

Abstract
Calcium (Ca2+) is a ubiquitous second messenger that regulates various activities in eukaryotic cells. Especially important role calcium plays in excitable cells. Neurons require extremely precise spatial-temporal control of calcium-dependent processes because they regulate such vital functions as synaptic plasticity. Recent evidence indicates that neuronal calcium signaling is abnormal in many of neurodegenerative disorders such as Alzheimer’s disease (AD), Huntington’s disease (HD) and Parkinson’s disease (PD). These diseases represent a major medical, social, financial and scientific problem, but despite enormous research efforts, they are still incurable and only symptomatic relief drugs are available. Thus, new approaches and targets are needed. This review highlight neuronal calcium-signaling abnormalities in these diseases, with particular emphasis on the role of neuronal store-operated Ca2+ entry (SOCE) pathway and its potential relevance as a therapeutic target for treatment of neurodegeneration.
Keywords: Alzheimer disease, Parkinson disease, Huntington disease, neurodegeneration, Ca2+ signaling, Ca2+ homeostasis, neuronal store-operated Ca2+ channels, neuronal store-operated Ca2+ entry
Graphical abstract
Introduction
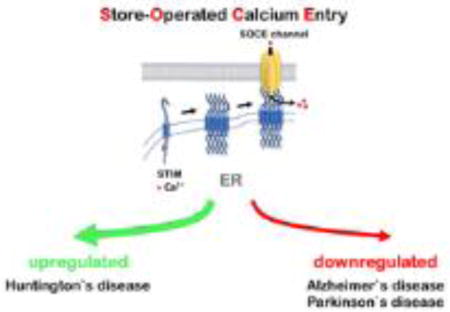
Calcium ions (Ca2+) are universal second messengers regulating wide range of important eukaryotic cells functions such as differentiation, proliferation, growth, survival, apoptosis, gene transcription and membrane excitability (Capiod, 2016; Clapham, 2007; La Rovere et al., 2016; Toth et al., 2016). Ca2+ plays an especially important role in neuronal cells, where it mediates multiple vital physiological processes and plays a central role in control of synaptic plasticity (Berridge, 1998). Neurons require extremely precise control of Ca2+ concentration in specific compartments for proper function. Organization of neuronal Ca2+ signaling machinery is complex. It includes various calcium-conducting channels and a great number of calcium-dependent proteins as downstream targets including kinases, phosphatases, transcription factors, enzymes and proteins that induce synaptic vesicle fusion (Brini et al., 2014; Clapham, 2007). Calcium-conducting channels in the plasma membrane are voltage-gated Ca2+ channels (VGCC), N-methyl-D-aspartate receptors (NMDAR), calcium-conducting α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR), canonical transient receptor potential (TRPC) channels and calcium release-activated channels (CRAC). Ryanodine receptors (RyR) and 1,4,5-triphosphate receptors (InsP3R) mediate calcium efflux from the endoplasmic reticulum (ER) - a cellular compartment performing a role of calcium depot. Stromal interacting molecules (STIMs) are calcium-sensor proteins that have an EF-hand Ca2+ binding domain located in the ER lumen (Williams et al., 2001). STIMs family includes STIM1 and STIM2 proteins. STIM2 binds Ca2+ with higher dissociation constant in comparison to STIM1 and it is more sensitive to luminal Ca2+ levels changes (Brandman et al., 2007). During ER calcium store depletion, STIMs oligomerize, subsequently translocate to the plasma membrane and interact with calcium-conducting channels to induce calcium influx and store refilling (Kraft, 2015; Soboloff et al., 2012). This process is called store-operated calcium entry (SOCE) (review in (Majewski and Kuznicki, 2015)). Subsequently calcium ions are transferred from the cytosol of the cell to the ER lumen with help of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). SOCE calcium fluxes are mediated by highly calcium-selective channels of Orai family (Kraft, 2015) encoding Ca2+ release-activated Ca2+ current (ICRAC), and non-selective cation channels of TRPC family encoding store-operated current (ISOC) (Parekh and Putney, 2005; Yuan et al., 2007) (Figure 1). It was suggested that Orai binds to TRPC and they form tertiary TRPC-Orai-STIM complex during SOCE activation (Liao et al., 2008; Liao et al., 2009; Zhang et al., 2016a) (Fig. 1). The exact contribution of Orai1 and TRPC channels to SOCE is under investigation (Chen et al., 2011a). SOCE plays an important signaling function in neurons (Majewski and Kuznicki, 2015). This review highlights neuronal SOCE deficiency observed during Alzheimer`s (AD), Huntington`s (HD) and Parkinson`s disease (PD).
Figure 1.

SOCE in neurons.
Complex calcium signaling system in neuronal cells allows activation of different spatially separated Ca2+-dependent processes at the same time. Neurons are extremely sensitive to calcium concentration levels and even subtle defects in Ca2+ homeostasis can lead to destructive consequences and alter normal neuronal activity. Multiple evidence suggest that Ca2+ dysregulation plays an important role in aging (Kumar et al., 2009) and neurodegeneration (Bezprozvanny, 2009). Some of the calcium signaling abnormalities that occur in AD, HD and PD neurons are highlighted in this review. Neurodegenerative diseases present an enormous humanity problem, but despite active research efforts, these disorders are still incurable, with most medicine offering only symptomatic relief (Cacabelos, 2017; Cummings et al., 2016; Mason and Barker, 2016). Restoring calcium signaling homeostasis presents an attractive target for drug discovery for neurodegenerative disease treatment.
