

Significance
CRISPR-Cas9 holds enormous potential for therapeutic genome editing. Effective therapy requires treatment to be efficient and safe with minimal toxicity. The sequence-based targeting for CRISPR systems necessitates consideration of the unique genomes for each patient targeted for therapy. We show using 7,444 whole-genome sequences that SNPs and indels can reduce on-target CRISPR activity and increase off-target potential when targeting therapeutically implicated loci; however, these occurrences are relatively rare. We further identify that differential allele frequencies among populations may result in population-specific alterations in CRISPR targeting specificity. Our findings suggest that human genetic variation should be considered in the design and evaluation of CRISPR-based therapy to minimize risk of treatment failure and/or adverse outcomes.
Keywords: CRISPR-Cas9, off-target specificity, on-target specificity, human genetic variation, therapeutic genome editing
Abstract
The CRISPR-Cas9 nuclease system holds enormous potential for therapeutic genome editing of a wide spectrum of diseases. Large efforts have been made to further understanding of on- and off-target activity to assist the design of CRISPR-based therapies with optimized efficacy and safety. However, current efforts have largely focused on the reference genome or the genome of cell lines to evaluate guide RNA (gRNA) efficiency, safety, and toxicity. Here, we examine the effect of human genetic variation on both on- and off-target specificity. Specifically, we utilize 7,444 whole-genome sequences to examine the effect of variants on the targeting specificity of ∼3,000 gRNAs across 30 therapeutically implicated loci. We demonstrate that human genetic variation can alter the off-target landscape genome-wide including creating and destroying protospacer adjacent motifs (PAMs). Furthermore, single-nucleotide polymorphisms (SNPs) and insertions/deletions (indels) can result in altered on-target sites and novel potent off-target sites, which can predispose patients to treatment failure and adverse effects, respectively; however, these events are rare. Taken together, these data highlight the importance of considering individual genomes for therapeutic genome-editing applications for the design and evaluation of CRISPR-based therapies to minimize risk of treatment failure and/or adverse outcomes.
The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system holds enormous potential for therapeutic genome editing to treat a wide spectrum of genetic diseases (1–8). Human CRISPR-Cas9 clinical trials have already been initiated (9, 10) and are likely to increase in number in the future. The development of successful therapies not only requires treatment efficacy but also requires that patient safety remain paramount. This requires assessing for toxicity related to reagent delivery, to the genome-editing reagents themselves, and to off-target effects. Significant progress has been made to aid in off-target prediction (11, 12) and for unbiased genome-wide off-target detection (13–19). From a therapeutic genome-editing perspective, these methods for unbiased genome-wide off-target detection are often limited by their reliance on the reference genome or the genome of the cells used for study to evaluate guide RNA (gRNA) efficiency, safety, and toxicity; however, newer methods circumvent limitations imposed by use of the reference genome through direct sequencing of target site regions to screen each individual patient (17).
Numerous efforts have been made to document human genetic variation. For example, the 1000 Genomes Project (1000G) database consists of 2,504 whole-genome sequences (WGSs) from 26 populations spanning Africa (AFR), East Asia (EAS), Europe (EUR), South Asia (SAS), and the Americas (AMR) (20). On average, individual genomes within the database deviated from the reference genome at 4.1–5.0 million sites. The majority of variants in an individual genome were common with only 1–4% of variants having a frequency <0.5%. Notably, these deviations included 2,100–2,500 structural variants per genome. The median number of variants varied across populations; however, the order of magnitude remained unchanged (Dataset S1). In total, across all individuals/populations studied, ∼64 million autosomal variants were identified with a frequency <0.5%, ∼12 million with a frequency between 0.5% and 5%, and ∼8 million with a frequency >5% (20).
Recent work has demonstrated the utility of considering variants when designing CRISPR genome-editing experiments (21). This included analysis evaluating the effect of variants on gRNA te or destroy protospacer adjacent motif (PAM) sequences (21). Therapeutic genome-editing reagents will encounter a unique genome for each patient seeking treatment. Therefore, we sought to evaluate whether variants should be considered for clinical translation of CRISPR-based therapies. We hypothesized that human genetic variation may alter CRISPR-Cas9 targeting on- and off-target specificity at therapeutically implicated loci. We further hypothesized that personalized off-target events could exist that would predispose patients to treatment failure and/or adverse outcomes. Finally, we investigated whether population-specific variants would also predispose patients to treatment failure and/or adverse outcomes.
Results
Therapeutic Loci and Off-Target Score Calculation.
To evaluate these hypotheses, we identified a comprehensive list of 23 human-genome and 7 viral-genome therapeutic targets based on literature mining for loci previously targeted by CRISPR-Cas9 for therapy (Table 1). These loci have been demonstrated to be amenable therapeutic targets for CRISPR-based strategies to elicit nonhomologous end joining (NHEJ) repair or homology-directed repair (HDR). A list of gRNAs was generated by designing all gRNAs targeting the indicated regions or using the gRNAs from previous studies (Dataset S2). gRNAs for both NHEJ and HDR applications were designed to include all gRNAs within the relevant exon(s) for coding region targets and ±100 bp for noncoding targets. It is important to note that HDR efficiency decreases as a function of the distance between the variant and the double-strand break site (22). We also identified gRNAs targeting viral genomes using the same approach (Table 1). This analysis resulted in a list of 2,481 gRNAs targeting human genomic regions and 484 gRNAs targeting viral genomic regions. In addition, 128 nontargeting gRNAs were included as negative controls. Using a previously published aggregate off-target score (23, 24), we calculated aggregate off-target scores using the reference genome for all gRNAs (Fig. 1A). For a given gRNA, a “local” off-target score is calculated for each genomic match (from 0 to four mismatches). The summation of all local off-target scores for a given gRNA results in a genome-wide off-target score, termed “aggregate off-target score” (see Materials and Methods for additional details). For this off-target scoring method (range, 0–100), higher scores indicate lower off-target cleavage potential and lower scores indicate higher off-target cleavage potential. Importantly, the probability of cleavage decreases with increased number of mismatches (25). Therefore, sites with higher numbers of mismatches, such as three or four mismatch sites, may not actually result in off-target cleavage despite prediction. To reflect this, sites with more mismatches (i.e., three or more) penalize the aggregate off-target score much less than sites with fewer (i.e., one, two) mismatches. Nontargeting gRNAs were designed without any perfect genomic matches and were also chosen based on having an aggregate off-target score >90%, suggesting that genomic cleavage is unlikely. While cleavage mediated by a nontargeting gRNA is still possible at sites with genomic mismatches, it is expected that these occurrences are rare.
Table 1.
