

Abstract
Germinal matrix hemorrhage is induced by stereotaxic injection of collagenase into the germinal matrix of P7 Sprague-Dawley rats. Hemoglobin assay, western blot, immunofluorescence and neurobehavioral tests were used to test the effects of BLVRA on hematoma resolution and anti-inflammatory response. We showed that BLVRA triggered a signaling cascade that ameliorated post-hemorrhagic neurological deficits in both short-term and long-term neurobehavioral tests in a GMH rat model. Specifically, BLVRA inhibited toll-like receptor 4 (TLR4) expression by translocating to the nucleus in an endothelial nitric oxide (eNOS)/nitric oxide (NO)-dependent manner. BLVRA also induced the upregulation of CD36 scavenger receptor level in microglia/microphages, of which the prominent role is to enhance hematoma resolution. However, the beneficial effects of BLVRA were abolished with the knockdown of eNOS, indicating that the eNOS/NO system is an important downstream factor of BLVRA. Our results demonstrate a mechanism of BLVRA modulating hematoma resolution and suppressing inflammation through eNOS/NO/TLR4 pathway in the GMH rat model.
Keywords: germinal matrix hemorrhage, innate immunity, hematoma resolution, inflammation
Graphical Abstract
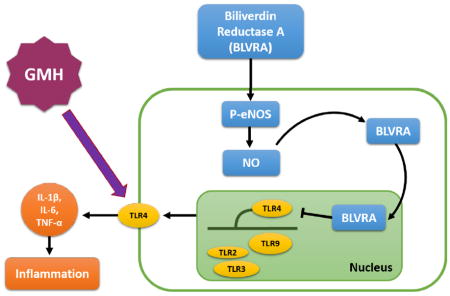
Introduction
Germinal matrix hemorrhage (GMH) results from the rupture of immature blood vessels in the subventricular region of premature infants 1. It occurs in up to 20% of infants delivered at <32 weeks gestation. Its common complications include significant developmental delay, mental retardation, hydrocephalus and cerebral palsy2. Current clinical management is limited to improving symptoms in GMH neonates but does not confer further protective or prophylactic effects. Thus, it is critically important to identify new approaches to modulate disease progression in GMH patients.
Hematoma toxicity is caused by the cytotoxic substances released from both activated neuroglia and hematogeous cells 3, 4,5. In GMH, the blood clot can also obstruct cerebrospinal fluid circulation, contributing to post-hemorrhagic hydrocephalus development 6, 7,8. Clinical studies use hematoma volume as a prognostic indicator, which negatively correlates with functional recovery 9
Biliverdin reductase (BVR) is a well-characterized enzyme in the heme degradation pathway that converts biliverdin to bilirubin. Two isoforms of BVR were named biliverdin reductase A (BLVRA) and biliverdin reductase B (BLVRB). Both of them can generate bilirubin but only BLVRA, also called Biliverdin-IX-alpha reductase, is a metabolic enzyme that converts biliverdin-IX-alpha into an antioxidant and antinitrosative molecule bilirubin-IX-alpha in this degradation pathway10–12. In addition, BLVRA decreased Toll-like receptor-4 (TLR4) expression and regulated inflammatory cytokines secretion in monocytes13. Although the anti-inflammatory role of BLVRA is more well-established, the role of bilirubin in the brain remains controversial. 14, 15,16. It is possible that bilirubin, in the presence of BLVRA, can confer more protective effects rather than toxic effects. However, more studies are warranted to understand the underlying mechanisms of bilirubin in brain diseases.
Nitric Oxide (NO) is an important intercellular messenger in the central/peripheral nervous system. Endothelial nitric oxide synthase (eNOS), neuronal nitric oxide synthase (nNOS), and inducible nitric oxide synthase (iNOS) are three isoforms of NOS. NO formed via eNOS is involved in maintaining microcirculation17, preventing platelet aggregation and inhibiting leukocyte migration and adhesion18. NO generated from nNOS acts as a neurotransmitter, contributing to neuronal plasticity and memory formation19. iNOS exists in macrophages and glial cells, generates NO and evokes nitrosative stress in response to pro-inflammatory cytokines20. The amount of NO from iNOS is 100–1000 times greater than that from eNOS and nNOS21. Innate eNOS activity is an indicator of brain injury. eNOS with greater activity plays a protective role against secondary neuronal insult. Currently, a study on acute liver injury shows that biliverdin administration inhibits TLR4 by biliverdin reductase translocation to the nucleus through NO generated from eNOS13. Yet, no studies have evaluated the potential protective effects of BLVRA in GMH.
In this study, we investigated if exogenous BLVRA enhances hematoma resolution and reduces inflammation through eNOS in a GMH rat model. We hypothesize BLVRA stimulation will increase microglia/macrophage phagocytosis of blood clots, alleviate post-hemorrhagic neurological deficits, ventricular dilation, oxidative stress and inflammation through eNOS/NO signaling pathway.
Materials and methods
Animals and surgeries
All experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee at Loma Linda University. All studies were conducted in accordance with the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. P7 Sprague-Dawley neonatal rat pups (weighing 12–15g, brain development is comparable to 30–32 week gestation infants) were purchased from Envigo (Livermore, CA). GMH was induced as previously described22. Animals were anesthetized with 3% isoflurane. The following stereotactic coordinates were located using Bregma as a reference point: 1.6 (rostral), 1.5 mm (lateral), and 2.7 (depth). A 27-gauge needle was inserted into a 1mm burr hole with a rate of 1μl/min. A stereotaxic-assisted infusion needle was used to mimic preterm right ganglionic eminence bleeding by injecting 3μl of 0.3U bacterial collagenase at 1μl/min. After the infusion, the needle was left at the place for 10 mins to prevent “back leakage”. Once the needle was removed, bone wax was used to seal the burr hole and the incision site was sutured. Animals were allowed to recover on a 37°C heated blanket. Animals were put back with their dams after recovering from anesthesia. Sham animals were operated with needle insertion without collagenase infusion.
