

Abstract
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase involved in the regulation of protein synthesis and degradation, longevity and cytoskeletal formation. The mTOR pathway represents a key growth and survival pathway involved in several diseases such as cancer, obesity, cardiovascular disease and neurodegenerative diseases. Numerous studies linked the alterations of mTOR pathway to age-dependent cognitive decline, pathogenesis of Alzheimer disease (AD) and AD-like dementia in Down syndrome (DS). DS is the most frequent chromosomal abnormality that causes intellectual disability. The neuropathology of AD in DS is complex and involves impaired mitochondrial function, defects in neurogenesis, increased oxidative stress, altered proteostasis and autophagy networks as a result of triplication of chromosome 21(chr 21). The chr21 gene products are considered a principal neuropathogenic moiety in DS. Several genes involved respectively in the formation of senile plaques and neurofibrillary tangles (NFT), two main pathological hallmarks of AD, are mapped on chr21. Further, in subjects with DS the activation of mTOR signaling contributes to Aβ generation and the formation of NFT. This review discusses recent research highlighting the complex role of mTOR associated with the presence of two hallmarks of AD pathology, senile plaques (composed mostly of fibrillar Aβ peptides), and NFT (composed mostly of hyperphosphorylated tau protein). Oxidative stress, associated with chr21-related Aβ and mitochondrial alterations, may significantly contribute to this linkage of mTOR to AD-like neuropathology in DS.
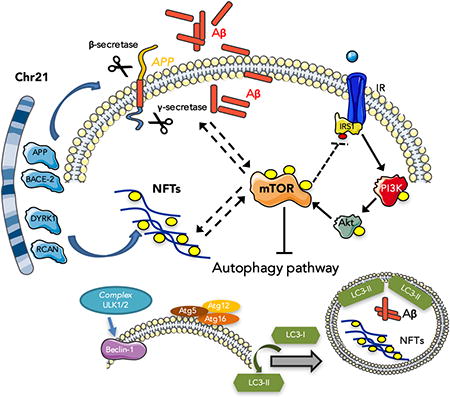
Graphical abstract
1. Introduction
The mammalian target of rapamycin (mTOR) is a 289 kDa serine/threonine protein kinase, highly conserved in all eukaryotes, and plays a key role in maintaining energy balance in many tissues of the body. The upstream signal components, which can interact and modulate the mTOR activity, are growth factors, 5′ AMP-activated protein kinase (AMPK), phosphoinositide 3-kinase (PI3K)/Akt, and glycogen synthase kinase 3 (GSK-3) [1]. Phosphorylation at Thr-2446, Ser-2448 and Ser-2481 within the kinase catalytic domain (KIN) domain of mTOR are correlated with an increase in mTOR activity. Adjacent to the KIN domain is the FKBP12 rapamycin-binding domain (FRB), the site of inhibitory interaction between rapamycin, a main mTOR inhibitor, and mTOR. The binding of rapamycin to FKBP12 disturbs mTOR–protein complex formation, thus impairing mTOR activity [2]. The mTOR interaction with several components led to insights that mTOR dysregulation is involved in many diseases, such as aging [3], cancer [4], obesity [5], cardiovascular disease [6] and neurodegenerative diseases [7].
