

Abstract
Neurofibromatosis type 1 (NF1) is a multisystem genetic disorder associated with reduced lifespan attributed largely to malignancy and vascular causes. One of the tumours associated with NF1 is phaeochromocytoma. The phaeochromocytoma has earned the moniker, a ‘great mimicker’, due to its varied means of presentation. We present a patient with NF1 who was diagnosed with a giant 20 cm phaeochromocytoma after suffering from an ischaemic stroke. Current guidelines do not advocate surveillance of phaeochromocytoma in asymptomatic patients with NF1, unlike other genetic syndromes associated with phaeochromocytoma. However, there is increasing evidence that this approach may not help in the early detection and treatment of this potentially life-threatening disease. Our patient remained hypertensive after surgery despite achieving biochemical cure. The suggested chronicity of the underlying tumour in our patient is a reminder to practising clinicians to rethink our strategy in identifying phaeochromocytoma in adults with NF1.
Keywords: adrenal disorders, hypertension, genetic screening / counselling, stroke, endocrine cancer
Background
Phaeochromocytoma is known as a great mimicker of diseases. Although largely a sporadic tumour, underlying genetic mutations have been identified in up to one-third of non-syndromic patients. Genetic syndromes, such as multiple endocrine neoplasia type 2, von-Hippel Lindau disease (vHL) and type 1 neurofibromatosis (NF1) harbour genetic mutations with a predilection for this tumour. In patients with these syndromes, the crux lies in detecting the tumour in its infancy to maximise the chance of cure and reduce complications. In our case presentation, the patient with NF1 did not receive any routine surveillance for the tumour and presented only when she suffered an ischaemic stroke. We would like to bring attention to the increased morbidity of untreated phaeochromocytoma, and to highlight the importance of considering this tumour in appropriate settings when encountering patients with NF1.
Case presentation
A 49-year-old Chinese woman with NF1 was admitted complaining of a 3-day history of right-sided weakness. She was previously well and did not attend any regular medical check-ups with her primary healthcare provider. She did not consume alcohol and was a non-smoker. Four out of nine of her siblings suffered from NF1, but none suffered from cardiovascular disease.
On physical examination, the patient was alert and vital signs revealed a blood pressure (BP) of 165/70 mm Hg and a pulse rate of 100 beats per min. Neurological examination confirmed right hemiparesis. Peripheral pulses were bilaterally equal. There were no carotid bruits and cardiorespiratory examination was unremarkable. Significantly, a large and firm ballotable mass measuring 15 cm over the left flank of the abdomen was found.
Investigations
Routine blood investigations showed normal full blood count and renal function. Low-density lipoprotein (LDL) was elevated at 4.6 mmol/L. Fasting blood glucose was normal. Evaluation for other causes of young hypertension and young stroke were negative (table 1).
Table 1.
Investigations for young stroke were performed and were all normal
| Results | Units | Reference intervals | |
| Haematology | |||
| Haemoglobin | 13.5 | g/dL | 11–15 |
| Haematocrit | 41.0 | % | 35–45 |
| Total white cell count | 8.4 | x 109/L | 3.6–9.3 |
| Platelet count | 405 | x 109/L | 170–420 |
| Electrolytes | |||
| Sodium | 138 | mmol/L | 134–144 |
| Potassium | 4.3 | mmol/L | 3.5–5.0 |
| Creatinine | 69 | μmol/L | 40–75 |
| Calcium, adjusted | 2.25 | mmol/L | 2.15–2.58 |
| Atherosclerosis risk factors | |||
| Glucose, fasting | 5.5 | mmol/L | 3.0–6.0 |
| HbA1c | 5.7 | % mmol/L | 54.5–6.4 |
| Total cholesterol | 6.7 | mmol/L | <6.2 |
| High-density lipoprotein | 1.5 | mmol/L | >1.0 |
| Low-density lipoprotein | 4.6 | mmol/L | <4.1 |
| Triglycerides | 1.4 | μmol/L | <2.3 |
| Homocysteine, S | 8 | 5–15 | |
| Coagulability profile | |||
| PT | 11.8 | s | 11.7–14.0 |
| PTT | 27.6 | s | 25.0–36.0 |
| Lupus anticoagulant | Absent | – | – |
| Vasculitis screen | |||
| Anti-dsDNA | Negative | IU/mL | <25 |
| Anticardiolipin IgM | 1 | MPL U/mL | <10 |
| Anticardiolipin IgG | 2 | GPL/mL | <10 |
| ECG | Normal sinus rhythm | ||
| 2D echocardiography | Ejection fraction >55%, no valvular abnormalities, no thrombus, no intracardiac shunt | – | |
| Inpatient 48 hours Holter monitoring | Negative for atrial fibrillation | – | |
| Ultrasound of carotid arteries | Bilateral internal carotid arteries <40% stenosis Common carotid arteries, external carotid arteries and vertebral arteries normal |
– | |
| Transcranial Doppler with bubble study | Negative for cardiac right-to-left shunt | – | |
2D, two dimensional; dsDNA, double-stranded DNA; PT, thrombin time; PTT, partial thromboplastin time, MPL- IgM Phospholipid GPL- IgG Phospholipid.