Animal treatments and experimental groups
BLVRA recombinant protein (BLVRA, Abnova), eNOS inhibitor (Iromycin A, 1mg/kg, Abcam), iNOS inhibitor Mercaptoethylguanidine dihydrobromide (MEG, 1mg/kg, Abcam), nNOS inhibitor, Nω-Propyl-L-arginine (NPA, 2mg/kg, Abcam) and NO donor (Sodium nitroprusside, SNP, 0.1nmol, Sigma-Aldrich) were administered intranasally (i.n.) to experimental animals at 1 h post-GMH and then once daily for 3 days. The scrambled siRNA (Origene) and BLVRA siRNA (Origene) were administered intracerebroventricularly (i.c.v.) to experimental animals at 24 h before GMH. We used 234 animals in this study and three pups died from the surgery. Animals were randomly divided into the following groups: sham-operated (sham; n=36), sham-operated + BLVRA (sham + BLVRA; n=6), GMH + PBS (GMH + vehicle; n=36), GMH + PBS + 2% DMSO (GMH + vehicle + DMSO; n=6), GMH + 0.03 μg/rat BLVRA (GMH + BLVRA; low dose; n=6), GMH + 0.1 μg/rat BLVRA (GMH + BLVRA; medium dose; n=36), GMH + 0.3 μg/rat BLVRA (GMH + BLVRA; high dose; n=6), GMH + scrambled siRNA (n=12), GMH + BLVRA siRNA (n=12), GMH + BLVRA + Iromycin A(n=30), GMH + BLVRA + MEG (n=18), GMH + BLVRA + NPA (n=18), sham-operated + SNP (sham + SNP; n=6), GMH + PBS + SNP (GMH + vehicle + SNP; n=6).
Neurobehavioral examination
The therapeutic effects of BLVRA on the neonates were evaluated by righting reflex, negative geotaxis and eye opening latency between 1 and 7 days post-GMH 22. Morris water maze, rotarod, foot fault tests and open-field were used to evaluate neurocognitive deficits and motor coordination in a blinded manner between 21 and 28 days post-GMH 22.
Histological volumetric analysis
National Institutes of Health ImageJ software was used to analyze ventricular volume, cortical thickness, white matter area, and grey matter loss in Nissl stained histological brain sections at 28 days post-GMH. Optical dissector principles were used to delineate cerebral structure borders following previously defined criteria from stereologic neuroanatomical studies 23–25. Volumes were calculated by the following formula: average (area of coronal section) × section interval × number of sections 26, 27.
Hemoglobin assay
Frozen extracted forebrains and 3 mL PBS were placed in separate glass tubes. Then we homogenized (Tissue Miser Homogenizer; Fisher Scientific, Pittsburgh, PA) the tissues for 60s and lysed erythrocyte membranes by ultra-sonication for 60s. We then centrifuged the homogenates for 30 min, immediately transferred the supernatant to a clean tube with a 4:1 ratio of Drabkin’s reagent (Sigma-Aldrich) and incubated them for 15 min. Absorbance was analyzed on a standard curve by spectrophotometric measurements (540 nm; Genesis 10uv; Thermo Fisher Scientific, Waltham, MA) using well-established protocols to measure hemorrhagic volume28–30.
Western Blot
The lysates from frozen tissues were separated by SDS-PAGE and transferred onto nitrocellulose membranes. After blocking with 5% milk for 2 h, membranes were incubated with the following primary antibodies at 4°C overnight: anti-BLVRA (1:1000, Santa Cruz), eNOS (1:1000, Santa Cruz), phospho-eNOS (S1177) (1:1000, Santa Cruz), iNOS (1:1000, Abcam), phospho-iNOS (Y151) (1:1000, Abcam), nNOS (1:1000, Abcam), phospho-nNOS (S1417) (1:1000, Abcam), CD36 (1:500, Santa Cruz), mannose receptor (CD206, 1:500, Santa Cruz), TLR4 (1:1000, Abcam), Actin (1:3000, Abcam), 3-Nitrotyrosine (3-NT, 1:1000, Abcam), 4-Hydroxynonenal (HNE, 1:1000, Abcam), dinitrophenol (DNP, 1:1000, Abcam), IL-1β (1:1000, Abcam), IL-6 (1:2000, Abcam), TNF-α (1:1000, Abcam), Lamin B (1:1000, Abcam), TLR2 (1:1000, Abcam), TLR3 (1:1000, Abcam), TLR9 (1:1000, Abcam), followed by respective secondary antibodies (1:5000, Santa Cruz) for 2 h then visualized by ECL plus Kit (GE Healthcare and Life Science). Relative density of the protein immunoblot images was analyzed by ImageJ software.
For cytosol-nuclei translocation study, NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) was used to isolate cytosolic and nuclear protein lysates according to manufacturer’s instructions. The following steps of Western blot were performed as described previously27.
Immunohistochemistry
Immunofluorescence
Tissues were embedded in Optimal Cutting Temperature Compound (OCT, Fisher Scientific) and then sectioned into 10 μm slices. Sections were stained with OX-42 (1:1000, Abcam), mannose receptor (CD206) (1:500, Abcam), GFAP (1:1000, Abcam), and NeuN (1:1000, Abcam) at 4°C overnight. Appropriate fluorescence conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA) were incubated for 1 h at room temperature. Leica DMi8 (Leica Microsystems, Germany) was used to image the peri-ventricular region. M2 microglia activated cells were counted on 6 sections in every group over a 20 × microscopic field and measured as cells/field, as described 31.