The catalytic mTOR subunit interacts with other proteins to form two distinct complexes: the mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The former, mTORC1, is formed by mTOR, the regulatory-associated protein of mTOR (Raptor), proline-rich Akt substrate 40 kDa (PRAS40) and the mammalian homolog of protein Lethal with Sec Thirteen (mLST8). The main functions of mTORC1 are to regulate protein synthesis, autophagy, mitochondrial function, lipogenesis, ketogenesis, and glucose homeostasis. mTORC2 is composed of mTOR, rapamycin-insensitive companion of mTOR (Rictor), mLST8 and mammalian stress-activated map kinase-interacting protein 1 (mSin1). The mTORC2 is able to control cellular shape by modulating actin function, but also phosphorylates Akt, which activates mTORC1 [8, 9]. Regulation of mTORC2 activity remains unclear; probably its activity is induced by PI3K/Akt signaling. On the other hand, mTORC1 is activated by growth factors (IGF1), insulin, LKB1/AMPK, PI3K/Akt, GSK-3β, IKKβ, MAPK, and p53 [10]. The best characterized downstream targets for mTORC1 are p70S6K and 4EBP1 that regulate translation, and Ulk1 and Atg13 that suppress autophagy. Several studies showed that mTORC1 also is able to regulate the insulin response by p70S6K activation, in particular in the brain. Phosphorylation of p70S6K by mTOR leads to phosphorylation of insulin receptor substrate-1 (IRS-1) and its degradation via the proteasome [11]. Clearly, the insulin/IGF-1/mTOR signaling pathway is a negative cycle loop that regulates a variety of pathophysiological features. Thus, the regulation of mTORC1 and mTORC2 activities are necessary for maintenance of brain homeostasis and in particular to restore the autophagic network [1].
The mTOR pathway and its upstream and downstream targets have a prominent role in the central nervous system (CNS); thus, mTOR regulates synaptic plasticity, memory retention and neuronal recovery [12, 13]. Recent studies from our laboratory have focused on the role of mTOR in the development of AD [14, 15]. mTOR signaling is closely associated with the presence of two hallmarks of AD pathology: senile plaques, composed mainly of fibrillar β-amyloid (Aβ) peptide, and neurofibrillary tangles (NFT), composed mostly of hyperphosphorylated tau protein. In fact, in subjects with AD and AD-like dementia, as in Down syndrome (DS), the activation of mTOR signaling contributes to Aβ generation and the formation of NFT. In light of these findings, our group started to investigate the status of mTOR pathway in the frontal cortex from DS autopsy cases [16]. DS, a genetic disorder characterized by the triplication of chromosome 21 (HAS21), is the most frequent chromosome abnormality that causes neurologic deficiencies. One of the main characteristics in individuals with DS is the early age of onset of AD pathology, with high incidence of clinical symptoms reminiscent of AD in the late 40- to early 50-years of age [17].
Our studies showed an early hyperactivation of mTOR signaling, which is correlated with tau hyperphosphorylation, in the brains of young DS subjects in comparison to healthy individuals. The sustained activation of mTOR is maintained in adult DS, in which AD pathology is marked. Within this context, DS provides a model for studying important aspects of AD. The neuropathology of AD in DS is complex and involves impaired mitochondrial function, defects in neurogenesis, increased oxidative stress, altered proteostasis and autophagy networks as a result of triplication of HSA21 genes [18]. Mapping of the gene that encodes the precursor protein (APP) from which the Aβ present in the senile plaques in both AD and DS to chromosome 21 was strong evidence that this chr 21 gene product was a principal neuropathogenic contributor to AD as well as DS. In addition, aberrant activation of the mTOR pathway, acting in parallel with other kinases overexpressed in DS (DYRK1A and RCAN1), contributes to the hyperphosphorylation of tau and formation of NFT.
This review discusses the role of mTOR in DS, and in particular, how mTOR influences Aβ deposition and tau aggregation, setting the stage for future studies on potential therapeutic strategies of intervention to slow the neurodegenerative process in DS and in AD.
2. mTOR, App Processing and Aβ in Down Syndrome
As discussed above, the presence of three copies of genes on chr 21 results in a strongly enhanced risk of developing AD. Chr21 carries 233 coding genes, 299 long non-coding genes [19] and 29 microRNAs; it is likely that one or more of these genes contribute to development of AD. However, the phenotype resulting from a dosage-sensitive gene depends on the number of copies of the gene in the genome [20]. Further, not all genes are dosage sensitive, and regulatory mechanisms act to prevent a gene from being overexpressed, also in response to environmental changes.