An acute ischaemic stroke was confirmed on MRI of the brain, which showed an infarct in the left internal capsule contributed by a stenosis in the M1 segment of the left middle cerebral artery.
To evaluate for the abdominal mass, a CT of the abdomen was performed and revealed a heterogeneous 18 cm left adrenal mass with cystic areas of necrosis (figure 1A,B). The right adrenal was normal.
Figure 1.
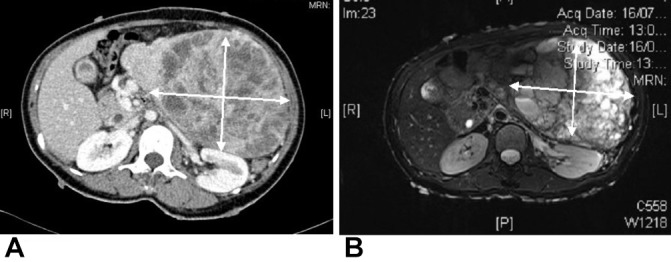
(A) CT abdomen showing a large 18 cm x 20 cm heterogeneous mass with areas of necrosis and mass effect on adjacent structures. (B) MRI abdomen of the same intra-abdominal mass showing hyperintensity on T2.
Differential diagnosis
While an acute ischaemic stroke in a patient with NF1 could be attributed to underlying atherosclerotic processes, some differential diagnoses deserve attention. Conditions such as Moya Moya disease (with associated renovascular hypertension) and adrenal pathologies such as phaeochromocytoma and primary hyperaldosteronism can contribute to both hypertension and strokes.
Further history revealed that the patient had spells of palpitations and diaphoresis lasting about 1 min each in the past few months, but had dismissed them as anxiety attacks. As this patient has underlying NF1 and a ballotable left flank mass, the diagnosis of phaeochromocytoma must be seriously considered given this suggestive history. Taken together with the CT images, this must be the diagnosis until proven otherwise.
Hormonal assessment for adrenal hypersecretion was performed. The only abnormality was marked elevation of the 24 hours urinary catecholamines and metanephrines at 111 500 nmol/day (reference intervals 480–2424 nmol/day) and 54 619 nmol/day (reference intervals 264–1729 nmol/day), respectively. Aldosterone and renin levels did not fulfil the required criteria for either primary or secondary hyperaldosteronism.
The diagnosis of phaeochromocytoma was, therefore, confirmed biochemically.
Treatment
The patient was seen in consultation with an endocrine surgeon and counselled for a left adrenalectomy. She received preoperative preparation with oral phenoxybenzamine 10 mg two times per day, followed by atenolol 25 mg subsequently for reflex tachycardia. After adequate adrenergic blockade and intravascular volume expansion, she underwent an elective open-left adrenalectomy where a giant 20 cm x 14 cm x 10 cm phaeochromocytoma was removed en bloc (figure 2). Despite adequate preoperative preparation, she still experienced intraoperative fluctuations in BP between systolic 70 and 170 mm Hg (figure 3).
Figure 2.
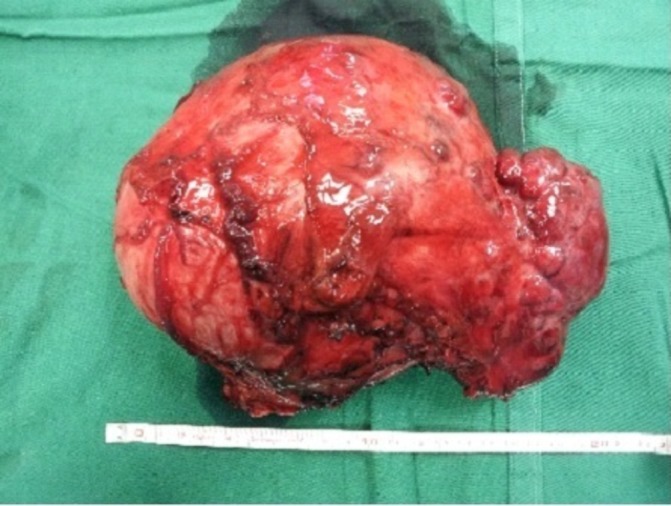
A large 20 cm phaeochromocytoma removed en bloc during surgery.
