

Abstract
In this article we introduce cellVIEW, a new system to interactively visualize large biomolecular datasets on the atomic level. Our tool is unique and has been specifically designed to match the ambitions of our domain experts to model and interactively visualize structures comprised of several billions atom. The cellVIEW system integrates acceleration techniques to allow for real-time graphics performance of 60 Hz display rate on datasets representing large viruses and bacterial organisms. Inspired by the work of scientific illustrators, we propose a level-of-detail scheme which purpose is two-fold: accelerating the rendering and reducing visual clutter. The main part of our datasets is made out of macromolecules, but it also comprises nucleic acids strands which are stored as sets of control points. For that specific case, we extend our rendering method to support the dynamic generation of DNA strands directly on the GPU. It is noteworthy that our tool has been directly implemented inside a game engine. We chose to rely on a third party engine to reduce software development work-load and to make bleeding-edge graphics techniques more accessible to the end-users. To our knowledge cellVIEW is the only suitable solution for interactive visualization of large bimolecular landscapes on the atomic level and is freely available to use and extend.
1. Introduction
Computational biology already offers the means to model large structural models of cell biology, such as viruses or bacteria on the atomic level [JGA*14] [JAAA*15]. Visualization of macromolecular structures plays an essential role in this modelling process of such organisms. The most widely known visualization softwares are: VMD [HDS96], Chimera [PGH*04], Pymol [DeL02], PMV [S*99], ePMV [JAG*11]. These tools, however, are not designed to render a large number of atoms at interactive frame-rates and with full-atomic details (Van der Walls or CPK spherical representation). Megamol [GKM*15] is a state-of-the-art prototyping and visualization framework designed for particle-based data and which currently outperforms any other molecular visualisation software or generic visualization frameworks such VTK/Paraview [SLM04]. The system is able to render up to 100 million atoms at 10 fps on commodity hardware, which represents, in terms of size, a large virus or a small bacterium. Larger bacteria, however, such as the well known E. coli, made out of tens of billions of atoms, which is two orders of magnitude bigger than what the highest-end available solution is able to render.
According to our domain experts, responsive visual feedback is of a great value for the modelling process of such organisms. However, none of the currently available solutions are able to serve the ambitions of our domain experts, which is to model large macromolecular structures such as E. coli. Related works have already presented bleeding-edge techniques that can render large datasets with up to billions of atoms at interactive framerates on commodity graphics hardware [LBH12] [FKE13] [LMPSV14]. However, to our knowledge, the tools which implemented these techniques were either not publicly available, or remained in the prototyping stage. Indeed, a very cumbersome task for researchers is releasing and maintaining a usable version of the source code once the article has been published. The presented techniques are often a proof-of-concept that would require substantial software development work to ensure a maximum degree of accessibility. Unfortunately, this is often omitted because of a busy research schedule and is simply left in the hand of interested third party developers. Consequently, if this task remains unachieved, end-users are unlikely to use state-of-the-art techniques in their work.
cellVIEW is a new solution that enables fast rendering of very large biological macromolecular scene. Unlike Megamol, which is designed for generic particle-data, cellVIEW is primarily designed for large biomolecular landscapes, and thus, exploits the repetitive nature of such structures to improve the rendering performance. While the main function of this tool is to assist our domain experts in their modelling task, the visualization of these datasets could also serve an educational purpose. By interactively showcasing the machinery of life in science museums, for instance, we could also improve the understanding of basic cell biology of the laymen audience.
cellVIEW is built on top of state-of-the-art techniques, and also introduces new means to efficiently reduce the amount of processed geometries. The approach we demonstrate in cellVIEW improves rendering performance compared to related work by introducing efficient occlusion culling and robust level-of-detail schemes. Our level-of-detail scheme also abstracts the shape of macromolecules efficiently, thus reducing visual clutter, as seen on the artistic depictions of David Goodsell in Figure 2. We showcase our tool with real, large-scale scientific data such as the HIV virus and Mycoplasma bacterium, which were provided by our cooperating domain scientists. Their datasets, not only contain information relative to the location of individual macromolecules, but also provide the path of nucleic acids strands, which is stored in the form of control points. We additionally extend our method to procedurally generate DNA strands on-the-fly via the GPU tessellation shader, thus reducing the modelling effort as well as GPU transfer times and memory space. Our system is implemented using a user-friendly and popular game engine. The ease of use of the engine guarantees our tool a maximum degree of accessibility, thus bridging the gap between bleeding-edge techniques and actual use in real applications. Additionally, since game engines are gaining in popularity among the visualization community, we anticipate third-party users adopting our tool, and thereby breaking the barriers caused by heterogeneous toolset usage across research departments.
Figure 2.
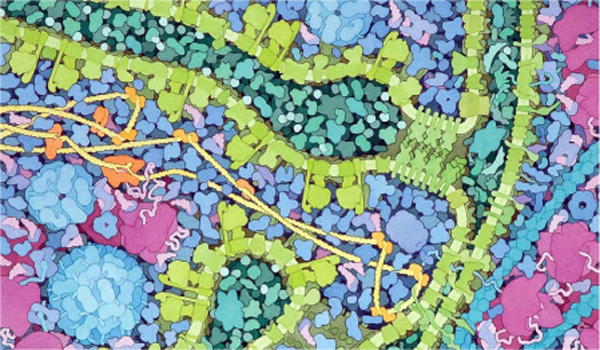
An illustration of David Goodsell depicting a cross section of a Mitochondrion. Given the complexity of the scene the artist deliberately chose to render molecules with highly abstracted shapes.