Among the triplicated genes, APP is considered the most toxic candidate that contributes to the pathogenesis of AD in the DS population. In addition, several other genes have been shown to contribute to the onset of AD such as SOD1, BACE2, S100B, RCAN1 and DYRK1A. However, the third copy of APP in DS is not directly correlated with Aβ levels, and Aβ accumulation becomes significant only in the second/third decade of life, with some exceptions in a few childhood post-mortem observations. Mouse and human studies suggest that APP is dosage sensitive as a function of aging [21-23].
Several studies demonstrated that the processing of APP is disturbed in AD. Genetic and cell biology studies have shed light on the role of α-, β- and γ-secretases that produce either non-amyloidogenic or amyloidogenic peptides [24]. However, β-secretase is considered to be the rate-limiting enzyme in Aβ generation. The amyloid cascade hypothesis claims that the production of Aβ play a key toxic role in the brain that ultimately result in AD pathology. In parallel, it has been suggested that the amyloidogenic processing of APP also disturbs the metabolism of tau and favors its aggregation [25].
Several other gene candidates encoded on Chr21 have been suggested to interfere with APP processing. Among these, the transcription factor ETS2 was demonstrated to transactivate the APP promoter, leading to its overexpression [26]. The small ubiquitin-related modifier 3 (SUMO3) and DYRK1A can modulate APP post-translational modifications, thus altering Aβ production [27, 28]. SUMO3 overexpression significantly increased Aβ40 and Aβ42 secretion, which was accompanied by an increase in full-length APP and its C-terminal fragments [27]. Another candidate is miR-155 that modulates γ-secretase activity through its effect on the expression of sorting nexin 27 [29]. Interestingly, the β-secretase homologue of BACE1, BACE2, is encoded on Chr21. However, BACE2 does not exert β-secretase activity, and in fact cleaves APP on the carboxy-terminal side of the β-secretase cut site, preventing generation of Aβ. While BACE2 mRNA is increased in DS, posttranscriptional regulatory mechanisms either prevent an increase in translation or the rate of degradation. These findings suggest that BACE2 is probably not responsible for AD pathology in the DS brain and may have a protective function [30].
Increasing evidence highlights that the activation of mTOR is able to push Aβ generation and deposition [31]. At least two main mechanisms can be discussed: i) mTOR, an inhibitor of autophagy, decreases the functionality of autophagy/lysosome system and therefore Aβ clearance; ii) mTOR may interact with several key signaling pathways including PI3K/Akt, GSK-3β, AMPK and insulin/IGF-1 and thus regulates Aβ generation/clearance [32].
2.1 mTOR, autophagy and Aβ production/clearance
Autophagy, a self-consumption mechanism, is a specialized degradative system for the removal of aggregated proteins and dysfunctional organelles [33]. This process is carried out within the lysosome, where digestion of removed material occurs by the activity of acidic hydrolases. Autophagy, one of the best-characterized downstream pathways regulated by mTOR, has been largely implicated in AD neurodegeneration [34]. Several studies suggested that reduced autophagy in AD brain and animal models, leading to the accumulation of protein aggregates, is likely caused by the hyperactivation of the PI3K/Akt/mTOR axis. The control of autophagocytosis is impaired in AD as well as in many other neurodegenerative diseases in which protein aggregation is one of the pathological hallmarks [35-37]. It is well established that aging, a major risk factor of neurodegeneration, is also associated with defects in autophagocytosis [38, 39]. On the other hand, recent studies have revealed that induction of autophagy can reduce Aβ accumulation and reduce cognitive decline in transgenic AD mice [40, 41].
As it occurs in AD brain, DS neurons are the earliest site of Aβ accumulation [42, 43], indicating that secretory and endosomal systems are central to Aβ generation. However, triplication of APP is sufficient to cause endosomal enlargement and alteration of intracellular trafficking defects [44, 45], independent from Aβ.