Figure 3.
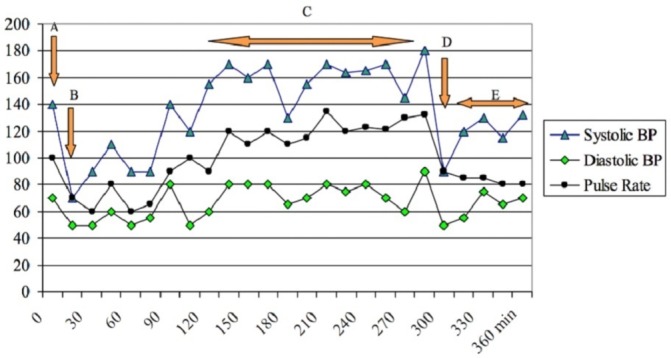
Blood pressure (BP) and pulse rate trends during operation. (A) At induction; (B) after intubation (intravenous phenylephrine given); (C) surgical manipulation of tumour (intravenous MgSO4 given); (D) surgical resection of tumour; (E) recovery.
It was histologically confirmed to be a composite phaeochromocytoma with ganglioneuroma elements (figure 4A,B).
Figure 4.
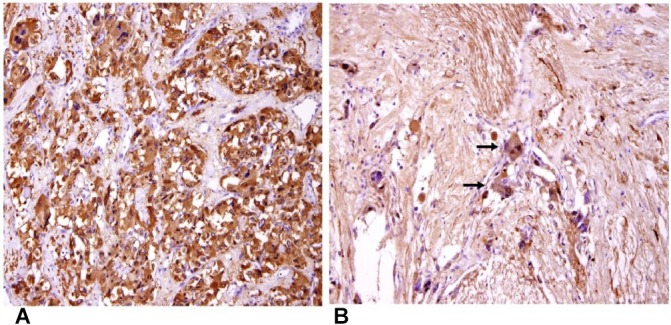
(A) Immunohistochemical staining for synaptophysin highlights the neoplastic cells in the phaeochromocytoma component of the tumour (original magnification ×200). (B) The neurofibrillary matrix and the scattered ganglion cells (arrows) in the ganglioneuroma component are highlighted by immunohistochemical staining for S100 protein (original magnification ×200).
Postoperatively, the urinary catecholamines and metanephrines were normalised (table 2). One month later, hypertension was recurred. Both biochemical and radiological tests were negative for recurrence. A 131-iodine-metaiodobenzylguanidine scintigraphy scan did not detect any tracer avid lesion. Screen for other secondary causes of hypertension was negative. She was started on nifedipine LA 30 mg daily and prazosin 1 mg two times per day and achieved good BP control.
Table 2.
Urinary catecholamines and metanephrines trend from preoperative to postoperative, showing normalisation of results after surgery
| Preoperative | Months postoperative | Reference intervals | |||||||
| 1 | 6 | 11 | 17 | 20 | 30 | 38 | |||
| U Catecholamines | |||||||||
| Total urine volume | 1800 | 900 | 1500 | 1400 | 1600 | 2000 | 1800 | 1773 | mL |
| Epinephrine, 24 hours | 265 | 20.2 | <10 | <10 | <10 | <10 | 94 | 21 | 9.3–122.0 nmol/day |
| Norepinephrine, 24 hours | 920 | 317 | 113 | 157 | 165 | 128 | 169 | 170 | 72–505 nmol/day |
| U Metanephrines | |||||||||
| Total urine volume | 1800 | 900 | 1500 | 1400 | 1600 | 2000 | 1800 | 1773 | mL |
| Metanephrines, 24 hours | 64 692 | 548 | 174 | 227 | 217 | 186 | 279 | 213 | 264–1729 nmol/day |
| Normetanephrine, 24 hours | 1 25 200 | 3370 | 737 | 1289 | 1538 | 836 | 1145 | 1073 | 480–2424 nmol/day |
Outcome and follow-up
Currently, it is 4 years postsurgery, and she remains well without any suggestive symptoms of recurrence. At home, her BP is well controlled, and her urinary catecholamines and metanephrines had remained normal during regular 6 monthly check-ups.
