

Abstract
The calcium-binding protein calbindin-D28k is critical for hippocampal function and cognition1-3, but its expression is markedly decreased in various neurological disorders associated with epileptiform activity and seizures4-7. In Alzheimer's disease (AD) and epilepsy, both of which are accompanied by recurrent seizures8, the severity of cognitive deficits reflects the degree of calbindin reduction in the hippocampal dentate gyrus (DG)4,9,10. However, despite the importance of calbindin in both neuronal physiology and pathology, the regulatory mechanisms that control its expression in the hippocampus are poorly understood. Here we report an epigenetic mechanism by which seizures chronically suppress hippocampal calbindin expression and impair cognition. We demonstrate that ΔFosB, a highly stable transcription factor, is induced in the hippocampus of mouse models of AD and seizures, where it binds and triggers histone deacetylation at the calbindin gene (Calb1) promoter, and downregulates Calb1 transcription. Notably, increasing DG calbindin levels, either by direct virus-mediated expression or inhibition of ΔFosB signaling, improves spatial memory in a mouse model of AD. Moreover, levels of ΔFosB and calbindin expression are inversely related in DG of patients with temporal lobe epilepsy (TLE) or AD, and correlate with performance on the Mini-Mental State Examination (MMSE). We propose that chronic suppression of calbindin by ΔFosB is one mechanism by which intermittent seizures drive persistent cognitive deficits in conditions accompanied by recurrent seizures.
Expression of calbindin-D28k in the hippocampal DG is indicative of cognitive function in both patients and mouse models of AD and epilepsy4,9-12. In addition, calbindin knockdown/knockout animals exhibit impaired synaptic plasticity and spatial memory1-3,13,14. These findings highlight calbindin's role as a critical regulator of neuronal calcium signaling and hippocampal function15. However, little is known about the regulatory mechanisms that modulate calbindin expression in normal or pathologic conditions. Considering how crucial calbindin is for synaptic function and cognition1-3,13,14, elucidating these mechanisms is essential as it may aid the development of novel therapeutics to ameliorate cognitive deficits in AD and other disorders associated with seizures.
To identify mechanisms that control hippocampal calbindin expression, we examined long-term gene regulation in the hippocampus of a transgenic AD mouse model expressing mutant human amyloid precursor protein (APP)16. APP mice of both sexes were examined at 2-4 months of age, an age when they begin to exhibit spontaneous recurrent seizures and cognitive deficits similar to AD patients, but prior to plaque deposition16-19. By immunohistochemical analysis, we found that hippocampal calbindin expression was lower in APP mice compared to non-transgenic littermates (NTG), and that levels of calbindin expression inversely correlated with the frequency of electroencephalographic (EEG) seizures (Fig. 1a,b), similar to patients with seizures5,10. Notably, even APP mice with relatively infrequent seizures exhibited reduced calbindin expression, suggesting that downregulation of hippocampal calbindin was mediated by long-lasting, activity-dependent mechanisms. After surveying the literature to determine what activity-dependent factors might regulate calbindin expression over extended periods of time, we focused on ΔFosB, a truncated splice variant of the transcription factor FosB. ΔFosB is a unique activity-dependent immediate early gene (IEG) product, possessing an unusually long half-life (∼8 days) that allows it to exert persistent control over neuronal gene expression20. The actions of ΔFosB in epigenetic gene regulation are well studied in the nucleus accumbens20,21, and a recent study suggests that it may also have functions in the hippocampus22. We found that seizures were associated with elevated hippocampal expression of ΔFosB, and higher levels of ΔFosB expression were correlated with lower calbindin expression in APP mice (Fig. 1b,c). Therefore, we hypothesized that ΔFosB is a key regulatory factor involved in the suppression of calbindin following epileptiform activity in AD and other seizure-associated disorders.
Figure 1. Epigenetic regulation of Calb1 in the hippocampus of APP and pilocarpine mice.
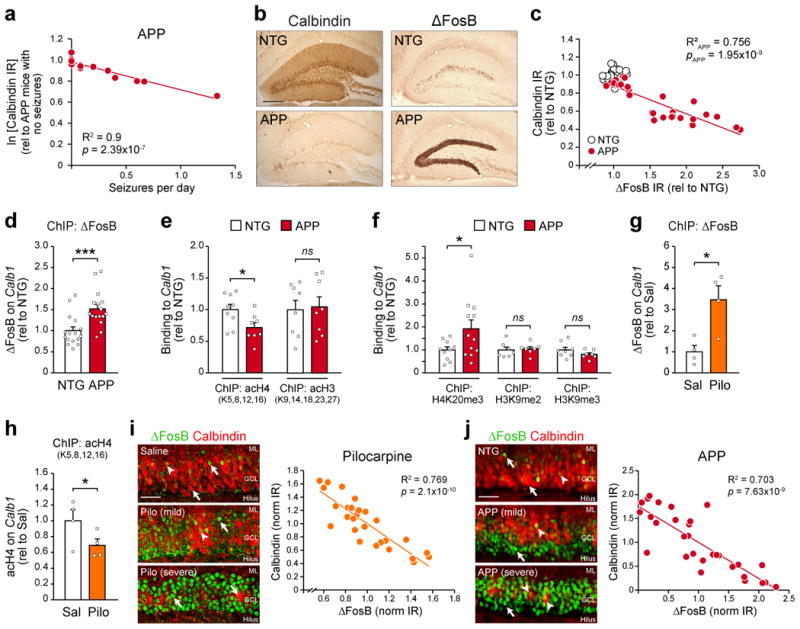
(a) Regression analysis of the relationship between hippocampal calbindin protein immunoreactivity (IR) and seizure frequency in 2-4 month-old APP mice (n=14 mice). IR values were transformed via natural log. (b) Representative images of hippocampal calbindin and ΔFosB IR in NTG and APP mice. Scale bar = 250 μm. (c) Regression analysis of calbindin and ΔFosB IR in APP mice (n=28 mice). NTG data points are also displayed (n=30 mice). (d) Binding of ΔFosB to the Calb1 promoter in the hippocampus of NTG and APP mice (n=16 mice/genotype, t30=3.72, ***p=8.3×10-4). (e) Levels of histone 4 (nNTG=9 mice, nAPP=8 mice, t15=-2.46, *p=0.026) and histone 3 (n=8 mice/genotype, t14=0.21, p=0.84) lysine acetylation on the Calb1 promoter of NTG and APP mice. (f) Levels of Calb1 histone 4 lysine 20 trimethylation (nNTG=10 mice, nAPP=12 mice, t20=2.09, *p=0.049), histone 3 lysine 9 dimethylation (n=7 mice/genotype, t12=0.31, p=0.76), and histone 3 lysine 9 trimethylation (nNTG=8 mice, nAPP=7 mice, t13=-1.38, p=0.19) in NTG and APP mice. (g) ΔFosB bound to Calb1 (n=4 mice/treatment, t6 =3.4, *p=0.015) and (h) Calb1 histone 4 lysine acetylation (n=4 mice/treatment, t6 =-1.94, one-tail *p=0.05) 3 days after pilocarpine-induced status epilepticus versus saline control. (i) Left, representative high-magnification images of ΔFosB and calbindin IR in the DG of pilocarpine-treated mice with mild (<2 stage five convulsions) or severe (>2 stage five convulsions) seizures during status epilepticus. Scale bar = 50 μm. Right, cell-by-cell regression analysis of ΔFosB and calbindin IR in a pilocarpine-treated mouse (n=30 cells). (j) Left, high-magnification images of hippocampal ΔFosB and calbindin IR in APP mice with varying severities of epilepsy. Right, cell-by-cell regression analysis of ΔFosB and calbindin IR in an APP mouse (n=30 cells). Scale bar = 50 μm. Arrowheads, granule cells double-labeled with both ΔFosB and calbindin. Arrows, granule cells single-labeled with either ΔFosB or calbindin. GCL, granule cell layer. ML, molecular layer. ns, non-significant. Data in panels (d-g) were analyzed using Student's t-tests. Error bars represent SEM.