Summary of therapeutically implicated loci
| Gene/virus | Target | Coordinates (hg19) | Disease | Repair | Refs. |
| Gene | |||||
| ALCAM | All exons | chr3:105085753–105295744 | HIV-1 infection | NHEJ | 63 |
| BCL11A | Enhancer | chr2:60722309–60722472 | β-Hemoglobinopathies | NHEJ | 64 |
| B2M | All exons | chr15:45003686–45010357 | Hypoimmunogenic cells for transplantation | NHEJ | 65 |
| CCR5 | Exon 1 | chr3:46414395–46415452 | HIV infection | NHEJ | 65 |
| CEP290 | Intron 26 | chr12:88494861–88495060 | Leber’s congenital amaurosis type 10 | NHEJ | 2, 66 |
| CXCR4 | Exon 2 | chr2:136872440–136873482 | HIV-1 infection | NHEJ | 67 |
| HLA-A | Exon 3 | chr6:29911046–29911320 | Hypoimmunogenic cells for transplantation | NHEJ | 68 |
| PCSK9 | Exons 1–2 | chr1:55505512–55509707 | Cardiovascular disease | NHEJ | 69 |
| PDCD1 | Exon 1 | chr2:242800916–242800990 | Tumor immunotherapy | NHEJ/HDR | 70, 71 |
| PSIP1 | Exons 2, 12, 14 | chr9:15468629–15510186 | HIV-1 infection | NHEJ | 72 |
| TPST2 | All exons | chr22:26921712–26992681 | HIV-1 infection | NHEJ | 63 |
| TRAC,TRBC1, TRBC2 | Exon 1 | chr14:23016448–23016719; chr7:142498738–142499111; chr7:142498726–142499111 | T cell immunotherapy | NHEJ | 73–75 |
| SLC35B2 | All exons | chr6:44221838–44225308 | HIV-1 infection | NHEJ | 63 |
| ADA | Intron 6/exon 7 | chr20:43251649–43251819 | Adenosine deaminase severe combined immunodeficiency (ADA-SCID) | HDR | 76 |
| ALB | Intron 1 | chr4:74270125–74270832 | Lysosomal storage disease, hemophilia A, B | HDR | 77 |
| CFTR | Exon 10 | chr7:117199519–117199709 | Cystic fibrosis | HDR | 78 |
| COL7A1 | Exons 2, 3, 14, 15, 54, 117 | chr3:48602217–48631981 | Epidermolysis bullosa | HDR | 79 |
| CYBB | Exon 7 | chrX:37658207–37658337 | X-linked chronic granulomatous disease | HDR | 80 |
| DMD | Exons/intron 45–55 | chrX:31533884–32250573 | Duchenne’s muscular dystrophy | HDR | 81, 82 |
| FANCC | Intron 4 | chr9:97934216–97934415 | Fanconi anemia | HDR | 83 |
| F9 | Intron 1 | chrX:138613012–138619169 | Hemophilia B | HDR | 84 |
| FAH | Exon 8/intron 8 | chr15:80464492–80464690 | Hereditary tyrosinemia type I | HDR | 85 |
| HBB | Exon 1 | chr11:5248162–5248251 | Sickle cell disease | HDR | 86, 87 |
| IL2RG | Exon 5 | chrX:70329079–70329240 | X-linked severe combined immunodeficiency (X-SCID) | HDR | 88 |
| SERPINA1 | Intron 4/exon 5 | chr14:94844848–94845047 | α-1-Antitrypsin deficiency | HDR | 89 |
| Virus | |||||
| Cytomegalovirus | Viral genome | — | Congenital defects, disease in immuno-compromised individuals | NHEJ | 49 |
| Epstein bar virus | Viral genome | — | Infectious mononucleosis, malignancies | NHEJ | 49 |
| Hepatitis B virus | Viral genome | — | Hepatitis B | NHEJ | 51, 90–92 |
| Herpes simplex virus type 1 | Viral genome | — | Cold sores, keratitis | NHEJ | 49 |
| HIV-1 | Viral genome (LTR) | — | HIV-1 infection | NHEJ | 50 |
| Human papilloma virus | E6–E7 oncogenes | — | Cervical carcinoma | NHEJ | 93 |
| JC virus | T antigen | — | Progressive multifocal leukoencephalopathy | NHEJ | 52 |
Fig. 1.

Off-target scores using the ambiguous genome approach. (A) Distribution of aggregate off-target scores in the reference and ambiguous genomes for human-genome–targeting, viral-genome–targeting, and nontargeting gRNAs. (B) Change in aggregate off-target score between ambiguous and reference genomes. (C) Distribution of off-target sites by number of mismatches. (D) Ratio of the number of off-target sites in ambiguous genomes compared with the reference genome stratified by the number of off-target sites in the reference genome. The y axis shows the ratios for each gRNA, whereas the x axis shows the number of off-target sites in the reference genome.
Single-Nucleotide Polymorphisms Can Create Novel Off-Target Sites.
We first investigated whether single-nucleotide polymorphisms (SNPs) altered the number of off-target sites in the genome using 7,444 WGSs from three different datasets: 1000 Genomes Project phase 3 (1000G) (n = 2,504) (20), a subset of the gnomAD database [an updated and expanded version of the ExAC dataset (26)] (n = 2,938), and a French Canadian (FC) dataset (n = 2,002) (27) (see Materials and Methods for additional details). Notably, the FC dataset is a founder population with increased genetic homogeneity. Fewer variants suggested a decreased probability to create or alter off-target sites a priori.
SNPs can alter off-target sites by increasing or decreasing the number of mismatches between a genomic region and the gRNA sequence. In addition, SNPs can create (alter NHG or NGH from reference genome to become an NGG motif) or destroy (alter reference genome NGG motif to NHG or NGH sequence) PAM sequences (H = A, C, or T). Creation of PAM sequences may generate new loci for off-target cleavage while destruction of PAM sequences potentially removes loci for off-target cleavage. SNPs present within the 1000G database led to the creation of 11,585,879 new NGG PAM sequences (4.1% of total PAMs in the reference genome, 11,585,879/281,005,914) and led to the destruction of 22,182,468 PAM sequences (7.9% of total PAMs in the reference genome, 22,182,468/281,005,914). To determine the number of PAMs per haploid genome within the 1000G dataset, the number of created PAMs was added and the number of destroyed PAMs was subtracted from the total number of NGG motifs in the reference autosomal genome (n = 281,005,914). Interestingly, the number of NGG motifs per haploid genome was similar to the reference genome, with a mean increase of 34 NGG motifs (median, −42 NGG motifs). However, the number of NGG motifs varied across individual haploid genomes (SD, ±1,559 NGG motifs). The number of NGG motifs also varied across populations, with individuals of European descent showing the largest reduction (−1,327 ± 922, mean ± SD) and individuals of African ancestry displaying the largest increase as compared to the reference genome (1,429 ± 1,142, mean ± SD, Fig. S1).
