

Abstract
Mutagenesis in model organisms following exposure to chemicals is used as an indicator of genotoxicity. Mutagenesis assays are also used to study mechanisms of DNA homeostasis. The present article focuses on detection of mutagenesis in prokaryotes, which boils down to two approaches: reporter inactivation (forward mutation assay) and reversion of an inactivating mutation (reversion mutation assay). Both methods are labor-intensive, involving visual screening, quantification of colonies on solid media, or determining a Poisson distribution in liquid culture. Here we present two reversion reporters for in vivo mutagenesis that produce a quantitative output, and thus have the potential to greatly reduce the amount of test chemical and labor involved in these assays. This output is obtained by coupling a TEM β lactamase-based reversion assay with GFP fluorescence, either by placing the two genes on the same plasmid or by fusing them translationally and interrupting the N-terminus of the ORF with a stop codon. We also describe a reporter aimed at facilitating the monitoring of continuous mutagenesis in mutator strains. This reporter couples two reversion markers, allowing the temporal separation of mutation events in time, thus providing information about the dynamics of mutagenesis in mutator strains. Here, we describe these reporter systems, provide protocols for use, and demonstrate their key functional features using error-prone Pol I mutagenesis as a source of mutations.
1. INTRODUCTION
Mutagenesis following exposure to chemicals is used to detect genotoxicity, which is an indicator of potential to cause cancer and birth defects (1,2). Mutagenesis assays are also used as a readout to study processes of DNA replication, DNA repair and DNA damage tolerization (3-6).
The present article focuses on direct detection of mutagenesis in prokaryotes. Genotoxicity can also be monitored in other ways. It can be detected indirectly, through transcriptional fusion of a reporter gene to a promoter that is induced following DNA damage such as alkA or nrdA, and genes belonging to the SOS response (umuDC, sulA, recN, recA) (reviewed in (1)). Genotoxicity can also be detected physically through visualization of DNA damage (breaks or rearrangements) (7). In addition to prokaryotic systems, a variety of eukaryotic organisms, notably yeast, Drosophila, and mouse, have also been used as reporters for genotoxocity. These eukaryotic systems have been reviewed extensively elsewhere (8-11).
Relative to indirect methods of mutagenesis detection, mutagenesis assays have the advantage of being more specific because they detect changes in DNA sequence rather than DNA damage-induced alterations in gene expression. Relative to eukaryotic model systems, bacterial assays are fast and cheap, although they can’t report on non-conserved targets (cytoskeleton, nucleotide excision repair to name two) or account for bioactivation as accurately. Still, direct mutagenesis assays in bacteria constitute one of three assays mandated for demonstration of safety for compounds in the pipeline for clinical development (the other two being a eukaryotic cell culture one and an animal test) and because of their relative low cost, are typically the first ones used to explore the safety of a compound (2,9).
Mutagenesis detection in prokaryotes boils down to two approaches: reporter inactivation (forward mutation assay) and reversion of an inactivating mutation (reversion mutation assay). Both are labor-intensive, involving visual screening, quantification of colonies on solid media, or obtaining a Poisson distribution for growth in a large number of parallel cultures.
Forward mutation assays are based on inactivation of a reporter. Reporters can produce colorimetric, luminescent, fluorescent or electrochemical signals (reviewed in (1)). Inactivation can be the result of a variety of mutations. Thus, compared to reversion assays, forward mutation assays detect events that are more frequent, which allows screening. Forward mutation assays also provide a more accurate representation of the range of genetic changes induced by the relevant mutagen because they are not dependent on specifically pre-determined mutations. In some cases, the readout for these assays is a selection, greatly increasing sensitivity. RpoB (RNA polymerase) is an example, as mutations in a variety of loci produce resistance to the antibiotic rifampin (3,12). Ara D is another example. The cells used in this assay have a mutation in the araD gene, which leads to accumulation of a toxic intermediate when arabinose is present. Mutations upstream of araD that inactivate the operon prevent the metabolism of arabinose, making cells resistant to arabinose (13).
Reversion assays detect the return to wild-type of an inactivating mutation in a pre-determined site, typically through a selection (auxotrophy, antibiotic resistance, FACS sorting, etc.). Availability of selection for reversion assays increase their sensitivity, but their dependence on specific mutations at predetermined sites makes them susceptible to sequence context effects and limits the range of genetic changes that can be detected.
The Ames Test was one of the first of these assays to be described and still by far the most widely-used prokaryotic testing method, in part because it is mandatory for regulatory compliance. This assay is based on reversion of a mutation preventing the biosynthesis of histidine, producing colonies on solid agar in the presence of trace amounts of histidine (14). A set of six strains have been developed to detect a broad range of point mutations and frameshifts. Two variations of Ames facilitate high-throughput formatting and reduce the amount of sample needed: Mini-Ames (which follows the standard Ames Test protocol, except at 1/5 the size) (15), and the Ames Fluctuation Test (which is performed in liquid culture, with growth detected through a chromophore) (16).
Reversion assays based on LacZ (17) and TEM β-lactamase have also been described (18,19), and one of the latter includes a set of six point mutations reporting on each type of point mutation that is possible in double-stranded DNA(19). Reversion, however, produces a binary output i.e. growth vs. no growth. This means that the generation of a single data point requires fine-tuning of the dose and of the dilution to obtain countable colonies (on solid plates) or a number positive wells that follows a Poisson distribution (in liquid).
A special type of reversion assay is the papillation assay, which is used to detect alterations in mutagenesis rates in vivo (20). This assay is based on a mutation in the gal2K gene, which makes cells unable to ferment galactose. Cells are grown on Mac-Conkey-galactose plates, producing white colonies. Spotting the surface of these colonies, each colored papilla (sectors) represents a microcolony derived from a single Gal+ mutant capable of galactose fermentation. The output is semiquantitative, though, as it depends on mutation events occurring early enough to allow for visual detection (21).
Here we present two sets of reporters for in vivo mutagenesis that produce a quantitative output, and thus have the potential to greatly reduce the amount of test chemical and labor involved in these assays. This output is obtained by coupling a TEM β-lactamase-based reversion assay with GFP fluorescence, either by placing the two genes on the same plasmid or by fusing them translationally.
As mentioned above, mutator strains, i.e. strains consistently exhibiting an elevated mutation frequency, can be identified by their ability to produce sectored colonies (for reviews on mutator strains see (22,23)). There are some indications that mutation rates in these strains are not constant, as there is a counterselection against high mutation rates due to the deleterious effects of mutations and possibly because of additional physiological adaptations to the stress caused by accumulation of deleterious mutations. In addition, studying the dynamics of mutagenesis in mutator strains using reporters is difficult because mutations can inactivate the reporter regardless of its forward or reversion status with a probability that grows exponentially with the number of mutations present. Here we describe a third reporter system aimed at facilitating monitoring mutagenesis in mutator strains. This reporter couples two reversion assays: TEM β-lactamase reversion and GFP reversion. This double set of markers allows the detection of sequential hits, separating mutation events in time and thus facilitating the detection of changes in mutation rates over time. mutations, S70T, which detects A:T→T:A mutations, S70R1, which detects A:T→C:G and A:T→T:A mutations.
Here, we describe these reporter systems, provide protocols for use, and demonstrate their key functional features using error-prone Pol I mutagenesis as a source of mutations.