2. Related Work
Large-scale Molecular Visualization
Lindow et al. [LBH12] have first introduced a method capable of quickly rendering large-scale atomic data consisting of several billions of atoms on commodity hardware. Rather than transferring the data from CPU to GPU every frame, they store the structure of each type of molecule only once and utilize instancing to repeat these structures in the scene. For each type of protein a 3D grid structure containing all the atoms is created and then stored on the GPU memory. Upon rendering, the bounding boxes of the instances are drawn and individually raycasted, similar to volumetric billboards [DN09]. Subsequently Falk et al. [FKE13] presented a similar approach with improved depth culling and hierarchical ray casting for impostors that are located far away and do not require a full grid traversal. Although this implementation features depth culling, their method only operates on the fragment level, while they could have probably benefited from a culling on the instance level too. With their new improvement they managed to obtain 3.6 fps in full HD resolution for 25 billion atoms on a NVidia GTX 580, while Lindow et al. managed to get around 3 fps for 10 billions atoms in HD resolution on a NVIDIA GTX 285. Le Muzic et al. [LMPSV14], introduced another technique for fast rendering of large particle-based datasets using the GPU rasterization pipeline instead. They were able to render up to 30 billions of atoms at 10 fps in full HD resolution on a NVidia GTX Titan. They utilize tessellation shaders to inject atoms on-the-fly into the GPU pipeline similar to the technique of Lampe et al. [LVRH07]. In order to increase the rendering speed they dynamically reduce the number of injected atoms according to the camera depth. To simplify the molecular structures they discard atoms uniformly along the protein chain and increase the radius of remaining atoms to compensate for the volume loss. This level-of-detail scheme offers decent results for low degrees of simplification, but it does not guarantee preserving the initial shape of the molecules, resulting in poor image quality with highly simplified shapes.
Occlusion Culling
A key aspect when rendering large and complex scenes is efficient occlusion culling. Grottel et al. [GRDE10] presented a method to perform coherent occlusion culling for particle-based datasets, which is closely related to Deferred Splatting [GBP04] and relies on temporal coherency. Their particle data is stored in a uniform grid, and they operate the culling at two-levels: at the level of grid cells first, and at the atomic level afterwards. Individual atoms are rendered via 2D depth impostors, because they have a much lower vertex count than sphere meshes for the same results. At the beginning of each frame they render an early depth pass with atoms that were visible during the previous frame. This pass results in an incomplete depth buffer that they utilize to determine the visibility of the remaining particles. For the coarse-level culling they determine the visibility of the grid cells by testing their bounding boxes against the incomplete depth buffer via hardware occlusion queries (HOQ). For the fine-level culling they test the visibility of individual atoms in the final render using the well known hierarchical Z-buffer (HZB) visibility technique [GKM93]. They construct the HZB from the incomplete depth buffer beforehand, and during the final render, they discard fragment operations from the vertex shader if the visibility test fails, thus compensating for the lack of early fragment rejection with depth impostors.
Illustrative Molecular Visualization
When rendering large structures the speed of execution is not the only concern. As the structures increase in size, they are also increasing in complexity, and it is necessary to display the data in the most suitable way. Ambient occlusion, for instance, has been shown to play an essential role when dealing with large molecular structures, as it provides important depth cues which increase shape perception [GKSE12, ESH13]. But the rendering style is not the only means to define visual encoding, geometric abstraction should be applied as well. Parulek et al. [PJR*14], demonstrated a continuous level-of-detail scheme for molecular data. Their object-space approach offers detail-on-demand in the focus area while applying gradual shape simplification schemes elsewhere. At the finest level of detail they were showcasing solvent excluded surface (SES) representation and abstracted molecular shape for distant molecule. They introduced an interesting abstraction approach, other than molecular surfaces, based on union of spheres obtained via clustering methods. Several common clustering methods are compared and evaluated.
Modelling of Nucleic Acids Chains
DNA plays a key role in cell biology, and thus is an important part of our datasets. Therefore, as with protein data, we shall also provide the means for efficient rendering of this type of structure. There are several scientific modeling tools [MC98, LO08, HLLF13] designed to generate DNA strands from a simple set of control points. These techniques, however, are all performed on the CPU, which means that geometry data must be uploaded on the GPU prior to the rendering. Because of the cost of transferring data from CPU to GPU, such approach would likely perform poorly when rendering and animating long DNA strands. Therefore, we introduce a new GPU-based approach which relies on dynamic instancing of DNA base-pairs along a curve. This approach is similar to the work of Lampe et al. [LVRH07], who use the geometry shader to dynamically instantiate residues along the protein backbone. The major difference here is the introduction of procedural building rules based on scientific data and the use of the tessellation shader, which offer a much greater bandwidth of injected primitives. Moreover, by changing the building rules, our approach can also be extended and applied to fibres or repetitive objects that are present in cellular environment (actin filaments, microtubules, lipoglycane, etc.).