A pathological link between APP and endosomal function was demonstrated in the Ts65Dn mouse model of DS, which carries an extra copy of ∼185 genes located on a region of mouse chromosome 16 (MMU16) orthologous to the “DS critical region” of chr21 that is required for development of DS [46]. These mice display key gross morphological features resembling human DS, and adult mice develop AD-related endosomal pathology, such as intraneuronal Aβ accumulation and degeneration of basal forebrain cholinergic neurons [44]. Nixon and co-workers showed that elevated APP expression in fibroblasts from DS individuals is necessary and sufficient to cause morphological and functional abnormalities of early endosomes, also observed in AD patients [47]. Interestingly, these endocytic abnormalities can be reversed by reducing the expression of APP or BACE-1.
These results indicate that the processing of APP to Aβ peptide occurs in acidic compartments, either in the late endosomes or autolysosomes. In addition, multiple APP metabolites, not exclusively Aβ, are likely to contribute to the neuropathology of AD in DS. For example, secreted APPβ, formed by β-secretase cleavage of APP is reportedly neurotoxic [48].
Studies from our group demonstrated a decreased LC3 II/I ratio, an index of autophagosome formation, early in DS brain (before development of AD neuropathology) that persists in DS with AD in comparison to their respective age-matched controls. We also found that specific components of the protein degradative pathways, both proteasome and autophagy, are oxidatively modified in DS cases [49]. Specifically, our results showed that oxidative damage targets GRP78, UCH-L1, V0-ATPase, cathepsin D and GFAP and that these irreversibly modifications, that likely disturb protein functions, are associated with reduced proteasome activity. We demonstrated that oxidative damage targets specifically different components of the intracellular quality control system such as GRP78, UCH-L1, V0-ATPase, cathepsin D and GFAP that couples with decreased activity of the proteasome and autophagosome formation in DS brain [49]. Furthermore, a link between protein oxidative damage and aberrant mTOR/autophagy axis induction was shown in Ts65Dn mice by our group [50].
2.2 mTOR and intracellular signaling regulates Aβ levels
As noted above, mTOR integrates multiple signaling pathways in response to growth factors, energy metabolism, nutrients and different stresses [51]. Among these pathways, the PI3K/Akt axis is the major upstream activator of mTOR in response to growth factors such as insulin or insulin-like growth factors [51] (figure 1). Interestingly, a number of in vitro studies have shown that Aβ is able to activate the PI3K/Akt pathway. For example, treatment of PC12 cells with Aβ (25-35) leads to increased Akt activity, in a PI3K-dependent manner [52], likely due to an increase of mTOR signaling. Similarly, treatment with Aβ(25-35) in N2A cells leads to a transient and significant increase in p70S6K, a downstream target of mTOR [53]. Data further supporting the link between Aβ accumulation and the up-regulation of mTOR signaling indicate that in primary neurons the levels of pAKT (Ser473), pmTOR (Ser2448), and its downstream target p4EBP1 were significantly increased following exposure to Aβ oligomers but not to Aβ monomers [54]. Conversely, Hugon and colleagues reported that 20 μM of Aβ42 decreased the steady-state levels of phosphorylated mTOR and p70S6K [55]. This apparently contradictory data may be reconciled based on the evidence that the high concentration of Aβ used in their study is extremely cytotoxic, thus affecting multiple pathways not just mTOR.
Figure 1.

A schematic overview of the interactions of mTOR signaling with Aβ deposition in Down syndrome brain.