Discussion
The association between NF1 and phaeochromocytoma is well established. Its incidence in NF1 is 0.1%–5.7%, roughly 10 times that of the general population.1-6Among hypertensive NF1 patients, the tumour is present between 20% and 56%. Phaeochromocytoma in NF1 is unilateral and intra-adrenal in more than 80%, and has a malignant rate of about 10%.7–11 Available guidelines suggest screening for the condition with urinary or serum metanephrines only when typical symptoms are present.12–14
The most common symptoms of phaeochromocytoma are hypertension, headaches, palpitations and diaphoresis.10 15 Hypertension is common in NF1. It increases with age and occurs between 2% and 15.8%.16 17 Secondary causes of hypertension are associated with NF1, including renal artery stenosis, coarctation of aorta and phaeochromocytoma. In a preadolescent child, the most common secondary cause is renovascular disease and this must be sought thoroughly as secondary hypertension can be found in up to 85%.18 19 In an adult, phaeochromocytoma becomes more common although no cause is found in the majority. The likelihood of an underlying secondary cause becomes higher in the event of refractory high BP or when hypertension occurs in pregnancy.
Phaeochromocytoma has been given the moniker ‘a great mimicker’ and can present with symptoms from almost any other system. Asymptomatic disease has also been reported and the incidence ranges between 11% and 48% in retrospective studies.20–22 In NF1 populations, recent retrospective studies agreed with the above findings, where symptoms of catecholamine excess were absent in 22%–42%, and hypertension was absent in 23%–83%.8 9 11 It is important to note that 31%–100% of phaeochromocytomas were diagnosed incidentally in these studies. Among the genetic syndromes associated with phaeochromocytoma, NF1 remains the only one where routine surveillance for the condition is currently not practised.23–25 Based on the above evidence, the approach of screening only hypertensive or symptomatic patients with NF1 may result in missed or delayed diagnosis in NF1.
Zinnamosca et al and Kepenekian et al evaluated a different approach to the detection of phaeochromocytoma in NF1.26 27 In both studies, asymptomatic NF1 patients were recruited for screening. Zinnamosca et al assessed 48 consecutive NF1 patients using urinary metanephrines and vanillylmandelic acid excretion while Kepenekian et al screened 156 consecutive NF1 patients with a combination of abdominal imaging (ultrasound or CT) and urinary fractionated metanephrines. This was followed by a functional scan with either 123I-metaiodobenzylguanidine scintigraphy or 18F-fluoro-dihydroxyphenylalanine if the screen was positive. The prevalences of phaeochromocytoma in the two studies were 14.6% and 7.7%, respectively.26 27 More than half of the subjects in both studies were asymptomatic. The smallest tumour was 1 cm while the largest was 5 cm. Interestingly, half of the cases in the Kepenekian study were non-secretory with tumour sizes smaller than the secretory tumours (mean 1.40 vs 2.52 cm).
In this case study, stroke was the initial presentation that led to the discovery of a giant 20 cm phaeochromocytoma and hypertension. The patient was not diagnosed with hypertension prior to this. Both NF1 and phaeochromocytoma have been associated with increased risk of cerebrovascular disease.28–30 In addition to hypertension, underlying NF1 vasculopathy may be a contributing factor. NF1 vasculopathy is a less discussed but well described complication. It involves a spectrum of vascular disorders affecting both peripheral and cerebral vessels, with Moya Moya syndrome being the pathognomonic description. The abnormalities seen in NF1 vasculopathy can range from stenosis, aneurysms to arterio-vascular malformations. The underlying pathophysiology is related to disorganised endothelial repair associated with a defective NF1 gene.31–33 The risk of an ischaemic stroke in catecholamine-producing tumours may be attributed to a few factors—uncontrolled hypertension, vascular spasm, dilated cardiomyopathy with a risk of left ventricular thrombus and labile BP with hypotensive episodes causing increased susceptibility in watershed regions. There is also evidence that catecholamines are involved in vessel remodelling and associated with increased intima media thickness of carotids.34 35
Given the size of the tumour, it is likely that she had been harbouring it for much longer than the duration of her reported symptoms. It is unfortunate that she was diagnosed only after presenting with a complication related to both phaeochromocytoma and hypertension. As she is not cured of hypertension even after surgery, her cardiovascular risks remain appreciable.