2. DESCRIPTION OF THE REPORTER SYSTEMS
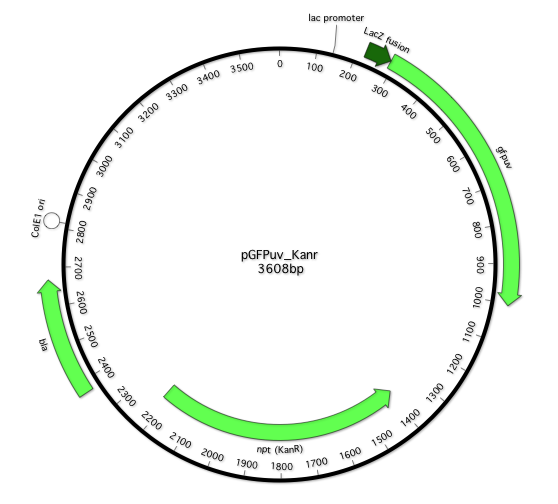
Our plasmid-based reporter systems are diagrammed in Fig. 1. Figure legends describe the annotation for each of these reporters in detail. The sequences, in MacVector and FASTA formats are provided as supplemental materials.
Fig 1. Reporter constructs.
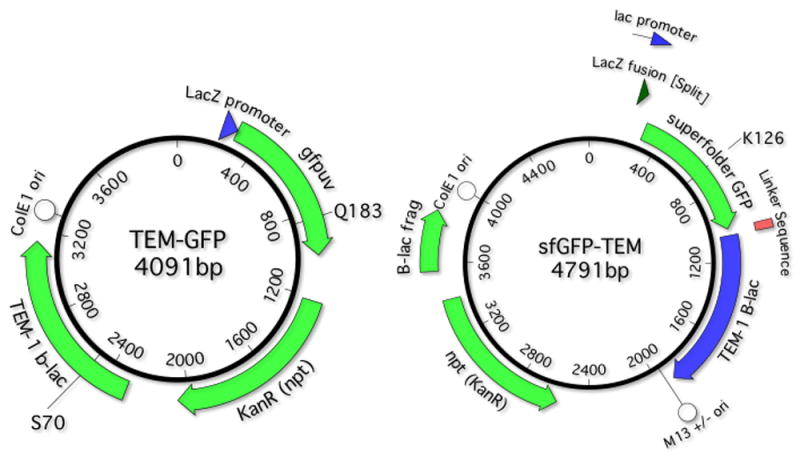
A. TEMrev-GFP reporter. Main features: Lac promoter: 143-172; LacZ fusion: 217-288; Cycle 3 GFP: 289-1005; Kan. Res. 1219-2004; β-lactamase: 2291-3151 (2492-2494 S70X reversion reporter); ColE1-like (pMB1) plasmid origin of replication 3299-4091. In the TEMrev-GFPrev variant, the Q183R mutant codon is at positions 835-837. The mutant codon is CGA (R), which requires a G→A transition to revert to CCA (Q). B sfGFP-TEM reporter. pMB1 ori 3999-479; B sfGFP lac promoter 143-172; lacZ fusion 223-259, sfGDP: 201-1014; 12 aa serine/glycine-rich linker 1015-1050; lactamase 1051-1911; the K126stop codon is at positions 579-81; M13 ori: 1953-2462; Kan resistance (opposite orientation) 3400-2575; lactamase fragment 3553-3851.
We selected TEM-1 β-lactamase, a gene that confers resistance to carbenicillin, as one of the reversion markers. TEM-1 was inactivated through mutations in the S70 position, the serine residue that polarizes the carbonyl group of the β-lactam amide bond in the β-lactam ring of β-lactamase antibiotics (24) and is completely intolerant to amino acid changes (25). We engineered point mutations at this position to be one nucleotide away from a serine-coding codon so that we can detect each of the 6 pairs of nucleotide substitutions that are possible in duplex DNA. Table 1 shows each of the six reporter codons, the single nucleotide changes that produce a serine codon and what that “nearest” codon is. The primers used to introduce these point mutations at the S70 position are described in Suppl. Table 1.
Table 1.
Reporter strategy: each S70 reporter mutation
| original codon | original AA | reporter codon | reporter AA | mutation needed for reversion | reversion codon |
|---|---|---|---|---|---|
| AGC | S | CCA | P | C:G →T:A | TCA |
| AGC | S | ACA | T | A:T→T:A | TCA |
| AGC | S | AGA | R | A:T→C/T:G/A | AGC or AGT |
| AGC | S | TGA | * | G:C→ C:G | TCA |
| AGC | S | AAC | N | A:T→ G:C | AGC |
| AGC | S | CGC | R | C:G→A:T | AGC |
The six reporters are: S70P, which detects C:G→T:A mutations; S70T, which reports A:T→T:A mutations; S70R1, which reports A:T→C:G and A:T→T:A mutations; S70stop, which detects G:C→C:G mutations; S70N, which detects A:T→G:C mutations; and S70R2, which detects C:G→A:T mutations.
2.1 TEMrev-GFP
We coupled a GFP fluorescent marker to the S70X TEM β-lactamase reporter set described above. Cycle 3 GFP, a variant of GFP optimized for fluorescence in E. coli (26), is placed the same plasmid downstream of TEM so that it is co-transcribed with it (TEMrev-GFP series, Fig. 1A). This way, growth in the presence of carbenicillin can be detected through fluorescence, a signal with much higher sensitivity and wider dynamic range than turbidity. GFP doesn’t require cell lysis, thus facilitating monitoring growth over time.
2.2 sfGFP-TEM
Another way in which we coupled reversion of TEM to GFP was through a translational fusion, placing GFP in the same open reading frame as β-lactamase, with a 12 serine and glycine-rich linker in between (sfGFP-TEM, Fig. 1B).
To obtain a functional translational fusion, we had to use superfolder GFP (sfGFP), an evolved form of cycle 3 GFP with two additional mutations selected for robustness to translational fusions (27). In addition, we found that sfGFP is only functional if located at the N-terminus, not at the C-terminus. Even this fusion is not stable in all strains: while it emits a high level of fluorescence in Top10 cells, we found that it produced inconsistent fluorescence JS200 cells and no fluorescence in AB1157 and its GW7101 (ada alkB∷CAT) derivative.
The introduction of a stop codon truncating GFP expression inactivates both GPF and β-lactamase, resulting in a non-fluorescent, carbenicillin-sensitive phenotype. As proof of principle, we introduced a TGA stop codon at three different positions (Q69, K113, and K126). As expected, the reversion of the stop codon at these positions resulted in fluorescence and carbenicillin resistance (stop codon reversions for sfGFP-TEM K126stop shown in Fig. 2).
Fig. 2. sfGFP-TEM K126stop reporter on solid plates.
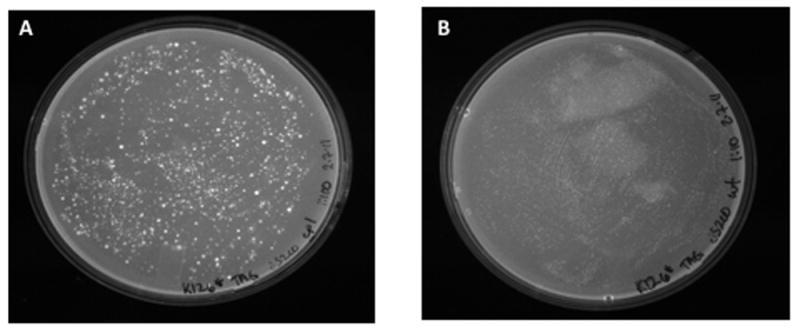
Following Pol I mutagenesis, JS200 cells were plated on LB carbenicillin. A. Cells expressing LF-Pol I. B. Cells expressing WT Pol I (control).