To further investigate the effect of SNPs within the 1000G, gnomAD, and FC datasets, we created an “ambiguous genome” by replacing each SNP position by an International Union of Pure and Applied Chemistry (IUPAC) ambiguity code to account for all possible SNP alleles. For example, an A > C SNP would be replaced by the ambiguity code “M” (M = A or C). With this replacement strategy, both alleles can map to the SNP locus without penalty. Therefore, all possible matches upstream of an NGG motif were identified in the reference and ambiguous genomes (up to four mismatches), which were used to calculate off-target scores. Interestingly, we observed a reduction in aggregate off-target scores for all three datasets when comparing the reference and ambiguous genomes, which suggested increased off-target cleavage potential (Fig. 1 A and B). The largest decrease in aggregate off-target scores was associated with the 1000G dataset (Fig. 1A and Dataset S3). For human genome-targeting gRNAs, the mean reductions in aggregate off-target scores were 9.3%, 7.8%, and 3.1% for the 1000G, gnomAD, and FC datasets, respectively (Fig. 1 A and B). For viral-genome–targeted gRNAs, the mean reductions of aggregate off-target scores were 9.1%, 7.5%, and 3.0%, respectively (Fig. 1 A and B). These data were consistent with the reduced genetic diversity within the FC founder population. The decreased mean aggregate off-target scores predominantly resulted from an increased number of low-scoring off-target sites, which are those with two to four mismatches from the reference gRNA sequence (Fig. 1C).
Notably, the ratio of the number of ambiguous genome to reference genome off-target sites suggested that gRNAs with fewer off-target sites in the reference genome displayed higher ratios (i.e., increased number of off-target sites in the ambiguous genome compared with the reference genome); however, this may reflect increased noise in the ratio because new off-target sites contribute more to the ratio for gRNAs with fewer off-target sites in the reference genome (Fig. 1D). As expected, nontargeting gRNAs showed the smallest decrease in aggregate off-target score (Fig. 1 A and B). Of the 2,327 (2,327/2,481, 93.8%) human-genome–targeted gRNAs with only one predicted perfect genomic match (single on-target site with zero mismatches) in the reference genome, 14 gRNAs (0.6%) were predicted to have new perfect off-target matches in the ambiguous genome. Of note, HLA-A_gRNA_0422 had an off-target score of 80.7% in the reference genome, but only 27.5% when considering gnomAD SNPs with the number of off-target sites being increased by 75 including three off-target sites with 0 or one mismatch. HLA-A_gRNA_0422 showed the largest score reduction when considering gnomAD and 1000G SNPs followed by consideration of the FC dataset. The total number of human-genome–targeted gRNAs with a reduction in aggregate off-target score of >30% was 23 (23/2,327, 1.0%), 17 (17/2,327, 0.7%), and 5 (5/2,237, 0.3%), respectively, for the 1000G, gnomAD, and FC datasets. For viral-genome–targeted gRNAs, these reductions were 3 (3/484, 0.6%), 5 (5/484, 1.0%), and 2 (2/484, 0.4%), respectively, for the same datasets. Overall, these results suggest that SNPs can create novel off-target sites and reduce the number of mismatches in existing off-target sites, thus increasing the potency of the associated off-target site. Notably, reduced alteration of off-target potential among the FC dataset suggested that the effect of genetic variation is likely dependent on the extent of the individual genetic diversity. Taken together, these data suggest that SNPs can increase the off-target cleavage potential for gRNAs and further suggest that an increased number of SNPs is likely to increase the off-target cleavage potential.
Variants Alter gRNA On-Target Specificity for Human-Genome–Targeting gRNAs.
The ambiguous genome approach offered an initial assessment of the effect of variants on targeting specificity; however, the ambiguous genome analysis approach is limited because it does not consider haplotypes present in the population, it does not discriminate allele frequencies, and it does not include insertion/deletions (indels). To address these limitations, we selected the subset of gRNAs with an aggregate off-target score of ≥80% in the reference genome and tested these against every possible haplotype in the 1000G dataset including both SNP and indel variants. This analysis was restricted to gRNAs with ≥80% aggregate off-target scores because gRNAs with low aggregate off-target scores are unlikely to be considered for therapeutic applications. Therefore, this subset included 481 human-genome–targeting, 150 viral-genome–targeting, and 128 nontargeting gRNAs.
Using this approach, we first investigated on-target sites for each human-genome–targeting gRNA, which identified 263 gRNAs (263/481, 54.7%) with on-target sites harboring variants from the 1000G dataset (Fig. 2 A–F and Dataset S4). These gRNAs targeted 83 different regions after aggregating SNPs based on proximity into local haplotypes (Fig. 2G offers an example of a single region with multiple SNPs in close proximity). These regions were composed of 310 unique haplotypes from the 1000G dataset with a mean of 3.7 different haplotypes per region. In total, 58.6% (n = 793/1,353) of gRNA–target haplotype pairs were predicted to yield a perfect match (perfect local targeting score) and perfect cutting frequency determination (CFD) score, which is another score for the assessment of gRNA activity and off-target cleavage potential (25). On the other hand, 27.8% (n = 376/1,353) of gRNA–haplotype pairs yielded a local on-target score below 50%, and 20.9% (n = 283/1,353) of gRNA–haplotype pairs resulted in a CFD below 50% (Fig. 2 A and D). These affected sites (i.e., sites with SNPs at their target site resulting in local on-target score <50% or CFD <50%) belonged to 176 (176/263, 66.9%) and 139 (139/263, 52.9%) of human-genome–targeting gRNAs, respectively. In total, 16.3% (n = 43/263) of gRNAs had at least one target haplotype yielding a null local on-target score or null CFD, where null signifies a local on-target score or CFD of zero. The frequency of null on-target haplotypes in the 1000G ranged from 0.02% (n = 1/5,008) to 39.4% (n = 1,973/5,008) with a mean of 2.0% (median, 0.06%). Similarly, the frequency of imperfect haplotypes [mismatch(es) at on-target site] was highly biased toward singletons (Fig. 2 B and E). Nonetheless, 15.6% (n = 41/263) of gRNAs with SNPs at their on-target sites were predicted to have a local on-target score or local CFD <100% in 50 samples/individuals or more. For instance, TPST2_gRNA_2070 (chr22:26,937,299–26,937,349; hg19) had a local on-target score of 38.7% (CFD, 13.6%) in 4,439 (88.6%) haploid genomes (Fig. 2 C and F). Six gRNAs targeted the HLA-A region (chr6:29,910,958–29,911,176; hg19; Fig. 2G). This region included nine SNPs implicated in 20 unique haplotypes (excluding the reference). Of note, a haplotype (haplotype #10, Fig. 2G) present in 39.4% (n = 1,973/5,008) of samples abrogated the target site for all six gRNAs. In 71 of 92 (77%) null haplotypes, the null score was due to an altered PAM site.
Fig. 2.

Variants can reduce gRNA targeting efficiency. (A) Distribution of on-target scores for human-genome–targeting gRNAs for each possible target haplotype. (B) Distribution of samples/individuals carrying haplotypes predicted to be targeted with a local on-target score of <100%. (C) Distribution of local on-target scores for the gRNA TPST2_gRNA_2070. (D) Distribution of on-target CFDs for human-genome–targeting gRNAs for each possible target haplotype. (E) Distribution of samples/individuals carrying haplotypes predicted to be targeted with a CFD of <100%. (F) Distribution of CFDs for the gRNA TPST2_gRNA_2070. (G) Example of haplotypes at the HLA-A locus. Inset plots with a restricted y-axis range are shown for A, B, D, and E for easier visualization of data.