The current approach to phaeochromocytoma screening in NF1 is fraught with shortcomings and pitfalls. Delayed or missed diagnoses of phaeochromocytoma would contribute to increased morbidity and mortality of this dangerous condition.
Screening for phaeochromocytoma only when patients with NF1 develop hypertension or typical symptoms is likely inadequate. This traditional method exposes the patient to the deleterious effects of catecholamine excess as seen in this case. We await more studies to recommend a cost-effective method of phaeochromocytoma screening in NF1 populations. Meanwhile, physicians should be cognisant of this rare but dangerous tumour associated with NF1, and have a low threshold for screening until clearer guidelines are available.
Learning points.
Delayed or missed diagnosis of phaeochromocytoma contributes to increased morbidity and mortality.
NF1 is associated with both essential and secondary hypertension. Regular surveillance for hypertension in this group of patients is important as vascular causes contribute to an increased mortality in NF1 patients.
When diagnosed with hypertension, tests to exclude secondary causes, including phaeochromocytoma must be performed in NF1 patients.
Physicians should be aware of the association of this rare and dangerous tumour with NF1 and have a low threshold for screening while awaiting further guidelines.
Footnotes
Contributors: YL conceived the idea for the manuscript, analysed the data and wrote the majority of the manuscript. LYRT assisted in literature research and writing of the case. YHH provided critical review of the manuscript as well as assisting in the elucidation of the pathological diagnosis of the case. MKSL provided invaluable guidance in intellectual input, critical review and editing of the manuscript for scientific content validity and accuracy.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet 2001;68:1110–8. 10.1086/320121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duong TA, Sbidian E, Valeyrie-Allanore L, et al. Mortality associated with neurofibromatosis 1: a cohort study of 1895 patients in 1980-2006 in France. Orphanet J Rare Dis 2011;6:18 10.1186/1750-1172-6-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masocco M, Kodra Y, Vichi M, et al. Mortality associated with neurofibromatosis type 1: a study based on Italian death certificates (1995-2006). Orphanet J Rare Dis 2011;6:11 10.1186/1750-1172-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zöller M, Rembeck B, Akesson HO, et al. Life expectancy, mortality and prognostic factors in neurofibromatosis type 1. A twelve-year follow-up of an epidemiological study in Göteborg, Sweden. Acta Derm Venereol 1995;75:136–40. [DOI] [PubMed] [Google Scholar]
- 5.Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123:124–33. 10.1542/peds.2007-3204 [DOI] [PubMed] [Google Scholar]
- 6.Walther MM, Herring J, Enquist E, et al. von Recklinghausen’s disease and pheochromocytomas. J Urol 1999;162:1582–6. 10.1016/S0022-5347(05)68171-2 [DOI] [PubMed] [Google Scholar]
- 7.Bausch B, Borozdin W, Neumann HP. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N Engl J Med 2006;354:2729–31. 10.1056/NEJMc066006 [DOI] [PubMed] [Google Scholar]
- 8.Shinall MC, Solórzano CC. Pheochromocytoma in Neurofibromatosis Type 1: When Should it Be Suspected? Endocr Pract 2014;20:792–6. 10.4158/EP13417.OR [DOI] [PubMed] [Google Scholar]
- 9.Moramarco J, El Ghorayeb N, Dumas N, et al. Pheochromocytomas are diagnosed incidentally and at older age in neurofibromatosis type 1. Clin Endocrinol 2017;86:332–9. 10.1111/cen.13265 [DOI] [PubMed] [Google Scholar]
- 10.Cotesta D, Petramala L, Serra V, et al. Clinical experience with pheochromocytoma in a single centre over 16 years. High Blood Press Cardiovasc Prev 2009;16:183–93. 10.2165/11530430-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 11.Gruber LM, Erickson D, Babovic-Vuksanovic D, et al. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol 2017;86:141–9. 10.1111/cen.13163 [DOI] [PubMed] [Google Scholar]
- 12.Hersh JH. American Academy of Pediatrics Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics 2008;121:633–42. 10.1542/peds.2007-3364 [DOI] [PubMed] [Google Scholar]
