of 2) in R, and clustering accuracy was determined using Rand Index (RI) (Rand, 1971), RI=(TP+TN)/(TP+TN+FP+FN). Random gene sets, comprised of an equivalent number of unique genes to our defined gene lists, were selected to establish the ability of random classifiers to stratify patients and applied 1000 times. Distribution frequencies were plotted using 0.1 bin sizes.
Statistical Analyses Performed
Students T-Test was used as part of our mouse RNA-Seq analyses, Mann-Whitney test was used on blind assessment of human tissue samples, Kruskal Wallis Test was used as part of our human RNA-Seq data analysis, and K-S Test was used on GSEA.
Data Availability
RNA-Seq files: GSE106330 and GEO102171
Human RNA-Seq Samples: GSE76987
Supplementary Material
Highlights.
BRAFV600E poorly initiates colon cancer, instead triggering tissue differentiation
BRAFV600E causes stem cell loss; reduced differentiation restores stem cells
Mutations that reduce differentiation permit BRAF-driven serrated tumorigenesis
Patients with reduced differentiation exhibit higher serrated tumor susceptibility
Acknowledgments
This work was supported by NCI R01CA190558 and a grant from the Human Genetics Institute of New Jersey (MPV). KT, OPC, and AOP were supported by New Jersey Commission on Cancer Research grants. (KT - DHFS16PPC036, OPC -DHSFS17PPC020, AOP – DFHS13PPC034). VS was supported by the Human Genetics Institute of New Jersey, Summer Undergraduate Research Fellowship. OAK was supported by the Aresty Research Center for Undergraduates. MPV is a member of the Intestinal Stem Cell Consortium, supported by NIDDK and NIAID, U01DK103141, and a member of the Cancer Institute of New Jersey, P30CA072720. The work was supported by an Initiative for Multidisciplinary Research Teams award from Rutgers University, Newark, NJ (to MPV and LZ). Authors would like to thank members of the Verzi lab, Dr. David Axelrod, Dr. Ronald Hart, and Dr. Ramesh Shivdasani for their constructive feedback and discussions.
Footnotes
The authors declare no conflicts of interest
Author Contributions: Experimental planning and execution were done by MPV, OPC, and KT. Writing was done by MPV, OPC, and KT. All authors contributed to benchwork. Animal husbandry was performed by KT, OPC, AOP, VRS, BNW, and OAK. Computational analysis was performed by MPV, OPC, KT, AZ, JX, JKT, and RLF. Human tissue samples were provided by LZ, and blind assessment of the KRT20 staining by MPV, OPC, KT, VRS, BNW, and OAK. Human staining data was analyzed by LZ and KT.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bae JM, Lee TH, Cho NY, Kim TY, Kang GH. Loss of CDX2 expression is associated with poor prognosis in colorectal cancer patients. World J Gastroenterol. 2015;21:1457–1467. doi: 10.3748/wjg.v21.i5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher LA, Snell KR, Giblett SM, Aldridge VS, Patel B, Cook SJ, Winton DJ, Marais R, Pritchard CA. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med. 2010;2:458–471. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JL, Wenzel PL, Saenz-Robles MT, Nair V, Ferrey A, Hagan JP, Gomez YM, Sharma N, Chen HZ, Ouseph M, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462:930–934. doi: 10.1038/nature08677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–284. doi: 10.1016/j.cell.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Sahoo D, Paik S, Guo X, Yothers G, Song N, Wilcox-Fogel N, Forgo E, Rajendran PS, Miranda SP, et al. CDX2 as a Prognostic Biomarker in Stage II and Stage III Colon Cancer. The New England journal of medicine. 2016;374:211–222. doi: 10.1056/NEJMoa1506597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes & development. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sousa EMF, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nature medicine. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- Delker DA, McGettigan BM, Kanth P, Pop S, Neklason DW, Bronner MP, Burt RW, Hagedorn CH. RNA sequencing of sessile serrated colon polyps identifies differentially expressed genes and immunohistochemical markers. PloS one. 2014;9:e88367. doi: 10.1371/journal.pone.0088367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Durand A, Donahue B, Peignon G, Letourneur F, Cagnard N, Slomianny C, Perret C, Shroyer NF, Romagnolo B. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1) Proceedings of the National Academy of Sciences of the United States of America. 2012;109:8965–8970. doi: 10.1073/pnas.1201652109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV, de Punder K, Angers S, Peters PJ, Maurice MM, et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature. 2016;530:340–343. doi: 10.1038/nature16937. [DOI] [PubMed] [Google Scholar]
- Fearon ER. Molecular genetics of colorectal cancer. Annual review of pathology. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Guo RJ, Funakoshi S, Lee HH, Kong J, Lynch JP. The intestine-specific transcription factor Cdx2 inhibits beta-catenin/TCF transcriptional activity by disrupting the beta-catenin-TCF protein complex. Carcinogenesis. 2010;31:159–166. doi: 10.1093/carcin/bgp213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazewinkel Y, Lopez-Ceron M, East JE, Rastogi A, Pellise M, Nakajima T, van Eeden S, Tytgat KM, Fockens P, Dekker E. Endoscopic features of sessile serrated adenomas: validation by international experts using high-resolution white-light endoscopy and narrow-band imaging. Gastrointest Endosc. 2013;77:916–924. doi: 10.1016/j.gie.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Kabiri Z, Greicius G, Madan B, Biechele S, Zhong Z, Zaribafzadeh H, Edison, Aliyev J, Wu Y, Bunte R, et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development. 2014;141:2206–2215. doi: 10.1242/dev.104976. [DOI] [PubMed] [Google Scholar]
