

Abstract
Primary adrenal disorders contribute 20%–30% of patients with endogenous Cushing's syndrome. Most of the primary adrenal diseases are unilateral and include adenoma and adrenocortical carcinoma, whereas bilateral adrenal lesions are uncommon and include primary pigmented nodular adrenocortical disease, primary bilateral macronodular adrenocortical hyperplasia, isolated micronodular adrenocortical disease, bilateral adenomas or carcinomas, and rarely pituitary adrenocorticotropic hormone-dependent adrenal nodular disease. Cyclic adenosine monophosphate-dependent protein kinase A signalling is the major activator of cortisol secretion in primary adrenal nodular disorders. We report two cases of bilateral adrenal nodular disease with endogenous Cushing's syndrome, including one each of primary pigmented nodular adrenocortical disease and primary bilateral macronodular adrenocortical hyperplasia.
Keywords: adrenal disorders, endocrine cancer
Background
Primary adrenal disorders are responsible for 20%–30% of patients of endogenous Cushing’s syndrome (CS).1 Unilateral adrenal adenoma causing CS contributes to approximately 75%–95% of the cases, while in 5% it is caused by adrenocortical carcinoma. Disorders with primary bilateral adrenal lesions causing CS contribute to 10% of cases and include primary pigmented nodular adrenocortical disease (PPNAD), primary bilateral macronodular adrenocortical hyperplasia (PBMAH), isolated micronodular adrenocortical disease (iMAD) and rarely bilateral adenomas or carcinomas.1–3 The development, proliferation and functions of adrenocortical cells are mainly regulated by the cyclic adenosine monophosphate-protein kinase A (cAMP–PKA) pathway. Mutations in cAMP–PKA signalling pathway (GNAS, PRKAR1A PRKACA, PDE11A, PDE8B), β-catenin pathway (CTNNB1) and mutations of ARMC5, MEN-1, APC, FH, MC2R lead to the tumour development in majority of the patients with adrenal adenoma, PPNAD and PBMAH leading to CS.1 4 PPNAD occurs sporadically in half of the cases, and in the rest, familial as a part of Carney’s complex (CNC), which is an autosomal dominant disorder associated with lentigens, cardiac myxoma and multiple endocrine and non-endocrine tumours. PPNAD usually presents in young adults with mild hypercortisolemia. It can also present as cyclical CS or may be asymptomatic in familial forms like CNC.5 Sometimes these patients may have paradoxical increase in cortisol in response to high-dose dexamethasone suppression test (HDDST). Diagnosis is confirmed on histopathological findings and genetic analysis.6 PBMAH usually presents in the fifth and sixth decades with equal gender distribution. It may be sporadic or familial with autosomal dominant inheritance and can present as overt or subclinical CS. However, PBMAH associated with McCune-Albright syndrome (MAS) presents in the first year of life. PBMAH is characterised by aberrant expression or function of one or several G-protein-coupled receptors (GPCR) leading to cell proliferation and abnormal regulation of steroidogenesis, or mutations of different genes leading to increased cortisol production resulting in CS.6–9
Case presentation
Case 1