- 13.Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44:81–8. 10.1136/jmg.2006.045906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol 2006;13:2–7. 10.1016/j.spen.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 15.Goldstein RE, O’Neill JA, Holcomb GW, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg 1999;229:755–64. 10.1097/00000658-199906000-00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med 1981;305:1617–27. 10.1056/NEJM198112313052704 [DOI] [PubMed] [Google Scholar]
- 17.Virdis R, Balestrazzi P, Zampolli M, et al. Hypertension in children with neurofibromatosis. J Hum Hypertens 1994;8:395–7. [PubMed] [Google Scholar]
- 18.Flynn JT. Evaluation and management of hypertension in childhood. Prog Pediatr Cardiol 2001;12:177–88. 10.1016/S1058-9813(00)00071-0 [DOI] [PubMed] [Google Scholar]
- 19.Fossali E, Signorini E, Intermite RC, et al. Renovascular disease and hypertension in children with neurofibromatosis. Pediatr Nephrol 2000;14:806–10. 10.1007/s004679900260 [DOI] [PubMed] [Google Scholar]
- 20.Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol 2004;150:681–6. 10.1530/eje.0.1500681 [DOI] [PubMed] [Google Scholar]
- 21.Noshiro T, Shimizu K, Watanabe T, et al. Changes in clinical features and long-term prognosis in patients with pheochromocytoma. Am J Hypertens 2000;13:35–43. 10.1016/S0895-7061(99)00139-9 [DOI] [PubMed] [Google Scholar]
- 22.Mannelli M, Ianni L, Cilotti A, et al. Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol 1999;141:619–24. 10.1530/eje.0.1410619 [DOI] [PubMed] [Google Scholar]
- 23.Binderup ML, Bisgaard ML, Harbud V, et al. Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J 2013;60:B4763. [PubMed] [Google Scholar]
- 24.Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658–71. 10.1210/jcem.86.12.8070 [DOI] [PubMed] [Google Scholar]
- 25.Kirmani S, Young WF. Hereditary paraganglioma-pheochromocytoma syndromes : Pagon RA, Adam MP, Ardinger HH, GeneReviews®. Seattle, WA: University of Washington, 1993–2017. [Google Scholar]
- 26.Zinnamosca L, Petramala L, Cotesta D, et al. Neurofibromatosis type 1 (NF1) and pheochromocytoma: prevalence, clinical and cardiovascular aspects. Arch Dermatol Res 2011;303:317–25. 10.1007/s00403-010-1090-z [DOI] [PubMed] [Google Scholar]
- 27.Képénékian L, Mognetti T, Lifante JC, et al. Interest of systematic screening of pheochromocytoma in patients with neurofibromatosis type 1. Eur J Endocrinol 2016;175:335–44. 10.1530/EJE-16-0233 [DOI] [PubMed] [Google Scholar]
- 28.Zelinka T, Petrák O, Turková H, et al. High incidence of cardiovascular complications in pheochromocytoma. Horm Metab Res 2012;44:379–84. 10.1055/s-0032-1306294 [DOI] [PubMed] [Google Scholar]
- 29.Terry AR, Jordan JT, Schwamm L, et al. Increased risk of cerebrovascular disease among patients with neurofibromatosis type 1: population-based approach. Stroke 2016;47:60–5. 10.1161/STROKEAHA.115.011406 [DOI] [PubMed] [Google Scholar]
- 30.Petramala L, Cavallaro G, Polistena A, et al. Multiple catecholamine-secreting paragangliomas: diagnosis after hemorrhagic stroke in a young woman. Endocr Pract 2008;14:340–6. 10.4158/EP.14.3.340 [DOI] [PubMed] [Google Scholar]
- 31.Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 cardiovascular task force. Genet Med 2002;4:105–11. 10.1097/00125817-200205000-00002 [DOI] [PubMed] [Google Scholar]
- 32.Oderich GS, Sullivan TM, Bower TC, et al. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg 2007;46:475–84. 10.1016/j.jvs.2007.03.055 [DOI] [PubMed] [Google Scholar]
- 33.Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet 2000;58:341–4. 10.1034/j.1399-0004.2000.580501.x [DOI] [PubMed] [Google Scholar]
- 34.Bernini G, Franzoni F, Galetta F, et al. Carotid vascular remodeling in patients with pheochromocytoma. J Clin Endocrinol Metab 2006;91:1754–60. 10.1210/jc.2005-2199 [DOI] [PubMed] [Google Scholar]
- 35.Holaj R, Rosa J, Zelinka T, et al. Long-term effect of specific treatment of primary aldosteronism on carotid intima-media thickness. J Hypertens 2015;33:874–82. 10.1097/HJH.0000000000000464 [DOI] [PMC free article] [PubMed] [Google Scholar]