To test this hypothesis, we performed a promoter analysis of Calb1 to identify potential regulatory regions where ΔFosB might bind. Our analysis revealed a region 320 bp upstream of the transcription start site containing cAMP and TPA response elements (CRE and TRE, respectively) in close proximity to one another (Supplemental Fig. 1a). Since both elements are known to recruit IEG proteins23, we performed chromatin immunoprecipitation (ChIP) on hippocampal tissue from APP as well as NTG mice to test whether ΔFosB enrichment occurred at this promoter region. Indeed, ChIP results confirmed binding of ΔFosB to the promoter of Calb1, and ΔFosB enrichment at this site was greater in APP mice relative to NTG littermates (Fig. 1d). ΔFosB enrichment on Calb1 was validated by comparison to IgG control ChIP and promoter amplicon sequencing (Supplemental Fig. 1b,c). These data highlight the potential ability of ΔFosB to regulate calbindin expression via actions at the Calb1 promoter gene.
We therefore assessed whether ΔFosB binding to Calb1 was associated with chromatin modifications that would impact Calb1 expression. Previous studies have demonstrated that reduction of calbindin protein levels in AD and epilepsy patients and rodent models occurs downstream of mRNA reduction4,7,9,24. In line with these findings, ΔFosB binds histone deacetylase 1 (HDAC1)21, suggesting that it suppresses Calb1 transcription via histone deacetylation. Indeed, we found hypoacetylated histone 4 lysine residues on the Calb1 promoter in APP mice relative to NTG controls (Fig. 1e and Supplemental Fig. 2a,b). We did not detect changes in acetylation on histone 3. Because long-term histone hypoacetylation can trigger histone methylation to further suppress gene expression21,25, we also assessed histone methylation at the Calb1 promoter. We found that the Calb1 promoter was hypermethylated in APP mice at histone 4 lysine 20, but not histone 3 lysine 9 (Fig. 1f and Supplemental Fig. 2c-e). Together, these results demonstrate Calb1 regulation by specific histone 4 modifications.
The activity-driven nature of ΔFosB expression20 and prevalence of calbindin reduction across many disorders accompanied by seizures4-7,9,10 suggest that epigenetic regulation of Calb1 by ΔFosB occurs in not just APP mice but also other rodent models with seizures. Therefore, we examined the relationship between ΔFosB and calbindin in a mouse model of epilepsy in which severe seizures lasting several hours (status epilepticus) are induced by pilocarpine. Similar to APP mice, we found elevated binding of ΔFosB to the Calb1 promoter, and lower Calb1 promoter histone 4 acetylation, in pilocarpine-treated mice relative to saline-treated controls (Fig. 1g,h and Supplemental Fig. 3a,b). Moreover, DG granule cells in pilocarpine-treated mice exhibited higher ΔFosB and lower calbindin protein expression that was evident on a cell-by-cell basis (Fig. 1i). The inverse relationship between ΔFosB and calbindin was also evident on a cell-by-cell basis in APP mice (Fig. 1j).
To demonstrate that a reduction in calbindin expression can be a direct result of ΔFosB-mediated epigenetic regulation, we tested whether adenoassociated virus (AAV)-mediated overexpression of ΔFosB in the hippocampus of wild-type C57BL/6 mice (AAV-ΔFosB mice) could suppress calbindin expression. Virus carrying CMV promoter-driven ΔFosB/eGFP or eGFP alone was stereotaxically infused into the DG of wild-type C57BL/6 mice (see Methods). This infusion resulted in robust ΔFosB and eGFP expression throughout the rostral-caudal extent of the DG (Fig. 2a,b and Supplemental Figs. 4,5). Viral overexpression of ΔFosB in wild-type mice did not affect neuronal survival or expression of neuron-specific genes such as NeuN (Supplemental Fig. 4c,e), and there was no detectable epileptiform activity in the hippocampus (Supplemental Fig. 6). Notably, ΔFosB overexpression did enhance the binding of ΔFosB to Calb1 (Fig. 2c and Supplemental Fig. 3c), and triggered Calb1 promoter histone 4 deacetylation (Fig. 2d and Supplemental Fig. 3d), consistent with its ability to recruit HDAC1 to gene promoters21. Furthermore, we found lower levels of calbindin mRNA and protein in the DG of AAV-ΔFosB mice (Fig. 2e-h). Reduced calbindin protein expression was apparent in both molecular and granule cell layers of the DG, reflecting the dendrites and cell bodies of granule cells, respectively (Supplemental Fig. 7a-d). Overall, these results demonstrate direct epigenetic control of Calb1 by ΔFosB. We also found that ΔFosB overexpression was sufficient to suppress granule cell expression of cFos, another gene target of ΔFosB (Supplemental Fig. 7e,f and21). These findings are consistent with prior studies showing that cFos expression in granule cells is increased acutely, but decreased chronically, by seizures26.
Figure 2. ΔFosB mediates transcriptional repression of Calb1 expression and causes spatial memory deficits.