A 3-year-old male child, who was born full term by emergency caesarean section for cord around the neck, had birth weight of 3.5 kg and was apparently normal until the age of 2.5 years. He presented with weight gain of approximately 7.5 kg over 5 months, facial plethora, roundening of face, voracious appetite, appearance of facial and pubic hairs, and hypertension. On examination, his height was 88 cm (between the 3rd and 10th percentile), his weight was 19.2 kg (>97th centile), had roundening of face, facial plethora, dorsocervical fat pad, central obesity, knuckle hyperpigmentation, acanthosis nigricans (grade III) and pubic vellus hairs. In addition, he had features of protein catabolism, including proximal muscle weakness, easy bruisability and cuticular atrophy; however, there were no striae. His blood pressure (BP) was 130/90 mm Hg (>95th percentile). He did not have any neurocutaneous markers like cafe-au-lait macule, lentigens, neurofibroma, bony swelling or other stigmata of CNC. There was no family history suggestive of similar illness. Haemogram, electrolytes, blood glucose, liver and renal function tests were normal. Hormonal evaluation revealed 08:00 hour plasma cortisol 1203 nmol/L (N: 171–536) and ACTH <1 pg/mL (N: 5–60), midnight plasma cortisol 1017 nmol/L and ACTH <1 pg/mL, dehydroepiandrosterone sulphate (DHEAS) 30.31 µg/dL (N:0.47–19.14), T3 1.38 ng/mL (N: 0.8–2.0), T4 8.6 µg/dL (N: 4.8–12.7), thyroid stimulating hormone (TSH) 0.95 µIU/mL (N: 0.27–4.2), luteinizing hormone (LH) 0.1 mIU/mL (N: <0.3 in prepubertal children), follicle stimulating hormone (FSH) 0.1 mIU/mL (N: <1 prepubertal children), prolactin 9.7 ng/mL (N: 4.7–23), testosterone 1.44 nmol/L (N:<1.7), 1 mg overnight dexamethasone suppression test (1mg-ONDST) 1107 nmol/L (N: <50), low-dose dexamethasone suppression test (LDDST) 1137 nmol/L (N: <50) and HDDST 925.7 nmol/L (N: >50% suppression). Contrast-enhanced CT (CECT) scan of the abdomen showed bulky medial limb of the left adrenal gland and nodular ‘string of beads’ appearance of the right adrenal gland. A diagnosis of primary pigmented nodular adrenal disease (PPNAD) was considered. Bilateral laparoscopic adrenalectomy was performed. On gross examination, right adrenal measured 3×2×1.5 cm and weighed 3 g, left adrenal gland measured 4×4×3 cm and weighed 20 g, and the cut surface of both the glands showed focal yellowish areas and blackish pigmentation. Histopathological examination of the adrenals showed multiple pigmented nodules of size 2–4 mm and hyperplastic cells with intervening atrophic cortical tissue, suggestive of PPNAD (figure 1A–D). Genetic analysis could not be done due to non-availability of the test. The child was started on hydrocortisone and fludrocortisone. He is doing well for the last 3 years with plasma ACTH in reference range.
Figure 1.
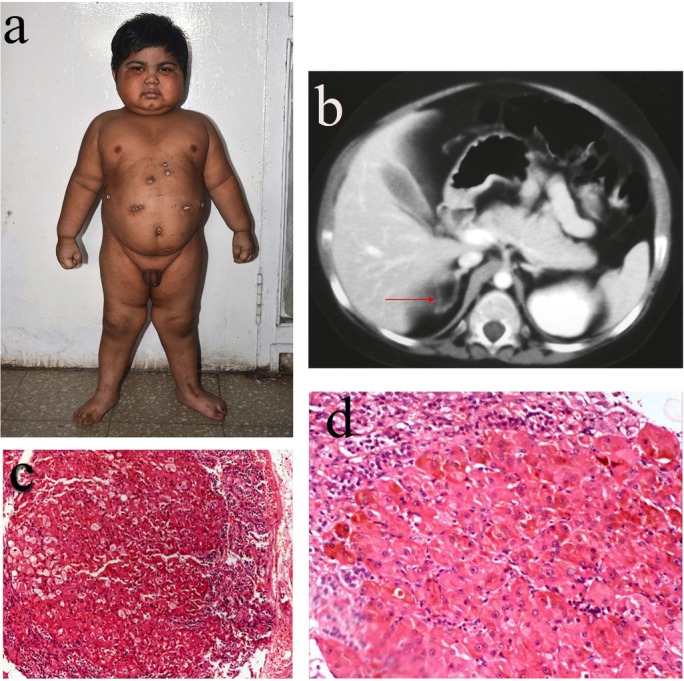
(A) Clinical picture of the patient with primary pigmented nodular adrenocortical disease. (B) Contrast-enhanced CT of the abdomen showing ‘string of beads’ appearance of the right adrenal gland. (C) Microphotograph showing large nodule comprising cells with abundant eosinophilic cytoplasm and compressed adrenal at periphery (H&E 10×). (D) Higher magnification showing abundant brownish pigment within the cytoplasm (H&E 20×).