Dihydroethidium (DHE) and hydroxyphenyl fluorescein (HPF) staining
Slides were incubated with DHE (2 μmol/L, Thermo Scientific) for 30 min at 37°C, protected from light. HPF staining was performed according to the manufacturer’s protocol (Thermo Scientific). Slides were incubated with HPF for 1 h at 37°C in the dark. Slides were imaged using a fluorescence microscope (Leica). ImageJ software was used to analyze relative fluorescence intensity of DHE staining. HPF positive cells were counted in microscopic field of 20 × and expressed as cells/field.
Nitric oxide assay
The nitric oxide assay kit (Abcam) was used to measure NO according to the manufacturer’s protocol. After standard curve preparation, fresh tissues were homogenized with a homogenizer (Tissue Miser Homogenizer; Fisher Scientific, Pittsburgh, PA) on ice. Supernatant was collected and transferred to a clean tube. A final concentration of 1 M perchloric acid (PCA, Fisher Scientific) was added to the supernatant tubes and then incubated for 5 min. The supernatant was transferred to a fresh tube after centrifuge. An equal PCA volume of potassium hydroxide (KOH, Fisher Scientific) was added to the supernatant. The supernatant was collected after centrifuge for 15 min. After reaction wells were assigned, a microplate colorimetric reader (OD540 nm, Bio-Rad, Hercules, CA) was used to measure the absorbance, converted into NO volumes on a nitrite standard calibration curve.
Statistical analysis
In this study, all animals were randomly assigned to different groups. All the experimental tests were blinded to the surgeons and researchers who did the experiments and analyze the research data. All tests for exploratory studies were performed two-sided. GraphPad was used to exclude outliers. Samples size were estimated using a type I error rate of 0.05 and a power of 0.8 on a 2-sided test by power analysis. For parametric data, analysis was using one-way ANOVA followed by the Tukey’s post-hoc test. For non-parametric data, Kruskall-Wallis with Dunn’s post-hoc tests was used to analysis. Longitudinal data were analyzed by two-way ANOVA with Tukey’s post-hoc test. Data were expressed as mean ± SD. P values of <0.05 were considered statistically significant. GraphPad Prism 6 (La Jolla, CA, USA) and SigmaPlot 11.0 (SysStat, Germany) were used for graphing and analyzing all the data.
Results
Exogenous BLVRA ameliorated short-term neurological deficits and accelerated hematoma resolution
To determine whether the administration of BLVRA is protective in GMH rat, short-term neurobehavioral tests, including righting reflex, negative geotaxis and eye-opening latency tests, were performed. Test results showed that BLVRA treatment attenuated GMH-induced neurological deficits and developmental delay (Fig. 1A–1C). Medium-dose BLVRA ameliorated GMH-induced neurological deficits to the greatest extent in short-term behavior tests during 1 to 3 days post-GMH. Eye opening was monitored daily from Day 1 up to Day 9 after GMH. GMH induced a significant delay in eye opening compared with the medium-dose treatment group after GMH. Thus, medium-dose BLVRA was used for the following studies.
Figure 1.

BLVRA ameliorated GMH-induced developmental delay in short-term neurobehavioral tests and enhances hematoma absorbance. From 1 to 7 days after GMH, neurological deficits evaluated by righting reflex (A), negative geotaxis (B), and eye opening latency (C). Hemoglobin content was evaluated at 24 h (D) and 72 h (E). Co-localization of activated microglia (OX-42; scale bar: 100 μm) with mannose receptor and DAPI at 72 h after GMH (F) and quantified activated M2 microglia (G). Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group, &P < 0.05 compared with BLVRA group, %P < 0.05 compared with scramble siRNA group. N = 6 each group.
Since the improvement in neurological outcomes can be associated with blood clot clearance, hemoglobin assay was performed in rat pups at 24 h and 72 h post-GMH. All groups showed significantly higher hemoglobin content compared with the sham group and no treatment had any effect on the hemoglobin content at 24 h post-GMH (Fig. 1D). However, at 72 h, BLVRA-treated group showed less hemoglobin content compared to the vehicle group. Meanwhile, BLVRA siRNA-administered animals presented significantly higher hemoglobin content compared to all other groups (Fig. 1E). Previous studies show that M2 microglia/macrophages are more effective in clearing blood clots.27, 32 Thus, 72 h was chosen for immunostaining of the co-localization of activated microglia/macrophage (OX-42) and M2 phenotype microglia/macrophage (mannose receptor) (Fig. 1F). The immunostaining results showed that GMH induced microglia activation and macrophage infiltration. BLVRA contributed to more M2 polarization in microglia/macrophages compared with GMH+vehicle group (Fig. 1G).
BLVRA inhibited inflammatory response in GMH and co-localized with all cell types in the brain
Since hematoma clearance contributes to decreased inflammatory response, western blot was conducted at 3 days after GMH to determine the anti-inflammatory effects of BLVRA (Fig. 2A). BLVRA expression level was significantly upregulated after BLVRA administration but downregulated by siRNA (Fig. 2B). TLR4 expression was increased in the vehicle group compared with the sham group, and BLVRA significantly reduced TLR4 expression. The opposite effects were found in BLVRA siRNA group (Fig. 2C). Similar patterns were found for the expression of interleukin 1 beta (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNF-α). GMH significantly induced these cytokine levels and BLVRA attenuated the induction. However, the knockdown of BLVRA siRNA restored the cytokine levels (Fig. 2D–2F). Furthermore, double immunofluorescence staining of BLVRA with Iba-1, GFAP, or NeuN showed that BLVRA was expressed on microglia, astrocytes and neurons at 3 days post-GMH (Fig. 2G–2I).