To further elucidate the effect of Aβ on mTOR signaling, a stable transfected cell line (Chinese hamster ovary cells) with a cDNA encoding APP751 containing the Val-717-Phe familial AD mutation, 7PA2 cells, was used [32, 56]. These cells produce high levels of Aβ oligomers, which have been shown to cause alteration of LTP and also learning and memory deficits [57, 58]. These transfected cell line display increased mTOR activity and signaling compared to nontransfected CHO cells as determined by measuring mTOR enzymatic activity and by measuring the steady-state levels of p70S6K [31]. In parallel, treatment with a gamma-secretase inhibitor, that blocks Aβ production, was able to prevent mTOR hyperactivation [31]. Taken together these in vitro data suggest a dose-dependent effect of Aβ on mTOR signaling, where low levels of Aβ cause an increase in mTOR signaling, while Aβ overload decreases mTOR signaling.
Recent data from our group analyzed the functionality of the PI3K/AkT/mTOR axis in the frontal cortex from DS autopsy cases without AD neuropathology (typically under the age of 40 years) and DS with AD neuropathology [16]. Our results showed a hyperactivation of the PI3K/Akt/mTOR axis in the brains of subjects with DS with or without AD pathology in comparison to healthy individuals. The hyperactivation was demonstrated by increased levels of the phosphorylated forms of PI3K (p85 subunit at Tyr-508), Akt (at Ser-473), and mTOR (at Ser-2448) and as well as alteration of both a mTOR downstream signaling target, p70S6K, which is increasingly phosphorylated at Thr-389, and autophagy [16].
These data were further supported by findings showing the prenatal upregulation of pS6 and p70S6K, that persisted throughout postnatal development, while the upregulation of p4EBP1 and mTOR in DS hippocampi also were detected postnatally [59]. This study also confirmed the upregulation of mTORC1 components and downstream signals in DS-AD patients, showing a positive correlation with total tau and p-tau [59].
AMP-activated protein kinase (AMPK), a key energy enzyme, regulates cellular metabolism to maintain energy homeostasis in response to decreased intracellular ATP levels. The structure and function of AMPK is mainly regulated by ADP levels [60]; AMPK is activated when cellular ADP levels increase coping with changes in cellular energy status. AMPK activity decreases in AD brain, indicating decreased mitochondrial biogenesis and function [61].
Of note AMPK and mTOR act as a common regulator of autophagy through direct phosphorylation of Ulk since the Atg1/Ulk complex plays an essential role in the initiation of autophagy [62]. Specifically, AMPK directly modulates Ulk1 through phosphorylation of Ser-317 and Ser-777, which results in activation of autophagy. Conversely, the activation of mTOR inhibits phosphorylation of Ulk1 Ser-757 and cuts off the interaction between Ulk1 and AMPK, thereby reducing autophagy.
Moreover, both AMPK and mTOR participate in the regulation of Aβ levels: 1) through induction of autophagy, the activation of AMPK limits the generation of Aβ [61] while the activation of mTOR, i.e., inhibition of autophagy, likely facilitates Aβ accumulation [40]. It has been proposed that Aβ levels in the AD brain are determined by the overall functional status of autophagy and that AMPK activation inhibits mTOR signaling activity to facilitate autophagy and to promote lysosomal degradation of Aβ [12, 63]. Further, AMPK is a physiological tau kinase and can increase the phosphorylation of tau at Ser-262. AMPK can also directly phosphorylate tau at Thr-231 and Ser-396/404.
In adult neuronal progenitor cells (NPC) isolated from the hippocampus of Ts65Dn mice, the ratio of pAMPK/AMPK is reduced [64]. These results support the notion of the failure of AMPK signaling to cope against reduced energy metabolism, i.e., reduced intracellular ATP levels. Why and how this crucial metabolic sensor is not activated in DS and the analysis of its upstream targets deserves further investigation. Valenti et al. suggested a positive correlation between activation of AMPK and increased mitochondrial ATP synthesis, and this beneficial effect may be secondary to treatment of NPC with resveratrol and EGCG on mitochondrial function [64]. According to this hypothesis, the specific activation or inhibition of AMPK by AICAR or compound C, respectively, was able to activate or inhibit ATP synthesis by oxidative phosphorylation.