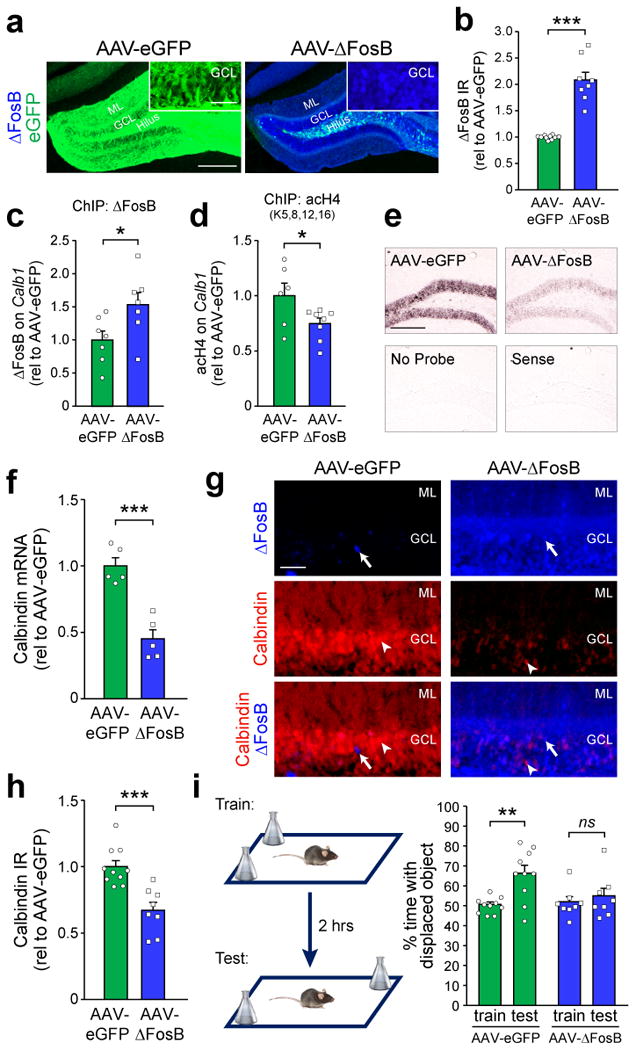
(a,b) Representative images and quantification of ΔFosB IR in the DG of wild-type mice that received bilateral hippocampal infusion of AAV carrying either ΔFosB/eGFP (AAV-ΔFosB) or eGFP alone (AAV-eGFP) (neGFP=10 mice, nΔFosB=8 mice, t16=8.23, ***p=3.83×10-7). eGFP expression is also displayed and can be observed in the cell bodies (GCL), axons (hilus), and dendrites (ML) of AAV-eGFP granule cells. Scale bars = 250 μm and 50 μm (inset). (c) Binding of ΔFosB (n=7 mice/treatment, t12=2.36, *p=0.036) and (d) histone 4 lysine acetylation (neGFP=6 mice, nΔFosB=8 mice, t12=-2.21, *p=0.047) on Calb1 in AAV-eGFP and AAV-ΔFosB mice. (e,f) In situ hybridization of calbindin mRNA (n=5 mice/treatment, t8=-5.94, ***p=3.44×10-4). Scale bar = 250 μm. (g,h) Calbindin protein IR (neGFP=10 mice, nΔFosB=8 mice, t16=-4.46, ***p=4×10-4) in DG granule cells of AAV-eGFP and AAV-ΔFosB mice. In panel (g), arrows indicate ΔFosB-expressing granule cells, and arrowheads indicate calbindin-expressing granule cells. Scale bar = 50 μm. (i) Left, object location test procedure. Right, performance of AAV-eGFP and AAV-ΔFosB mice in the object location test (paired t-tests: AAV-eGFP, n=10 mice, t9=4.6, **p=0.0013; AAV-ΔFosB, n=8 mice, t7=1.24, p=0.25). Data in panels (a-h) were analyzed using Student's t-tests. Error bars represent SEM.
To determine whether ΔFosB-mediated gene changes directly impact cognition, we tested spatial memory in AAV-ΔFosB mice using the object location test. This test is hippocampus-dependent27, and has previously been used to demonstrate spatial memory impairments in mouse models of AD and epilepsy28,29. We found that overexpression of ΔFosB in the hippocampus was sufficient to impair spatial memory (Fig. 2i).
To demonstrate that ΔFosB is not only sufficient but also necessary for regulating hippocampal calbindin expression and memory function, we tested whether inhibition of ΔFosB signaling in APP mice could normalize calbindin expression and ameliorate spatial memory deficits. To block ΔFosB signaling, we infused AAV carrying ΔJunD/eGFP into the DG of APP mice. ΔJunD is a truncated mutant of the transcription factor JunD, a predominant endogenous binding partner of ΔFosB in the brain20. This truncation removes the transactivation domain of JunD while leaving its dimerization and DNA-binding domains intact30. Therefore, dimerization of ΔJunD with ΔFosB inhibits ΔFosB in a dominant negative fashion. Notably, consistent with its truncation, ΔJunD does not preclude ΔFosB from binding its gene targets, but instead prevents the interaction of ΔFosB with downstream transcriptional co-regulators at target gene promoters20,30. Expression of ΔJunD in different brain regions effectively antagonizes ΔFosB-mediated gene regulation20,31. Using the object location test, we found that expression of ΔJunD in the hippocampus improved spatial memory in APP mice (Fig. 3a). We also demonstrate that ΔFosB is required for epigenetic suppression of Calb1 in APP mice, since ΔJunD expression restored Calb1 promoter histone acetylation (Fig. 3b and Supplemental Fig. 3e) and improved calbindin protein expression in the DG (Fig. 3c,d). Given that ΔJunD-mediated amelioration of spatial memory in APP mice was accompanied by improved calbindin expression, we then asked whether direct restoration of hippocampal calbindin expression in APP mice could also improve memory. To bypass endogenous control of Calb1 by ΔFosB, we infused AAV carrying CMV promoter-driven calbindin/eGFP into the hippocampus to elevate calbindin expression in APP mice (Fig. 3e,f). We found that directly elevating hippocampal expression of calbindin in APP mice improved object location memory (Fig. 3g), similar to hippocampal ΔJunD expression.
Figure 3. Both blockade of ΔFosB signaling and direct rescue of calbindin expression ameliorate spatial memory deficits in APP mice.