Figure 2.

BLVRA inhibited inflammatory response after GMH and its expression on brain cells. Western blots were analyzed for TLR4, IL-1β, IL-6 and TNF-α expression at 3 days after GMH (A–F). Co-localization of BLVRA with microglia (Iba-1) (G), astrocyte (GFAP) (H), neuron (NeuN) (I), and DAPI at 72 h after GMH. Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group, &P < 0.05 compared with BLVRA group, %P < 0.05 compared with scramble siRNA group. N = 6 each group. Scale bar = 10 μm.
GMH suppresses endogenous expression of BLVRA
It has been reported that biliverdin administration could induce the phosphorylation of eNOS depending on biliverdin reductase13. However, whether BLVRA plays a role in the phosphorylation of eNOS has not been reported. We hypothesized that the phosphorylation of eNOS is subject to BLVRA. The expression profile of endogenous BLVRA and three isoforms of NOS was analyzed at 0, 3, 6, 12 h and 1, 3, 5, 7 days post GMH (Fig. 3A). Time course results showed that GMH induced a significant decrease in BLVRA expression at all time points compared to 0 h (Fig. 3B). Phosphorylation of eNOS (S1177) level was decreased until 3 days post-GMH and then increased on Day 5 and Day 7 (Fig. 3C). GMH induced the phosphorylation of nNOS (S1417) from 3 h and phosphorylation of iNOS (Y151) from 3 days post-GMH, respectively (Fig. 3D and 3E). TLR4 expression was significantly increased at 3 h after GMH and still remained at a higher level compared to sham on Day 7 (Fig. 3F).
Figure 3.

Endogenous BLVRA, TLR4, phosphorylation of three isoforms NOS, and three isoforms NOS in the brain post GMH. Western blots anaylysis of BLVRA, phosphorylation of eNOS (S1177), eNOS, phosphorylation of nNOS (S1417), nNOS, phosphorylation of iNOS (Y151), iNOS, and TLR4 expression at 3, 6, 12 h and 1, 3, 5, 7 days after GMH (A–F). Values are expressed as mean ± SD. *P < 0.05 compared with sham group. N = 6 each group.
eNOS knockdown abolished the beneficial effects of BLVRA in GMH
To further investigate whether three isoforms of NOS were involved in BLVRA-induced protective mechanisms in GMH, we showed BLVRA-treated group significantly lower hemoglobin content compared to the vehicle group at 72 h post-GMH, which was reversed by iromycin A stimulation, an eNOS selective inhibitor. MEG (iNOS inhibitor) + BLVRA and NPA (nNOS inhibitor) + BLVRA groups had significantly less hemoglobin content compared to vehicle and iromycin A + BLVRA group (Fig. 4A). To assess the association between BLVRA and M2 phenotype microglia, double immunofluorescence staining showed that the number of activated M2 microglia was significantly increased in BLVRA-treated group, but iromycin A administration did not induce M2 phenotype microglia activation in GMH+BLVRA group compared with vehicle group (Fig. 4B and 4C). Additionally, eNOS knockdown abolished BLVRA-induced mannose receptor upregulation (Fig, 4D–4F). CD36 expression was induced in BLVRA-treated group but decreased with eNOS inhibition (Fig, 4G). Moreover, western blot analysis showed that BLVRA treatment significantly decreased the expression levels of TLR4 and proinflammatory cytokines compared with vehicle-treated group, which was again, abrogated by iromycin A administration (Fig. 4H–4L). Since 2% DMSO was used to dissolve iromycin A, a vehicle group with DMSO was added. Analysis showed that 2% DMSO alone had no effect on inflammatory response.
Figure 4.

Endothelia nitric oxide synthase abrogated BLVRA benefits. Hemoglobin content was evaluated with eNOS inhibitor (iromycin A), iNOS inhibitor (MEG), and nNOS inhibitor (NPA) co-administered with BLVRA at 72 h after GMH (A). Co-localization of BLVRA (scale bar: 100 μm), mannose receptor and DAPI at 72 h after GMH (B) and quantified activated M2 microglia (C). The expression levels of BLVRA, CD206, and CD36 were measured by western blot assay (D–F). Western blots revealed expression of TLR4, IL-1β, IL-6 and TNF-α expression at 3 days after GMH in eNOS inhibitor group (G–K). Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group, &P < 0.05 compared with BLVRA. N = 6 each group.
Translocation of BLVRA to the nucleus decreased TLR4 expression, which required NO generated from eNOS
As BLVRA interacted with eNOS expression and the knockdown eNOS reversed the beneficial effects of BLVRA, we used three selective isoforms NOS inhibitors to further assess the underlying mechanism of BVLRA in GMH. Phosphorylation of eNOS was elevated in BLVRA-treated group compared with vehicle group, and this expression was reversed by iromycin A (Fig. 5A and 5B). No difference was seen in phosphorylation of nNOS between vehicle and BLVRA treatment group (Fig. 5C). BLVRA decreased phosphorylation of iNOS compared with vehicle group (Fig. 5D). Western blot analysis also indicated expression of three isoforms NOS were significantly decreased in the respective inhibitor groups.
Figure 5.

BLVRA translocated into nucleus depending on NO generated from eNOS. Phosphorylation of three isoforms NOS were evaluated by Western blot (A–D). No assay was conducted at 72 h after GMH (E and F). Western blot analysis of BLVRA and TLR4 in nucleus and cytoplasm (G–M). Lamin B was used as a nucleus loading control. Actin was used as a cytoplasm loading control. Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group, &P < 0.05 compared with BLVRA. N = 6 each group.