Case 2
A 32-year-old female presented with complaints of excessive hair growth over the face for 7 years, oligomenorrhea, weight gain and proximal muscle weakness of 3-year duration. She was a known case of diabetes mellitus for 7 years and was hypertensive for the last 5 years. On examination, her weight was 64 kg, with body mass index of 23.5 kg/m2 and waist circumference of 94 cm. She had roundening of face, hirsutism (Ferriman-Gallwey score 15/36), dorsocervical fat pad, striae, ecchymosis, cuticular atrophy, proximal muscle weakness and easy bruisability. Her BP was 160/100 mm Hg. Haemogram, electrolytes, blood glucose, renal and liver function tests were normal. Hormonal evaluation showed 08:00 hour plasma cortisol 958 nmol/L, ACTH <1 pg/mL, midnight plasma cortisol 1073 nmol/L (N: <207 nmol/L), DHEAS 30.31 µg/dL (N: 98–340), testosterone 2.01 nmol/L (N: 0.2–2.9), 1mg-ONDST 1298 nmol/L (N : <50), LDDST 804 nmol/L (N : <50) and HDDST 1043 nmol/L. CECT of the abdomen revealed bilateral enlarged bulky nodular adrenal glands with a nodular lesion in the right adrenal gland. A diagnosis of PBMAH was considered and she was evaluated for aberrant stimulators. Serum cortisol samples were drawn from an indwelling cannula at baseline, 30, 60, 90 and 120 min after administering the suspected stimulant (Lacorix protocol) 17 l10 An increment in cortisol of ≥50% was considered as positive response, while an increase between 25% and 49% from baseline was considered as partial response. The patient had a 100% increase in serum cortisol from baseline after intravenous administration of 0.5 mg of terlipressin (table 1).
Table 1.
Stimulation tests with various agents for aberrant receptor expression
| Test/Cortisol levels | 0 min | 30 min | 60 min | 90 min | 120 min | Max increase from baseline (%) |
| MMT (10 kcal/kg) (Ensure, Abbott, India) | 986 | 1034 | 984 | 978 | 953 | 4.8 |
| ITT (0.1 U/kg) | 951 | 835 | 841 | 785 | 802 | No increase |
| Posture stimulation | 910 | 1020 | 1086 | 1052 | 1099 | 20.8 |
| Triptorelin test (0.1 mg intravenous, Ferring) | 949 | 983 | 1065 | 1114 | 1180 | 24.3 |
| hCG stimulation test (5000 IU intramuscular) | 1074 | 1044 | 1072 | 1084 | 1133 | 5.5 |
| Metoclopramide test (10 mg intravenous) | 1201 | 1172 | 1161 | 1141 | 1146 | No increase |
| Terlipressin (0.5 mg intravenous) | 935 | 1468 | 1785 | 1879 | 1751 | 100 |
Response to terlipressin suggested that the aberrant stimulator was antidiuretic hormone (ADH), and it resulted in CS via acting through V1 receptors, which are overexpressed in the adrenal cortex. Bilateral laparoscopic adrenalectomy was performed and the patient was started on physiological replacement with hydrocortisone and mineralocorticoids. Histological examination of the resected tissue demonstrated non-pigmented nodule measuring >5 mm, and diffuse and nodular hyperplasia of compact and clear cells of the adrenal cortex with preservation of internodular tissue (figure 2A–C).
Figure 2.