Game Engines and Biomolecular Visualization
Game engines are becoming increasingly popular in the molecular visualization community. Shepherd et al. [SZA*14] have developed an interactive application to showcase 3D genome data using a game engine. Their visualization is multi-scale and is able to render a large amount of data thanks to the implementation of a level-of-detail scheme. Various works on interactive illustration of biological processes have also mentioned using game engines to interactively visualize biomolecular processes in 3D, such as polymerization [KPV*14] and membrane crossings [LMWPV15]. Similarly to our work, Baaden et al. [LTDS*13] developed a molecular viewer which offers artistic and illustrative rendering methods based on the Unity3D game engine. Their primary intention was to democratize biomolecular visualization thanks to the use of a more intuitive and user friendly framework. Their tool has managed to prove that game engines are also useful in serious visualization projects. One noticeable technical difference between cellVIEW and UnityMol, other than the scale of the supported datasets, is that our tool is fully integrated in the “What you see is what you get” (WYSIWYG) editor of the Unity3D engine. Thus, our tool coexists with the engine toolset which provides a rich set of functionalities that can be directly used to enhance the quality of our visualization.
3. Efficient Occlusion Culling
The overwhelmingly increasing size of structural biology datasets calls for efficient means for reducing the amount of processed geometries. Our rendering pipeline is based on the work of Le Muzic et al. [LMPSV14], which relies on the tessellation shader to dynamically inject sphere primitives in the pipeline for each molecule. However, without proper occlusion culling, the injection of sphere primitives would still be performed, even if a molecule is completely hidden behind occluders. The presented occlusion culling method is inspired by the work of Grottel et al. [GRDE10]. We have revisited their technique to provide efficient occlusion culling for macromolecular datasets that are several orders of magnitude larger than the ones showcased with their method.
3.1. Temporal coherency
We developed a custom visibility technique, implemented with compute shaders and using the well-known hierarchical Z-buffer (HZB) occlusion culling. This solution has the advantage to reduce GPU driver overhead compared to HOQ used by Grottel et al. [GRDE10], since multiple queries can be performed in a single call. The approach rely on the use of an item-buffer to precisely determine the visibilty of the molecules at the end of a frame. Then at the beginning of the next frame, the previously visible molecules are firstly drawn. This will result in an partially complete frame, in case of eventual camera motion. The next step is to determine the remaining visible elements in order to complete the frame. We generate the HZB from the partially complete depth buffer and we compute the visibility information for the remaining molecules. The remaining visible molecules are finally drawn and we use the item buffer to determine which molecules are present on the screen at the end of this frame. The sequential steps of our occlusion culling method for a given frame are laid down as follows:
Clear HZB and depth buffer
Draw visible molecules at the previous frame
Generate HZB from the depth buffer obtained in step 2
Compute HZB-visibility for the remaining molecules
Draw HZB-visible molecules from step 4
Find visible molecules via item-buffer for the next frame
3.2. Accelerating Texture Writes
Individual atoms are rendered via 2D sphere impostors, because they have a much lower vertex count than sphere meshes for the same results. The depth of the sphere impostors is corrected in the fragment shader in order to mimic a spherical volume. Upon drawing the atoms, many of them are actually occluded by other atoms of the same or surrounding molecules. These atoms would normally be processed, as a well-known limitation of graphics hardware, so far, has been the lack of early depth fragment rejection for depth impostors. Thanks to advances in graphics hardware however, it is now possible to activate early depth rejection when a fragment is modifying the output depth value. Hence, thanks to this feature, we may now easily avoid fragment computation for hidden atoms. This feature is called conservative depth output. Once activated, in order for conservative depth output to work, we must output a depth which is greater than the depth of the 2D billboard. This way the GPU is able to tell if a fragment will be occluded beforehand by querying the visibility internally. A description of the depth conservative output sphere impostor is given in Figure 3. Additionally, to limit the number of texture writes, we only output the id of the molecules to the render texture upon rendering. The colors are fetched afterwards in post-processing by reading the molecules properties from the id.
Figure 3.
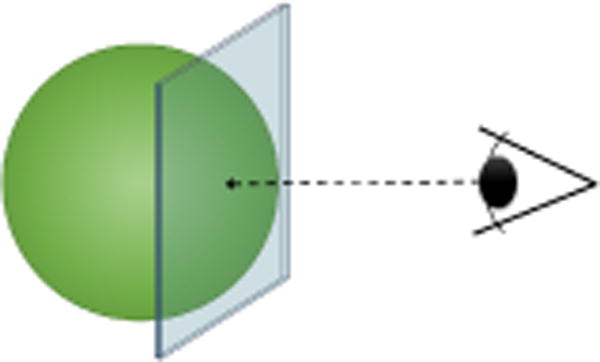
Depth conservative sphere impostors, in order to benefit from early depth culling for depth impostors we must guaranty that the output depth will be greater than the depth of the billboard.