Some studies have indicated that BLVRA directly binds to the TLR4 promoter to inhibit its transcription and expression. Therefore, NO assay was performed and the results showed that vehicle group had greater NO generation compared with sham group, which was reduced by BLVRA treatment (Fig. 5E). The sham + BLVRA group induced NO generation compared with sham group, but administration of iromycin A reversed these effects. iNOS and nNOS were not involved in the BLVRA-induced NO generation, because there were no differences in NO generation between MEG + BLVRA or NPA + BLVRA treatment and sham animals (Fig. 5F).
BLVRA accumulation in the nucleus was observed in BLVRA-treated group. Inhibition of NO generation with iromycin A resulted in less BLVRA translocated to the nucleus (Fig. 5G and 5J). A similar trend was found in the stimulation with SNP (NO donor) in the absence of BLVRA, which also led to significant accumulation of BLVRA in the nucleus both in sham + SNP and vehicle + SNP groups post-GMH (Fig. 5H and 5K). In addition, the suppressive effect of BLVRA on TLR4 expression was diminished by iromycin A administration (Fig. 5I and 5L–5M).
BLVRA decreased oxidative and nitrosative stress and function in TLR family
With regard to oxidative/nitrosative stress induced by the degradation of heme after GMH, DHE staining showed BLVRA decreased GMH-induced superoxide anion (O2−) both at 3 days and 28 days after GMH (Fig. 6A–6C). Furthermore, HPF staining showed peroxynitrite (ONOO) was significantly decreased in BLVRA-treated group (Fig. 6D and 6E). Oxidative and nitrosative stress biomarkers (3-nitrotyrosine, 4-hydroxynonenal, and dinitrophenol) were significantly decreased with BLVRA treatment compared with vehicle group (Fig. 6F–6I). GMH-induced upregulation of TLR2 expression was decreased in BLVRA-treated group (Fig. 6J and 6K). Similar patterns in decreased levels of TLR3 and TLR9 were also observed in BLVRA-treated animals (Fig. 6L and 6M).
Figure 6.

BLVRA attenuated oxidative stress and nitrosative stress and regulated TLR family expression. DHE staining evaluated superoxide anion (O2−) at 3 days and 28 days after GMH in the brain (A–C). HPF staining was used to measure peroxynitrate (ONOO) at 72 h after GMH (D and E). Expression of 3-NT, HNE and DNP was analyzed by Western blot (F–I). TLR2, TLR3, and TLR9 expression were conducted at 72 h after GMH (J–M). Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group. N = 6 each group.
BLVRA treatment improves long-term brain morphology
As BLVRA contributes to accelerating hematoma cleanup, we performed Nissl staining to analyze morphology. The hemorrhagic ventricular dilation of ipsilateral hemisphere was significantly decreased in BLVRA-treated groups (Fig. 7A, 7C). Brain weight was measured at 28 days after GMH, BLVRA treatment significantly reduced GMH-induced brain tissue loss (Fig. 7B). Cortical thickness and grey matter loss were presented as a ratio to the mean of sham (Fig. 7D). The medium dose of BLVRA significantly decreased loss in the cortex tissue and grey matter compared with vehicle group (Fig. 7D, 7E). White matter loss was also significantly reduced with medium-dose BLVRA treatment, compared to the vehicle-treated group (Fig. 7F).
Figure 7.

BLVRA attenuated ventricular dilation and brain tissue loss. Nissl staining evaluated at 28 days after GMH (A). Quantification of brain weight (B), ventricular volume (C), cortical thickness (D), grey matter loss (E), and white matter (F) at 28 days after GMH. Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group. N = 6 each group.
BLVRA improved long-term neurological function recovery
To evaluate the importance of BLVRA in attenuating long-term neurological deficits, we performed Morris water maze from Block 1 to Block 4 with one block on each day, followed by a probe trial. In Block 1 and Block 2, BLVRA-treated animals did not demonstrate a significant improvement in cognitive function compared to the vehicle-treated group. It might be because animals were still learning the location of the platform. However, in Block 3 and Block 4, GMH+vehicle animals swam significantly greater distances when finding the platform in Morris water maze (Fig. 8A) and less time in the target quadrant in the probe trials (Fig. 8B) compared with all other groups. These results indicated that GMH led to significant spatial learning deficit. Medium-dose BLVRA-treated animals demonstrated the shortest swimming distance and the least time spent in the target quadrant. In addition, medium dose of BLVRA improved cognitive and motor function in most of GMH-operated animals, as evaluated by foot fault test and Rotarod (Fig. 8C and 8D). Hyperactivity was also successfully decreased in medium-dose BLVRA-treated group in the open field test (Fig. 8E). Significant weight loss was observed in the vehicle-treated group compared with sham group. GMH-induced weight loss was attenuated in all BLVRA-treated groups, especially the medium dose group (Fig. 8F).
Figure 8.

BLVRA ameliorated long-term neurological deficits after GMH. Neurobehavioral assessments of Morris water maze swim distance (A), probe trial (B), Rotarod (C), foot fault test (D) and open-field (E) were analyzed at 21 to 28 days after GMH. Body weight (F) was measured at 0 to 28 days after GMH. Values are expressed as mean ± SD. *P < 0.05 compared with sham group, #P < 0.05 compared with sham group. N = 6 each group.