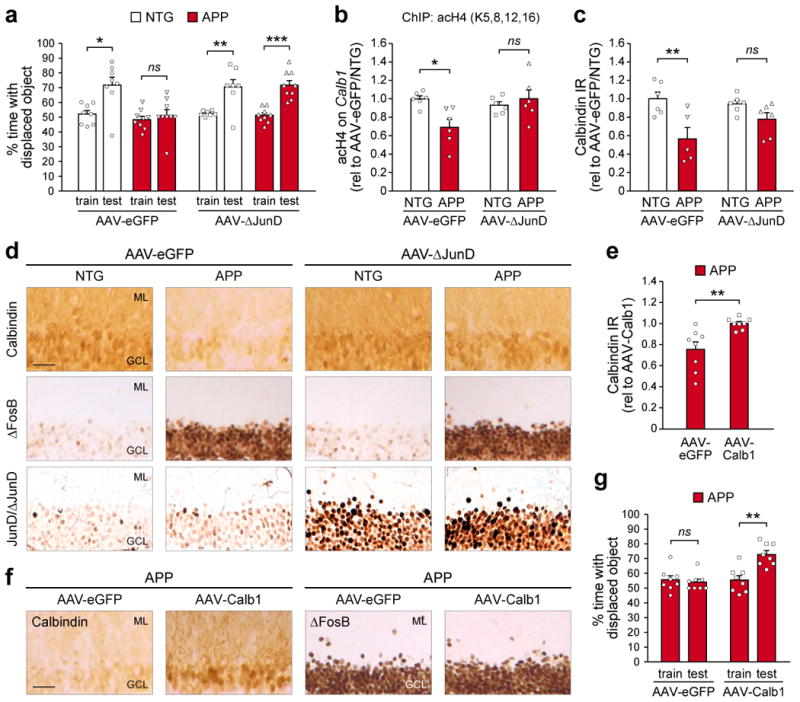
(a) Object location test performance of NTG and APP mice treated with bilateral hippocampal infusion of AAV carrying either ΔJunD/eGFP (AAV-ΔJunD) or eGFP alone (AAV-eGFP) (paired t-tests: AAV-eGFP/NTG, n=8 mice, t7=2.9, *p=0.023; AAV-eGFP/APP, n=9 mice, t8=0.55, p=0.6; AAV-ΔJunD/NTG, n=7 mice, t6=3.63, **p=0.01; AAV-ΔJunD/APP, n=9 mice, t8=5.84, ***p=3.86×10-4). (b) Histone 4 lysine acetylation on the Calb1 promoter of NTG and APP mice treated with AAV-eGFP or AAV-ΔJunD (n=6 mice/group; two-way ANOVA: genotype F1,20=3.07 p=0.095, treatment F1,20=2.98 p=0.1, interaction F1,20=7.73 p=0.012; Tukey's HSD: AAV-eGFP/NTG vs. APP *p=0.021, AAV-ΔJunD/NTG vs. APP p=0.89). (c,d) Quantification and representative images of calbindin IR in NTG and APP mice treated with AAV-eGFP or AAV-ΔJunD (n=6 mice/group except AAV-eGFP/APP n=5 mice; two-way ANOVA: genotype F1,19=13.82 p=0.0015, treatment F1,19=0.58 p=0.46, interaction F1,19=3.45 p=0.079; Tukey's HSD: AAV-eGFP/NTG vs. APP **p=0.0056, AAV-ΔJunD/NTG vs. APP p=0.44). In panel (d), corresponding images of ΔFosB IR (middle row) and JunD/ΔJunD IR (bottom row) are also shown. Scale bar = 50 μm. (e,f) Quantification and images of calbindin IR in APP mice treated with either AAV carrying either CMV promoter-driven calbindin/eGFP (AAV-Calb1) or AAV-eGFP (n=8 mice/group; Student's t-test: t14=3.36, **p=0.0046). In panel (f), corresponding images of ΔFosB IR (right) are also shown. Scale bar = 50 μm. (g) Object location test performance of APP mice treated with AAV-eGFP or AAV-Calb1 (n=8 mice/group; paired t-tests: AAV-eGFP, t7=-0.45, p=0.67; AAV-Calb1, t7=3.89, **p=0.0059). Error bars represent SEM.
Our findings in APP mice suggest that regulation of calbindin by ΔFosB may be a potential therapeutic target in AD and other seizure-associated disorders. To determine whether the relationship between ΔFosB and calbindin also exists in human patients, we examined expression patterns of both proteins in the DG of patients diagnosed with mild cognitive impairment (MCI) or AD. Indeed, higher ΔFosB expression corresponded with lower calbindin expression in MCI and AD patients (Fig. 4a-d), similar to APP mice. In addition, we observed a correlation between levels of both proteins and MMSE scores in MCI but not AD patients, presumably because of the numerous deficits in AD relative to MCI (Fig. 4e-h). There was also a significant correlation between ΔFosB or calbindin expression and MMSE scores when all patients (control, MCI, AD) were analyzed together (Supplemental Figs. 8 and 9). Lastly, to determine whether the relationship between ΔFosB and calbindin expression also exists in non-AD related conditions in which recurrent seizures occur, we examined DG tissue resected from TLE patients. Consistent with findings in MCI and AD patients, we observed an inverse relationship between ΔFosB and calbindin expression in TLE patients (Fig. 4i,j).
Figure 4. Increased hippocampal ΔFosB expression corresponds with decreased calbindin expression in patients diagnosed with MCI, AD, or TLE.
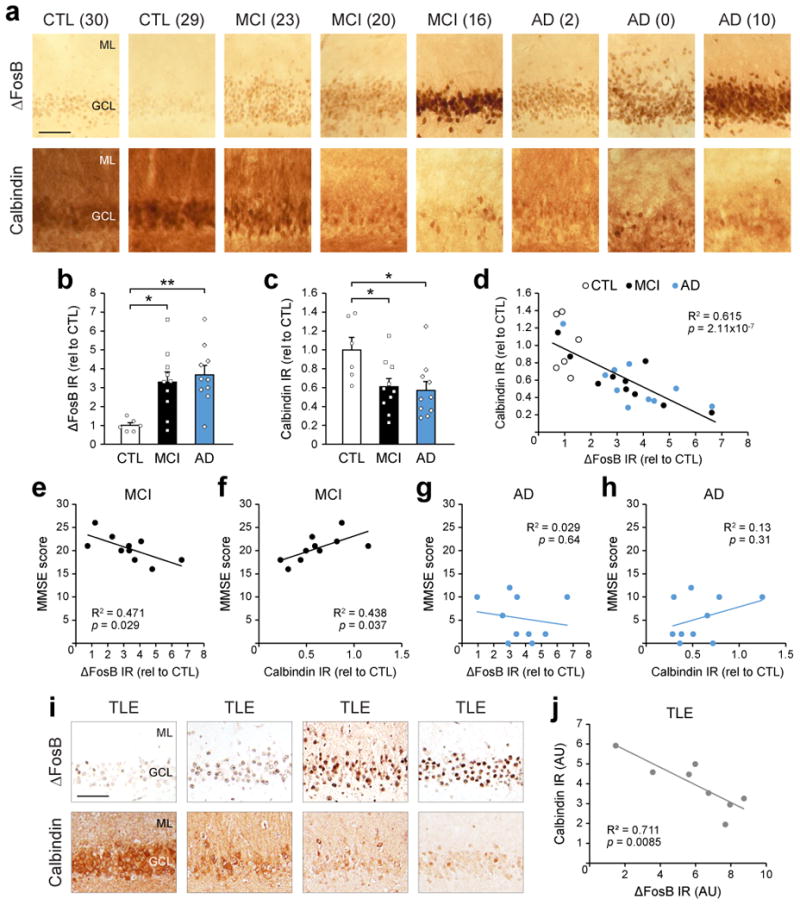
(a-d) Images, quantification, and regression analysis of ΔFosB and calbindin IR in DG of postmortem control (CTL, n=6 patients), MCI (n=10 patients), and AD (n=10 patients) (ΔFosB one-way ANOVA: F2,23=6.893, p=0.0045; Tukey's HSD: CTL vs. MCI *p=0.0153, CTL vs. AD **p=0.0045; calbindin one-way ANOVA: F2,23=4.461, p=0.023; Tukey's HSD: CTL vs. MCI *p=0.045, CTL vs. AD *p=0.026). In panel (a), numbers in parentheses represent MMSE scores. Scale bar = 100 μm. (e-h) Regression analyses of ΔFosB/calbindin IR and MMSE performance in MCI and AD patients. (i,j) Representative images and regression analysis of ΔFosB and calbindin IR in surgically resected DG from TLE (n=8 patients). Scale bar = 100 μm. AU, arbitrary units. Additional control, MCI, AD and TLE patient information is provided in Supplemental Fig. 9. Error bars represent SEM.