4. Twofold Level-of-Detail
Proteins are key elements of biological organisms, and thus it is important to visualize them in order to understand how these work. They are also present in fairly large quantities, which is challenging to render interactively without proper level-of-detail schemes (LOD). Additionally, their complex shapes might cause a high degree of visual clutter, which may render overly complex images. We propose a twofold LOD scheme which provides rendering acceleration and offers a clearer depiction of the scene using smoothly abstracted shapes. Our technique also offer a seamless continuum between the different levels of abstractions from highly detailed to highly abstracted.
Our rendering pipeline is based on the work of Le Muzic et al. [LMPSV14], where LOD was dynamically determined during the tessellation stage. To reduce the number of spheres, atoms were periodically skipped along the protein backbone, and the radii of remaining atoms were increased to compensate the volume loss. This technique, although fully dynamic, offers poor results for highly decimated molecules since it does not guarantee to preserve the overall shape. We employ clustering methods instead, similar to the technique of Parulek et al. [PJR*14], to simplify the shape of the molecules and reduce the number of primitives to render. Atoms corresponding to one cluster are replaced by a single sphere with a radius that approximates the size of the cluster. Clustering offers a very good decimation ratio as well as accurate shape abstraction, because it tends to preserve low-frequency details. With higher shape accuracy we are also able to switch to simpler LOD proxies closer to the camera, thus gaining in render speed without compromising image quality.
The clustering of the molecules is precomputed and results in a set of spheres which are stored in the GPU memory. We compute our LOD levels using a GPU-based K-means clustering algorithm. In our tests, we deemed that four levels were sufficient with our current datasets. The compression factor of each level was manually chosen to obtain the best performance/image quality ratio. The results of the clustering of our four levels is shown in Figure 4. These parameters can be easily changed via the editor interface. A side-by-side comparison between our illustrative LOD compared to full atomic detail is provided in Figure 5.
Figure 4.
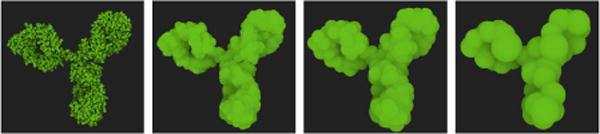
Our level-of-detail scheme allows to reduce the number of sphere primitives from 10182 to 50 while preserving the overall shape of the protein. From left to right, the protein is shown with (i) full-atomic detail, (ii) only 15 percent of the overall sphere count, (iii) 5 percent and (iv) 0.5 percent.
Figure 5.

Side-by-side comparison of our illustrative LOD compared with full atomic details. Our illustrative LOD provides smoother and elegant shapes, while also reducing the processing load.
5. Dynamic DNA Generation
Animating individual molecules is fairly straightforward because modifying the atomic structure may not be required. In the case of DNA, the positions of the control points of the DNA path highly influence its structure, namely the positions and rotations of the individual nucleic acids. As a result, each modification of the control points of the DNA path requires a new computation of the strand. Current approaches are only performed on the CPU, [HLLF13, LO08, MC98] which means that the whole nucleic acids chain has to be transferred to the GPU upon re-computation. While this approach is viable for low to mid sized DNA strands, it is likely to perform poorly for large and dynamic DNA paths featuring a large number of control points.
We propose to use the dynamic tessellation to leverage the generation of nucleic acid strands. So far we have only used tessellation to instantiate data stored in the GPU memory. However it is possible to include building rules characteristic to the DNA’s well known geometry to procedurally generate a double helix structure simply based on control points. Thus, data transfers as well as GPU memory space can be dramatically reduced.
Similar to GraphiteLifeExplorer [HLLF13], our goal is more illustrative than strict biomolecular modeling. Therefore we privilege rendering performance over accuracy, and we provide only a limited array of folding types. Although the study of DNA structures has revealed many different types of folding, requiring complex modeling algorithms, the most commonly recognizable shape that of B-DNA that exhibits a regular structure which is simple to model: a spacing of 3.4Å and a rotation of 34.3° between each base. Based on these rules we are able to procedurally generate B-DNA strands based on path control points via GPU dynamic tessellation. The workflow which we employ is described as follow:
Resample control points (on the CPU).
Compute smooth control point normals (on the CPU).
Upload control point data to the GPU
Draw all the path segments in one pass, one vertex shader per segment
Read the control points and adjacent points needed for smooth cubic interpolation. (In vertex shader, for each segment)
Do uniform sampling along the cubic curve segment to determine the positions of the bases. (In vertex shader, for each segment)
Pass the position of the bases to the tessellation shader. (In vertex shader, for each segment)
Compute normal vector of each base using linear interpolation between the control points normals (In tessellation shader, for each base)
Inject atom, then translate and rotate accordingly (In tessellation shader, for each atom of each base)
Render sphere impostor from injected atom (In geometry & fragment shader, for each atom of each base)
5.1. Smooth Normals Computation
A well known challenge when dealing with 3D splines is to determine smooth and continuous frames along the whole curve. Any twists or abrupt variation in frame orientation would cause visible artifacts due to irregularities in the DNA structure, which should be avoided at all costs. We perform the computation of the smooth and continuous frames primarily on the CPU. We first determine the normal direction for every control point of the path. Then, we sequentially browse the control points and rotate the normal direction vector around the tangent vector in order to minimize the variation in orientation compared to the previous control point normal. The recalculated normals are then uploaded to the GPU along with the control points positions. Then, during the instantiation of the nucleic acids, we obtain the normal vector of a nucleic acid by linear interpolation between the two normal vectors of the segment.