Calcium dysregulation in Alzheimer`s disease
Alzheimer`s disease (AD) is a neurodegenerative disorder, which affects memory formation and storage processes. In most cases AD appears sporadically and affects people over 60 years of age. Small portion of cases (approximately 1%–2%) refers to familial form of AD, which is characterized by an earlier onset and more severe pathogenesis (Hardy and Selkoe, 2002). Familial form of AD is caused by mutations in genes encoding Presenilin 1 (PS1), Presenilin 2 (PS2) and amyloid-precursor protein (APP) (Bergmans and De Strooper, 2010; Hardy, 2009; Hardy and Selkoe, 2002). Presenilins form the catalytic subunits of the γ-secretase protease complex, which is together with β-secretase are responsible for APP protein cleavage and subsequent production of toxic Aβ peptides (Jellinger, 2009). Several hypothesis about causes of AD have been proposed, but so-called “amyloid cascade hypothesis” is a dominant model of AD pathogenesis. It states that increased production of amyloidogenic Aβ42 peptide (or an increase in Aβ42:Aβ40 ratio) is driving AD, causing reduced number of synapses and neuronal death (Fleming, 2017; Hardy, 2009; Hardy and Selkoe, 2002). Based on this idea, major efforts were spent by the industry on developing agents which can reduce Aβ production or eliminate Aβ from the brain. However, so far these agents did not show benefit in AD clinical trials (Cummings et al., 2014; Karran and Hardy, 2014; Karran et al., 2011). As alternative point of view, “calcium hypothesis of AD” was suggested (Alzheimer's Association Calcium Hypothesis, 2017; Bezprozvanny and Mattson, 2008; Briggs et al., 2016; Khachaturian, 1989). This hypothesis speculates that dysregulation in cellular calcium homeostasis is the main driving force of neurodegeneration in AD (Alzheimer's Association Calcium Hypothesis, 2017; Bezprozvanny and Mattson, 2008; Briggs et al., 2016; Khachaturian, 1989). Several lines of experimental evidence support this idea. ER Ca2+ levels are elevated in AD and in aging neurons (Bezprozvanny and Mattson, 2008). Rise in ER Ca2+ concentration results in subsequent compensatory alterations and defects in neuronal Ca2+ signaling. Altered Ca2+ signals shift the balance between Ca2+ -dependent phosphatase calcineurin (CaN) and its opponent Ca2+ /calmodulin-dependent protein kinase II (CaMKII), which are extremely abundant in synaptic locations. Shift in the balance of CaMKII and CaN activity occludes longterm potentiation and facilitates long-term depression, causing synaptic and memory impairments and leading to synaptic loss and neurodegeneration (Berridge, 2011; Bezprozvanny and Hiesinger, 2013; Popugaeva et al., 2017).
What is a reason for increased calcium content during AD? How do AD-causing mutations induce calcium signaling dysregulation? One possible explanation is that Aβ peptides form Ca2+ -permeable pore in the plasma membrane (Arispe et al., 2007; Arispe et al., 1993). Indeed, a part of neurites surrounding β-amyloid plagues have elevated steady-state Ca2+ levels (Kuchibhotla et al., 2008). Aβ may induce increased calcium influx via L-type VGCCs (Ueda et al., 1997), but it was also reported that Aβ oligomers suppress P/Q-type VGCCs calcium currents (Nimmrich et al., 2008). NMDAR is another potential source for intracellular calcium, playing important role in excitatory synaptic neurotransmission. Activation of synaptic NMDARs is required for synaptic plasticity and drives LTP generation, but sustained activation of NMDARs can cause subsequent calcium overload and toxicity (Hardingham and Bading, 2010). Role of NMDA receptor in AD and particularly Aβ effects on NMDA receptor were studied intensively (Foster et al., 2017; Mota et al., 2014a). It was proposed that at early AD stages NMDA receptor is overactivated (Parameshwaran et al., 2008; Zhang et al., 2016b). Indeed, recent reports indicate that Aβ oligomers applied on cultured cortical neurons activate GluN2B-containing NMDAR and induce an immediate Ca2+ rise (Ferreira et al., 2012). Potentially neuroprotective drugs which block NMDAR has intolerable side effects, presumably because of extreme importance of this receptor for normal neuronal function. An exception is a non-competitive NMDAR inhibitor memantine, which is currently approved for AD treatment (Lipton, 2006). In contrast to other NMDAR blockers, positive effects of memantine administration are likely observed because it preferentially blocks excessively activated NMDARs (Mota et al., 2014b). New generation of drugs which selectively bind to and inhibit only excessivly activated NMDARs are considered for AD treatment (Lipton, 2007). Another adverse effect of Aβ is a reduction in NMDAR expression and its enhanced endocytosis (Snyder et al., 2005). Enhanced endocytosis of NMDARs is in part due to Aβ-mediated activation of STEP61 phosphatase (Snyder et al., 2005). Downregulation of GluN1 subunit of NMDAR was observed in postmortem AD patient’s samples (Jacob et al., 2007). It was shown that the specific N-terminal splice cassette containing GluN1 is decreased drastically in AD, suggesting that neurons bearing this isoform are more vulnerable (Hynd et al., 2004). Some data indicate that Aβ may directly bind to and modulate activity of NMDA receptors (De Felice et al., 2007; Lacor et al., 2007; Sinnen et al., 2016; Texido et al., 2011). Reduction of NMDAR activity in AD may also be induced by an oxidative stress, most likely due to oxidation of extracellular NMDAR cysteins and intracellular targets such as calmodulin (Foster et al., 2017). Taking together, we may conclude that Aβ causes dysregulation of NMDAR expression and activity by multiple mechanisms. Disrupted NMDAR signaling further leads to impaired synaptic plasticity, reduced LTP, enhanced LTD and synaptic loss (Foster et al., 2017; Mota et al., 2014a).
In addition to adverse Aβ effects, mutated presenilins directly cause Ca2+ dysregulation in AD. Different cellular models expressing AD mutant presenilins show overfilling of the ER with Ca2+ and the excessive Ca2+ release through the InsP3R (Ito et al., 1994; Leissring et al., 1999a; Leissring et al., 1999b; Nelson et al., 2007; Stutzmann et al., 2004; Stutzmann et al., 2006; Tu et al., 2006a). To explain these results it has been suggested that mutant presenilins directly affect InsP3R1 gating (Cai et al., 2006; Cheung et al., 2010; Cheung et al., 2008), store-operated Ca2+ influx (Leissring et al., 2000; Yoo et al., 2000), RyR (Chan et al., 2000; Hayrapetyan et al., 2008; Rybalchenko et al., 2008; Stutzmann et al., 2006) or SERCA ER Ca2+ pump (Green et al., 2008). Another possible explanation is that presenilin by itself forms ER calcium leaking pore, and most of the AD-causing mutations collapse this pore and induce ER Ca2+ overfilling (Nelson et al., 2011; Nelson et al., 2007; Tu et al., 2006a; Zhang et al., 2010b). This idea was initially controversial (Shilling et al., 2012) but it was supported by an unbiased screen for ER Ca2+ leak channels (Bandara et al., 2013). Interestingly, presenilins share the fold with chloride channels (Theobald, 2016) and the high resolution crystal structure of archaeal presenilin homologue PSH1 has a hole that traverses through the entire protein and is large enough to allows passage of small ions (Li et al., 2013).