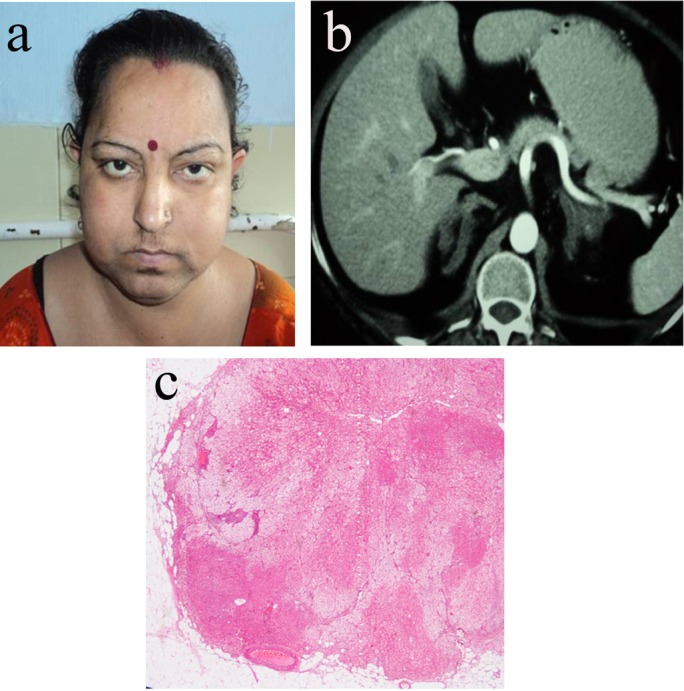
(A) Clinical picture of the patient with primary bilateral macronodular adrenocortical hyperplasia (PBMAH). (B) Contrast-enhanced CT of the abdomen showing bilateral bulky adrenals suggestive of PBMAH and (C) showing multiple non-pigmented nodules, and nodular hyperplasia of compact and clear cells of the adrenal cortex with preservation of internodular tissue seen as eosinophilic areas suggestive of PBMAH.
Discussion
ACTH-independent endogenous CS accounts for 20%–30% of patients with CS. Among these, functioning unilateral adrenal adenoma or carcinoma constitutes majority of the cases, while 10% of these have bilateral lesions and the causes include PBMAH, PPNAD, iMAD and rarely bilateral adrenal adenoma or carcinoma.1–3
The cAMP–PKA pathway is involved in the development, proliferation, function and survival of adrenocortical cells, and this common pathway has been implicated in the tumourigenesis of many adrenal cortical tumours. Mutations in cAMP–PKA signalling pathway (GNAS, PRKAR1A PRKACA, PDE11A, PDE8B) and β-catenin pathway (CTNNB1) and mutations of ARMC5, MEN-1, APC, FH, MC2R lead to the tumour development in majority of the patients with adrenal adenoma, PPNAD and PBMAH leading to CS.1 4
PPNAD is a rare form of ACTH-independent CS. It is more common in older children and young adults and have higher female-to-male ratio. However, PPNAD has been reported in older individuals as well.11 12 It can occur sporadically or as a familial autosomal dominant disorder or as a part of CNC, which occurs due to the inactivating mutation of PRKA1A gene located on the long arm of chromosome 17 (17q22-24). Familial PPNAD occurs in 50%-90% of the cases and PRKA1A is the most common mutation, while mutation of PRKAACA, PDE11A and PDE8B is found in rest of the cases. CNC is characterised by lentigens over the skin and mucosa, cardiac myxoma and multiple endocrine and non-endocrine tumours; however, the stigmata of CNC were not present in the index patient. These features may be absent early in the evolution of the disease in patients with PPNAD, which are diagnosed on screening of affected family members, and it has been suggested that in sporadic cases genetic analysis for PRKA1A should be done.13 PPNAD presents with mild hypercortisolemia or as cyclical CS, and imaging of the adrenals may be normal, thereby establishing that the diagnosis is difficult in initial stages. The index patient has florid features of CS and had typical ‘string of beads’ appearance on CECT.
PBMAH manifests in the fifth and sixth decades of life with equal gender distribution; however, PBMAH associated with MAS presents in the first year of life.