5.2. Double Helix Instancing
When instancing individual pairs of nucleic acids in the tessellation shader, we first fetch the nucleic acid atoms, position them along the curve, orient them toward the normal direction and then rotate then around the tangent vector in order to generate the double helix. We always orient the first base of a segment according to the normal direction only, while the subsequent bases are all oriented towards the normal direction first and then rotated with an increasing angular offset of 34.3° around the tangent of the curve. The angular offset of a given base is defined as follows: α = i × 34.3, where i corresponds to the index of the base inside a segment. The last base of a segment must therefore always perform an offset rotation of (360 − 34.3)° around the tangent vector. This way it connects smoothly to the first base of the next segment, which is oriented towards the normal vector only. The result of the procedural generation of B-DNA is given in Figure 6 as well as a visual explanation of the different steps.
Figure 6.
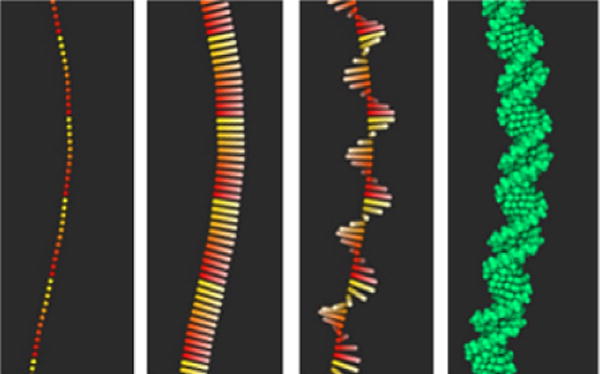
Procedural generation of B-DNA structures via GPU dynamic tessellation. In the first image we can see the position of the individual bases. The color gradient highlights the individual segments. In the second image we draw the smooth normals along the curve, the color desaturation shows the direction of the vector. The third image shows the rotation offset of the normal vector along the tangent, and the last image shows the final result.
5.3. Control Points Resampling
Given that the bases of a segment must perform a revolution to connect smoothly to the next segment, it is trivial to determine the number of bases per segment as follows: n = 360 ÷ 34,3. From the number of bases per segment we can easily deduce the required size of a segment as follows: s = n × 3.4 Å, which results in a segment length of approximately 35 Å. This constraint implies that all control points be spaced uniformly with a distance of 35 Å. However, it may be the case that control points obtained via modelling software have arbitrary spacing. Therefore, we must resample the control points along the curve to ensure a uniform spacing before uploading it to the GPU. Although we resample the control points according to the B-DNA build rules, the length of the interpolated curve segments will always be slightly greater because of the curvature. We did not find this to be visually disturbing, mostly because consecutive segments in our dataset did not showcase critically acute angles, so the overall curvature of individual curve segments remained rather low.
6. Results
We have tested our tool with several datasets of different nature and sizes. The datasets were modelled with cellPACK [JAAA*15], a modelling tool for procedural generation of large biomolecular structures. cellPACK is developed and used by our domain experts, it is publicly available and offers to anyone the means for experimenting and creating their own models. Our program reads the files that are generated by cellPACK and is able to reconstruct and display the scene in a multiscale approach. The generated files comprise of a list of elements with their properties such as name, position, rotation, and PDB identifier that indicates the atomic structure [SLJ*98]. The structural data is directly fetched online from the Protein Data Bank via the PDB identifier. In case an entry is not present or refers to a custom PDB file, we load the protein information from a dedicated repository provided by the domain experts. The generated files also include control points for the linear or repetitive type of structures such as DNA, unfolded peptide, lypoglycane, etc.
6.1. Use cases
HIV Virus + Blood Plasma
The first dataset we showcase is a combination of two datasets: the HIV virus [JGA*14] surrounded by blood plasma. The HIV is a retrovirus and thus only contains RNA, which features much more complex modeling rules than DNA and forbids dynamic procedural generation. For this specific case the atomic structure of RNA would have to be modelled ad-hoc with a third party tool before being loaded in cellVIEW. Without the genomic information, the dataset comprises a total of 60 millions atoms consisting in 40 different types of molecules. For the purpose of benchmarking, we periodically repeat this dataset to reach an overall number of 15 billion atoms.
Mycoplasma
To demonstrate the use of our dynamic building rules for DNA, we use the data from Mycoplasma mycoide, one of the smallest bacteria with a genome of 1,211,703 baise pairs. Mycoplsama has been widely studied by biologists, and was the first organism to be fully synthetized. For this dataset we only showcase a preliminary model built with cellPACK and containing only a quarter of the total genome. This dataset comprises a set of 9617 control points defining the overall path of the DNA and the PDB reference of the nucleic acid base pairs. The pairs are instanced along the path resulting in an overall number of 11,619,195 atoms. We were able to procedurally generate and render the entire dataset at 70 fps without any culling nor LOD schemes. With this test we simply wanted to show the raw computation time in order to demonstrate the efficiency of our technique. Naturally, when using LOD and culling schemes like with protein data, the performance would considerably increase and would not impact the rest of the computation. The results of the two datasets are shown in Figure 7, and a preliminary render of the Mycoplasma is shown in Figure 8.