Dysregulation of Ca2+ signaling causes excessive Ca2+ release via RyR (Briggs et al., 2016). RyR-mediated Ca2+ release is enhanced in neurons from presenilin mutant mice (Stutzmann et al., 2006). RyR2 expression levels are elevated in AD brains (Bruno et al., 2012) and activity of RyR is enhanced due to increased ER Ca2+ levels. Enhanced Ca2+ release via RyR2 affects synaptic plasticity, compensating for changes in LTP and LTD (Chakroborty et al., 2012b). In the spines of AD neurons, NMDA receptor-mediated Ca2+ influx triggers supranomal Ca2+ responses mediated by RyR2 (Goussakov et al., 2010). Excessive Ca2+ release triggers overactivation of Ca2+-dependent SK channels and impairs the induction of synaptic plasticity changes (Chakroborty et al., 2012b). Indeed, SK channels overactivation was at least partially responsible for destablization of mushroom spines and late-phase LTP defects in presenilin knockin (PS1-M146V-KI) neurons (Zhang et al., 2015a). These results suggest that inhibition of RyR2 activity may helps to alleivate AD symptoms. Indeed, previous studies showed that short term treatment with RyR inhibitor dantrolene was able to stabilize Ca2+ signals, ameliorate cognitive decline and reduce neuropathology, amyloid load and memory impairments in various AD mouse models (Chakroborty et al., 2012a; Oules et al., 2012; Peng et al., 2012). However, in our previous studies we observed that long-term feeding of the RyR inhibitor dantrolene exacerbated amyloid plaque formation and resulted in the loss of hippocampal synaptic markers and neuronal deterioration in AD mice (Zhang et al., 2010a). One potential problem with interpreting these conflicting results is that specific RyR inhibitors do not exist and the drug dantrolene, used in most studies, has additional targets such as store-operated Ca2+ channels (Zhao et al., 2006). Moreover, dantrolene is specific for RyR1 (Krause et al., 2004), and does not block RyR2 and RyR3 effectively. Using genetic knockout approach we found that RyR3 appears to play a dual role in the context of AD pathology, rather than an invariable positive or negative effect (Liu et al., 2014). Our results indicated that RyR3 plays an important protective role in early stages of AD by helping to reduce neuronal excitability and activity-dependent Aβ production. However, in older AD mice deletion of RyR3 resulted in beneficial effects (Liu et al., 2014). Thus, although RyR3 appears to play a protective role in younger mice, these results suggested that in aging brain RyR3 may contribute to AD pathogenesis by amplifying ER Ca2+ release through calcium-induced calcium release (CICR) mechanism and by enhancing the dysregulation of intracellular Ca2+. These results consistent with reports that dantrolene exerted beneficial effects in several AD mouse models (Chakroborty et al., 2012a; Oules et al., 2012; Peng et al., 2012).
Recent results suggest a significant role for neuronal store-operated calcium entry (SOCE) pathway in AD pathogenesis (Fig. 2). SOCE pathway is activated following depletion of ER Ca2+ levels. Reduced expression levels of SOCE ER Ca2+ sensor STIM2 were discovered in experiments with AD patient fibroblasts (Bojarski et al., 2009). Levels of STIM2 were also reduced in hippocampal samples from PS1-M146V-KI mouse model and in cortical samples from sporadic AD patients (Sun et al., 2014). Hippocampal neurons from PS1-M146V-KI mice displayed reduced mushroom dendritic spines density and almost complete absence of STIM2-gated store-operated calcium entry (SOCE) in dendritic spines (Sun et al., 2014; Zhang et al., 2016a). Following store depletion, STIM2 translocates to neuronal plasma membrane and drives calcium influx into cell through interaction with calcium conducting channels on the plasma membrane. In case of hippocampus, TRPC6 and Orai2 were recently identified as SOCE-forming channels in hippocampal neurons (Zhang et al., 2016a). Orai1 was also implicated as a SOCE channel in developing hippocampal synaptic spines (Korkotian et al., 2017). Reduction of synaptic SOCE was demonstarted in APP mutant mice and following application of Aβ peptides, although effects were less dramatic than for presenilins mutations (Popugaeva et al., 2015; Zhang et al., 2015b). Activation of neuronal SOCE with pharmacological agents or overexpression of SOCE pathway components restored mushroom spines deficiency in hippocampal neurons from presenilin and APP-based AD mice models (Popugaeva et al., 2015; Sun et al., 2014; Zhang et al., 2016a; Zhang et al., 2015b). We proposed that proper SOCE functioning is necessary for the stability of mushroom hippocampal dendritic spines, and that initially protective downregulation of this pathway became toxic with time (Popugaeva et al., 2017; Zhang et al., 2016a). Recent results suggested that presenilin-mediated cleavage of STIM1 may contribute to SOCE dysregulation in AD (Tong et al., 2016). Pharmacological restoration of spine SOCE pathway is one potential way for drug development in AD (Zhang et al., 2016a).
Figure 2.

SOCE impairment in AD.
In summary, various calcium signaling mechanisms are dysregulated in AD. The main question for future investigation is to determine the importance of these signaling abnormalities for synaptic and memory loss in AD. These pathways also constitute potential therapeutic targets for treatment of AD.
Calcium signaling alterations in Huntington’s and Parkinson`s diseases
Huntington’s disease is a monogenic autosomal dominant inherited neurodegenerative disorder, caused by CAG trinucleotide repeat expansion in huntingtin (Htt) gene (The. et al., 1993). Htt gene encodes huntingtin protein, which in mutant form contains expanded polyglutamine tract near the N-terminus. Expansion longer than 35 repeats results in HD, with longer polyglutamine tract leading to the earlier disease onset (Langbehn et al., 2004). GABAergic medium-sized spiny projection neurons (MSNs) of the caudate and putamen nuclei in the striatum are most affected in HD (Vonsattel and DiFiglia, 1998). Huntington’s disease is characterized by movement disorganization, mood and cognition impairments (Bates et al.). Similar to other neurodegenerative disorders Ca2+ signaling is dysregulated in HD (Raymond, 2017).