- Kanth P, Bronner MP, Boucher KM, Burt RW, Neklason DW, Hagedorn CH, Delker DA. Gene Signature in Sessile Serrated Polyps Identifies Colon Cancer Subtype. Cancer Prev Res (Phila) 2016;9:456–465. doi: 10.1158/1940-6207.CAPR-15-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Escudero S, Shivdasani RA. Intact function of Lgr5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3932–3937. doi: 10.1073/pnas.1113890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegl L, Neumann J, Vieth M, Greten FR, Reu S, Jung A, Kirchner T. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:1015–1022. doi: 10.1038/modpathol.2011.43. [DOI] [PubMed] [Google Scholar]
- Landau MS, Kuan SF, Chiosea S, Pai RK. BRAF-mutated microsatellite stable colorectal carcinoma: an aggressive adenocarcinoma with reduced CDX2 and increased cytokeratin 7 immunohistochemical expression. Hum Pathol. 2014;45:1704–1712. doi: 10.1016/j.humpath.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang X, Zhan Q, Brock MV, Herman JG, Guo M. CDX2 serves as a Wnt signaling inhibitor and is frequently methylated in lung cancer. Cancer Biol Ther. 2012;13:1152–1157. doi: 10.4161/cbt.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh H, Hakamata Y, Sato K, Eda A, Yanaka I, Honda S, Osawa H, Kaneko Y, Sugano K. Conversion of gastric mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice. Biochemical and biophysical research communications. 2002;294:470–479. doi: 10.1016/S0006-291X(02)00480-1. [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, Constantino-Casas F, Eser S, Hieber M, Seidler B, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell. 2013;24:15–29. doi: 10.1016/j.ccr.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand WM. Objective Criteria for the Evaluation of Clustering Methods. Journal of the American Statistical Association. 1971;66:846–850. [Google Scholar]
- Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012;107:1315–1329. doi: 10.1038/ajg.2012.161. quiz 1314, 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer P, Sreekumar A, Reinke S, Rad R, Schafer R, Sers C, Blaker H, Herrmann BG, Morkel M. Transgenic expression of oncogenic BRAF induces loss of stem cells in the mouse intestine, which is antagonized by beta-catenin activity. Oncogene. 2015;34:3164–3175. doi: 10.1038/onc.2014.247. [DOI] [PubMed] [Google Scholar]
- Sakamoto N, Feng Y, Stolfi C, Kurosu Y, Green M, Lin J, Green ME, Sentani K, Yasui W, McMahon M, et al. BRAFV600E cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. Elife. 2017;6 doi: 10.7554/eLife.20331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Sekine S, Yamashita S, Tanabe T, Hashimoto T, Yoshida H, Taniguchi H, Kojima M, Shinmura K, Saito Y, Hiraoka N, et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J Pathol. 2016;239:133–138. doi: 10.1002/path.4709. [DOI] [PubMed] [Google Scholar]
- Silberg DG, Sullivan J, Kang E, Swain GP, Moffett J, Sund NJ, Sackett SD, Kaestner KH. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology. 2002;122:689–696. doi: 10.1053/gast.2002.31902. [DOI] [PubMed] [Google Scholar]
- Stringer EJ, Duluc I, Saandi T, Davidson I, Bialecka M, Sato T, Barker N, Clevers H, Pritchard CA, Winton DJ, et al. Cdx2 determines the fate of postnatal intestinal endoderm. Development. 2012;139:465–474. doi: 10.1242/dev.070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh E, Traber PG. An intestine-specific homeobox gene regulates proliferation and differentiation. Molecular and cellular biology. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Du H, Fu X, Li K, Li A, Zhang Y. Smad4 restoration leads to a suppression of Wnt/beta-catenin signaling activity and migration capacity in human colon carcinoma cells. Biochemical and biophysical research communications. 2009;380:478–483. doi: 10.1016/j.bbrc.2009.01.124. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife. 2015;4:e06907. doi: 10.7554/eLife.06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Flier LG, Sabates-Bellver J, Oving I, Haegebarth A, De Palo M, Anti M, Van Gijn ME, Suijkerbuijk S, Van de Wetering M, Marra G, et al. The Intestinal Wnt/TCF Signature. Gastroenterology. 2007;132:628–632. doi: 10.1053/j.gastro.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Verzi MP, Shin H, He HH, Sulahian R, Meyer CA, Montgomery RK, Fleet JC, Brown M, Liu XS, Shivdasani RA. Differentiation-specific histone modifications reveal dynamic chromatin interactions and partners for the intestinal transcription factor CDX2. Dev Cell. 2010;19:713–726. doi: 10.1016/j.devcel.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–81. doi: 10.1002/gene.10029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq files: GSE106330 and GEO102171
Human RNA-Seq Samples: GSE76987