Figure 7.
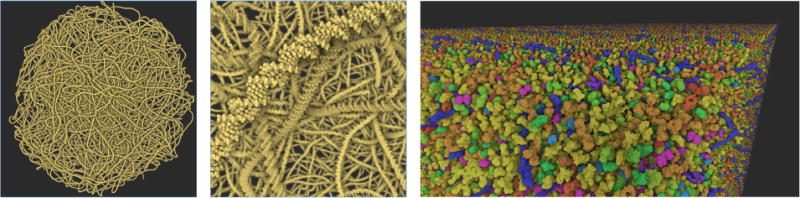
The results of our rendering test, showing DNA from Mycoplasma on the left and HIV in blood plasma on the right. The first dataset has approximatively 11 million atoms and the second one approximatively 15 billion
Figure 8.

Preliminary results of the Mycoplasma model. The model additionally features proteins, RNA and lycosomes.
6.2. Performance Analysis
It is rather challenging to precisely evaluate the performance of our tool, as the speed of execution depends on many factors, such as camera position or level-of-detail parameters, and which are arbitrarily chosen. It is also worth mentioning that available software solutions are not able to deal with the amount of data presented in our largest datasets. Therefore we did not perform a thorough comparison with available software solutions and related work. We perform an intra-performance evaluation instead, using our different datasets.
Table 1 provides a descriptive listing of the rendering performance for each dataset, with and without our optimizations. The rendering tests were performed on an Intel Core i7-3930 CPU 3.20 GHz machine coupled with a GeForce GTX Titan X graphics card with 12GB of video RAM. During our tests we have monitored the rendering speed with various camera settings, from far-out to close-up and from many angles. The measured performance represents the slowest render speed obtained, in frame per seconds at full HD resolution. The LOD parameters were carefully tuned in order to obtain the best ratio between performance and image quality. From these results we can clearly see the impact of the LOD in terms of performance for all datasets. We can also observe that the culling greatly improves the rendering speed when displaying a very dense dataset. Additionally, our tool is able to render datasets which are equivalent in size to the ones showcased in related work at higher framerates (> 60fps).
Table 1.
Performance comparison for each dataset used in our study. During our tests we have monitored the rendering speed with various camera settings, from far-out to close-up and from many angles. The measured performance represents the slowest render speed obtained, in frame per seconds at full HD resolution. The first column shows the size of the dataset in terms of number of atoms, then from left to right: without optimizations, with LOD only, with occlusion culling only and finally with LOD and occlusion culling.
| Dataset | Size | Raw | LOD | O. Culling | LOD + O. Culling |
|---|---|---|---|---|---|
| HIV | 15M | 80 | 130 | 110 | 140 |
| HIV + blood plasma | 60M | 25 | 90 | 60 | 120 |
| HIV + blood plasma × 250 | 15B | <1 | 15 | <1 | 60 |
| Mycoplasma DNA | 12M | 70 | n/a | n/a | n/a |
It is worth mentioning that it would always be possible to render larger datasets at more than 60 fps using more aggressive LOD settings and thus trading image quality. However, one could question the utility of this approach to display datasets that would be one or several orders of magnitude larger. Indeed, when viewing our largest dataset in its entirety, the view starts to exhibit graining artefacts due to the very small screen-size of individual molecules. These arte-facts create unwanted visual clutter, and therefore another type of approach rather than the particle-based one should be considered in this case.
7. Expert Feedback & Discussion
Domain experts who have experimented with cellVIEW have responded favorably and with great enthusiasm. One of our domain experts, a core actor of the cellPACK project, wrote:
Prior to cellVIEW, visualizing this type of data was cumbersome for the experts and as the scale increased, it was often not possible to view large models with all structures turned on with a standard computer. cellVIEW now provides state-of-the-art techniques to accomplish this task. Some experts were dismayed that cellVIEW could not yet be implemented in their lab’s preferred or homemade visualization toolsets (i.e., not simply a python or C++ library they could access), but most had some experience working with the Unity 3D framework, so the transition to this standalone tool was sufficient. For large biological structures, such as Mycoplasma mycoides, the cellPACK viewers are currently unable to visualize the complete models produced by the packing algorithm. Because cellVIEW can handle Mycoplasma and larger models in atomic detail and with ease, it is evident that cellVIEW will become a critical tool for cellPACK users who wish to explore multi-scale modeling extremes such whole bacterial cells and ultimately whole mammalian cells.
cellVIEW is open source, free to use, and available online, as well as the datasets modelled with cellPACK (https://github.com/illvisation/cellVIEW). With cellVIEW we wanted to guarantee the maximum degree of accessibility as possible. Therefore, we opted for a well-known, generic, and universal development framework to encourage third-party users to experiment and also to contribute to our project, such as visualization scientists, scientific illustrators, biologists or students. Although this solution might not have been the most preferred one for our main users at first, the ease of use of the tool has shown to be very valuable to them. The main advantage to us is that the development and maintenance of the core platform is already taken care of. This allows small teams of researchers to allocate their resources more efficiently and to focus on developing the actual technologies more quickly. However, the engine also presents a few drawbacks which would need to be addressed in the future in order to become a stronger contender as a visualization framework. Firstly, the advanced GPU programming features we use to develop cellVIEW are based on DirectX 11, which makes our tool only available to Windows platforms, at least until Unity3D supports advanced GPU programming with OpenGL. Another major drawback is that the source code of the core of the engine is not yet publicly available, which may be critical in case a missing core feature would need to be manually coded.