It has been reported that mutant huntingtin protein (mHtt) expression causes increased surface expression and currents of NR2B-bearing NMDARs, by accelerating delivery of receptors to the plasma membrane (Chen et al., 1999; Fan et al., 2007; Zeron et al., 2002). As a result, a balance between synaptic and extrasynaptic NMDAR is shifted in MSNs (Levine et al., 2010; Milnerwood et al., 2010; Plotkin and Surmeier, 2015). The insertion of extrasynaptic NMDARs depends on activation of calpain protease and STEP phosphatase (Gladding et al., 2014; Gladding et al., 2012). Recent report demonstrated differential changes in function of NMDARs in direct and indirect striatal pathways (Botelho et al., 2014). All these findings lead to the hypothesis that imbalance in activity of synaptic (pro-survival) and extrasynaptic (pro-death) NMDARs is a key pathogenic event in HD (Levine et al., 2010; Milnerwood et al., 2010; Plotkin and Surmeier, 2015). Consistent with this idea, NMDAR inhibitor memantine displayed neuroprotective effect in experiments with HD MSN cultures (Wu et al., 2006) and treatment of HD mice with low doses of memantine ameliorated neuropathological and behavioral phenotype by suppressing activity of extra-synaptic NMDAR (Dau et al., 2014; Okamoto et al., 2009). Overall, it appears that excessive function of extrasynaptic NMDARs is one of the important reasons for synaptic loss in early HD. Indeed, some benefit of memantine was observed in a small scale trial with HD patients (Ondo et al., 2007).
Besides effects on NMDAR, mHtt is reported to bind directly to InsP3R1 (Kaltenbach et al., 2007; Tang et al., 2003) and VGCC (Kaltenbach et al., 2007; Swayne et al., 2005). mHtt binding to InsP3R1 increases its affinity for InsP3 and enhances Ca2+ release from ER stores in response to activation of metabotropic glutamate receptors, mGluR1/5 (Tang et al., 2003). Inhibition of InsP3R1 diminishes adverse effects of mHtt and protects striatal neurons (Tang et al., 2009; Tang et al., 2005). In addition to effects of mHtt on InsP3R1, it may cause excessive Ca2+ leak through RyR, which also leads to depletion of internal Ca2+ stores (Suzuki et al., 2012a). In agreement with this observation, RyR inhibitors showed neuroprotective effects in vitro and improved motor behavior in YAC128 HD mice model (Chen et al., 2011b; Suzuki et al., 2012a). Recently our group demonstrated neuronal SOCE enhancement in MSNs from YAC128 HD mice, which likely results from continuous depletion of ER Ca2+ stores (Wu et al., 2016b; Wu et al., 2011) (Fig. 3). Moreover, expression of ER-resident protein STIM2 that controls synaptic SOCE pathway is elevated in aged YAC128 striatal cultures and in YAC128 mouse striatum. TRPC1 is a potential SOC channel subunit upregulated in response to mHtt expression (Wu et al., 2011). Inhibition of SOCE or knockdown of STIM2 resulted in neuroprotective effects and rescued dendritic spines deficiency observed in YAC128 HD mice model (Wu et al., 2016b; Wu et al., 2011). Therefore, SOCE inhibitors may constitute a potential new approach for HD treatment.
Figure 3.

SOCE impairment in HD.
Parkinson`s disease is a movement disorder, affecting preferentially people of advanced age and characterized by selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) (Lees et al., 2009). The pathological hallmark of PD is the presence of alfa-synuclein intraneuronal inclusions called Lewy bodies and reduced dopamine secretion due to cell death in substania nigra (Jellinger, 2009). Similar to AD, PD is mostly sporadic disease, but approximately about 10–15% of PD cases are familial (Fleming, 2017). The genes responsible for familial PD are called PARKs, with both recessive and dominant mutations identified (Kumar et al., 2011). Pathogenic mechanisms of sporadic and familial PD are still elusive. Similar to AD and HD, effective disease-modifying therapy for PD is absent (Cacabelos, 2017). Genes associated with PD are implicated in synaptic exocytosis and endocytosis, endosomal trafficking, autophagy and mitochondrial maintenance processes (Kumar et al., 2011; Trinh and Farrer, 2013). Strongest genetic link is with the mitochondrial biology, suggesting that mitochondrial dysfunction plays a key role in PD pathogenesis (Cieri et al., 2016). Recent data indicate that Ca2+ signaling dysregulation is also implicated in PD pathology (Cali et al., 2014; Zaichick et al., 2017).
SNc dopaminergic neurons display spontaneous pacemaking activity in the absence of synaptic input (Guzman et al., 2009; Puopolo et al., 2007). This pacemaking activity leads to sustained Ca2+ influx via L-type CaV1.3 Ca2+ channels (Chan et al., 2007). It has been proposed that this continuous Ca2+ influx makes SNc neurons selectively vulnerable by leading to basal mitochondrial oxidative stress (Chan et al., 2007; Surmeier et al., 2012; Surmeier et al., 2016). As a result, even subtle changes in intracellular Ca2+ homeostasis or in mitochondrial function due to advanced age or environmental factors may lead to neuronal degeneration. L-type Ca2+ channels play enhanced role in driving pacemaking activity of aging SNc neurons. Pharmacological inhibition of CaV1.3 L-type Ca2+ channels by izradipine restores Ca2+ - independent ‘juvenile’ pacemaking activity and protects SNc neurons in animal models of PD (Chan et al., 2007; Ilijic et al., 2011). Clinical use of L-type Ca2+ channel inhibitors dihydropyridines (DHPs) to treat hypertension has been linked to significant reduction of PD risk in retrospective studies (Becker et al., 2008; Gudala et al., 2015; Pasternak et al., 2012). Phase 3 controlled trial of izradipine in PD patients is currently in progress.