In summary, we have discovered a novel regulatory mechanism in which epileptiform activity recruits ΔFosB to epigenetically suppress calbindin expression in the DG. In doing so, we highlight ΔFosB's role as a transcription factor capable of chronically suppressing a gene that regulates synaptic transmission, plasticity, cognition, and seizure-related pathology. Given the prevalence of seizures across various disorders such as autism and schizophrenia32,33 in addition to epilepsy and AD, the identification of ΔFosB's role in the hippocampus may have broad implications for understanding why cognitive deficits are co-morbidities of many disorders accompanied by seizures. Furthermore, nuclear accumulation of ΔFosB due to its unusually long half-life provides a mechanism to explain why cognitive deficits persist even during seizure-free periods. Our finding that elevated ΔFosB expression correlates with reduced calbindin expression in both AD and TLE patients suggests that patients and rodent models of human disease share similar mechanisms of epigenetic regulation induced by seizures. By restoring memory function in APP mice either via ΔFosB inhibition or virus-driven calbindin expression, we have identified a molecular pathway that can potentially be targeted to improve cognition in AD and other seizure-associated disorders. Moreover, our finding that alterations in ΔFosB and calbindin expression correlate with MMSE scores in MCI, but not AD, patients suggests that possible therapeutic interventions targeting either protein should be initiated as early as possible to maximize effectiveness. The association of MMSE scores with ΔFosB and calbindin in MCI patients might be explained by the fact that hippocampal dysfunction, to which alterations in ΔFosB and calbindin expression greatly contribute, is particularly prominent in early disease. However, as AD progresses, disease pathophysiology in additional brain areas also contributes to overall cognitive dysfunction. The results of our study are consistent with a growing body of evidence showing that seizures occurring in early stages of AD impair memory and exacerbate cognitive decline18,19. The prevalence of seizures in AD may also be much greater than previously thought, as routine scalp EEG has been shown to miss “silent” hippocampal seizures detected via foramen ovale electrodes in AD patients34. Finally, some evidence suggests that calbindin reduction in granule cells following epileptiform activity may confer neuroprotection against excitotoxicity35,36 (but also see37,38). Demonstration of calbindin suppression by ΔFosB may provide further support for a neuroprotective role of calbindin reduction in granule cells, since previous studies have shown that both ΔFosB and HDAC1 can protect neurons from neurotoxic injury39,40. Interestingly however, we did not find obvious neuronal loss when we blocked ΔFosB signaling or virally expressed calbindin in APP mice (see Fig. 3d,f), which may be related to the time frame of our experiments or the possible existence of redundant neuroprotective mechanisms. Regardless, a potential trade-off between neuroprotection and synaptic function may emphasize caution for therapeutics targeting either pathway, as efforts to augment one pathway may generate side effects by simultaneously altering the other. To validate the therapeutic potential of targeting ΔFosB and/or calbindin, future studies will need to further characterize their functions in the hippocampus.
Methods
Mice
Heterozygous APP transgenic mice expressing human APP carrying the Swedish (K670N, M671L) and Indiana (V717F) familial AD mutations (hAPP770 numbering) driven by the platelet-derived growth factor (PDGF) β chain promoter (Line J20)16 were used in this study. The line was crossed for >10 generations onto a C57BL/6 background, and heterozygosity was maintained by breeding with wild-type C57BL/6 mice from The Jackson Laboratory (Bar Harbor, ME). Male and female mice from this line were used for experiments, and were evaluated at 2-4 months of age. At this age, many APP mice exhibit both recurrent seizures and cognitive deficits, but no plaque deposits17. Age- and sex-matched nontransgenic (NTG) wild-type animals from the same line were used as controls.
Wild-type C57BL/6 mice from The Jackson Laboratory were used at 2-4 months of age for AAV-ΔFosB overexpression experiments (see AAV-mediated gene transfer). Wild-type C57BL/6 mice from Charles River Laboratories (Wilmington, MA) were used at 2 months of age for pilocarpine experiments (see Pilocarpine-induced seizures).
For sacrifice, mice were anesthetized and perfused transcardially with PBS. Hemibrains were either fixed by immersion in 4% phosphate-buffered paraformaldehyde and sectioned at 30 μm (in preparation for in situ hybridization or immunohistochemistry) or frozen at -80°C (for chromatin immunoprecipitation). All procedures were approved by the Institutional Animal Care and Use Committees of Thomas Jefferson University, Baylor College of Medicine, and The Nathan Kline Institute.