Discussion
A previous study showed that BLVRA converted biliverdin-IX into bilirubin-IX which plays an important role in brain hemorrhage. Unbound bilirubin easily crosses the blood-brain barrier and triggers neurotoxicity 33. Other studies revealed that bilirubin functions as a powerful antioxidant after hemorrhagic stroke 34, 35. However, most studies were done in adult stroke models and there have been only a few studies so far to look at the role of biliverdin-bilirubin system in neonatal hemorrhagic stroke. It is possible that BLVRA, as the main isoform of BVR, maintains bilirubin as an antioxidative cytoprotectant, which would attenuate neurological deficits after hemorrhage 36. In this study, we presented that BLVRA played a crucial role in hematoma absorption, thereby inhibiting innate immunity-mediated inflammation and attenuating hydrocephalus in a GMH rat model. Furthermore, we demonstrated that BLVA reduced inflammatory response by translocating to the nucleus in an NO-dependent manner.
GMH leads to significant hydrocephalus and brain atrophy in the long term. Morphological features, such as the ventricular volumes, cortical thickness, grey matter loss, and white matter area, were all significantly altered after GMH. We also demonstrated that BLVRA attenuated GMH-induced hyperactivity and spatial memory deficits in the long-term studies. Thus, together with short-term behavioral results, a significant improvement of developmental profile was observed in BLVRA-treated animals after GMH.
Efficient removal of hematoma is crucial in GMH since hematoma obstructs normal CSF circulation and contributes to hydrocephalus development after the hemorrhage. Microglia are a distinct cell type of immune/myeloid cells in the central nervous system. It only takes a few minutes to activate resident phagocytes and microglia in the brain after hemorrhage 3,37. The activated microglia in neonates might have a beneficial innate immune function by recruiting hematogenous phagocytes to the peri-hematoma region 27, 38, 39. Some evidence suggested that activated microglia/macrophages in the central nervous system are heterogeneous, which can exist broadly into polarization phenotypes. The classically activation state, M1, involves the production of inflammatory cytokines and reactive oxygen species. The alternative activation state, M2, is characterized as an anti-inflammatory regulator associated with debris clearance and injury recovery 40. Although more research has shown that the nature of microglia is far more complex than simple classification of M1/M2 polarization, several reports have demonstrated that M2-like microglia are responsible in hematoma removal. 27, 41, 42 In this study, we found that more M2-like microglia in the BLVRA-treated group than other groups, which indicates the protective effect of BLVRA treatment might be dependent on phagocytosis of M2-like microglia.
We also evaluated whether BLVRA mitigated inflammation to facilitate hematoma resolution. We chose to focus on innate immunity because neonates have limited antigen exposure and distinct adaptive immunity compared to the adult. Thus, innate immune system function is particularly critical in infants in the defense of any inflammatory challenges. TLR family are pattern-recognition receptors that are strongly associated with the innate immune system through activating pro-inflammatory signaling pathways 43. TLR4 is the best characterized TLR that triggers downstream signaling cascade, contributing to the transcription of several pro-inflammatory genes 43. It has not been investigated whether inhibition of TLR4 is involved in the beneficial effects of BLVRA in brain injuries and its related neurological deficits. We showed that BLVRA treatment exerted its repressive effect on inflammatory mediators and cytokines.
A previous report shows that biliverdin upregulates phosphorylated eNOS expression and contributes to anti-inflammatory response, which is dependent on biliverdin reductase in mouse primary macrophage 13, 42. In agreement with this study, our time course results also showed that BLVRA expression level was strongly correlated with phosphorylation of eNOS and TLR4. BLVRA co-administration with iromycin A (a selective eNOS inhibitor) reversed the beneficial effects of BLVRA on hematoma clearance at 3 days. This effect was independent of iNOS and nNOS. We believe the anti-inflammatory effect of BLVRA is regulated, at least in part, through eNOS activation, since iromycin A successfully diminished the phagocytotic effects of BLVRA. eNOS knockdown also reversed the tendency in the BLVRA treatment group- abolishing BLVRA-induced M2 polarization of microglia. Furthermore, the beneficial effect of BLVRA in inducing CD36, a scavenger receptor, was again abolished by eNOS inhibition. BLVRA attenuated the expression of TLR4 and inflammatory cytokines through regulating eNOS expression. Based on the abovementioned data, BLVRA regulates hematoma resolution and inflammation by modulating eNOS activity.
Based on the NO assay and time course data, it is deducible that iNOS might be the primary source of NO immediately after GMH because 1) iNOS expression was significantly elevated from Day 3 after GMH while eNOS expression was lower than the sham group up to Day 5 2) iNOS was shown to be able to generate more NO than eNOS and nNOS44. Thus, NO generation was upregulated after GMH, possibly due to the significantly elevated iNOS level at 3 days after GMH. However, corresponding with the dramatic increase in phosphorylation of eNOS by BLVRA, concomitant NO generation was found to be nNOS and iNOS independent after GMH induction. Moreover, through eNOS knockdown and NO donor administration, the BLVRA accumulation in the nucleus was found to be positively associated with NO produced by eNOS. Based on the nitrasative and oxidative stress data, we postulated that following brain injury, iNOS-derived NO formed peroxynitrite (ONOO) with superoxide generated from uncoupled eNOS, causing further oxidative stress and nitrosative stress to the brain tissues, in spite of more initial NO generation 45–48. Thus, eliminating resources of superoxide, such as uncoupled eNOS, would be essential to reducing oxidative and nitrosative stress. Some studies, together with our current data, suggested a possible role for BLVRA in recoupling eNOS to eliminate oxidative stress after brain injury49,50. The significant relationship between BLVRA, superoxide anion (O2−) and peroxynitrite formed by GMH revealed a crucial anti-oxidative role for BLVRA.
In conclusion, for the first time we identified that BLVRA improved both short-term and long-term neurobehaviorial outcomes through inducing microglial phagocytosis after GMH. Activated M2 microglia contributed to hematoma clearance through eNOS. We elucidated mechanisms by which eNOS-derived NO translocated BLVRA to nucleus and suppressed TLR4 expression. Our studies indicated that BLVRA could be a promising therapeutic target for GMH patients.