We repeated this type of haplotype-based analysis using human-genome–targeted gRNAs in samples/individuals from the FC dataset and identified 155 human-genome–targeting gRNAs targeting 243 different haplotypes in 26 unique genomic regions (Fig. S2 and Dataset S5). Here, we found 430 (430/2,844, 15.1%) and 317 (317/2,844, 11.1%) gRNA–haplotype pairs that reduced the local on-target score or local CFD below 50%, respectively. In total, 9.7% (n = 15/243) of gRNAs had null local on-target scores in at least one haplotype. These haplotypes had a mean frequency of 1.5% (n = 59.4/4,004; median = 2.5%). In total, 2.9% (n = 7/243) of gRNAs targeted at least one null haplotype seen in more than 40 samples (∼1% frequency) clustered in five unique regions. The most common null haplotype was present in 22.6% of haploid genomes (n = 906/4,004; chr22:26936744–26936919; hg19; TPST2_gRNA_2144). In total, 69% (n = 87/126) of null haplotypes were due to an altered PAM sequence. Overall, these results suggest that genetic variants in the on-target site can dramatically affect gRNA targeting specificity and efficiency, particularly if the variants are located within PAM sequences.
Characteristics of Off-Target Sites.
We then assessed the global characteristics of off-target sites for all human-genome–targeting, viral-genome–targeting, and nontargeting gRNAs with an aggregate off-target score ≥80% in the reference genome. The 1000G and FC dataset variants altered 21,981 and 10,348 off-target sites, respectively, targeted by gRNAs from Dataset S2 in the reference genome (Datasets S6 and S7). 1000G- and FC-derived variants created an additional 23,316 and 8,773 unique off-target sites, respectively. In both cases, ∼8% (n = 1,767/23,316 and 699/8,773) were solely due to novel PAM sites created by variants. On the other hand, variants overlapping PAMs destroyed matches at 13.8% (n = 3,039/21,981) and 10.5% (n = 1,084/10,348) of reference sites. When variants altered the underlying off-target site sequence, the median change in local off-target score due to variants was ±0.03% in the 1000G and FC datasets and the mean difference in local CFD was ±2.0% in both datasets. These small changes in local off-target and CFD scores were predominantly due to the large number of off-target sites with four mismatches, which accounted for >92% of off-target sites in both the 1000G and FC datasets. In the 1000G dataset, 73.2% (n = 556/759) of the gRNAs with an aggregate off-target score ≥80% in the reference genome had new off-target sites with less than four mismatches, 10.1% (n = 77/759) with less than three mismatches, and 0.5% (n = 4/759) with less than two mismatches. The new mismatches were at 1,531, 84, and 5 unique sites, respectively (Dataset S8). In the FC dataset, 49.3% (n = 374/759) of the gRNAs with an aggregate off-target score ≥80% in the reference genome had new off-target sites with less than four mismatches, 4.9% (n = 37/759) with less than three mismatches, and 0.7% (n = 5/759) with less than two mismatches. The new mismatches were at 599, 45, and 5 unique sites, respectively (Dataset S8).
Indels can theoretically have a higher impact on reference sequence than SNPs, potentially resulting in the creation of novel/altered off-target sites. To investigate the effect of indels, we examined regions with off-target sites consisting of indel-only haplotypes in the 1000G dataset to consider indels independently from SNPs. This analysis identified 184 sites with 69.6% (128/184) of them with novel off-target sites. This was a modest enrichment compared with haplotypes consisting of SNPs-only (n = 21,169/34,636; 61.1%; Fisher exact test, P = 0.019), although this was not significant in the FC dataset (indels-only: 357/522, 68.4%; SNPs-only: 7,777/11,897, 65.4%; P = 0.16). However, 7.6% (n = 27/357) of indel-mediated off-target site alteration in the FC dataset had less than four mismatches, whereas that ratio was 0.8% for SNPs (n = 66/7,777; Fisher exact test, P = 1.1 × 10−15), suggesting that indels are more likely to create more potent novel off-target sites. Similarly, sites in the reference genome with less than four mismatches were more likely to be completely destroyed by indels than by SNPs (1000G odds ratio = 10.5, P = 9.5 × 10−4; FC odds ratio = 20.6, P = 4.1 × 10−12).
Notably, of the 45,297 (21,981 + 23,316) different off-target sites, 5,633 (12.4%) were covered by structural variants annotated in the 1000G dataset, suggesting that these could also modify the probability of off-target effects. For instance, an HLA-B gene haplotype (chr6:31,238,852–31,238,965; hg19) was a strong off-target site for HLA-A_gRNA_0422 in 29.1% (n = 1,459/5,008) of haplotypes (local off-target score, 60.5–68.3%; local CFD, 100). In total, 6.9% (n = 344/5,008) haploid genomes had a deletion of this region, completely removing this target site in the 1000G dataset. On the other hand, three samples (3/5,008, 0.06%) had a duplication covering this site (chr6:31,131,451–31,272,307; hg19), thus increasing the number of off-target sites.
Assessing Off-Target Effects Using Personal Genomes.
We calculated aggregate off-target scores for all gRNAs (human-genome–targeting, viral-genome–targeting, and nontargeting) in the 1000G and FC datasets (Fig. 3). In the vast majority of cases (n = 3,753,205/3,796,064 gRNA–haploid genome pairs, 98.9%), the individual gRNA aggregate off-target score was ≥80%. Nonetheless, this accounted for 42,859 haploid genome–gRNA pairs with a score <80%, implicating 62 gRNAs and virtually all samples (Fig. 3A). The FC dataset showed similar statistics, with 99.3% (n = 2,823,851/2,842,840) of haploid genome–gRNA having a score ≥80% (Fig. 3B). Again, all samples had an aggregate off-target score of <80% for any one of 23 gRNAs. Consistently, the mean reduction in aggregate off-target score was −0.03% and −0.01% for the 1000G and FC datasets, respectively, when examining 758 and 710 gRNAs with at least one overlapping variant at an off-target site, respectively (Fig. 3 C and D). Only seven gRNAs for the 1000G dataset and one gRNA for the FC dataset had a reduction in aggregate off-target score of more than 5% in at least one individual. Four gRNAs showed a very strong reduction in score (>15% reduction in aggregate off-target score) in at least one haplotype in the 1000G dataset (Table 2). ALB_gRNA_0837 had an aggregate off-target score between 82.4% and 84.2% in 83% of haplotypes (4,153/5,008). However, the aggregate off-target score was reduced below 46% in the remaining 17% samples (855/5,008). This was primarily due to a single off-target site on chromosome 11 (chr11:100,402,414–100,402,433; hg19) (Table 2). In the reference genome, this region on chromosome 11 contains two mismatches (local off-target score, 2.4%), one of which is rescued by rs11560892 (C > G) matching to position 18 of the gRNA sequence. The remaining mismatch is not predicted to alter targeting (local off-target score, 100%), thus creating a potent off-target site.
Fig. 3.