Pilocarpine-induced seizures
Mice were initially administered scopolamine methyl nitrate (2 mg/kg sc; Sigma-Aldrich) and terbutaline hemisulfate salt (2 mg/kg sc; Sigma-Aldrich) to block peripheral effects of pilocarpine and dilate the respiratory tract, respectively. They also received 150 mg/kg ethosuximide sc (Sigma-Aldrich), as we have found that it improved mortality in C57BL/6 mice41. Thirty min later, pilocarpine hydrochloride (260 mg/kg sc; Sigma-Aldrich) was administered, and mice were placed in a standard mouse cage without bedding on a heating pad set at 37°C. Acute seizures were behaviorally monitored using a modified Racine's scale: stage 1, mouth and facial movement; stage 2, head nodding; stage 3, rearing with unilateral forelimb tonic-clonic movements; stage 4, rearing with bilateral forelimb tonic-clonic movements; stage 5, rearing and falling with tonic-clonic movements of the forelimbs. Once status epilepticus began (defined by the first stage 3-5 seizure that was not followed by resumption of normal behavior), mice were placed in a new cage at room temperature for 2 hrs and returned to the heated cage after seizure activity was reduced with diazepam (10 mg/kg sc; Henry Schein). Mice were then administered 5% dextrose-lactated Ringer's solution (1 mL ip; Henry Schein) while sedated by diazepam. After 2 hrs animals were then returned to the home cage, which was placed on the heating pad until sacrifice 3 days later.
Human tissue
Fixed dentate gyrus samples from AD and MCI patients were obtained from the Alzheimer's Disease Research Center at the University of California San Diego (San Diego, CA), sectioned at 60 μm, and stained for ΔFosB and calbindin (see Diaminobenzidine staining) after antigen retrieval with citrate buffer and formic acid. Fixed dentate gyrus samples from temporal lobe epilepsy patients were obtained and used with informed consent under IRB protocol H-10255, using epilepsy surgery resection specimens derived from adult patients treated at Baylor College of Medicine (Houston, TX). Sections were deparaffinized, subjected to antigen retrieval as above, and then stained for ΔFosB and calbindin.
AAV-mediated gene transfer
AAV serotype 2 carrying CMV-ΔJunD-IRES2-eGFP (AAV-ΔJunD), CMV-ΔFosB-IRES2-eGFP (AAV-ΔFosB), or CMV-eGFP (AAV-eGFP) was developed and characterized by the Nestler lab20. Previous experiments demonstrate that AAV2 is neurotropic and achieves stable neuronal gene expression within 18-22 days of infusion into the brain. The presence of IRES2 in AAV-ΔFosB and AAV-ΔJunD allows independent expression of ΔFosB or ΔJunD and eGFP, although there is preferential expression of the gene closest to the promoter. AAV serotype 2 carrying CMV-mCalb1-IRES2-eGFP (AAV-Calb1) was synthesized by Vector Biolabs (Philadelphia, PA).
One μL of virus solution was stereotaxically infused unilaterally or bilaterally into the hippocampus at rostral (-1.7 mm A/P, 1.2 mm M/L, 2 mm D/V from bregma) and caudal (-2.7 mm A/P, 2 mm M/L, 2.1 mm D/V from bregma) coordinates. Approximately 2×108 infectious particles were infused into each hippocampus. Mice were allowed to recover for 22-28 days post-surgery before further experimentation or sacrifice. Virus expression was assessed in all mice. Mice that did not exhibit expression in the hippocampus were excluded from analysis.
Object location
Object location is a dentate gyrus-dependent spatial memory test27 that requires that mice learn and remember the positions of two objects in an arena. Extra-arena spatial cues exist to help orient the mice during the train and test phases. For training, two identical flasks were placed at adjacent far corners of the arena (Fig. 2h), and animals were allowed to explore both flasks in 3 blocks of 3 min each with 3 min breaks in between. The amount of time mice spent exploring each flask was recorded by the experimenter. After a delay of 2 hrs, mice were returned to the arena for the test phase. In this phase, we displaced one flask to the adjacent empty corner, causing the two flasks to be diagonal from one another. Mice were given 3 minutes to explore both flasks, and the amount of time spent exploring each flask was recorded. Mice that remember the original locations of the two flasks will spend more time exploring the displaced flask vs. the non-displaced flask during the test phase. Previous studies have shown that both animal models of AD and epilepsy are impaired in this task28,29.
Electroencephalogram (EEG) recording
Mice were stereotaxically implanted with a 6-electrode array headcap for EEG monitoring: Two EEG screws were placed bilaterally over the left and right frontal cortices (AP: +1.5, ML: +/- 1.4), and a depth electrode was placed in right hippocampus (AP: -2.2, ML: -2, DV: -1.8). In addition, a reference screw and a depth ground electrode were placed over and inside cerebellum, respectively. Finally, an EEG screw was placed over parietal cortex (AP: -2.2, ML: -2) for additional anchor. All EEG screws were wrapped with silver wires connected to a pedestal (Plastics One, Roanoke, VA), and the entire assembly was secured on the skull with dental cement (Ortho-Jet, LangDental, Wheeling, IL). Animals were allowed to recover for at least 4 days prior to commencement of recordings.
For EEG recordings, a flexible cable connected the animals' headcaps to a commutator thus allowing them to move freely during the recordings. Video-monitored EEG recordings were performed in the animals' home cage using a Stellate Harmonie (Natus Medical, Pleasanton, CA) interface. Spike count analysis was performed using LabChart Pro (AD Instruments, Dunedin, NZ).
Chromatin immunoprecipitation (ChIP)
Whole hippocampi were subdissected, fixed in 1% formaldehyde for 15 min, and homogenized in a 0.5% NP40 cell lysis buffer using Potter-Elvehjem PTFE tissue grinders. Nuclei were subsequently pelleted and reconstituted in 1% SDS. Nuclear lysis was accomplished via repetitive freeze-thaw, and chromatin shearing followed via the Digital SLP probe sonifier (Branson Ultrasonics) or Q800R bath sonicator (QSonica). Both methods of sonication were calibrated to produce fragment sizes 100-600 bp (with a primary peak at 200 bp), detected via ethidium bromide gel. Afterwards, ChIP was performed using the Magna ChIP A Kit (Millipore). ChIP antibodies were used at 2 μg, and included rabbit anti-ΔFosB (Cell Signaling 9890 and D3S8R), rabbit anti-H3K9+K14+K18+K23+K27ac (Abcam ab47915), rabbit anti-H4K5+K8+K12+K16ac (Millipore 06-866), rabbit anti-H3K9me2 (Abcam ab1220), rabbit anti-H3K9me3 (Millipore 07-442), rabbit anti-H4K20me3 (Millipore 07-463), and normal rabbit IgG (Millipore 12-370). Prior to the addition of antibody, 2% of the sheared chromatin was set aside as input. Following ChIP, immunoprecipitated chromatin was eluted using 0.1M NaHCO3 and 1% SDS. Next, protein was digested using 0.1 mg/mL proteinase K, and DNA was separated using phenol-chloroform extraction. DNA was then pelleted and washed twice using 70% ethanol, and reconstituted in MilliQ H2O. Calb1 promoter enrichment was analyzed using qPCR of ChIP DNA versus input. Primers for Calb1 promoter amplification are as follows: 5′-TTCAAATACTCAACTGCCTCG-3′ (forward) and 5′-GGAGGCTTTCACTCCTGAATGT-3′ (reverse).