However, it is still unclear whether AMPK could serve a potential therapeutic target for AD or DS with AD. Hence, further studies will be needed to clarify the role of AMPK in AD and DS with AD.
3. TAU Hyperphosphorylation and NFT Pathology in Down Syndrome
Tau pathological modifications have been detected in the outer molecular layer of hippocampus in DS individuals of 30-40 years of age, followed later by NFT in the CA1 region of the hippocampus and neuron loss in the entorhinal cortex [65]. Aberrant phosphorylation of tau has also been reported in trisomic Ts65Dn and Ts1Cje, mouse models of DS [66, 67], which however do not develop tangles.
Genetic variants in the microtubule associated protein tau (MAPT) gene are a risk factor for the age of onset of AD in people with DS [68], promoting the formation of NFT [69]. The phosphorylation of tau plays a physiological role in microtubule dynamics, the aberrant hyperphosphorylation of tau prejudices its ability to bind microtubules, leading to self- assembly and formation of tau aggregates. At least 45 phosphorylation sites have been identified in the tau protein, most being serine and threonine residues [70].
A number of studies have suggested that trisomy of DYRK1A, an Hsa21-encoded protein expressed in fetal and adult brain, may contribute to the aberrant phosphorylation of tau [71]. People trisomic for Hsa21 express elevated levels of DYRK1A and exhibit increased DYRK1A kinase activity in their brains [66, 72-74]. DYRK1A is found in the cytosol of cells and colocalization between NFT and DYRK1A has been reported [75]. In vitro studies indicate that tau is phosphorylated by DYRK1A at Thr212 [66, 76] and the phosphorylation at this site is also able to prepare tau for further phosphorylation at additional sites by other kinases, such as glycogen synthase kinase3-β (GSK-3β) [76]. Moreover, Dyrk1A transgenic mouse brains displayed an increase in tau phosphorylation also at Ser-202 and Ser-404 residues [77]. Tau hyperphosphorylation occurring in the brains from both Ts65Dn mice and DS patients correlates with DYRK1A hyperactivation [78]. In addition, tau phosphorylation on Thr-212 occurs in patients with AD, and may contribute to disease pathogenesis [79]. Overexpression of DYRK1A may also contribute indirectly to the early onset of NFT through phosphorylation of alternative splicing factors, leading to an imbalance between 3R-tau and 4R-tau. The correct balance of 3R/4R is emerging as critical for normal tau function, and its disruption may lead to the typical NFT pathology seen in DS, AD, and various tauopathies [80].
The regulator of calcineurin 1 (RCAN1), formerly known as Down syndrome critical region 1 (DSCR1, Adapt78), also is encoded on HSA21, and its overexpression might result in increased p-tau levels. RCAN1 acts as an endogenous regulator of calcineurin (Caln), a multifunctional calcium-activated serine/threonine protein phosphatase [81]. RCAN1 overexpression results in the inhibition of signaling pathways that are controlled by the nuclear factor of activated T cells (NFAT) transcription factor [82, 83]. In addition, Tau is dephosphorylated by Caln [84]. In AD the chronic overexpression of the RCAN1, inhibits Caln and is associated with pathology progression [85-88]. In DS fetal brains, Caln activity is lower than in normal subjects [89], together with hyperphosphorylated NFAT [90]. Dyrk1A interacts with and phosphorylates RCAN1 at Ser-112 and Thr-192 residues. Phosphorylation of RCAN1 at Thr-192 enhances the ability of RCAN1 to inhibit the phosphatase activity of calcineurin (Caln), leading to reduced NFAT transcriptional activity and enhanced Tau phosphorylation [81]. Therefore, the overexpression of Dyrk1A and RCAN1 in DS may lead to an increase in Tau hyperphosphorylation, contributing to the early onset of the pathological features of AD in DS patients (figure 2).
Figure 2.

A schematic overview of the interactions of mTOR signaling with tau pathology in Down syndrome brain.