2.3 TEMrev-GFPrev
The goal of this reporter is to facilitate monitoring mutagenesis in mutator strains in a more quantitative manner than papillation assays. TEMrev-GFPrev is identical to the TEMrev-GFP reporter described above but it carries a mutant GFP inactivated through a point mutation replacing Q183 with an R (CAA to CGA), which reverts with C:G→T:A mutations (Fig. 1A). The method is diagrammed in Fig. 3. Colonies are first grown on carbenicillin to identify S70 reversion events. A few non-fluorescent carbenicillin-resistant colonies are picked, and grown in liquid culture. The plasmid DNA from these cultures is recovered and retransformed into a readout strain (Top10 or DH5α) to identify R183Q (fluorescent) revertants (Fig. 3). Under these conditions, fluorescent colonies have to be the result of a mutation event that occurred after carbenicillin reversion, unless the reversion was already present in one of the copies of the plasmid pool when the first mutation occurred. This alternate explanation can be ruled out if the frequency of reversion is lower than one divided by the copy number of the reporter plasmid.
Fig. 3. Method for detection of two mutations separated in time.
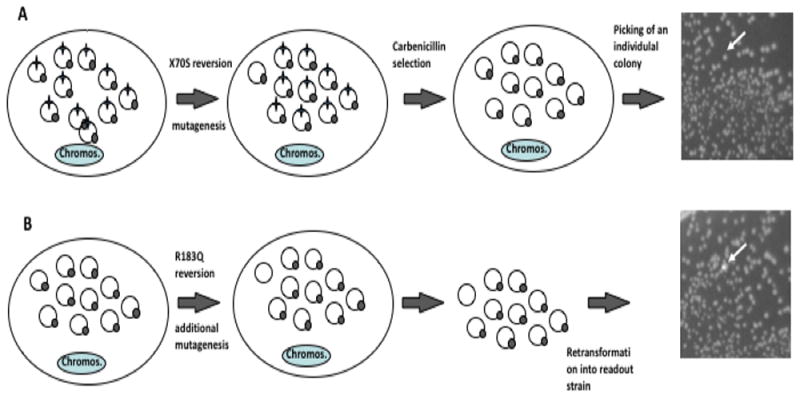
Colonies representing reversions in TEM1 β-lactamase are expanded under restrictive (mutagenic) conditions, their plasmid pools recovered through miniprep and retransformed. The two reversion reporters are shown as star (S70X) and circle (Q183R). A. Detection of first mutation: once reversion at the S70X site occurs, under selective pressure, the reversion events get amplified, representing a majority of the plasmid population, and leading to carbenicillin resistance. B. Detection of second mutation: single, carbenicillin-resistant colonies (white arrow) are grown. Plasmids from these cultures are recovered. Reversions at the Q183 site of the GFP reporter are detected by retransformation of recovered plasmids into a readout strain, producing fluorescent colonies on a background on non-fluorescent ones (white arrow). Reversion cannot have already been present in one of the copies of the plasmid pool when the first mutation occurred if the frequency of reversion is lower than one divided by the copy number of the reporter plasmid (in this example, one in 10).
3. EXPERIMENTAL VALIDATION
To validate our reporter system, we used error-prone Pol I plasmid replication, as previously described in detail by our group (28,29). This system is based on expression of an error-prone variant of DNA polymerase I (low fidelity Pol I or LF-Pol I) in JS200, a polA12 (temperature-sensitive) strain of E. coli (30). LF-Pol I bears three mutations synergistically decreasing its replication fidelity: I709N in motif A (broadening its active site), A759R in motif B (favoring its closed conformation) and D424A (inactivating its proofreading domain) (31).Shift of this strain to 37 °C makes J200 cells dependent on the activity of LF-Pol I for survival. Pol I performs ColE1 plasmid replication (32) and processes of Okazaki fragments during lagging-strand replication in plasmid and chromosomal DNA (30,32).
Overnight culture under restrictive conditions (37 °C) leads to an increased mutation frequency by over three orders of magnitude in ColE1 plasmids, about 1 nucleotide substitution per 1.5 kb. This is true for most of the plasmid sequence, where Pol I appears to be competing with Pol III (32). These loads are higher in areas replicated exclusively by Pol I: the 150 nucleotides immediately downstream of the RNA/DNA switch (leading-strand synthesis by Pol I), ~500 nucleotides upstream of the RNA/DNA switch (gap-filling of lagging-strand synthesis by Pol I), and ~20nt patches corresponding to areas of Okazaki fragment processing by Pol I (29,33). It is worth noting that LF-Pol I is partially dominant in vivo, as expression of this polymerase still produces Col E1 plasmid mutagenesis at permissive temperature or in polA WT strains, albeit with a 3- to 5-fold lower frequency relative to JS200 at restrictive temperature (28).
In terms of mutation spectrum, we were able to estimate the mutation frequency of LF-Pol I on a single strand in vivo (32). The vast majority of mutations (>95%) are point mutations and can be grouped in four groups: most frequent: C→T transitions (60%); frequent: A→G and A →T (20 and 10% of the total, respectively); rare: G→T, G →A, and G→T, and extremely rare: T→C, T→A, A→C and C→G. Note that, based on the very low frequency of T→C transitions in vivo, mismatch repair appears to be intact in these cells (32). Given that our reporter detects mutations in double-stranded DNA, i.e. in pairs of complementary mutations, we expect the following ranking based on frequency: C:G →T:A > A:T→G:C ~ A:T→T:A > G:C→T:A>T:A→ G:C>G:C → C:G
3.1 Mutagenesis
We transformed our six TEMrev-GFP reporters, a TEM-GFP positive control, and a negative control not bearing the TEM1 gene (supplemental file and Suppl. Fig. 1) into JS200 E. coli cells expressing LF-Pol I (31). As an additional control, we also transformed these reporter and control plasmids into a JS200 cells expressing WT Pol I. After recovery at 30 °C, cells were plated onto LB agar plates pre-warmed to 37°C containing kanamycin, thus switching our transformants to restrictive conditions. Mutagenesis occurred during growth overnight at 37°C.
3.2 Read-out on Solid Media
Transformants produced a high density of colonies (near-lawn). These colonies were harvested from the plate into ~ 1.5 ml of LB broth. Absorbance at 600 nm was determined to normalize the washes to OD600 = 1. These normalized stocks were used to plate kanamycin (at further dilution of 1:107) and carbenicillin plates at different dilutions, depending on reversion frequencies (between neat and 1:103 dilutions). This time, plates were incubated overnight at 30°C (i.e. under permissive conditions) to minimize additional mutagenesis. Following incubation, the number of colonies on each plate were counted, and this number was used to calculate the reversion rate for each reporter (Figure 4). Interestingly, fluorescence was not uniform across all carbenicillin-resistant colonies, possibly due to the presence of additional mutations affecting GFP expression and/or function (not shown).
Fig. 4. LF-Pol I reversion profile.