It usually presents as subclinical CS and less commonly as overt CS.6 Previously, PBMAH was thought to be a sporadic disorder, but recent studies have shown that it is more of a genetic disorder with familial clustering.9 ARMC5 is a tumour suppressor gene; cells carrying inactivating mutations of this gene lose capacity to induce apoptosis and subsequent tumour formation.1 Loss of heterozygosity of ARMC5 at 16p11.2 has been found in many patients with PBMAH.9 Mutations of PDE11A, PDE8B, PRKACA, MC2R and aberrant expression of other GPCRs are involved in pathogenesis of PBMAH. Bilateral macronodular adrenal hyperplasia is also described in hereditary leiomyomatosis, familial adenomatous polyposis, multiple endocrine neoplasia-1 and renal cell carcinoma. In majority of these patients, it is bilateral non-functioning adrenomegaly.14–16
Steroidogenesis in PBMAH can be regulated by aberrant stimulators (other than ACTH) due to aberrant expression of their respective receptors in adrenocortical nodular glands. Aberrant GPCRs for glucose-dependent insulinotropic peptide, β-adrenergic agonist, vasopressin (V1–V3R), angiotensin II, glucagon, LH/human chorionic gonadotropin (hCG), 5-HT4 and leptin have been demonstrated in adrenals, and these regulate steroidogenesis by mimicking GPCR activity of ACTH.17 18
V1R is normally expressed in the adrenal cortex but is not a major regulator of steroidogenesis, and it is the most common aberrant receptor documented in patients with PBMAH. In ADH-related PBMAH, V1R is overexpressed in the adrenal cortex, thus leading to exaggerated steroidogenic response to V1-agonist (terlipressin) but not to V2-agonist (desmopressin). Case 2 in this series showed cortisol response of >50% from baseline on stimulation with terlipressin, suggesting aberrant expression of V1 receptor. Medical therapy with vaptans (relcovaptan, conivaptan-V1R antagonist) is a theoretical option in these patients; however, we did not consider this therapy due to the lack of evidence in the literature of their use in patients with PBMAH.19 20
Diagnosis of both PBMAH and PPNAD is suspected in a patient of CS with low or undetectable ACTH with hypercortisolemia and characteristic findings on adrenal imaging. ACTH levels are low or undetectable in patients with PPNAD and PBMAH; however, sometimes ACTH levels may be in low normal range in PBMAH, as adrenocortical cells have been shown to produce ACTH in hyperplastic adrenal nodules.21 Paradoxical rise of cortisol on HDDST has been described in patients with PPNAD due to the overexpression of glucocorticoid receptors; however, it was not seen in our patient. Patients with PPNAD and with PBMAH and virilising adrenal adenoma may show paradoxical rise in cortisol after HDDST, as was seen in our patient with PBMAH.22–24
On CECT, adrenal glands are enlarged bilaterally with multiple hypodense contrast-enhancing nodules >5 mm in diameter in patients with PBMAH. Further on MRI, adrenals appear hypointense compared with the liver and isointense relative to the muscle on T1-weighted images, while on T2-weighted images they appear hyperintense compared with the liver. However, diffuse adrenal enlargement without nodules does not rule out the diagnosis of PBMAH.24 Typical CECT characteristics of PPNAD include ‘string of beads’ appearance of adrenals but this is rarely observed. Usually multiple hyperplastic small nodules not exceeding 5 mm in size are found; however, nodules up to 1–2 cm sometimes may be seen in older patients with PPNAD.25 In this case study, case 1 had typical ‘string of beads’ appearance, while case 2 had nodular appearance of the gland.
On gross examination, paired adrenal weight ranges from 60 to 400 g, and nodules appear yellow in colour due to high lipid content. Histological examination shows pigmented hyperplastic nodules >5 mm in diameter with variable internodular cortical atrophy in PBMAH, while PPNAD is usually characterised by normal to bulky adrenals containing multiple black or brown nodules of size 2–4 mm, most often with internodular cortical atrophy.25 In this series, both the cases had typical features on histopathology as stated earlier.