Variants can increase the risk of off-target effects. (A) Distribution of aggregate off-target scores for each 1000G haplotype. (B) Distribution of aggregate off-target scores for each FC haplotype. (C) Difference in aggregate off-target scores for each 1000G haploid genome and the reference genome. The x axis corresponds to different gRNAs, and each dot represents the difference in score of each haploid genome in the 1000G dataset. The figure includes 758 gRNAs with at least one match with overlapping variants. (D) Difference in aggregate off-target score for each FC haploid genome and the reference genome. The x axis corresponds to different gRNAs and each dot represents the difference in score of each haploid genome in the FC dataset. The figure includes 710 gRNAs with at least one match with overlapping variants.
Table 2.
Representative example of off-target sites created by variants present in the 1000 Genomes database
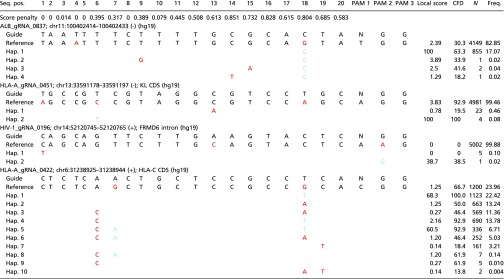 |
Variants included in the chr11:100402414–100402433 (hg19) haplotype are rs566289682, rs555981507, rs181027193, and rs11560892. Variants included in the chr13:33591178–33591197 (hg19) haplotype are rs200611452 and rs116289670. Variants present in the chr14:52120745–52120765 (hg19) haplotype are rs532153306 and rs552139758. Sites displaying mismatches with the gRNA sequence are shown in red, whereas sites where variants rescue the gRNA sequence are highlighted in blue. CFD, cutting frequency determination; Freq., frequency; Hap., haplotype; PAM, protospacer adjacent motif; Seq. pos., sequence position.
Similarly, HLA-A_gRNA_0451 had an aggregate off-target score of >88.6% in the majority of haplotypes (n = 5,004/5,008, 99.8%) (Table 2). However, four haplotypes showed an aggregate off-target score of 48%, which was mainly due to a rescued off-target site on chromosome 13 (chr13:33,591,178–33,591,197; hg19; Table 2). Notably, this region falls in the coding region (exon 1) of the KL gene, a gene associated with hyperphosphatemic familial tumoral calcinosis (28); these haplotypes belonged to four unique haplotypes. Although all of the four samples/individuals had one copy of the on-target site that had a perfect match, they also carried a copy whereby the on-target site was predicted to have very low (n = 3/4; local on-target score, 0.4%) or reduced (n = 1/4; local on-target score, 55.5%) activity, making these individuals at potentially increased risk of both treatment failure and adverse effects due to off-target cleavage (Fig. 2G).
One individual was a carrier of the G allele of rs552139758 (A > G), which created a novel PAM sequence on chromosome 14 (chr14:52,120,745–52,120,765; hg19) and a novel off-target site for HIV-1_gRNA_0196 (Table 2). This PAM-creation site on chromosome 14 falls within intron 1 of the FRDM6 gene and results in reduction of the aggregate off-target score for HIV-1_gRNA_0196 from >80.8 to 61.9%. In addition, HLA-A_gRNA_0422 had an off-target site in the coding sequence of HLA-C (Table 2) whereby two haplotypes showed local off-target scores >60% in 1,459 haploid genomes. This is also an example where the CFD score (66.7) was a better predictor of off-target potential than the local off-target score (1.25%), in that the former was less affected by the presence of SNPs (Table 2).
Additionally, instances were identified with an improved mean aggregate off-target score due to variants within the 1000G dataset (mean Δ aggregate off-target score of >0; Fig. 3C). The distribution of gRNAs with a Δ aggregate off-target score of >0 was shifted toward 0, suggesting that variants were more likely to decrease the aggregate off-target score within each haploid genome (Fig. S3). Notably, F9_gRNA_1349 had a 4.8% increase in aggregate off-target score albeit only in one haploid genome. In total, 727 (14.5%, 727/5,008) haploid genomes displayed a Δ aggregate off-target score of >0 for HSV-1_gRNA_0057, with 38 (0.8%, 38/5,008) displaying a Δ aggregate off-target score of >4%. HSV-1_gRNA_0079 had a Δ aggregate off-target score of >0 in all haploid genomes, although the gain was limited due to the gRNA already having an aggregate off-target score of 98.9 in the reference genome (0.89 mean Δ aggregate off-target score). Overall, the magnitude of off-target score increase was likely blunted in this analysis since we selected for gRNAs with high aggregate off-target scores (≥80%). Finally, we investigated whether some populations were more at risk for off-target effects than other populations in the 1000G dataset. We calculated the difference in scores between each sample and the reference for each gRNA. In total, 721 of 758 (95.1%) of gRNAs showed significant differences in scores between populations after adjusting for multiple comparisons (Kruskal–Wallis, P < 6.6 × 10−5). Overall, African-ancestry populations showed the largest reduction in scores compared with the reference population (Δ aggregate off-target score, −0.0346; SD, 1.0745), while Europeans populations displayed the smallest changes (Δ aggregate off-target score, −0.0216; SD, 0.9882) (Dataset S9). This is consistent with increased genetic diversity observed in populations of African ancestry. Taken together, variants may predispose a subset of individuals to adverse events for CRISPR-mediated therapeutic genome editing.
Discussion
CRISPR technology holds enormous potential for clinical translation as therapy for a wide array of genetic disorders. Historically, gene therapy clinical trials have demonstrated that a small subset of patients may experience adverse events (29). Our data suggest that variants may contribute to both treatment failure of CRISPR-based therapies as well as predispose individuals to adverse outcomes due to personalized off-target effects; however, the effect of variants on on- and off-target specificity is not unique to CRISPR genome editing, but also extends to other genome-editing platforms including zinc finger nucleases and TAL effector nucleases. Notably, we identified variant-induced off-target sites in coding sequence. This type of situation potentially offers an adverse clinical outcome if such sites are located within genes with important roles for cellular function (e.g., tumor suppressor genes). It may be advisable for safety considerations to exclude gRNAs with predicted off-target sites within or near important genes such as tumor suppressors even if they have three or four mismatches. As such, these data may suggest the utility of WGS for patients before therapeutic genome-editing treatments. WGS data would allow for in silico on- and off-target analysis, which may identify patients predisposed to treatment failure and/or adverse outcomes before therapy initiation. Notably, given the creation/alteration of off-target sites in noncoding sequence, WGS would likely be required for this analysis as opposed to whole-exome sequencing. Minimally, our results suggest that on-target sites should be investigated by conventional Sanger sequencing to assure maximal gRNA efficiency. Alternatively, in vitro unbiased genome-wide off-target detection methods can be employed (13–19). It is also possible to overcome adverse events by using enhanced-specificity/high-fidelity versions of SpCas9 (30–32), by using other methodologies to enhance targeting specificity (25, 33–36), and/or by furthering the understanding of cleavage kinetics to help minimize nuclease exposure to reduce off-target potential (37). However, variants that create potent off-target sites (e.g., novel zero or one mismatch sites) are likely to be problematic even in the setting of improved specificity techniques. Furthermore, enhanced-specificity/high-fidelity nucleases are only available for SpCas9 at present.