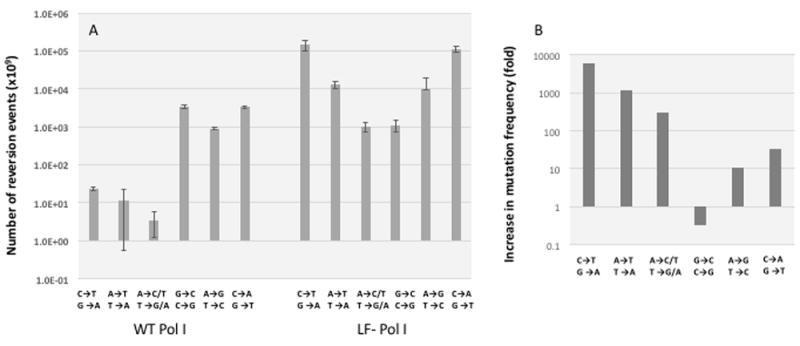
The set of six TEMrev-GFP reporters was transformed in JS200 cells expressing LF-Pol I and error-prone reporter plasmid replication was performed as described in methods. As a control, the same reporters were transformed into JS200 cells expressing WT Pol I. A Original reversion frequencies, in log scale. Error bars represent standard deviation of triplicates. Reversion frequencies for LF-Pol I samples were normalized for viability (these cells exhibited a 50% decrease in viability) and for plasmid copy number (at least 10-fold lower than WT) (31). B. Reversion frequency relative to control cells expressing WT polymerase l (fold, in log scale)
For the sfGFP-TEM K126stop reporter, plating a transformation of JS200 cells expressing LF-Pol I at the restrictive temperature produced a semi-lawn of carbenicillin-resistant, fluorescent colonies (Fig. 2 A). Sequencing of 10 these colonies showed point mutations at the stop codon in all cases, producing an L (three times), W (three times), Q (twice), Y (twice). In eight of these cases, the WT signal was still detectable, suggesting that the plasmid carrying the K126 point mutations had not replaced all the copies of the original K126 stop reporter. Cells expressing WT- Pol I had practically no colonies (Fig. 2B)
3.3 Read-out in Liquid Media
Following mutagenesis, plate washes were normalized to OD600 = 1 and used to inoculate 96-well plates in a 1:20 dilution. The plates were deep-well round-bottom plates with glass beads (to facilitate oxygenation) and a final volume of 1mL (see Methods). The plates were then covered with AirPore breathable sheets, in order to protect against cross-contamination and evaporation effects, while still allowing for microbe growth under aerobic conditions, and grown at 30°C shaking at 325 rpm. At different time-points 200 μl of each culture were transferred to a set of black-walled flat-bottomed 96-well microtiter plates and kept at 4°C. At the end of the experiment, we determined growth by reading absorbance at 600 nm. Fluorescence readings were also obtained on a fluorescence-enabled spectrophotometric plate reader, with excitement λ = 395 nm, and emission λ = 509 nm. Results were then used to plot growth kinetics curves for each construct under each antibiotic selection. Fig. 5 shows the growth and fluorescence emission kinetics for two of our reporters, S70P (which detects C:G→T:A mutations)(Fig. 5 A,C) and for S70R1, which detects (A:T→C:G and A:T →T:A mutations) (Fig. 5 B,D)
Fig. 5. Mutagenesis assay in 96-well format.
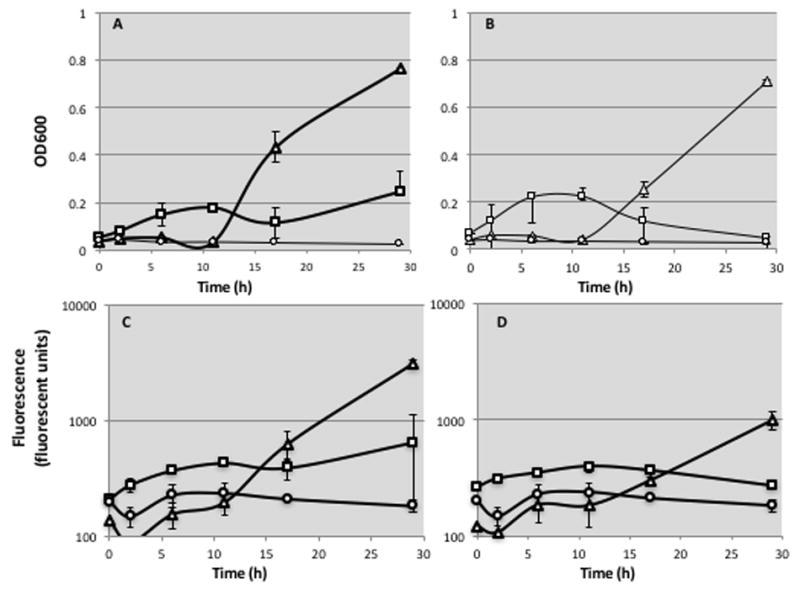
Cells bearing two sample reporters, S70P (which detects C:G→T:A transitions) or S70R1 (which detects A:T → C:G and A:T → T:A mutations) underwent error-prone plasmid replication as described in Methods, recovered by washing the plates, and inoculated into 96 deep-well plates to a final OD600 of 0.5. At different time points (shown in the X-axis) samples were drawn and kept at 4°C. After completion of the time-course, fluorescence and optical density (OD600) were measured. LF-Pol I mutagenesis, triangles; WT Pol I control, squares, negative control with no β-lactamase gene, circles A. S70P reporter, OD600, in log scale. B. S70R1 reporter, OD600, in log scale. C. S70P reporter, fluorescence. D. S70R1 reporter, fluorescence. Error bars represent standard deviation between duplicates.
3.4 Continuous Mutagenesis Detection
Colonies expressing LF- Pol I and bearing the TEMrev-GFPrev S70P reporter were plated under restrictive conditions as described in the “readout on solid plates” section above but at a higher dilution in order to obtain individual carbenicillin-resistant colonies. Three non-fluorescent, carbenicillin-resistant colonies were picked, and grown in liquid culture under restrictive conditions. The DNA from these cultures was recovered and retransformed into DH5α cells to identify R183Q (fluorescent) revertants (Fig. 3B diagram). The results of these experiments are listed in Table 2. Using the GFP reporter to quantify “second hit” mutagenesis, we found a mutation frequency of 137 in 106 cells, in line with frequencies seen by carbenicillin reversion. Three colonies were also grown under permissive conditions and retransformed into reporter cells. Here we find a frequency that is 3.7-fold lower, about 37 fluorescent colonies per 106 transformants. Given that the average plasmid copy number for the reporter plasmid in LF-Pol expressing cells is less than ten plasmids per cell (31),these results confirm that the observed fluorescent colonies are most likely the result of mutations at the 183 position of GFP that occurred after the P70S reversion.
Table 2.
GFP reversion frequencies, compared to those of S70P
| Pol I | Temperature | Total colonies screened (×104) | Total number of transformations | Average transformation efficiency | Number of fluorescent colonies | Frequency of GFP reversion (×106) | S70P reversion frequency (×106) |
|---|---|---|---|---|---|---|---|
| WT | 30 °C | 60.4 | 6 | 10.1 | 0 | <1.7 | N/A |
| WT | 37 °C | 18.1 | 6 | 3.0 | 3 | 16.5 | 0.004 |
| LF | 30 °C | 13.4 | 8 | 1.6 | 5 | 37.2 | N/A |
| LF | 37 °C | 5.1 | 8 | 0.6 | 7 | 137.0 | 180.9 |
4. Discussion
Here we present three reporter systems to detect and quantify point mutations. These reporters are on a plasmid bearing a pMB1 (ColE1-like) plasmid origin of replication. This has several advantages over a chromosomal location: (1) a plasmid reporter increases the number of targets for mutagenesis by at least one order of magnitude, since ColE1 plasmids are multicopy plasmids (34,35); (2) the fact that plasmids are present in multiple copies also allows amplification of reporter signal through selection; (3) a plasmid reporter facilitates exposure to mutagens ex vivo; in this scenario, transformation would be performed only to obtain a readout. It needs to be kept in mind, though, that most mutagens require biotransformation (36), restricting the applications of such an ex-vivo assay.
The first reporter system, TEMrev-GFP is based on reversion of an inactivating mutation in TEM β-lactamase and co-transcription of GFP.A similar approach was previously reported by Schmid et al., although in their case reversion of TEM β-lactamase was coupled to β-galactosidase (18).
One limitation of reversion-based reporter systems is that they look at a limited number of sequence changes in a pre-determined site. To be able to characterize the complete spectrum of point mutations, we generated a panel of mutations in the S70 of β-lactamase position reporting for all possible point mutations. A similar approach that has been previously described also for TEM β-lactamase (19)and for LacZ (17). The spectrum results obtained through these sets of reversion reporters is still subject to sequence-context and strand bias effects, though.