It was recently demonstrated that TRPC1 regulates the L-type Ca2+ channels activity in adult dopaminergic neurons in the SNc region (Sun et al., 2017). Store depletion and subsequent activation of TRPC1 via STIM1 inhibits the frequency and amplitude of the rhythmic activity in dopaminergic neurons (Sun et al., 2017). The death of dopaminergic neurons was induced by loss of either TRPC1 or STIM1 and it was prevented by inhibition of L-type Ca2+ channels (Sun et al., 2017). Moreover, application of PD-mimicking neurotoxins induced downregulation of TRPC1 and thapsigargin-mediated Ca2+ influx (Arshad et al., 2014; Bollimuntha et al., 2006; Bollimuntha et al., 2005). Activation or overexpression of TRPC1 protects cells against neurotoxin-mediated cytotoxicity (Arshad et al., 2014; Bollimuntha et al., 2006; Bollimuntha et al., 2005). These results suggested that impaired SOC may play a role in PD pathogenesis. Supporting the important role of SOCE for dopaminergic neurons, it was shown that expression of mutant dominant-negative form of Orai1 channel in Drosophila leading to downregulation of the tyrosine hydroxylase, an enzyme essential for dopamine synthesis, and the dopamine transporter, which is required for dopamine uptake after synaptic release (Pathak et al., 2015). Even more direct evidence for the role of SOC in PD was provided by the recent studies of PARK14 (Zhou et al., 2016). PARK14 is encoding Ca2+- independent phospholipase A2 group 6 (PLA2g6). It was shown that skin fibroblasts from idiopathic PD patients and patients bearing familial R747W mutation in PLA2g6 gene exhibit depleted stores and reduced SOCE (Zhou et al., 2016). It was previously suggested that Pla2g6 acts as an essential component of signal transduction from the intracellular ER Ca2+ stores to the plasma membrane SOCE channels (Bolotina, 2008; Csutora et al., 2006; Smani et al., 2016). Impaired store-operated activation of Pla2g6 leads to SOCE downregulation and subsequent depletion of intracellular ER Ca2+ stores during inherited and sporadic PD (Zhou et al., 2016). ER depletion triggers autophagic dysfunction, leading to progressive SNc neuronal loss and age-dependent PD (Zhou et al., 2016). Based on these findings, it was suggested that that Pla2g6-mediated SOCE pathway constitutes a novel potential therapeutic target for PD treatment (Zhou et al., 2016). Therefore, SOCE pathway present at attractive target for PD drug discovery.
Concluding remarks
Proper work of calcium-signaling machinery is essential for neuronal function. It is clearly demonstrated that calcium signaling dysregulation is a common feature of neurodegenerative diseases. Abnormal Ca2+ signaling leads to mitochondrial dysfunction and synaptic instability, and restoration of normal calcium homeostasis is a potential strategy for neurodegenerative diseases treatment. Interestingly, SOCE impairment is observed in AD, HD and PD but the nature of this disruption is unique for each disease. HD and PD both exhibit excessively depleted neuronal ER calcium stores, in contrast ER calcium stores are overfilled in AD neurons. In HD SOCE is overactivated due to ER depletion, while in PD reduced calcium levels in ER is a consequence of impaired SOCE. In AD neurons SOCE is downregulated as a compensatory response to ER Ca2+ overfilling. Our previous investigation indicated that synaptic spines in hippocampal and striatal neurons have an opposite sensitivity to SOCE impairments (Sun et al., 2014; Wu et al., 2016b). These differences are probably due to different downstream SOCE targets in different neuronal types. These findings have direct implications for drug discovery. It appears that inhibition of SOCE in HD and activation of SOCE in AD and PD is potentially beneficial strategy, but this approach may result in toxic effects on neuronal cells in non-target region. Molecular identity of SOCE channels in striatal and SNc neurons will be determined in the future. Development of drugs targeting particular SOCE channel isoforms may help to achieve high selectivity and to overcome a potential toxicity.
Highlights.
Calcium (Ca2+) signaling is dysregulated in aging neurons and in neurons affected by neurodegenerative disorders
Elevated endoplasmic reticulum (ER) Ca2+ levels and reduced neuronal store-operated Ca2+ entry (SOCE) are linked with synaptic loss in Alzheimer’s disease
Reduced ER Ca2+ levels and enhanced SOCE are linked with synaptic loss in Huntington’s disease
Reduced SOCE and reduced ER Ca2+ levels are linked with neuronal cell death in Parkinson’s disease
Acknowledgments
Ilya Bezprozvanny is a holder of the Carl J. and Hortense M. Thomsen Chair in Alzheimer’s Disease Research. This work was supported by the R01NS080152, R01NS056224, R01AG055577 and by the Russian Science Foundation Grant 14-25-00024 (IB) (chapter Calcium dysregulation in Alzheimer`s disease, Figure 1), by the state grant 17.1360.2014/K (IB) (Calcium signaling alterations in Huntington’s and Parkinson`s diseases, Figure 2), and by the grant of President of Russian Federation 14.Y30.17.1043-MK (EP) (Figure 3).
After ER Ca2+ depletion, STIMs oligomerize in the ER and translocate toward PM where they activate calcium entry trough interaction with Orai, TRPC channels or Orai/TRPC channels complex. Calcium current passing through Orai channels is called Ca2+ release-activated Ca2+ current (ICRAC), non-selective cation current passing through TRPC channels or TRPC-ORAI channels complexes is called store-operated current (ISOC). Activation of G-protein coupled receptor (GPCR) by extracellular ligand induces activation of phospholipase C (PLC), which subsequently hydrolysis phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol-1,4,5-trisphospahte (InsP3) and diacylglycerol (DAG). InsP3 activates InsP3R in the ER, which induces calcium release from the intracellular store. DAG can activate TRPC channels composed of TRPC3/6/7 isoforms directly (Jellinger, 2009). Presenilins (PS) support ER calcium leak (Bezprozvanny, 2009). Ca2+ can also be released from ER via ryanodine receptors (RyR) in the process of Ca2+ -induced Ca2+ release (CICR). Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) transports Ca2+ ions from the cytoplasm into the ER using energy of ATP hydrolysis.
In hippocampal dendritic spines SOCE is mediated by TRPC6/Orai2 channels complex (Zhang et al., 2016a). SOCE channels play a critical role in maintenance of hippocampal mushroom dendritic spines (Zhang et al., 2016a). AD-causing mutations in presenilins impair ER Ca2+ leak function and lead to elevation of ER Ca2+ levels (Tu et al., 2006b). Overfilling of ER Ca2+ stores causes excessive Ca2+ release through the InsP3R1 and RyR2. It also causes compensatory downregulation of STIM2 expression, SOCE impairment and loss of mushroom spines in AD neurons.