It is important to note that our study only considered NGG-restricted gRNAs compatible with SpCas9 (or enhanced-specificity versions such as SpCas9-HF1/eSpCas9/HypaCas9) (30–32); however, the effect of variants altering on- and off-targeting specificity is unlikely to be restricted to SpCas9 and will likely affect all CRISPR nucleases considered for therapeutic genome-editing applications (38). In addition, given the wide array of genetic or viral diseases that could be targeted by genome-editing approaches, we have evaluated only a small subset of the possible therapeutic loci; however, we have identified gRNAs with variant-induced reduction in predicted gRNA efficacy at on-target sites and variant-induced creation of potent off-target sites. This finding appears unlikely to be specific to the chosen loci and more likely to be a generalized phenomenon. However, it is important to note that our data suggest that these findings are rare, which is consistent with previous work (38).
The FC dataset was included to evaluate for population-specific effects due to novel variants present and/or variants present at differential allele frequencies within a specific population. Deleterious population-specific effects were not overtly observed in this dataset; however, stratification of the 1000G dataset by population demonstrated population-specific effects for on- and off-target specificity. The minimal population-specific effects observed in the FC dataset are consistent with its increased genetic homogeneity as a founder population and thus fewer differences with the reference genome. Notably, founder populations are associated with fewer variants; however, the variants are often more frequent. The increased frequency of particular variants may become problematic for therapeutic genome editing if certain high-frequency variants alter on-target sites and/or create high-potency off-target sites. Taken together, differential variant frequencies within populations are likely to contribute to population-specific effects for CRISPR-based therapeutic targeting.
As the understanding of Cas9 binding continues to unfold and aid in determination of off-target loci (39) and factors affecting accessibility of these sequences [e.g., nucleosomes (40)], it may be possible to refine in silico off-target analysis beyond sequence-only to further predict if off-target sites and/or variant-induced off-target sites are likely to result in off-target cleavages. In particular, future analysis would benefit from differentiating between the requirements for Cas9 binding vs. Cas9 cleavage (32, 41); incorporation of this type of information would likely increase the reliability of identifying off-target sites with a high probability of cleavage.
To minimize the possibility of variants affecting gRNAs in development for clinical translation, it may be useful to consider variants at the gDNA design stage. For example, publicly available variant databases [e.g., dbSNP, dbVAR, ExAC (26), 1000G (20)] may be examined during gRNA design to create variant-aware gRNAs (21). In silico analysis, such as presented in this manuscript, can also be used to aid gRNA selection for clinical translation. In addition, gRNAs derived from the reference genome or variant-aware gRNAs can be tested in diverse cell lines or primary cells to evaluate for toxicity. One might also evaluate a therapy-optimized CRISPR gRNA using patient-derived induced pluripotent stem cells differentiated to the relevant lineage, which could represent a viable paradigm for empiric evaluation of variant-induced effects on CRISPR targeting; however, this approach could be compromised by somatic mosaicism, which has been detected in many individuals across numerous tissue types (42). The somatic mutation rate has been estimated to be ∼10−9/nucleotide/cell division (43, 44). Further estimates suggested 3,500–8,900 cell divisions for cells such as lymphocytes, lymphoblastoid cell lines, or colonic mucosae in ∼65-y-old individuals (44). Therefore, it is conceivable that somatic variants may limit the ability to evaluate on- and off-target sites using any of the suggested methods, particularly for individuals with advanced age. Of note, estimates of the germline mutation rate have varied widely with estimates above and below the somatic mutation rate (43, 45, 46). Interestingly, somatic mosaicism could also be exploited for CRISPR-based therapy, such as for cancers with genomic amplifications, through induction of apoptosis due to numerous double-strand breaks (47, 48).
Taken together, our analysis suggests the necessity for preclinical studies to consider variants at the gDNA design stage and/or to validate more than one gRNA for clinical translation to increase the likelihood of providing safe, effective, and personalized therapeutic options for all patients regardless of genotype. In summary, our data suggest that human genetic variation alters on- and off-target specificity for CRISPR-based therapeutic genome editing. Therefore, it will be prudent to account for patient-specific genomes in on- and off-target analyses as CRISPR-based therapies approach the clinic.
Materials and Methods
gRNA Design.
gRNAs were designed using publicly available tools (11) and/or identified in previously published studies (Table 1 and Dataset S2). gRNAs for both NHEJ and HDR applications were designed to include all gRNAs within the relevant exon(s) for coding region targets and ±100 bp for noncoding targets. Human genome (hg19) was used to obtain gene-based sequences. Viral sequences utilized were as follows: EBV (49): KC207813.1 human herpesvirus 4 strain Akata, complete genome;
CMV (49): KF297339.1 human herpesvirus 5 strain TB40-E clone Lisa, complete genome;
HSV1 (49): JN555585.1 human herpesvirus 1 strain 17, complete genome;
HPV E6E7: LC193821.1 human papillomavirus type 16 DNA, complete genome, isolate: FT001;
HIV1 (50): AF105229.1 cloning vector pHR′-CMVLacZ, complete sequence;
HBV (51): AF305422.1 synthetic construct hepatitis B virus 1.28-mer overlength sequence; EU570069.1 hepatitis B virus isolate 1-B24, complete genome; FJ899793.1 hepatitis B virus isolate C122-2, complete genome; V01460.1 hepatitis B virus (strain ayw) genome;
JCV (52): NC_001699.1 JC polyomavirus, complete genome.
CRISPOR (11) was used to obtain gRNA efficiency scores from Fusi et al. (53), Chari et al. (54), Xu et al. (55), Doench et al. (25, 56), Wang et al. (57), Moreno-Mateos et al. (58), Housden et al. (59), Prox. GC (60), -GG (61), and Out-of-Frame (62).
Calculation of Off-Target and CFD Scores.
Off-target scores were calculated as previously described (11, 23, 24). Briefly, the number and position of mismatches between gRNA–DNA were calculated with scores ranging from 0 (nontargeting) to 1 (perfect match), which was termed the “local off-target score.” Based on this analysis, sequences with a score >0 were considered potential off-targets. For sequences with more than four mismatches, a score of 0 was assigned. An aggregate off-target score from all possible local off-targets was calculated according to Sanjana et al. (23):
In this equation, n signifies the number of potential off-target “hits” and Shit(hi) is the targeting score of the possible off-target sequence hi. Therefore, a “local” off-target score was calculated for each genomic match (from 0 to four mismatches) for a given gRNA. The summation of all local off-target scores for a given gRNA resulted in a genome-wide off-target score, termed “aggregate off-target score.” For this off-target scoring method (score range, 0–100), higher scores indicate lower off-target cleavage potential and lower scores indicate higher off-target cleavage potential.
CFD scores were calculated as previously described (21, 25). Briefly, percent activity values are provided in Doench et al. (25) for all possible gRNA–DNA mismatches. These percent activity values can be multiplied together in the setting of multiple gRNA–DNA mismatches (25). When a local off-target score or local CFD was calculated at the on-target site, it is referred to as an “on-target score.”