Supplementary Material
Highlights.
BLVRA revolves hematoma and attenuates inflammation after GMH.
BLVRA improves both short-term and long-term neurological outcomes.
This protective effect of BLVRA is mediated by eNOS/NO/TLR4 signaling pathway.
Acknowledgments
The authors thank for Desislava M. Doycheva and Jay Malaguit their technical assistance on this project. This study was supported R01 grant form National Institute of Neurological Diseases and Stroke to JHZ (R01-NS078755).
Abbreviations
- GMH
germinal matrix hemorrhage
- BLVRA
biliverdin reductase-A
- NOS
nitric oxide synthase
Footnotes
Conflict of Interest: The authors state no conflict of interests exists.
Author Contributions
Yiting Zhang, Yan Ding, Jiping Tang and John H. Zhang contributed to concept and design the project. Yiting Zhang, Tai Lu, Yixin Zhang, Ningbo Xu, Lingyan Yu performed the experiments. Yiting Zhang, Yixin Zhang and Ningbo Xu analyzed the data. Devin W. McBride and Jerry J. Flores assisted in experimental design, data acquisition and analysis. Yiting Zhang and Yan Ding wrote the manuscript. All authors read and approved the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010 Jan;67(1):1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heron M, Sutton PD, Xu J, Ventura SJ, Strobino DM, Guyer B. Annual summary of vital statistics: 2007. Pediatrics. 2010 Jan;125(1):4–15. doi: 10.1542/peds.2009-2416. [DOI] [PubMed] [Google Scholar]
- 3.Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005 Apr;27(3):268–79. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- 4.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006 Jan;5(1):53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 5.Egawa N, Lok J, Washida K, Arai K. Mechanisms of Axonal Damage and Repair after Central Nervous System Injury. Transl Stroke Res. 2017 Feb;8(1):14–21. doi: 10.1007/s12975-016-0495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aquilina K, Chakkarapani E, Love S, Thoresen M. Neonatal rat model of intraventricular haemorrhage and post-haemorrhagic ventricular dilatation with long-term survival into adulthood. Neuropathol Appl Neurobiol. 2011 Feb;37(2):156–65. doi: 10.1111/j.1365-2990.2010.01118.x. [DOI] [PubMed] [Google Scholar]
- 7.Cherian S, Whitelaw A, Thoresen M, Love S. The pathogenesis of neonatal post-hemorrhagic hydrocephalus. Brain Pathol. 2004 Jul;14(3):305–11. doi: 10.1111/j.1750-3639.2004.tb00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang G, Yang Y, Wu G, Hua Y, Keep RF, Xi G. Early Erythrolysis in the Hematoma After Experimental Intracerebral Hemorrhage. Transl Stroke Res. 2017 Apr;8(2):174–82. doi: 10.1007/s12975-016-0505-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keep RF, Xi G, Hua Y, Hoff JT. The deleterious or beneficial effects of different agents in intracerebral hemorrhage: think big, think small, or is hematoma size important? Stroke. 2005 Jul;36(7):1594–6. doi: 10.1161/01.STR.0000170701.41507.e1. [DOI] [PubMed] [Google Scholar]
- 10.Kapitulnik J, Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci. 2009 Mar;30(3):129–37. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Maines MD. New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology (Bethesda) 2005 Dec;20:382–9. doi: 10.1152/physiol.00029.2005. [DOI] [PubMed] [Google Scholar]
- 12.Franklin EM, Browne S, Horan AM, et al. The use of synthetic linear tetrapyrroles to probe the verdin sites of human biliverdin-IXalpha reductase and human biliverdin-IXbeta reductase. FEBS J. 2009 Aug;276(16):4405–13. doi: 10.1111/j.1742-4658.2009.07148.x. [DOI] [PubMed] [Google Scholar]
- 13.Wegiel B, Gallo D, Csizmadia E, et al. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc Natl Acad Sci U S A. 2011 Nov 15;108(46):18849–54. doi: 10.1073/pnas.1108571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002 Feb;96(2):287–93. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 15.Loftspring MC, Johnson HL, Feng R, Johnson AJ, Clark JF. Unconjugated bilirubin contributes to early inflammation and edema after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011 Apr;31(4):1133–42. doi: 10.1038/jcbfm.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fereshtehnejad SM, Poorsattar Bejeh Mir K, Poorsattar Bejeh Mir A, Mohagheghi P. Evaluation of the possible antioxidative role of bilirubin protecting from free radical related illnesses in neonates. Acta Med Iran. 2012;50(3):153–63. [PubMed] [Google Scholar]
- 17.Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev. 2009 Mar;61(1):62–97. doi: 10.1124/pr.108.000547. [DOI] [PubMed] [Google Scholar]
- 18.Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K, Deckmyn H. Platelets at work in primary hemostasis. Blood Rev. 2011 Jul;25(4):155–67. doi: 10.1016/j.blre.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003 Jun;55(2):271–324. doi: 10.1124/pr.55.2.3. [DOI] [PubMed] [Google Scholar]
- 20.Marletta MA. Mammalian synthesis of nitrite, nitrate, nitric oxide, and N-nitrosating agents. Chem Res Toxicol. 1988 Sep-Oct;1(5):249–57. doi: 10.1021/tx00005a001. [DOI] [PubMed] [Google Scholar]
- 21.Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010 Sep 15;23(2):75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Lekic T, Manaenko A, Rolland W, et al. Rodent neonatal germinal matrix hemorrhage mimics the human brain injury, neurological consequences, and post-hemorrhagic hydrocephalus. Exp Neurol. 2012 Jul;236(1):69–78. doi: 10.1016/j.expneurol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ekinci N, Acer N, Akkaya A, Sankur S, Kabadayi T, Sahin B. Volumetric evaluation of the relations among the cerebrum, cerebellum and brain stem in young subjects: a combination of stereology and magnetic resonance imaging. Surg Radiol Anat. 2008 Aug;30(6):489–94. doi: 10.1007/s00276-008-0356-z. [DOI] [PubMed] [Google Scholar]