The definitive treatment of ACTH-independent macronodular hyperplasia (AIMAH) and PPNAD is bilateral adrenalectomy. However, medical treatment according to identification of aberrant receptor has been tried with variable results in many case studies. Postoperatively, supplementation with hydrocortisone and fludrocortisone is required lifelong. Signs of CNC should be actively searched in patients with sporadic cases of PPNAD on follow-up, as it may manifest later with passage of time.
Learning points.
Primary bilateral macronodular adrenocortical hyperplasia (PBMAH) and primary pigmented nodular adrenocortical disease (PPNAD) are rare causes of ACTH-independent Cushing’s syndrome.
PPNAD occurs in children as part of Carney’s complex but can also present sporadically.
Work-up for aberrant stimulators should be performed in patients with PBMAH to find the overexpression of eutopic/ectopic receptor.
Paradoxical rise in cortisol to high-dose dexamethasone suppression test can be seen in patients with PBMAH.
Diagnoses of PBMAH and PPNAD rely on histopathological findings.
Footnotes
Contributors: KDS wrote the manuscript and managed the patients. RW edited the manuscript. UN did histopathological examination. AB identified the cases, edited the manuscript and managed the patients.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Lodish M, Stratakis CA. A genetic and molecular update on adrenocortical causes of cushing syndrome. Nat Rev Endocrinol 2016;12:255–62. 10.1038/nrendo.2016.24 [DOI] [PubMed] [Google Scholar]
- 2.Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab 2003;88:5593–602. 10.1210/jc.2003-030871 [DOI] [PubMed] [Google Scholar]
- 3.Lacroix A, Bourdeau I. Bilateral adrenal Cushing's syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin North Am 2005;34:441–58. 10.1016/j.ecl.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 4.Mazzuco TL, Durand J, Chapman A, et al. Genetic aspects of adrenocortical tumours and hyperplasias. Clin Endocrinol 2012;77:1–10. 10.1111/j.1365-2265.2012.04403.x [DOI] [PubMed] [Google Scholar]
- 5.Gunther DF, Bourdeau I, Matyakhina L, et al. Cyclical cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab 2004;89:3173–82. 10.1210/jc.2003-032247 [DOI] [PubMed] [Google Scholar]
- 6.Mermejo LM, Mazzuco TL, Grunenwald S, et al. ACTH-Independent macronodular adrenal hyperplasia. Endocrinol Metab 2011;26:1–11. 10.3803/EnM.2011.26.1.1 [DOI] [Google Scholar]
- 7.Findlay JC, Sheeler LR, Engeland WC, et al. Familial adrenocorticotropin-independent Cushing's syndrome with bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab 1993;76:189–91. 10.1210/jcem.76.1.8380604 [DOI] [PubMed] [Google Scholar]
- 8.Nies C, Bartsch DK, Ehlenz K, et al. Familial ACTH-independent Cushing's syndrome with bilateral macronodular adrenal hyperplasia clinically affecting only female family members. Exp Clin Endocrinol Diabetes 2002;110:277–83. 10.1055/s-2002-34590 [DOI] [PubMed] [Google Scholar]
- 9.Assié G, Libé R, Espiard S, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med 2013;369:2105–14. 10.1056/NEJMoa1304603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung DJ. Letter: Association of the Parathyroid Adenoma Volume and the Biochemical Parameters in Primary Hyperparathyroidism (Endocrinol Metab 26:62-66, 2011, Yul Hwang-Bo et al.). Endocrinol Metab. 2011;26:185 10.3803/EnM.2011.26.2.185 [DOI] [Google Scholar]