Nontargeting gRNA Design.
In total, 128 gRNAs were used as negative controls. The 128 gRNAs were previously designed to lack perfect matches within the genome and have an aggregate off-target score of >90% based on the calculation described in Calculation of Off-Target and CFD Scores (21).
PAM Creation and Destruction Analysis.
To determine the total number of PAMs in the genome for SpCas9, all NGG motifs were identified on the sense and antisense strands using the matchPattern function from the Biostrings package for all 22 autosomes. Destroyed PAMs were defined as GG sites that were overlapped by a SNP (this analysis was performed on both strands). In these cases, the reference allele was G so alternative alleles destroyed the GG motif (i.e., altered reference genome NGG motif to NHG or NGH sequence). Created PAMs were defined by identification of all SNPs with an alternative allele of a G and that were preceded or followed by a G nucleotide, thus creating a GG motif (i.e., altered NHG or NGH sequence to become an NGG motif; this analysis was performed on both strands). To determine the number of PAMs per haploid genome, we generated all possible haploid genomes from the 1000 Genomes dataset by inserting the alternative allele of SNPs at each site carried by the samples. We then counted the total number of NGG motifs for each haploid genome.
Genomic Coordinates.
All genomic coordinates displayed are hg19. Coordinates for viral genomes are not displayed.
WGS Data.
In total, 7,444 WGSs were obtained for analysis. These data were obtained from the 1000G database (n = 2,504) (20), a subset from the genome aggregation database [the gnomAD dataset, an updated and expanded version of the ExAC dataset (26); n = 2,939], and Low-Kam et al. (27) (n = 2,002). All three datasets were sequenced at low coverage (<15×).
Ambiguous Genome Analysis.
Variants were downloaded from the 1000G phase 3 dataset (n = 2,504) (20). Data from two other whole-genome sequencing datasets were also accessed: an FC (n = 2,002) dataset from the Montreal Heart Institute biobank (27) and a subset of the GnomAD dataset (n = 2,938). An ambiguous genome was built using a custom R (version 3.2.0) script based on the R package Biostrings (version 2.38.4). Human genome sequences were obtained using the BSgenome (version 1.38.0) package BSgenome.Hsapiens.UCSC.hg19.masked (version 1.3.99), applying the default masks (assembly gaps and intracontig ambiguities). Each nucleotide was replaced at the SNP positions by an IUPAC ambiguity code to account for all possible SNP alleles. For example, an A→C SNP would be replaced by the ambiguity code “M,” so that both alleles can map to the SNP location without penalty.
For each gRNA, all possible matches were identified in the reference and ambiguous genomes using the Biostring matchPDict function allowing up to four mismatches. Only matches upstream of an NGG motif and that had less than five ambiguities were considered. The restriction of less than five ambiguities was imposed so that ambiguities did not overinflate the number of matches. For each match, the targeting score was calculated as described in Sanjana et al. (23) using mismatch penalties from Hsu et al. (24), as well as the CFD score (25) (see Calculation of Off-Target and CFD Scores for more detail). For each gRNA, we reported the number of matches for each mismatch category, the aggregated score, and mean, median, SD, and 10th, 25th, 75th, and 90th percentiles of the CFD score.
On-Target Haplotype Analysis.
To measure on-target effects in personal genomes, each SNP and indel in the 1000G and FC datasets overlapping the predicted on-target sites (including the PAM) was considered. The sequences 22 bp on either side of each variant were identified and overlapping target sites were merged to create local haplotypes. Genomic sequences were created based on existing haplotypes in the datasets and tested whether they were targeted by gRNAs using the Biostring matchPDict function (up to four mismatches). In total, 481 human-genome–targeting, 150 viral-genome–targeting, and 128 nontargeting gRNAs with aggregate off-target scores ≥80% in the reference genome were investigated. Only matches upstream of an NGG PAM were considered valid matches. The number of mismatches, off-target scores, and CFD scores were calculated as above for each match.
Δ Aggregate Off-Target Score.
The “Δ aggregate off-target score” was calculated as the difference between the reference aggregate off-target score and each sample’s aggregate off-target score.
Off-Target Haplotype Analysis.
To measure off-target effects in personal genomes, each SNP and indel in the 1000G and FC datasets was considered. Sequences 22 bp on either side of each variant were identified and overlapping sequences were merged to create local haplotypes. Genomic sequences were created based on existing haplotypes in the datasets and tested whether they were targeted by gRNAs using the Biostring matchPDict function (up to four mismatches). Only matches upstream of an NGG PAM were considered valid matches. The number of mismatches, off-target scores, and CFD scores were calculated as above for each match. Each sample (individual) was then separated into haploid genomes and the aggregate off-target score was calculated given the individual’s haplotypes:
where sg,i,j is the off-target site score of the jth of n off-target sites of gRNA g in haplotypes of the haploid genome i. represents the sum of all local off-target scores in nonvariable regions of the genome (not overlapped by variants) for gRNA g. The aggregate off-target score of guide g (Zg,i) in the haploid genome i is given by the following:
Off-Target Analysis Computational Tool.
The computational tool (“CRISPR Off-Target Tool,” version 2.0.1) used to perform the off-target analysis as well as its source code are available for download at www.mhi-humangenetics.org/en/resources.
Supplementary Material
Acknowledgments
We thank Daniel E. Bauer for helpful discussions. We thank all participants and staff of the André and France Desmarais Montreal Heart Institute (MHI) Hospital Cohort. J.-C.T. holds the Canada Research Chair in Personalized Medicine and is funded by Genome Canada and Genome Quebec. G.L. is funded by Genome Canada and Genome Quebec, the Canada Research Chair Program, and the MHI Foundation. S.H.O. is supported by National Heart, Lung, and Blood Institute Award P01HL032262 and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Award P30DK049216 (Center of Excellence in Molecular Hematology). M.C.C. is supported by NIDDK Award F30DK103359.
Footnotes
The authors declare no conflict of interest.
Data deposition: A list of individual aggregate off-target scores for samples from the 1000 Genomes Project and their local scores and a list of individual aggregate off-target scores for samples from the French Canadian dataset and their local scores are available for download at the following link: www.mhi-humangenetics.org/en/resources.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1714640114/-/DCSupplemental.