8. Conclusions and Future Work
We have introduced cellVIEW, a tool for real-time multi-scale visualization of large molecular landscapes. Our tool is able to load files generated by cellPACK a powerful modeling tool for representing entire organisms at the atomic level. cellVIEW was engineered to work seamlessly inside the Unity3D game engine, which allows us to prototype and deploy quickly and to leverage performance via advanced GPU programming. The method which we presented also features notable improvements over previous works. We provide the means for efficient occlusion culling, which is crucial when dealing with such large scale datasets. We also implemented a level-of-detail scheme, which allows both acceleration of rendering times and provides a clear and accurate depiction of the scene. Finally, we demonstrated the use of dynamic tessellation to generate biolmolecular structures on-the-fly based on scientific modeling rules.
In future work we would like to tighten the collaboration with domain experts and achieve interactive viewing of more complex organisms and bacteria such as E. coli. As the scale increases the view exhibits highly grainy results due to the very small size of molecules. In the future we would like to focus on better representation for this case, and perhaps find new semantics that could be integrated in our level-of-detail continuum. We also would like to use our rendering to experiment with in-situ simulations as a visual exploration tool for scientists, and also as an educational tool to showcase the machinery of life to a lay audience.
Figure 1.
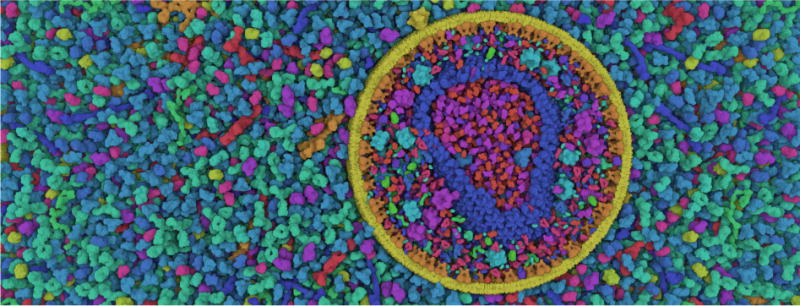
Real-time screen-shot of an illustrative cross-section of the HIV virus surrounded by blood plasma. Our rendering tool is directly integrated in the Unity3D game engine and is able to render datasets with up to 15 billion atoms smoothly at 60Hz and in high resolution. Because these datasets exhibit high visual complexity, we opted for an illustrative rendering style to improve shape perception, inspired by the style of scientific illustrators.
Acknowledgments
This project has been funded by the Vienna Science and Technology Fund (WWTF) through project VRG11-010 and also supported by EC Marie Curie Career Integration Grant through project PCIG13-GA-2013-618680. Additionally, this work has been carried out within the PhysioIllustration research project 218023, which is funded by the Norwegian Research Council. Autin, L. received support from the National Institutes of Health under award number P41GM103426.
References
- [DeL02].DELANO WL. The pymol molecular graphics system [Google Scholar]
- [DN09].DECAUDIN P, NEYRET F. Computer Graphics Forum. Vol. 28. Wiley Online Library; 2009. Volumetric billboards; pp. 2079–2089. [Google Scholar]
- [ESH13].EICHELBAUM S, SCHEUERMANN G, HLAWITSCHKA M. Pointao improved ambient occlusion for point-based visualization [Google Scholar]
- [FKE13].FALK M, KRONE M, ERTL T. Computer Graphics Forum. Vol. 32. Wiley Online Library; 2013. Atomistic visualization of mesoscopic whole-cell simulations using ray-casted instancing; pp. 195–206. [Google Scholar]
- [GBP04].GUENNEBAUD G, BARTHE L, PAULIN M. Computer Graphics Forum. Vol. 23. Wiley Online Library; 2004. Deferred splatting; pp. 653–660. [Google Scholar]
- [GKM93].GREENE N, KASS M, MILLER G. Proceedings of the 20th annual conference on Computer graphics and interactive techniques. ACM; 1993. Hierarchical z-buffer visibility; pp. 231–238. [Google Scholar]
- [GKM*15].GROTTEL S, KRONE M, MULLER C, REINA G, ERTL T. Megamol a prototyping framework for particle-based visualization. Visualization and Computer Graphics, IEEE Transactions on. 2015;21(2):201–214. doi: 10.1109/TVCG.2014.2350479. [DOI] [PubMed] [Google Scholar]
- [GKSE12].GROTTEL S, KRONE M, SCHARNOWSKI K, ERTL T. Pacific Visualization Symposium (PacificVis), 2012 IEEE. IEEE; 2012. Object-space ambient occlusion for molecular dynamics; pp. 209–216. [Google Scholar]
- [GRDE10].GROTTEL S, REINA G, DACHSBACHER C, ERTL T. Computer Graphics Forum. Vol. 29. Wiley Online Library; 2010. Coherent culling and shading for large molecular dynamics visualization; pp. 953–962. [Google Scholar]