- 11.Papanastasiou L, Fountoulakis S, Voulgaris N, et al. Identification of a novel mutation of the PRKAR1A gene in a patient with Carney complex with significant osteoporosis and recurrent fractures. Hormones 2016;15:129–35. 10.14310/horm.2002.1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aleksova J, Ward G, Taubman K, et al. Cushing's conundrum: an unusual case of primary pigmented nodular adrenal disease in a 60-year-old woman. Lancet Diabetes Endocrinol 2016;4:630 10.1016/S2213-8587(15)00262-4 [DOI] [PubMed] [Google Scholar]
- 13.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 2001;86:4041–6. 10.1210/jcem.86.9.7903 [DOI] [PubMed] [Google Scholar]
- 14.Burgess JR, Harle RA, Tucker P, et al. Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg 1996;131:699–702. 10.1001/archsurg.1996.01430190021006 [DOI] [PubMed] [Google Scholar]
- 15.Marchesa P, Fazio VW, Church JM, et al. Adrenal masses in patients with familial adenomatous polyposis. Dis Colon Rectum 1997;40:1023–8. 10.1007/BF02050923 [DOI] [PubMed] [Google Scholar]
- 16.Matyakhina L, Freedman RJ, Bourdeau I, et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 2005;90:3773–9. 10.1210/jc.2004-2377 [DOI] [PubMed] [Google Scholar]
- 17.Lacroix A, Ndiaye N, Tremblay J, et al. Ectopic and abnormal hormone receptors in adrenal cushing's syndrome. Endocr Rev 2001;22:75–110. 10.1210/edrv.22.1.0420 [DOI] [PubMed] [Google Scholar]
- 18.Lacroix A, Baldacchino V, Bourdeau I, et al. Cushing's syndrome variants secondary to aberrant hormone receptors. Trends Endocrinol Metab 2004;15:375–82. 10.1016/j.tem.2004.08.007 [DOI] [PubMed] [Google Scholar]
- 19.Hofland J, Hofland LJ, van Koetsveld PM, et al. ACTH-independent macronodular adrenocortical hyperplasia reveals prevalent aberrant in vivo and in vitro responses to hormonal stimuli and coupling of arginine-vasopressin type 1a receptor to 11β-hydroxylase. Orphanet J Rare Dis 2013;8:142 10.1186/1750-1172-8-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louiset E, Contesse V, Groussin L, et al. Expression of vasopressin receptors in ACTH-independent macronodular bilateral adrenal hyperplasia causing Cushing's syndrome: molecular, immunohistochemical and pharmacological correlates. J Endocrinol 2008;196:1–9. 10.1677/JOE-07-0413 [DOI] [PubMed] [Google Scholar]
- 21.Stratakis CA, Sarlis N, Kirschner LS, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med 1999;131:585–91. 10.7326/0003-4819-131-8-199910190-00006 [DOI] [PubMed] [Google Scholar]
- 22.Yotsumoto S, Aizawa T, Kotani M, et al. Virilizing adrenal adenoma stimulated by dexamethasone in a middle-aged woman. J Clin Endocrinol Metab 1979;48:660–3. 10.1210/jcem-48-4-660 [DOI] [PubMed] [Google Scholar]
- 23.Clouston WM, Cannell GC, Fryar BG, et al. Virilizing adrenal adenoma in an adult with the Beckwith-Wiedemann syndrome: paradoxical response to dexamethasone. Clin Endocrinol 1989;31:467–73. 10.1111/j.1365-2265.1989.tb01270.x [DOI] [PubMed] [Google Scholar]
- 24.Rockall AG, Babar SA, Sohaib SA, et al. CT and MR imaging of the adrenal glands in ACTH-independent cushing syndrome. Radiographics 2004;24:435–52. 10.1148/rg.242035092 [DOI] [PubMed] [Google Scholar]
- 25.Peppercorn PD, Reznek RH. State-of-the-art CT and MRI of the adrenal gland. Eur Radiol 1997;7:822–36. 10.1007/s003300050214 [DOI] [PubMed] [Google Scholar]