References
- 1.Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: Prospects and challenges. Nat Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol Ther. 2016;24:430–446. doi: 10.1038/mt.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White MK, Khalili K. CRISPR/Cas9 and cancer targets: Future possibilities and present challenges. Oncotarget. 2016;7:12305–12317. doi: 10.18632/oncotarget.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- 5.Prakash V, Moore M, Yáñez-Muñoz RJ. Current progress in therapeutic gene editing for monogenic diseases. Mol Ther. 2016;24:465–474. doi: 10.1038/mt.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll D. Genome editing: Progress and challenges for medical applications. Genome Med. 2016;8:120. doi: 10.1186/s13073-016-0378-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doudna JA. Genomic engineering and the future of medicine. JAMA. 2015;313:791–792. doi: 10.1001/jama.2015.287. [DOI] [PubMed] [Google Scholar]
- 8.Cornu TI, Mussolino C, Cathomen T. Refining strategies to translate genome editing to the clinic. Nat Med. 2017;23:415–423. doi: 10.1038/nm.4313. [DOI] [PubMed] [Google Scholar]
- 9.Cyranoski D. CRISPR gene-editing tested in a person for the first time. Nature. 2016;539:479. doi: 10.1038/nature.2016.20988. [DOI] [PubMed] [Google Scholar]
- 10.Sheridan C. CRISPR therapeutics push into human testing. Nat Biotechnol. 2017;35:3–5. doi: 10.1038/nbt0117-3. [DOI] [PubMed] [Google Scholar]
- 11.Haeussler M, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai SQ, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D, et al. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12:237–243, 1. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 15.Frock RL, et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan WX, et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun. 2017;8:15058. doi: 10.1038/ncomms15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai SQ, et al. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J, et al. Digenome-seq web tool for profiling CRISPR specificity. Nat Methods. 2017;14:548–549. doi: 10.1038/nmeth.4262. [DOI] [PubMed] [Google Scholar]
- 19.Cameron P, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods. 2017;14:600–606. doi: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 20.Auton A, et al. 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canver MC, et al. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet. 2017;49:625–634. doi: 10.1038/ng.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paquet D, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 23.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doench JG, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lek M, et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Low-Kam C, et al. Whole-genome sequencing in French Canadians from Quebec. Hum Genet. 2016;135:1213–1221. doi: 10.1007/s00439-016-1702-6. [DOI] [PubMed] [Google Scholar]
- 28.Hamosh A, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002;30:52–55. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosley KS, et al. CRISPR germline engineering—the community speaks. Nat Biotechnol. 2015;33:478–486. doi: 10.1038/nbt.3227. [DOI] [PubMed] [Google Scholar]
- 30.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinstiver BP, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JS, et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tycko J, Myer VE, Hsu PD. Methods for optimizing CRISPR-Cas9 genome editing specificity. Mol Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose JC, et al. Rapidly inducible Cas9 and DSB-ddPCR to probe editing kinetics. Nat Methods. 2017;14:891–896. doi: 10.1038/nmeth.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott DA, Zhang F. Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat Med. 2017;23:1095–1101. doi: 10.1038/nm.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyle EA, et al. High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proc Natl Acad Sci USA. 2017;114:5461–5466. doi: 10.1073/pnas.1700557114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horlbeck MA, et al. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife. 2016;5:e12677. doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sternberg SH, LaFrance B, Kaplan M, Doudna JA. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature. 2015;527:110–113. doi: 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Huallachain M, Karczewski KJ, Weissman SM, Urban AE, Snyder MP. Extensive genetic variation in somatic human tissues. Proc Natl Acad Sci USA. 2012;109:18018–18023. doi: 10.1073/pnas.1213736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milholland B, et al. Differences between germline and somatic mutation rates in humans and mice. Nat Commun. 2017;8:15183. doi: 10.1038/ncomms15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besenbacher S, et al. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat Commun. 2015;6:5969. doi: 10.1038/ncomms6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conrad DF, et al. 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aguirre AJ, et al. Genomic copy number dictates a gene-independent cell response to CRISPR-Cas9 targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munoz DM, et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 2016;6:900–913. doi: 10.1158/2159-8290.CD-16-0178. [DOI] [PubMed] [Google Scholar]
- 49.van Diemen FR, et al. CRISPR/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog. 2016;12:e1005701. doi: 10.1371/journal.ppat.1005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu W, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci USA. 2014;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Hao R, Chen S, Guo D, Chen Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J Gen Virol. 2015;96:2252–2261. doi: 10.1099/vir.0.000159. [DOI] [PubMed] [Google Scholar]
- 52.Wollebo HS, et al. CRISPR/Cas9 system as an agent for eliminating polyomavirus JC infection. PLoS One. 2015;10:e0136046. doi: 10.1371/journal.pone.0136046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fusi N, Smith I, Doench J, Listgarten J. 2015. In silico predictive modeling of CRISPR/Cas9 guide efficiency. bioRxiv:10.1101/021568.
- 54.Chari R, Mali P, Moosburner M, Church GM. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12:823–826. doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu H, et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25:1147–1157. doi: 10.1101/gr.191452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doench JG, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moreno-Mateos MA, et al. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Housden BE, et al. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal. 2015;8:rs9. doi: 10.1126/scisignal.aab3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren X, et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep. 2014;9:1151–1162. doi: 10.1016/j.celrep.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farboud B, Meyer BJ. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 2015;199:959–971. doi: 10.1534/genetics.115.175166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bae S, Kweon J, Kim HS, Kim J-S. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods. 2014;11:705–706. doi: 10.1038/nmeth.3015. [DOI] [PubMed] [Google Scholar]
- 63.Park RJ, et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat Genet. 2017;49:193–203. doi: 10.1038/ng.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandal PK, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruan GX, et al. CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital amaurosis 10. Mol Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilen CB, et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011;7:e1002020. doi: 10.1371/journal.ppat.1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Torikai H, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–1349. doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ding Q, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beane JD, et al. Clinical scale zinc finger nuclease mediated gene editing of PD-1 in tumor infiltrating lymphocytes for the treatment of metastatic melanoma. Mol Ther. 2015;23:1380–1390. doi: 10.1038/mt.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schumann K, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci USA. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fadel HJ, et al. TALEN knockout of the PSIP1 gene in human cells: Analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J Virol. 2014;88:9704–9717. doi: 10.1128/JVI.01397-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Torikai H, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–5705, and erratum (2015) 126:2527. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Provasi E, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eyquem J, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Joglekar AV, et al. Integrase-defective lentiviral vectors as a delivery platform for targeted modification of adenosine deaminase locus. Mol Ther. 2013;21:1705–1717. doi: 10.1038/mt.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharma R, et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Firth AL, et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015;12:1385–1390. doi: 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sebastiano V, et al. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci Transl Med. 2014;6:264ra163, and erratum (2014) 6:267er8. doi: 10.1126/scitranslmed.3009540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Ravin SS, et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med. 2017;9:eaah3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 81.Ousterout DG, et al. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maggio I, Liu J, Janssen JM, Chen X, Gonçalves MAFV. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci Rep. 2016;6:37051. doi: 10.1038/srep37051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Osborn MJ, et al. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum Gene Ther. 2015;26:114–126. doi: 10.1089/hum.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yin H, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeWitt MA, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dever DP, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Genovese P, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yusa K, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin S-R, et al. The CRISPR/Cas9 system facilitates clearance of the pntrahepatic HBV templates in vivo. Mol Ther Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong C, et al. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110–117. doi: 10.1016/j.antiviral.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 92.Ramanan V, et al. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep. 2015;5:10833. doi: 10.1038/srep10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kennedy EM, et al. Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J Virol. 2014;88:11965–11972. doi: 10.1128/JVI.01879-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.