- [HDS96].HUMPHREY W, DALKE A, SCHULTEN K. Vmd: visual molecular dynamics. Journal of molecular graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- [HLLF13].HORNUS S, LÉVY B, LARIVIÈRE D, FOUR-MENTIN E. Easy dna modeling and more with graphitelifeexplorer. PloS one. 2013;8(1):53609. doi: 10.1371/journal.pone.0053609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [JAAA*15].JOHNSON GT, AUTIN L, AL-ALUSI M, GOOD-SELL DS, SANNER MF, OLSON AJ. cellpack: a virtual mesoscope to model and visualize structural systems biology. Nature methods. 2015;12(1):85–91. doi: 10.1038/nmeth.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [JAG*11].JOHNSON GT, AUTIN L, GOODSELL DS, SANNER MF, OLSON AJ. epmv embeds molecular modeling into professional animation software environments. Structure. 2011;19(3):293–303. doi: 10.1016/j.str.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [JGA*14].JOHNSON GT, GOODSELL DS, AUTIN L, FORLI S, SANNER MF, OLSON AJ. 3d molecular models of whole hiv-1 virions generated with cellpack. Faraday discussions. 2014;169:23–44. doi: 10.1039/c4fd00017j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [KPV*14].KOLESAR I, PARULEK J, VIOLA I, BRUCKNER S, STAVRUM A-K, HAUSER H. Illustrating polymerization using three-level model fusion. arXiv preprint arXiv:1407.3757. 2014 doi: 10.1186/1471-2105-15-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [LBH12].LINDOW N, BAUM D, HEGE H-C. Computer Graphics Forum. Vol. 31. Wiley Online Library; 2012. Interactive rendering of materials and biological structures on atomic and nanoscopic scale; pp. 1325–1334. [Google Scholar]
- [LMPSV14].LE MUZIC M, PARULEK J, STAVRUM A-K, VIOLA I. Computer Graphics Forum. Vol. 33. Wiley Online Library; 2014. Illustrative visualization of molecular reactions using omniscient intelligence and passive agents; pp. 141–150. [Google Scholar]
- [LMWPV15].LE MUZIC M, WALDNER M, PARULEK J, VIOLA I. Visualization Symposium (PacificVis), 2015 IEEE Pacific. IEEE; 2015. Illustrative timelapse: A technique for illustrative visualization of particle-based simulations; pp. 247–254. [Google Scholar]
- [LO08].LU X-J, OLSON WK. 3dna: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nature protocols. 2008;3(7):1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [LTDS*13].LV Z, TEK A, DA SILVA F, EMPEREUR-MOT C, CHAVENT M, BAADEN M. Game on, science-how video game technology may help biologists tackle visualization challenges. PloS one. 2013;8(3):57990. doi: 10.1371/journal.pone.0057990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [LVRH07].LAMPE OD, VIOLA I, REUTER N, HAUSER H. Two-level approach to efficient visualization of protein dynamics. Visualization and Computer Graphics, IEEE Transactions on. 2007;13(6):1616–1623. doi: 10.1109/TVCG.2007.70517. [DOI] [PubMed] [Google Scholar]
- [MC98].MACKE TJ, CASE DA. Modeling unusual nucleic acid structures [Google Scholar]
- [PGH*04].PETTERSEN EF, GODDARD TD, HUANG CC, COUCH GS, GREENBLATT DM, MENG EC, FERRIN TE. Ucsf chimera a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [PJR*14].PARULEK J, JÖNSSON D, ROPINSKI T, BRUCKNER S, YNNERMAN A, VIOLA I. Computer Graphics Forum. Vol. 33. Wiley Online Library; 2014. Continuous levels-of-detail and visual abstraction for seamless molecular visualization; pp. 276–287. [Google Scholar]
- [S*99].SANNER MF, et al. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61. [PubMed] [Google Scholar]
- [SLJ*98].SUSSMAN JL, LIN D, JIANG J, MANNING NO, PRILUSKY J, RITTER O, ABOLA E. Protein data bank (pdb): database of three-dimensional structural information of biological macromolecules. Acta Crystallographica Section D: Biological Crystallography. 1998;54(6):1078–1084. doi: 10.1107/s0907444998009378. [DOI] [PubMed] [Google Scholar]
- [SLM04].SCHROEDER WJ, LORENSEN B, MARTIN K. The visualization toolkit. Kitware; 2004. [Google Scholar]
- [SZA*14].SHEPHERD JJ, ZHOU L, ARNDT W, ZHANG Y, ZHENG WJ, TANG J. Exploring genomes with a game engine. Faraday discussions. 2014;169:443–453. doi: 10.1039/c3fd00152k. [DOI] [PubMed] [Google Scholar]