The point mutation spectrum profile of LF-Pol I that we describe here (Fig. 4) is consistent with the one previously reported by Suzuki et al. (19). Compared with the profile that our group generated previously based on sequencing (32), only one pair exhibits a lower reversion frequency than expected: A:T→G:C. This could be the result of sequence-context dependent effects (37,38), which can be in part due to differential efficiency of mismatch repair (6). Our observation that S70R1, which detects A:T→C:G and A:T→T:A mutations, produces fewer reversions than S70T, which detects A:T→T:A alone directly confirms the impact of local sequence context on mutation rates. Overall, then, profiling mutation spectrum using our TEMrev-GFP mutation set gives a general idea of which types of point mutations are favored, particularly if there is as strong bias for a specific type, but the spectrum obtained is not as reliable as that obtained by sequencing a forward mutagenesis reporter or genomic sequence because it does not sample a variety of sequence contexts.
When the frequency of mutagenesis is expressed as ratio of LF-Pol I vs WT-Pol I, one of the values is below 1 (G:C→C:G Fig. 4B). This could be due to a conservative estimate of difference in plasmid copy number between WT-expressing and LF-Pol I-expressing cells (10-fold) or to a different mutagenic mechanism operating in WT-Pol I expressing cells, as suggested by the high mutation frequency seen in these cells, which is between 10 and 100-fold above that of other reporters. In any case, LF-Pol I appears to induce a very low frequency of G:C→C:G, consistent with our previous estimate of LF-Pol I mutation spectrum based on extensive sequencing (32).
It is also worth noting that two of the six reporters in the set produce between 10 and 100-fold higher background mutation frequencies in the control strain expressing WT Pol I relative to the other three (Fig. 4A). The two reporters are S70stop and S70T, which report G:C→C:G and A:T→T:A, respectively. We ignore the reason for this increased background in mutation frequency for two transversions in our reporter, as C:G→T:A transitions predominate instead in the spontaneous mutation spectrum of E. coli (37) and Pol I produces predominantly transitions as well (3). The replication fidelity of Pol I can be modulated by pols II and IV, though, which could result in an increased number of transversions (3).
GFP facilitates quantification of growth in vivo, producing a much stronger signal than turbidity. Note that in Fig. 5, the results of fluorescence (panels C and D) are shown in a logarithmic scale and that the ratio of background-to-signal is at least two orders of magnitude higher. Further, for a given time-point, the result of GFP fluorescence is quantitative. Our reversion assays report that LF-Pol I produces more C:G→T:A mutations relative to A:T→C:G and A:T→T:A mutations. In Fig. 5, the fluorescent signal is much stronger at 30h time-point for the C:G→T:A reporter so a measurement at this time-point would have been proportional to mutation frequency determined by plating. Relative to other quantitative reporters, GFP has several advantages that make it ideal for continuous measurement in culture: (1) it is highly stable, (2) no addition of an external substrate is necessary; (3) no cell lysis is required; (4) less susceptible to substrate interference. However, in genotoxicity sensors based on fusions with DNA-damage-inducible promoters, GFP has been found to have low sensitivity compared to enzyme-based reporters, even when GFP variants producing enhanced signal through mutagenesis were used (39). Here we rely on two levels of signal amplification: one is plasmid, which is present in multiple copies, increasing GFP expression; the other one is growth: TEM β-lactamase gives revertants a dramatic growth advantage, increasing the number of cells present in liquid culture at a given time in a mutator relative to a control (see Fig. 5A,B).
Our sfGFP-TEM K126stop reporter is based on generating a translational fusion between GFP and TEM β-lactamase and interrupting translation through a stop codon or a frameshift. Reversion of this codon results in both resistance to carbenicillin and fluorescence (Fig. 2A). A translational fusion represents a new approach for quantitative detection of reversions that should further increase the signal-to-noise ratio for GFP fluorescence, as cells without a reversion are not even fluorescent. Further, this cassette can be placed in any desired location and orientation. However, this fusion protein appears not to be very stable, and only works in some strains of E. coli.
Finally, our TEMrev-GFPrev reporter represents an alternative to papillation assays for the characterization of mutator strains. The main advantage is that the output in this case is quantitative rather than semi-quantitative, allowing head-to-head comparisons between different mutators and/or growth conditions. Different inactivating GFP mutations can be introduced, depending on the mutagenic profile of the mutator strain. The chromophore-containing cyclized hexapeptide (residues 64 through 69) is a good target for inactivating mutations, with narrow tolerance to alternative amino acids(40). Suppl. Table 2 shows a list of 12 mutations inactivating cycle 3 GFP fluorescence in E. coli outside the active site hexapeptide that can also be explored as possible reversion sites.
Position 183 is located within a 9Å shell around the chromophore without directly contacting it. A C:G→T:A mutation, which is by far the predominant nucleotide substitution introduced by LF-Pol I, reverts 183R back to Q. We confirmed that reversion to Q is the predominant mutation in fluorescent colonies: nine out of nine fluorescent colonies sequenced produced this reversion. We don’t know if other amino acid substitutions are allowed at this site, but C and A substitutions are not tolerated either because of a dramatic destabilizing effect on GFP (41).
A decreased plasmid copy number for LF-Pol I cells compared to WT-expressing cells, previously reported in (31) and factored in Fig. 4, is the likely cause for the observed transformation efficiency of DNA recovered from these cells (Table 2)
The mutation frequency for “second hit” mutagenesis that we obtained using our GFP reversion reporter slightly lower relative to the first round of mutagenesis (137, compared to 181 in 106 cells, Table 2) although given that two different reporters were used to measure “first hit” and second hit” mutagenesis and that the readout for carbenicillin resistance involved two rounds of growth on solid plate (at 37 °C and then at 30 °C), whereas GFP reversion only involved one round, it is hard to draw any conclusions regarding the frequency of mutagenesis of the second hit versus the first hit other that the two results are consistent between each other. We also found a significant amount of revertants in our WT-expressing control grown at 37 °C (16 in 106 cells) but not at 30 °C (Table 2) and don’t have an explanation at the moment for this observation.
For DNA from colonies grown under permissive conditions we find a frequency that is 3.7-fold lower relative to DNA from colonies grown under restrictive conditions, about 37 fluorescent colonies per 106 transformants. This result aligns well with a previous report of partial dominance for LF-Pol I (28). Given that the average plasmid copy number for the reporter plasmid in LF-Pol expressing cells is less than ten plasmids per cell (31), the very low rate of reversion that we observed confirms that the fluorescent colonies are we see are most likely the result of mutations at the 183 position of GFP that occurred after the P70S reversion.
This means that LF-Pol I-expressing cells continue to generate mutations after one passage in culture. Our TEMrev-GFPrev reporter system should be of use to fine-tune LF-Pol I-expressing cells and other existing mutator strain ssuch as XL-1 red (42), the MP6 mutagenesis system (43), or strains with altered dNTP pools (40) to identify conditions supporting constant mutation rates over time.