Diaminobenzidine (DAB) staining
Brain sections were avidin-biotin/immunoperoxidase labeled using the following primary antibodies: rabbit anti-ΔFosB (1:300, Cell Signaling 9890), rabbit anti-calbindin (1:15,000, Swant CB-38A), rabbit anti-JunD (1:1000, Santa Cruz Biotechnology sc-74). Biotinylated goat anti-rabbit (1:200, Vector BA-1000) was used as the secondary antibody. DAB was used as the chromagen. For analysis of immunoreactive (IR) structures, two coronal sections (300 μm apart) were selected per mouse between -2.54 and -2.88 mm from bregma. The integrated optical density (IOD) of immunostains was determined using the MetaMorph Image Analysis Software (Molecular Devices) and averaged in two areas (0.04 mm2 each) of either the molecular (calbindin) or granule cell layer (ΔFosB) of the dentate gyrus (DG) and stratum radiatum of CA1. Relative IR was thus expressed as the IOD ratio in the DG versus CA1. The mean ratio of nontransgenic (NTG) mice was defined as 1. For quantification of ΔFosB and calbindin in fixed human dentate gyrus, IR values were background corrected using comparison with nonspecific staining in a nearby acellular white matter tract.
Immunofluorescence staining
All brain sections within an experiment were processed, stained, and imaged at the same time, with the same parameters. Brain sections were fluorescently labeled using the following primary antibodies: rabbit anti-ΔFosB (1:100, Cell Signaling 9890; 1:1000, Cell Signaling D3S8R), mouse anti-calbindin (1:1000, Swant 300), mouse anti-NeuN (1:5000, Millipore MAB377), goat anti-cFos (1:300, Santa Cruz Biotechnology sc-52g). Fluorophore-conjugated secondary antibodies utilized in this study included donkey anti-rabbit AMCA (1:200, Jackson ImmunoResearch 711-155-152), donkey anti-goat Cy3 (1:200, Jackson ImmunoResearch 705-165-147), donkey anti-mouse Alexa Fluor 594 (1:500, Life Technologies A-21203), donkey anti-rabbit Alexa Fluor 488 (1:500, Life Technologies A-21206). For analysis of calbindin-IR, ΔFosB-IR, and NeuN-IR structures, fluorescence intensity (FI) was determined in the DG molecular (calbindin) or granule cell layer (ΔFosB, NeuN, and calbindin) in every 10th serial coronal section throughout the rostral-caudal extent of the hippocampus using MetaMorph Image Analysis Software. FI of CA1 stratum radiatum was obtained in the same sections. Relative IR was expressed as the FI ratio in the DG versus CA1. The mean ratio of NTG mice was defined as 1. For analysis of cFos-IR, fluorescently labeled cells were counted in the granule cell layer in every 10th serial coronal section throughout the rostral-caudal extent of the hippocampus. For cell-by-cell analysis of ΔFosB-IR and calbindin-IR, ΔFosB and calbindin FI were determined for individual cells via ImageJ (NIH). Random blocks of 8-10 adjacent cells from rostral hippocampal sections (-1.6 mm from bregma) of APP and pilocarpine-treated mice were chosen for quantification. The mean FI for both proteins was defined as 1.
In situ hybridization (ISH)
All solutions used to process brain sections designated for ISH were pretreated with DEPC and/or autoclaved to minimize RNA degradation. Non-sterile tools and surfaces were also pretreated with RNase Zap (Ambion). ISH-competent free-floating brain sections were digested with 1 ug/mL proteinase K for 12 min before overnight hybridization at 65°C with digoxygenin-labeled full-length antisense riboprobe for mouse Calb1 (synthesized from Calb1 cDNA, IMAGE cat# MMM1013-202766730). Sense and no probe controls were included. The sections were then washed once with 5× SSC/0.5% Tween20 and 7 times with 0.2× SSC/0.5% Tween20. This is followed by blocking with 10% heat-inactivated sheep serum and overnight incubation with 1:5000 alkaline phosphatase-conjugated sheep anti-digoxigenin antibody (Roche 11333089001) at 4°C. Development a blue/purple stain for colorimetric detection was achieved via incubation with the chromogen NBT/BCIP (Roche) for 3 hrs at room temperature. Sections were then washed in PBS/EDTA and fixed with 4% paraformaldehyde for 10 min before mounting onto slides. Determination of Calb1 mRNA signal was performed via IOD analysis of the DG granule cell layer, similar to IOD analysis for DAB staining.
Statistics
Statistical analyses were performed using SPSS-23 (IBM) and Prism 7 (GraphPad). Sample sizes for both biochemical and behavioral experiments were determined based on calculations performed on empirical data and power analyses. The number of animals used for each experiment was appropriate to detect biochemical or behavioral differences with 80% power and alpha set at 0.05. Unless otherwise stated, results are represented as sample means ± standard errors of the mean. These data are distributed normally as stipulated by the central limit theorem. Differences between experimental groups were assessed by Student's t-test (two-tailed except where indicated as one-tailed) when comparing means between two groups, paired t-test when comparing means within the same individuals, one-way ANOVA when comparing means between three or more groups, two-way ANOVA when performing multifactorial analyses, and the Welch's F test when data failed the Levene's test for homogeneity of variances. Post-hoc analyses were used where appropriate. Correlations were assessed by simple regression analysis. No specific method of randomization was used, but animals were semi-randomly assigned to experimental groups based on birth order after balancing for age, sex, and genotype. We found no differences in the ΔFosB/calbindin relationship between male and female mice, therefore both sexes were included in experiments (ΔFosB two-way ANOVA: genotype F1,55=24.41, ***p<0.0001; sex F1,55=0.39, p=0.53; interaction F1,55=1.35, p=0.25; calbindin two-way ANOVA: genotype F1,55=48.78, ***p<0.0001; sex F1,55=0.049, p=0.83; interaction F1,55=0.0014, p=0.97). For all analyses, the experimenters were blinded to the genotype and treatment type of each mouse. In vivo and ChIP experiments were replicated at least once, immunohistochemical experiments were replicated at least twice.
Further detailed information on experimental design and reagents can be found in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We thank Dr. Rohan Jagirdar for helpful comments on the manuscript. This work was supported by the Margaret Q. Landenberger Research Foundation (JC), the Hassel Family Foundation (JC), National Institutes of Health Grants NS085171 (JC), F30-AG048710 (JCY), NYS OMH (HES, JJF), and AG051848, BX003040, AG0051839, AG005131 (RAR).
Footnotes
Data availability: The data that support the findings of this study are available from the corresponding author upon request.