- 24.Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996 Mar 18;366(4):580–99. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Klebe D, Krafft PR, Hoffmann C, et al. Acute and delayed deferoxamine treatment attenuates long-term sequelae after germinal matrix hemorrhage in neonatal rats. Stroke. 2014 Aug;45(8):2475–9. doi: 10.1161/STROKEAHA.114.005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacLellan CL, Silasi G, Poon CC, et al. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab. 2008 Mar;28(3):516–25. doi: 10.1038/sj.jcbfm.9600548. [DOI] [PubMed] [Google Scholar]
- 27.Flores JJ, Klebe D, Rolland WB, Lekic T, Krafft PR, Zhang JH. PPARgamma-induced upregulation of CD36 enhances hematoma resolution and attenuates long-term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiol Dis. 2016 Mar;87:124–33. doi: 10.1016/j.nbd.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choudhri TF, Hoh BL, Solomon RA, Connolly ES, Jr, Pinsky DJ. Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke. 1997 Nov;28(11):2296–302. doi: 10.1161/01.str.28.11.2296. [DOI] [PubMed] [Google Scholar]
- 29.Lekic T, Manaenko A, Rolland W, Tang J, Zhang JH. A novel preclinical model of germinal matrix hemorrhage using neonatal rats. Acta Neurochir Suppl. 2011;111:55–60. doi: 10.1007/978-3-7091-0693-8_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang J, Liu J, Zhou C, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004 Oct;24(10):1133–45. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007 Jun;130(Pt 6):1643–52. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klebe D, McBride D, Flores JJ, Zhang JH, Tang J. Modulating the Immune Response Towards a Neuroregenerative Peri-injury Milieu After Cerebral Hemorrhage. J Neuroimmune Pharmacol. 2015 Dec;10(4):576–86. doi: 10.1007/s11481-015-9613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee YK, Daito Y, Katayama Y, Minami H, Negishi H. The significance of measurement of serum unbound bilirubin concentrations in high-risk infants. Pediatr Int. 2009 Dec;51(6):795–9. doi: 10.1111/j.1442-200X.2009.02878.x. [DOI] [PubMed] [Google Scholar]
- 34.Dohi K, Mochizuki Y, Satoh K, et al. Transient elevation of serum bilirubin (a heme oxygenase-1 metabolite) level in hemorrhagic stroke: bilirubin is a marker of oxidant stress. Acta Neurochir Suppl. 2003;86:247–9. doi: 10.1007/978-3-7091-0651-8_53. [DOI] [PubMed] [Google Scholar]
- 35.Pyne-Geithman GJ, Morgan CJ, Wagner K, et al. Bilirubin production and oxidation in CSF of patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005 Aug;25(8):1070–7. doi: 10.1038/sj.jcbfm.9600101. [DOI] [PubMed] [Google Scholar]
- 36.Dyrkaez GR, Libby RD, Hamilton GA. Letter: Trivalent copper as a probable intermediate in the reaction catalyzed by galactose oxidase. J Am Chem Soc. 1976 Jan 21;98(2):626–8. doi: 10.1021/ja00418a060. [DOI] [PubMed] [Google Scholar]
- 37.Atangana E, Schneider UC, Blecharz K, et al. Intravascular Inflammation Triggers Intracerebral Activated Microglia and Contributes to Secondary Brain Injury After Experimental Subarachnoid Hemorrhage (eSAH) Transl Stroke Res. 2017 Apr;8(2):144–56. doi: 10.1007/s12975-016-0485-3. [DOI] [PubMed] [Google Scholar]
- 38.Starossom SC, Mascanfroni ID, Imitola J, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012 Aug 24;37(2):249–63. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lapchak PA, Zhang JH. The High Cost of Stroke and Stroke Cytoprotection Research. Transl Stroke Res. 2017 Aug;8(4):307–17. doi: 10.1007/s12975-016-0518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003 Jan;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 41.Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013:746068. doi: 10.1155/2013/746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawanokuchi J, Shimizu K, Nitta A, et al. Production and functions of IL-17 in microglia. J Neuroimmunol. 2008 Feb;194(1–2):54–61. doi: 10.1016/j.jneuroim.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 43.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001 Nov;1(2):135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 44.Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KT. The role of the nitric oxide pathway in brain injury and its treatment--from bench to bedside. Exp Neurol. 2015 Jan;263:235–43. doi: 10.1016/j.expneurol.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998 Aug 04;95(16):9220–5. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001 May 04;276(18):14533–6. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 47.Drexler H, Hornig B. Endothelial dysfunction in human disease. J Mol Cell Cardiol. 1999 Jan;31(1):51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- 48.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000 Nov 10;87(10):840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 49.Sabri M, Ai J, Marsden PA, Macdonald RL. Simvastatin re-couples dysfunctional endothelial nitric oxide synthase in experimental subarachnoid hemorrhage. PLoS One. 2011 Feb 23;6(2):e17062. doi: 10.1371/journal.pone.0017062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barone E, Mancuso C, Di Domenico F, et al. Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J Neurochem. 2012 Jan;120(1):135–46. doi: 10.1111/j.1471-4159.2011.07538.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.