5. Materials
5.1 Transformation and Mutagenesis
-
Competent Cells
Top10
JS200-pHSG_WT
JS200-pHSG_LF
-
ColE1 Vectors
TEMrev-GFP
TEMrev-GFPrev
sfGFP-TEM K126stop
pGFPuv_KanR
500 mL centrifuge bottles
Eppendorf centrifuge 5810 R (Eppendorf)
50 mL conical tubes (Fisher Scientific, Cat.# 1443222)
15 mL culture tubes (E&K Scientific, Cat.# EK-62262)
LB broth (Fisher Scientific, Cat.# BP1426-2)
LB agar (Fisher Scientific, Cat.# BP1425-2)
100mm × 15mm disposable Petri dishes (Fisher Scientific, Cat.# FB0875713)
Kanamycin solution, 30 mg/mL, store at -20°C
Kanamycin (30 μg/m) LB agar and broth
1.5 mL microfuge tubes (E&K Scientific, Cat.# 280150)
TropiCooler, Model 260014 (Boekel Scientific)
MaxQ 4000 shaker/incubator (Barnstead International)
Water-jacketed incubator (Forma Scientific)
5.2 Washing Plates
Kanamycin (30 μg/m) LB broth
Plate spinner
Plate spreader
Ethanol (200 proof)
Bunsen burner
Spectrophotometer cuvettes (Fisher Scientific, Cat.# 14955127)
1.5 mL microfuge tubes (E&K Scientific, Cat.# 280150)
BioMate 3 Spectrophotometer (Thermo Scientific)
5.3. Readout (Plates)
Kanamycin (30 μg/m) LB agar and broth
Carbenicillin (100 μg/m) LB agar and broth
1.5 mL microfuge tubes (E&K Scientific, Cat.# 280150)
Plate spreader
Plate spinner
Ethanol (200 proof)
Bunsen burner
Water-jacketed incubator (Forma Scientific)
UV Light
5.4 Readout (Liquid Culture Assay)
Kanamycin (30 μg/m) LB broth
Carbenicillin (100 μg/m) LB broth
3mm diameter glass beads (Sigma-Aldrich, Cat.# Z265926)
AirPore tape sheets (Qiagen, Cat.# 2017-10-RP)
96-well round-bottomed deep-well plates (Fisher Scientific, Cat.# 10011-944)
96-well flat-bottomed black-walled plates (Fisher Scientific, Cat.# 82050-744)
Microtiter plate lids (Fisher Scientific, Cat.# 82050-829)
MaxQ 4000 shaker/incubator (Barnstead International)
SpectraMax M2e Fluorometric and Spectrophotometric plate reader. Dual monochromators, Absorbance 200-1000nm and excitation 250-850nm (Molecular Devices)
5.5 Plasmid Recovery
15 mL culture tubes (E&K Scientific, Cat.# EK-62262)
1.5 mL microfuge tubes (E&K Scientific, Cat.# 280150)
Nucleospin Plasmid (NoLid) kit (Macherey-Nagel, Cat.# 740499.250)
5.6 Sequencing Plasmids of Interest
NanoDrop ND-1000 Spectrophotometer for DNA quantification (Thermo Scientific)
0.6 mL microfuge tubes (E&K Scientific, Cat.# 280060-S)
MacVector version 12.7.5 for sequence analysis (MacVector Inc.)
6. Methods
6.1 Transformation and Mutagenesis
6.1.1 Preparation of Competent Cells for Chemical Transformation
Prepare a 5mL overnight culture in a 15mL culture tube in LB media for the strain of interest. If necessary, include selective antibiotic in the media for the desired strain.
Expand this culture into a sterile 1L Erlenmeyer flask containing 500mL of LB media with selective antibiotic.
Incubate this flask at 30°C or 37°C (depending on the cell line; incubate JS200 cell lines at 30°C, all others at 37°C) with shaking (225 rpm) until exponential phase is reached (OD600 = 0.4 - 0.6).
Chill the flask containing the cells on ice for 20 minutes a.
Transfer the liquid cultures to 500 mL plastic centrifuge bottles and centrifuge at 4000rpm for 20 minutes at 4°C.
Pour off supernatant, and resuspend the cell pellet in 50 mL of chilled calcium chloride solution (100 mM CaCl2, 10 mM HEPES, 15% Glycerol, pH 7).
Transfer the resuspended cells into a 50mL conical tube.
Centrifuge the cells at 4000rpm for 20 minutes at 4°C.
Pour off supernatant, and resuspend the cell pellet in 50 mL of chilled calcium chloride solution, and centrifuge the cells at 4000rpm for 20 minutes at 4°C.
Repeat step 9.
After 3rd wash with calcium chloride solution, pour off supernatant, and resuspend the cells in 5mL of chilled calcium chloride solution.
Keep cells on wet ice and use immediately, or aliquot into 1.5mL microfuge tubes and place on dry ice for storage at -80°C.
6.1.2 Chemical Transformation
Pipette 40μl of chemically competent cells into 1.5mL microfuge tube per transformation.
Pipette 100μg of plasmid DNA into the tube containing the competent cells and mix well by pipetting up and down.
Incubate on ice for 30 minutes.
Heat-shock the cells at 42°C for 90 seconds on Tropicooler block.
Place cells back on ice for 5 minutes.
Add 1 mL of LB Broth to the microfuge tube containing the cells and DNA.
Allow the cells to recover for 30 minutes - 1 hour at 30°C or 37°C b with shaking (225 rpm).
Plate transformed cells by spreading 100 μL with sterile plate spreader onto pre-warmed LB agar plates containing 30μg/mL Kanamycin.
Allow the cells to grow overnight at either 30°C or 37°C.
6.1.3 Mutagenesis
For ColE1-like ori plasmids which have been transformed into JS200-LF-Pol I expressing strains, incubate at 37°C to induce mutagenesis c.
Use the same plasmids transformed into JS200-pHSG_WT as control.
6.2 Washing Plates
Observe plate for “near-lawn”, high density of colonies.
Place plate on a plate spinner.
Add 1mL LB broth directly to the plate surface containing bacterial growth.
Use sterile plate spreader to collect colonies into LB Broth.
Tilt plate slightly of collect broth containing harvested colonies into one area, and transfer as much as possible into 1.5mL microfuge tube.
Repeat steps 3-5. Collect second wash into the same 1.5mL microfuge tube d.
Dilute plate washes 1:20 directly in spectrophotometer cuvettes (950 μL media + 50 μL plate wash), and mix by pipetting.
Measure OD600 of diluted plate wash using the BioMate 3 spectrophotometer, and multiply the measurement by 20 to obtain the actual OD600 of the undiluted plate wash.
Normalize all plate washes to OD600 = 1 prior to readout experiments.
6.3 Readout (Plates)
Pre-warm LB agar plates containing 30μg/mL kanamycin and LB agar plates containing 100μg/mL carbenicillin in incubator. For JS200 transformants, 30°C (permissive temperature) should be used.
Plate 100 μL of plate washes (all washes plated to both LB agar plates containing 30μg/mL kanamycin and LB agar plates containing 100μg/mL carbenicillin) at appropriate dilutions to yield countable colonies e.
Incubate overnight. For JS200 transformants, 30°C (permissive temperature) should be used.
Determine the number of colonies on each plate.
Use counts to determine CFU/ml of OD normalized cultures on each type of selective media.
- Determine fraction of TEM β-lactamase S70 revertants in 109 cells by the formula:
6.4 Readout (Liquid Culture Assay)
Aseptically place one sterile 3mm diameter glass bead into each well of a 96-well deep-well round-bottomed plate using sterilized forceps f.
Transfer 950 μL of LB broth containing 30μg/mL kanamycin to one well and 950 μL of LB broth containing 100μg/mL carbenicillin to another well for each construct to be tested at each time point g.
Inoculate wells with 50 μL of 1:10 diluted plate washes (final inoculation OD600 = 0.05), leaving one row of non-inoculated media as negative control h.
Cover with AirPore tape sheet.
Remove 200 μL of T0 time point to 96-well black-walled clear-bottomed flat-bottomed plates, cover with sterile plate lid, place at 4°C.
Cover deep-welled plate with sterile plate lid and place in incubator at 30°C or 37°Cb with shaking (325 rpm)i.
At appropriate time points, remove 200 μL of culture from pre-assigned well to 96-well black-walled plates and return culture to shaking incubatorj.
Between time points, the black-walled plates should be stored at 4°C, and the deep-welled plates should be incubated at the appropriate temperature with shaking.
At the last time point, remove culture and uninoculated blank wells to black-walled plates.
Read OD600 and fluorescence (ex. 395 nm, em. 509 nm) from black-walled plates on SpectraMax M2e Fluorometric and Spectrophotometric plate reader.
Plot OD600 versus time and fluorescence versus time for constructs under both kanamycin selection and carbenicillin selection to estimate relative rates of TEM β-lactamase S70 reversion.