Author Contributions: JCY and JC conceived the project. JCY, XZ, EJN, HES, and JC designed the experiments. JCY, MSP, IP, KM, BFC, JJF, YZ, JWP, HES, and JC performed the experiments and analyzed the data. CAM and DY collected and analyzed specimens derived from human epilepsy patients. RAR provided fixed AD brain samples and clinical information. All authors discussed results and JCY, RAR, HES, and JC wrote the manuscript.
Authors report no competing financial interests or conflicts.
References
- 1.Westerink RH, Beekwilder JP, Wadman WJ. Differential alterations of synaptic plasticity in dentate gyrus and CA1 hippocampal area of Calbindin-D28K knockout mice. Brain Res. 2012;1450:1–10. doi: 10.1016/j.brainres.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 2.Molinari S, et al. Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc Natl Acad Sci U S A. 1996;93:8028–8033. doi: 10.1073/pnas.93.15.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jouvenceau A, et al. Decrease in calbindin content significantly alters LTP but not NMDA receptor and calcium channel properties. Neuropharmacology. 2002;42:444–458. doi: 10.1016/s0028-3908(01)00202-7. [DOI] [PubMed] [Google Scholar]
- 4.Emmanuele V, et al. Decreased hippocampal expression of calbindin D28K and cognitive impairment in MELAS. J Neurol Sci. 2012;317:29–34. doi: 10.1016/j.jns.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abraham H, et al. Degree and pattern of calbindin immunoreactivity in granule cells of the dentate gyrus differ in mesial temporal sclerosis, cortical malformation- and tumor-related epilepsies. Brain Res. 2011;1399:66–78. doi: 10.1016/j.brainres.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Magloczky Z, Halasz P, Vajda J, Czirjak S, Freund TF. Loss of Calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76:377–385. doi: 10.1016/s0306-4522(96)00440-x. [DOI] [PubMed] [Google Scholar]
- 7.Iacopino AM, Christakos S. Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases. Proc Natl Acad Sci U S A. 1990;87:4078–4082. doi: 10.1073/pnas.87.11.4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin J, Scharfman HE. Shared cognitive and behavioral impairments in epilepsy and Alzheimer's disease and potential underlying mechanisms. Epilepsy Behav. 2013;26:343–351. doi: 10.1016/j.yebeh.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palop JJ, et al. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karadi K, et al. Correlation between calbindin expression in granule cells of the resected hippocampal dentate gyrus and verbal memory in temporal lobe epilepsy. Epilepsy Behav. 2012;25:110–119. doi: 10.1016/j.yebeh.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 11.Hall AM, et al. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease. J Neurosci. 2015;35:6221–6230. doi: 10.1523/JNEUROSCI.2552-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez PE, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klapstein GJ, et al. Calbindin-D28k fails to protect hippocampal neurons against ischemia in spite of its cytoplasmic calcium buffering properties: evidence from calbindin-D28k knockout mice. Neuroscience. 1998;85:361–373. doi: 10.1016/s0306-4522(97)00632-5. [DOI] [PubMed] [Google Scholar]
- 14.Jouvenceau A, et al. Glutamatergic synaptic responses and long-term potentiation are impaired in the CA1 hippocampal area of calbindin D(28k)-deficient mice. Synapse. 1999;33:172–180. doi: 10.1002/(SICI)1098-2396(19990901)33:3<172::AID-SYN2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 15.Brini M, Cali T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci. 2014;71:2787–2814. doi: 10.1007/s00018-013-1550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mucke L, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palop JJ, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vossel KA, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Ann Neurol. 2016;80:858–870. doi: 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vossel KA, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renthal W, et al. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eagle AL, et al. Experience-Dependent Induction of Hippocampal DeltaFosB Controls Learning. J Neurosci. 2015;35:13773–13783. doi: 10.1523/JNEUROSCI.2083-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter DS, Harrison AJ, Falenski KW, Blair RE, DeLorenzo RJ. Long-term decrease in calbindin-D28K expression in the hippocampus of epileptic rats following pilocarpine-induced status epilepticus. Epilepsy Res. 2008;79:213–223. doi: 10.1016/j.eplepsyres.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 26.Peng Z, Houser CR. Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J Neurosci. 2005;25:7210–7220. doi: 10.1523/JNEUROSCI.0838-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kesner RP, Taylor JO, Hoge J, Andy F. Role of the dentate gyrus in mediating object-spatial configuration recognition. Neurobiol Learn Mem. 2015;118:42–48. doi: 10.1016/j.nlm.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Cho KO, et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun. 2015;6:6606. doi: 10.1038/ncomms7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma T, et al. Suppression of eIF2alpha kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown PH, Kim SH, Wise SC, Sabichi AL, Birrer MJ. Dominant-negative mutants of cJun inhibit AP-1 activity through multiple mechanisms and with different potencies. Cell Growth Differ. 1996;7:1013–1021. [PubMed] [Google Scholar]
- 31.Berton O, et al. Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol Psychiatry. 2009;66:554–561. doi: 10.1016/j.biopsych.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hesdorffer DC, et al. Epilepsy, suicidality, and psychiatric disorders: a bidirectional association. Ann Neurol. 2012;72:184–191. doi: 10.1002/ana.23601. [DOI] [PubMed] [Google Scholar]
- 34.Lam AD, et al. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat Med. 2017;23:678–680. doi: 10.1038/nm.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagerl UV, et al. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagerl UV, Mody I. Calcium-dependent inactivation of high-threshold calcium currents in human dentate gyrus granule cells. J Physiol. 1998;509(Pt 1):39–45. doi: 10.1111/j.1469-7793.1998.039bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattson MP, Rychlik B, Chu C, Christakos S. Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons. Neuron. 1991;6:41–51. doi: 10.1016/0896-6273(91)90120-o. [DOI] [PubMed] [Google Scholar]
- 38.Lopez-Meraz ML, Wasterlain CG, Rocha LL, Allen S, Niquet J. Vulnerability of postnatal hippocampal neurons to seizures varies regionally with their maturational stage. Neurobiol Dis. 2010;37:394–402. doi: 10.1016/j.nbd.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurushima H, et al. Selective induction of DeltaFosB in the brain after transient forebrain ischemia accompanied by an increased expression of galectin-1, and the implication of DeltaFosB and galectin-1 in neuroprotection and neurogenesis. Cell Death Differ. 2005;12:1078–1096. doi: 10.1038/sj.cdd.4401648. [DOI] [PubMed] [Google Scholar]
- 40.Kim D, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iyengar SS, et al. Suppression of adult neurogenesis increases the acute effects of kainic acid. Exp Neurol. 2015;264:135–149. doi: 10.1016/j.expneurol.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.