In striatal MSN spines SOCE is gated by STIM2 (Wu et al., 2016b). Molecular identity of TRPC/Orai channels in MSN spines have not been identified yet, with TRPC1 and Orai1/2 are most likely candidates. In HD neurons mutant huntingtin protein (mHtt) binds directly to the InsP3R1 and sensitizes it to basal InsP3 levels (Tang et al., 2003). Ca2+ leak via RyR also enhanced in HD neurons (Suzuki et al., 2012b). Enhanced Ca2+ efflux from ER leads to chronic ER depletion and overactivation of SOCE pathway (Wu et al., 2016a; Wu et al., 2011), which appears to be toxic to striatal neurons and triggers dendritic spines loss (Wu et al., 2016b).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alzheimer's Association Calcium Hypothesis, W. Calcium Hypothesis of Alzheimer's disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13:178–182 e117. doi: 10.1016/j.jalz.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Arispe N, Diaz JC, Simakova O. Abeta ion channels. Prospects for treating Alzheimer's disease with Abeta channel blockers. Biochim Biophys Acta. 2007;1768:1952–1965. doi: 10.1016/j.bbamem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshad A, Chen X, Cong Z, Qing H, Deng Y. TRPC1 protects dopaminergic SH-SY5Y cells from MPP+, salsolinol, and N-methyl-(R)-salsolinol-induced cytotoxicity. Acta Biochim Biophys Sin. 2014;46:22–30. doi: 10.1093/abbs/gmt127. [DOI] [PubMed] [Google Scholar]
- Bandara S, Malmersjo S, Meyer T. Regulators of Calcium Homeostasis Identified by Inference of Kinetic Model Parameters from Live Single Cells Perturbed by siRNA. Science signaling. 2013;6:ra56. doi: 10.1126/scisignal.2003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ. Huntington disease. Nat Rev Dis Primers. 2015 Apr 23;1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2008;70:1438–1444. doi: 10.1212/01.wnl.0000303818.38960.44. [DOI] [PubMed] [Google Scholar]
- Bergmans BA, De Strooper B. gamma-secretases: from cell biology to therapeutic strategies. Lancet Neurol. 2010;9:215–226. doi: 10.1016/S1474-4422(09)70332-1. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling and Alzheimer's disease. Neurochem Res. 2011;36:1149–1156. doi: 10.1007/s11064-010-0371-4. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Hiesinger PR. The synaptic maintenance problem: membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol Neurodegener. 2013;8:23. doi: 10.1186/1750-1326-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska-Kijek A, Gruszczynska-Biegala J, Kuznicki J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer's disease. Biochim Biophys Acta. 2009;1793:1050–1057. doi: 10.1016/j.bbamcr.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Bollimuntha S, Ebadi M, Singh BB. TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 2006;12:141–149. doi: 10.1016/j.brainres.2006.04.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J Biol Chem. 2005;280:2132–2140. doi: 10.1074/jbc.M407384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotina VM. Orai, STIM1 and iPLA(2)β: a view from a different perspective. J Physiol. 2008;586:3035–3042. doi: 10.1113/jphysiol.2008.154997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho EP, Wang E, Chen JY, Holley S, Andre V, Cepeda C, Levine MS. Differential Synaptic and Extrasynaptic Glutamate-Receptor Alterations in Striatal Medium-Sized Spiny Neurons of Aged YAC128 Huntington's Disease Mice. PLoS Curr. 2014;6 doi: 10.1371/currents.hd.34957c4f8bd7cb1f5ec47381dfc811c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs CA, Chakroborty S, Stutzmann GE. Emerging pathways driving early synaptic pathology in Alzheimer's disease. Biochem Biophys Res Commun. 2016 doi: 10.1016/j.bbrc.2016.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Cali T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci. 2014;71:2787–2814. doi: 10.1007/s00018-013-1550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2012;33:1001 e1001–1006. doi: 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacabelos R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. International Journal of Molecular Sciences. 2017;18:551. doi: 10.3390/ijms18030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Lin P, Cheung KH, Li N, Levchook C, Pan Z, Ferrante C, Boulianne GL, Foskett JK, Danielpour D, Ma J. The presenilin-2 loop peptide perturbs intracellular Ca2+ homeostasis and accelerates apoptosis. J Biol Chem. 2006;281:16649–16655. doi: 10.1074/jbc.M512026200. [DOI] [PubMed] [Google Scholar]
- Cali T, Ottolini D, Brini M. Calcium signaling in Parkinson's disease. Cell Tissue Res. 2014;357:439–454. doi: 10.1007/s00441-014-1866-0. [DOI] [PubMed] [Google Scholar]
- Capiod T. Extracellular Calcium Has Multiple Targets to Control Cell Proliferation. Advances in experimental medicine and biology. 2016;898:133–156. doi: 10.1007/978-3-319-26974-0_7. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PloS one. 2012a;7:e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgo J, Stutzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer's disease mice. J Neurosci. 2012b;32:8341–8353. doi: 10.1523/JNEUROSCI.0936-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- Chen KT, Ong HL, Liu X, Ambudkar IS. Contribution of TRPC1 and Orai1 to Ca(2+) entry activated by store depletion. Advances in experimental medicine and biology. 2011a;704 doi: 10.1007/1978-1094-1007-0265-1003_1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- Chen X, Wu J, Lvovskaya S, Herndon E, Supnet C, Bezprozvanny I. Dantrolene is neuroprotective in Huntington's disease transgenic mouse model. Mol Neurodegener. 2011b;6:81. doi: 10.1186/1750-1326-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP(3) receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri D, Brini M, Cali T. Emerging (and converging) pathways in Parkinson's disease: keeping mitochondrial wellness. Biochem Biophys Res Commun. 2016 doi: 10.1016/j.bbrc.2016.08.153. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Csutora P, Zarayskiy V, Peter K, Monje F, Smani T, Zakharov SI, Litvinov D, Bolotina VM. Activation mechanism for CRAC current and store-operated Ca2+ entry: calcium influx factor and iPLA2beta -dependent pathway. J Biol Chem. 2006 doi: 10.1074/jbc.M606504200. [DOI] [PubMed] [Google Scholar]
- Cummings J, Aisen PS, DuBois B, Frölich L, Jack CR, Jones RW, Morris JC, Raskin J, Dowsett SA, Scheltens P. Drug development in Alzheimer’s disease: the path to 2025. Alzheimer's Research & Therapy. 2016;8:39. doi: 10.1186/s13195-016-0207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer's Research & Therapy. 2014;6:37-37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dau A, Gladding CM, Sepers MD, Raymond LA. Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol Dis. 2014;62:533–542. doi: 10.1016/j.nbd.2013.11.013. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Fan MM, Fernandes HB, Zhang LY, Hayden MR, Raymond LA. Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington's disease. J Neurosci. 2007;27:3768–3779. doi: 10.1523/JNEUROSCI.4356-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira IL, Bajouco LM, Mota SI, Auberson YP, Oliveira CR, Rego AC. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium. 2012;51:95–106. doi: 10.1016/j.ceca.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Fleming SM. Mechanisms of Gene-Environment Interactions in Parkinson's Disease. Curr Environ Health Rep. 2017;4:192–199. doi: 10.1007/s40572-017-0143-2. [DOI] [PubMed] [Google Scholar]