6.5 Plasmid Recovery
Pick reversion colonies from LB agar plates containing 100μg/mL carbenicillin generated in section 2.3.1, and inoculate into 3 mL of LB broth containing 100μg/mL carbenicillin.
Grow cultures overnight under permissive conditions (30°C) with shaking (225 rpm).
Harvest cells by centrifugation at 11,000 × g for 1 minute, pour off supernatant, and isolate plasmid DNA (miniprep) based on manufacturer’s instructions.
6.6 Sequencing Plasmids of Interest
-
Quantify plasmid DNA yield and purity using NanoDrop Spectrophotometer
Open NanoDrop software (ND-1000, version 3.8.1), and select nucleic acid quantification.
Place 2 μL of purified water on cleaned pedestal and lower arm to initialize spectrophotometer.
Place 2 μL of elution buffer on cleaned pedestal and lower arm to blank spectrophotometer.
Place 2 μL of sample to be quantified on cleaned pedestal and lower arm to measure absorption spectrum between 220 nm and 350 nm.
Transfer 0.5 - 1 μg of plasmid DNA to 0.6 μL microfuge tube with appropriate label.
Transfer 10 μL of 5 μM sequencing primer to 0.6 μL microfuge tube with appropriate label.
Send plasmid DNA and sequencing primer to sequencing facility.
Assemble and analyze sequences using the program MacVector.
Supplementary Material
{kind=link}
Fig. 6. Q183R reporter.
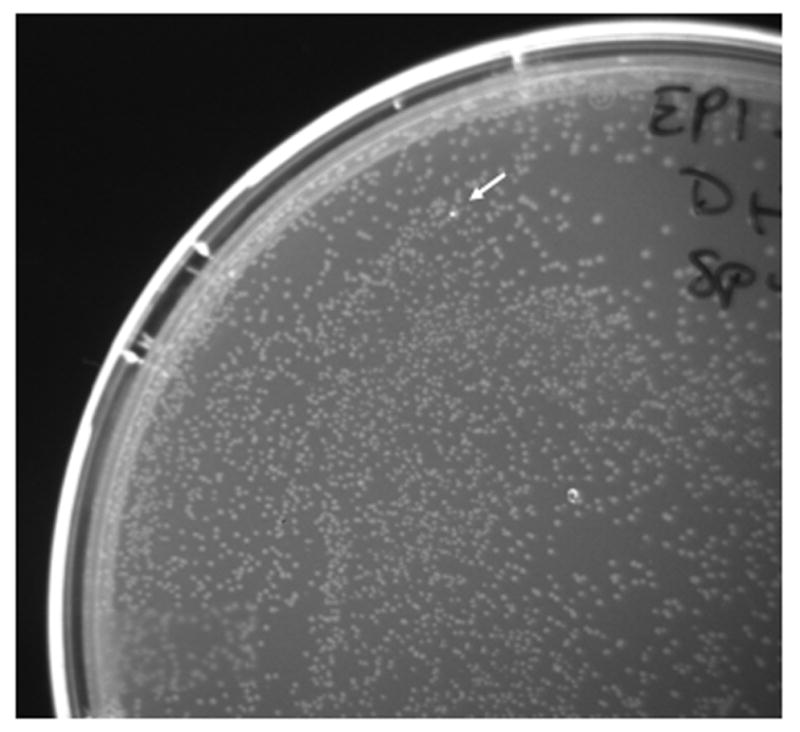
R183Q reversion on a plate containing 9400 colonies is shown. Transformed plasmids were recovered from expansion in liquid culture of a non-fluorescent carbenicillin-resistant colony and grown under continuous mutagenesis conditions (diagrammed in Fig. 3). No revertants were seen in 73,500 transformants from plasmids recovered in cells expressing WT polymerase grown under the same conditions.
Acknowledgments
This work was supported by R01ES019625-01A1 and by funds from UCSC’s Office of Research for support of intellectual property.
Footnotes
From this point forward, cells must be kept chilled at all times for best results.
JS200 transformants need to be grown at 30°C if permissive conditions are desired; for mutagenesis or in the case of non temperature-sensitive strains, cells need to be grown at 37°C
Pre-warming plates to 37°C prior to plating cells is essential for efficient mutagenesis.
Typical recovery per 2 mL of LB is ~1.5 mL of plate wash.
Experimenter must estimate dilution needed to achieve a total number of colonies on plate between 50-500. This may require some trial and error. Our results indicate that a dilution factor of 107 is effective for all constructs and controls on kanamycin plates and positive controls (WT β-lactamase) on carbenicillin plates and no dilution for negative controls on carbenicillin plates. For reporter constructs on carbenicillin plates, dilutions may vary depending on the expected reversion frequency, but generally range between a 1:10 dilution and a dilution factor of 103. Be sure to take note of the dilution factor used for each construct plated, as this will be used to calculate CFU/mL (see section 2.3.1 step 6).
Sterilize forceps by dipping in ethanol and holding over flame until red hot.
It is recommended to have triplicates at each time point.
Inoculate different time points and different constructs and controls into separate wells. Inoculate each culture into both kanamycin wells and carbenicillin wells. Leave at least three wells on each plate uninoculated, to be used as blanks during spectrophotometry/fluorimetry.
For cell lines growing at 30°C, culture growth will be slower. Plan time points accordingly.
Take 200 μL from fresh wells at each time point. Do not resample wells that have already been sampled at previous time points because the stirring of the culture that occurs when a sample is taken alters growth on a microtiter plate. Sample well by stabbing the micropipette tip through the AirPore sheet. Use caution to avoid disturbing/cross-contaminating wells containing later time points or blanks.