- Foster TC, Kyritsopoulos C, Kumar A. Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer's disease. Behav Brain Res. 2017;322:223–232. doi: 10.1016/j.bbr.2016.05.012. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Fan J, Zhang LY, Wang L, Xu J, Li EH, Lombroso PJ, Raymond LA. Alterations in STriatal-Enriched protein tyrosine Phosphatase expression, activation, and downstream signaling in early and late stages of the YAC128 Huntington's disease mouse model. Journal of neurochemistry. 2014;130:145–159. doi: 10.1111/jnc.12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladding CM, Sepers MD, Xu J, Zhang LY, Milnerwood AJ, Lombroso PJ, Raymond LA. Calpain and STriatal-Enriched protein tyrosine phosphatase (STEP) activation contribute to extrasynaptic NMDA receptor localization in a Huntington's disease mouse model. Hum Mol Genet. 2012;21:3739–3752. doi: 10.1093/hmg/dds154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca(2+) influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer's disease mice. J Neurosci. 2010;30:12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudala K, Kanukula R, Bansal D. Reduced Risk of Parkinson's Disease in Users of Calcium Channel Blockers: A Meta-Analysis. Int J Chronic Dis. 2015;2015:697404. doi: 10.1155/2015/697404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature reviews. Neuroscience. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hayrapetyan V, Rybalchenko V, Rybalchenko N, Koulen P. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium. 2008;44:507–518. doi: 10.1016/j.ceca.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Selective loss of NMDA receptor NR1 subunit isoforms in Alzheimer's disease. Journal of neurochemistry. 2004;89:240–247. doi: 10.1111/j.1471-4159.2003.02330.x. [DOI] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis. 2011;43:364–371. doi: 10.1016/j.nbd.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grunblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci. 2009;24:114–125. doi: 10.1159/000197890. [DOI] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha GH, Ukani L, Chepanoske CL, Zhen Y, Sahasrabudhe S, Olson J, Kurschner C, Ellerby LM, Peltier JM, Botas J, Hughes RE. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014;76:185–205. doi: 10.1002/ana.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann N Y Acad Sci. 1989;568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Oni-Biton E, Segal M. The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. The Journal of physiology. 2017;595:125–140. doi: 10.1113/JP272645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R. STIM and ORAI proteins in the nervous system. Channels. 2015;9:245–252. doi: 10.1080/19336950.2015.1071747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene--a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bodhinathan K, Foster TC. Susceptibility to Calcium Dysregulation during Brain Aging. Front Aging Neurosci. 2009;1:2. doi: 10.3389/neuro.24.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KR, Djarmati-Westenberger A, Grunewald A. Genetics of Parkinson's disease. Semin Neurol. 2011;31:433–440. doi: 10.1055/s-0031-1299782. [DOI] [PubMed] [Google Scholar]
- La Rovere RM, Roest G, Bultynck G, Parys JB. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium. 2016;60:74–87. doi: 10.1016/j.ceca.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. 2004;65:267–277. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999a;274:32535–32538. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer's presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999b;72:1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- Levine MS, Cepeda C, Andre VM. Location, location, location: contrasting roles of synaptic and extrasynaptic NMDA receptors in Huntington's disease. Neuron. 2010;65:145–147. doi: 10.1016/j.neuron.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Orai in TRPC-mediated Ca(2+) entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca(2+) entry. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- Liu J, Supnet C, Sun S, Zhang H, Good L, Popugaeva E, Bezprozvanny I. The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels (Austin) 2014;8:230–242. doi: 10.4161/chan.27471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski L, Kuznicki J. SOCE in neurons: Signaling or just refilling? Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2015;1853:1940–1952. doi: 10.1016/j.bbamcr.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Mason SL, Barker RA. Advancing pharmacotherapy for treating Huntington's disease: a review of the existing literature. Expert Opin Pharmacother. 2016;17:41–52. doi: 10.1517/14656566.2016.1109630. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Mota SI, Ferreira IL, Rego AC. Dysfunctional synapse in Alzheimer's disease - A focus on NMDA receptors. Neuropharmacology. 2014a;76(Pt A):16–26. doi: 10.1016/j.neuropharm.2013.08.013. [DOI] [PubMed] [Google Scholar]
- Mota SI, Ferreira IL, Rego AC. Dysfunctional synapse in Alzheimer's disease – A focus on NMDA receptors. Neuropharmacology. 2014b;76(Part A):16–26. doi: 10.1016/j.neuropharm.2013.08.013. [DOI] [PubMed] [Google Scholar]
- Nelson O, Supnet C, Tolia A, Horré K, De Strooper B, Bezprozvanny I. Mutagenesis Mapping of the Presenilin 1 Calcium Leak Conductance Pore. The Journal of Biological Chemistry. 2011;286:22339–22347. doi: 10.1074/jbc.M111.243063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease–linked mutations specifically disrupt Ca(2+) leak function of presenilin 1. Journal of Clinical Investigation. 2007;117:1230–1239. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C. Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–797. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto SI, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009 doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondo WG, Mejia NI, Hunter CB. A pilot study of the clinical efficacy and safety of memantine for Huntington's disease. Parkinsonism Relat Disord. 2007;13:453–454. doi: 10.1016/j.parkreldis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Oules B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Brechot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine Receptor Blockade Reduces Amyloid-beta Load and Memory Impairments in Tg2576 Mouse Model of Alzheimer Disease. J Neurosci. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameshwaran K, Dhanasekaran M, Suppiramaniam V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Experimental neurology. 2008;210:7–13. doi: 10.1016/j.expneurol.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Pasternak B, Svanstrom H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol. 2012;175:627–635. doi: 10.1093/aje/kwr362. [DOI] [PubMed] [Google Scholar]
- Pathak T, Agrawal T, Richhariya S, Sadaf S, Hasan G. Store-Operated Calcium Entry through Orai Is Required for Transcriptional Maturation of the Flight Circuit in Drosophila. J Neurosci. 2015;35:13784–13799. doi: 10.1523/JNEUROSCI.1680-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Liang G, Inan S, Wu Z, Joseph DJ, Meng Q, Peng Y, Eckenhoff MF, Wei H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci Lett. 2012;516:274–279. doi: 10.1016/j.neulet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JL, Surmeier DJ. Corticostriatal synaptic adaptations in Huntington's disease. Current opinion in neurobiology. 2015;33:53–62. doi: 10.1016/j.conb.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E, Pchitskaya E, Bezprozvanny I. Dysregulation of neuronal calcium homeostasis in Alzheimer's disease - A therapeutic opportunity? Biochem Biophys Res Commun. 2017;483:998–1004. doi: 10.1016/j.bbrc.2016.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E, Pchitskaya E, Speshilova A, Alexandrov S, Zhang H, Vlasova O, Bezprozvanny I. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Molecular Neurodegeneration. 2015;10:37. doi: 10.1186/s13024-015-0034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem Biophys Res Commun. 2017;483:1051–1062. doi: 10.1016/j.bbrc.2016.07.058. [DOI] [PubMed] [Google Scholar]
- Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Shilling D, Mak DO, Kang DE, Foskett JK. Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J Biol Chem. 2012;287:10933–10944. doi: 10.1074/jbc.M111.300491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Gibson ES, Kennedy MJ. Local and Use-Dependent Effects of beta-Amyloid Oligomers on NMDA Receptor Function Revealed by Optical Quantal Analysis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36:11532–11543. doi: 10.1523/JNEUROSCI.1603-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smani T, Dominguez-Rodriguez A, Callejo-Garcia P, Rosado JA, Avila-Medina J. Phospholipase A2 as a Molecular Determinant of Store-Operated Calcium Entry. Adv Exp Med Biol. 2016;898:111–131. doi: 10.1007/978-3-319-26974-0_6. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-[beta] Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nature reviews. Molecular cell biology. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL, 3rd, Bezprozvanny I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron. 2014;82:79–93. doi: 10.1016/j.neuron.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhang H, Selvaraj S, Sukumaran P, Lei S, Birnbaumer L, Singh BB. Inhibition of L-Type Ca2+ Channels by TRPC1-STIM1 Complex Is Essential for the Protection of Dopaminergic Neurons. J Neurosci. 2017;37:3364–3377. doi: 10.1523/JNEUROSCI.3010-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT. Physiological phenotype and vulnerability in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009290. doi: 10.1101/cshperspect.a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E. Calcium and Parkinson's disease. Biochem Biophys Res Commun. 2016 doi: 10.1016/j.bbrc.2016.08.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Nagai Y, Wada K, Koike T. Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochemical and biophysical research communications. 2012a;429:18–23. doi: 10.1016/j.bbrc.2012.10.107. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Nagai Y, Wada K, Koike T. Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem Biophys Res Commun. 2012b;429:18–23. doi: 10.1016/j.bbrc.2012.10.107. [DOI] [PubMed] [Google Scholar]
- Swayne LA, Chen L, Hameed S, Barr W, Charlesworth E, Colicos MA, Zamponi GW, Braun JE. Crosstalk between huntingtin and syntaxin 1A regulates N-type calcium channels. Mol Cell Neurosci. 2005;30:339–351. doi: 10.1016/j.mcn.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Tang TS, Guo C, Wang H, Chen X, Bezprozvanny I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J Neurosci. 2009;29:1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Texido L, Martin-Satue M, Alberdi E, Solsona C, Matute C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell calcium. 2011;49:184–190. doi: 10.1016/j.ceca.2011.02.001. [DOI] [PubMed] [Google Scholar]
- The., Huntington's., Disease., Collaborative., Research., and Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Theobald DL. Presenilin adopts the ClC channel fold. Protein Sci. 2016;25:1363–1365. doi: 10.1002/pro.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong BC, Lee CS, Cheng WH, Lai KO, Foskett JK, Cheung KH. Familial Alzheimer's disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci Signal. 2016;9:ra89. doi: 10.1126/scisignal.aaf1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AB, Shum AK, Prakriya M. Regulation of neurogenesis by calcium signaling. Cell Calcium. 2016;59:124–134. doi: 10.1016/j.ceca.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins Form ER Ca(2+) Leak Channels, a Function Disrupted by Familial Alzheimer's Disease-Linked Mutations. Cell. 2006a;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006b;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. Journal of neurochemistry. 1997;68:265–271. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochemical Journal. 2001;357:673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ryskamp DA, Liang X, Egorova P, Zakharova O, Hung G, Bezprozvanny I. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington's Disease Mouse Model. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016a;36:125–141. doi: 10.1523/JNEUROSCI.1038-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ryskamp DA, Liang X, Egorova P, Zakharova O, Hung G, Bezprozvanny I. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington's Disease Mouse Model. The Journal of Neuroscience. 2016b;36:125–141. doi: 10.1523/JNEUROSCI.1038-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem Biol. 2011;18:777–793. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Tang T, Bezprozvanny I. Evaluation of clinically relevant glutamate pathway inhibitors in in vitro model of Huntington's disease. Neurosci Lett. 2006;407:219–223. doi: 10.1016/j.neulet.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nature cell biology. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaichick SV, McGrath KM, Caraveo G. The role of Ca(2+) signaling in Parkinson's disease. Disease Models & Mechanisms. 2017;10:519–535. doi: 10.1242/dmm.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu J, Sun S, Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling, excitability, and synaptic plasticity defects in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2015a;45:561–580. doi: 10.3233/JAD-142427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010a;30:8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010b;30:8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun S, Wu L, Pchitskaya E, Zakharova O, Fon Tacer K, Bezprozvanny I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer's Disease Treatment. The Journal of Neuroscience. 2016a;36:11837–11850. doi: 10.1523/JNEUROSCI.1188-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, Bezprozvanny I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer's Disease. J Neurosci. 2015b;35:13275–13286. doi: 10.1523/JNEUROSCI.1034-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li P, Feng J, Wu M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurological Sciences. 2016b;37:1039–1047. doi: 10.1007/s10072-016-2546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Weisleder N, Han X, Pan Z, Parness J, Brotto M, Ma J. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J Biol Chem. 2006;281:33477–33486. doi: 10.1074/jbc.M602306200. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Yen A, Rymarczyk G, Asai H, Trengrove C, Aziz N, Kirber MT, Mostoslavsky G, Ikezu T, Wolozin B, Bolotina VM. Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson's disease. Nature communications. 2016;7:10332. doi: 10.1038/ncomms10332. [DOI] [PMC free article] [PubMed] [Google Scholar]