References
- 1.Biran A, Yagur-Kroll S, Pedahzur R, Buchinger S, Reifferscheid G, Ben-Yoav H, Shacham-Diamand Y, Belkin S. Bacterial genotoxicity bioreporters. Microb Biotechnol. 2010;3:412–427. doi: 10.1111/j.1751-7915.2009.00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krewski D, Acosta D, Jr, Andersen M, Anderson H, Bailar JC, 3rd, Boekelheide K, Brent R, Charnley G, Cheung VG, Green S, Jr, et al. Toxicity testing in the 21st century: a vision and a strategy. J Toxicol Environ Health B Crit Rev. 2010;13:51–138. doi: 10.1080/10937404.2010.483176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curti E, McDonald JP, Mead S, Woodgate R. DNA polymerase switching: effects on spontaneous mutagenesis in Escherichia coli. Mol Microbiol. 2009;71:315–331. doi: 10.1111/j.1365-2958.2008.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oller AR, Fijalkowska IJ, Dunn RL, Schaaper RM. Transcription-repair coupling determines the strandedness of ultraviolet mutagenesis in Escherichia coli. Proc Natl Acad Sci U S A. 1992;89:11036–11040. doi: 10.1073/pnas.89.22.11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul S, Million-Weaver S, Chattopadhyay S, Sokurenko E, Merrikh H. Accelerated gene evolution through replication-transcription conflicts. Nature. 2013;495:512–515. doi: 10.1038/nature11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahluwalia D, Schaaper RM. Hypermutability and error catastrophe due to defects in ribonucleotide reductase. Proc Natl Acad Sci U S A. 2013;110:18596–18601. doi: 10.1073/pnas.1310849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solanky D, Haydel SE. Adaptation of the neutral bacterial comet assay to assess antimicrobial-mediated DNA double-strand breaks in Escherichia coli. J Microbiol Methods. 2012;91:257–261. doi: 10.1016/j.mimet.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaivao I, Sierra LM. Drosophila comet assay: insights, uses, and future perspectives. Front Genet. 2014;5:304. doi: 10.3389/fgene.2014.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch AM, Sasaki JC, Elespuru R, Jacobson-Kram D, Thybaud V, De Boeck M, Aardema MJ, Aubrecht J, Benz RD, Dertinger SD, et al. New and emerging technologies for genetic toxicity testing. Environmental and molecular mutagenesis. 2011;52:205–223. doi: 10.1002/em.20614. [DOI] [PubMed] [Google Scholar]
- 10.Nohmi T, Suzuki T, Masumura K. Recent advances in the protocols of transgenic mouse mutation assays. Mutat Res. 2000;455:191–215. doi: 10.1016/s0027-5107(00)00077-4. [DOI] [PubMed] [Google Scholar]
- 11.Vogel EW, Nivard MJ. Model systems for studying germ cell mutagens: from flies to mammals. Adv Exp Med Biol. 2003;518:99–114. doi: 10.1007/978-1-4419-9190-4_9. [DOI] [PubMed] [Google Scholar]
- 12.Severinov K, Soushko M, Goldfarb A, Nikiforov V. RifR mutations in the beginning of the Escherichia coli rpoB gene. Mol Gen Genet. 1994;244:120–126. doi: 10.1007/BF00283512. [DOI] [PubMed] [Google Scholar]
- 13.Whong WZ, Stewart J, Ong T. Use of the improved arabinose-resistant assay system of Salmonella typhimurium for mutagenesis testing. Environmental mutagenesis. 1981;3:95–99. doi: 10.1002/em.2860030111. [DOI] [PubMed] [Google Scholar]
- 14.Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat Res. 2000;455:29–60. doi: 10.1016/s0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 15.Flamand N, Meunier J, Meunier P, Agapakis-Causse C. Mini mutagenicity test: a miniaturized version of the Ames test used in a prescreening assay for point mutagenesis assessment. Toxicol In Vitro. 2001;15:105–114. doi: 10.1016/s0887-2333(01)00003-0. [DOI] [PubMed] [Google Scholar]
- 16.Fluckiger-Isler S, Baumeister M, Braun K, Gervais V, Hasler-Nguyen N, Reimann R, Van Gompel J, Wunderlich HG, Engelhardt G. Assessment of the performance of the Ames II assay: a collaborative study with 19 coded compounds. Mutat Res. 2004;558:181–197. doi: 10.1016/j.mrgentox.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Cupples CG, Miller JH. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A. 1989;86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid C, Arndt C, Reifferscheid G. Mutagenicity test system based on a reporter gene assay for short-term detection of mutagens (MutaGen assay) Mutat Res. 2003;535:55–72. doi: 10.1016/s1383-5718(02)00282-6. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki T, Suzuki K, Tashiro Y, Saito K, Umeno D. Probing the mutation spectrum in E. coli. Nucleic Acids Symp Ser (Oxf) 2007:289–290. doi: 10.1093/nass/nrm145. [DOI] [PubMed] [Google Scholar]
- 20.Oller AR, Fijalkowska IJ, Schaaper RM. The Escherichia coli galK2 papillation assay: its specificity and application to seven newly isolated mutator strains. Mutat Res. 1993;292:175–185. doi: 10.1016/0165-1161(93)90145-p. [DOI] [PubMed] [Google Scholar]
- 21.Schaaper RM. Suppressors of Escherichia coli mutT: antimutators for DNA replication errors. Mutat Res. 1996;350:17–23. doi: 10.1016/0027-5107(95)00086-0. [DOI] [PubMed] [Google Scholar]
- 22.Marinus MG. DNA methylation and mutator genes in Escherichia coli K-12. Mutat Res. 2010;705:71–76. doi: 10.1016/j.mrrev.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JH, Michaels M. Finding new mutator strains of Escherichia coli--a review. Gene. 1996;179:129–132. doi: 10.1016/s0378-1119(96)00529-x. [DOI] [PubMed] [Google Scholar]
- 24.Matagne A, Lamotte-Brasseur J, Frere JM. Catalytic properties of class A beta-lactamases: efficiency and diversity. Biochem J. 1998;330(Pt 2):581–598. doi: 10.1042/bj3300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Firnberg E, Labonte JW, Gray JJ, Ostermeier M. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol Biol Evol. 2014;31:1581–1592. doi: 10.1093/molbev/msu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crameri A, Whitehorn EA, Tate E, Stemmer WP. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 27.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 28.Alexander DL, Lilly J, Hernandez J, Romsdahl J, Troll CJ, Camps M. Random mutagenesis by error-prone pol plasmid replication in Escherichia coli. Methods Mol Biol. 2014;1179:31–44. doi: 10.1007/978-1-4939-1053-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Troll C, Alexander D, Allen J, Marquette J, Camps M. Mutagenesis and functional selection protocols for directed evolution of proteins in E. coli. J Vis Exp. 2011 doi: 10.3791/2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camps M, Loeb LA. Critical role of R-loops in processing replication blocks. Front Biosci. 2005;10:689–698. doi: 10.2741/1564. [DOI] [PubMed] [Google Scholar]
- 31.Camps M, Naukkarinen J, Johnson BP, Loeb LA. Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proc Natl Acad Sci U S A. 2003;100:9727–9732. doi: 10.1073/pnas.1333928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Troll C, Yoder J, Alexander D, Hernandez J, Loh Y, Camps M. The mutagenic footprint of low-fidelity Pol I ColE1 plasmid replication in E. coli reveals an extensive interplay between Pol I and Pol III. Curr Genet. 2014;60:123–134. doi: 10.1007/s00294-013-0415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen JM, Simcha DM, Ericson NG, Alexander DL, Marquette JT, Van Biber BP, Troll CJ, Karchin R, Bielas JH, Loeb LA, et al. Roles of DNA polymerase I in leading and lagging-strand replication defined by a high-resolution mutation footprint of ColE1 plasmid replication. Nucleic Acids Res. 2011;39:7020–7033. doi: 10.1093/nar/gkr157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cesareni G, Helmer-Citterich M, Castagnoli L. Control of ColE1 plasmid replication by antisense RNA. Trends Genet. 1991;7:230–235. doi: 10.1016/0168-9525(91)90370-6. [DOI] [PubMed] [Google Scholar]
- 35.Million-Weaver S, Camps M. Mechanisms of plasmid segregation: have multicopy plasmids been overlooked? Plasmid. 2014;75:27–36. doi: 10.1016/j.plasmid.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guengerich FP. Metabolism of chemical carcinogens. Carcinogenesis. 2000;21:345–351. doi: 10.1093/carcin/21.3.345. [DOI] [PubMed] [Google Scholar]
- 37.Lee H, Popodi E, Tang H, Foster PL. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 2012;109:E2774–2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res. 2003;544:65–85. doi: 10.1016/s1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 39.Hakkila K, Maksimow M, Karp M, Virta M. Reporter genes lucFF, luxCDABE, gfp, and dsred have different characteristics in whole-cell bacterial sensors. Anal Biochem. 2002;301:235–242. doi: 10.1006/abio.2001.5517. [DOI] [PubMed] [Google Scholar]
- 40.Tse L, Kang TM, Yuan J, Mihora D, Becket E, Maslowska KH, Schaaper RM, Miller JH. Extreme dNTP pool changes and hypermutability in dcd ndk strains. Mutat Res. 2016;784-785:16–24. doi: 10.1016/j.mrfmmm.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain RK, Ranganathan R. Local complexity of amino acid interactions in a protein core. Proc Natl Acad Sci U S A. 2004;101:111–116. doi: 10.1073/pnas.2534352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muteeb G, Sen R. Random mutagenesis using a mutator strain. Methods Mol Biol. 2010;634:411–419. doi: 10.1007/978-1-60761-652-8_29. [DOI] [PubMed] [Google Scholar]
- 43.Badran AH, Liu DR. Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat Commun. 2015;6:8425. doi: 10.1038/ncomms9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.