The trisomy of Hsa21 encoded genes might also have an effect on the expression and activity of kinases or phosphatases encoded by chromosomes other than Hsa21 that controls tau phosphorylation. For example, increased abundance of cyclin-dependent kinase 5 (CDK5) has been reported in the brains of young Ts65Dn mice that model aspects of DS [91]. As well, decreased activity of phosphatases such as protein phosphatase 2A (PP2A) have been associated with the development of AD in people who have DS [92].
APP was one of the first postulated links between DS and AD pathology. APP and tau reportedly converge in cellular mechanisms that could greatly compromise neuronal function such as mitochondrial function [93]. Further, Aβ (1-42) oligomers contribute to hyperphosphorylated tau [94, 95], and both Aβ (1-42) oligomers and hyperphosphorylated tau synergistically contribute to cellular and cognitive impairment in the AD and DS [70]. Moreover, it has been shown that the overexpression of APP C-terminal in the CA1 region of hippocampus is sufficient to induce the translocation of SET (PP2A inhibitor 1 and 2) to the cytoplasm and thus to the increased phosphorylation of tau at Ser-202/Thr-205 as seen in the progression of the disease in AD and also in DS [96].
3.1 Role of mTOR signaling in tau pathology
Compelling evidence support the critical role of mTOR in the tau-related pathological progress in DS and AD, implying that the activity status of mTOR significantly influences, if not determines, the abnormal hyperphosphorylation of tau, the onset of paired helical filaments, and the formation of NFT [32]. Multiple studies, mainly in AD, fully elucidated that the activation of mTOR signaling promotes tau pathology, while inhibiting mTOR signaling decreases the progress of tau pathology [15, 97]. Recent studies in DS patients and DS mouse models confirmed that the alteration of mTOR signaling is an early degenerating event in the brain that contributes to the early development of AD hallmarks (Aβ and tau) and AD-like cognitive decline [13, 16, 49, 59]. The mechanisms by which hyperactive mTOR signaling can lead to tau hyperphosphorylation are various some of which include: i) direct regulation of tau phosphorylation; ii) decreased autophagy turnover, which is a known degradation pathway for tau; and iii) the enhancement of the translation of tau mRNA.
Results from our laboratory demonstrated the hyperactivation of the PI3K/Akt/mTOR axis in the frontal cortex of subjects with DS with or without AD pathology in comparison to healthy individuals [16]. PI3-K/Akt/mTOR signaling is considered a primary candidate to transmit pathophysiological responses from Aβ to tau. Our results suggested that the aberrant activation of the mTOR axis led to the alteration of its downstream signalling target p70S6K, that might directly cause tau hyperphosphorylation. In addition, other kinases overexpressed in DS (DYRK1A and RCAN1) acting in parallel with mTOR contributes, to the tau hyperphosphorylation and to NFTs deposition observed in DS and AD as noted above [16]. Unexpectedly, our results showed that GSK-3β, one of the major candidate kinases involved in tau hyperphosphorylation, was inhibited in DS brain [16]. Similar data were obtained from old TC1 mice, a model of trisomy21, supporting the inactivation of GSK-3β in DS and consistent with the notion of a less preeminent role of this kinase in tau hyperphosphorylation in DS [1, 78]. However, intriguingly, it was previously demonstrated a mutual relationship between RCAN1, calcineurin and GSK-3β. When the equilibrium between these three players is altered, such as in AD and DS, aberrant tau phosphorylation may occur [98]. Furthermore, a crosstalk between RCAN1 and mTOR may exists. Previous studies on neuronal cells showed that RCAN1 overexpression lead to mitochondrial degeneration, observed in DS, and lower cellular levels of ATP, which in turn can inhibit mTOR signaling and activate autophagy [99]. Within this context, the interaction between RCAN1, mTOR and GSK-3β may have a key role in the progression of tau pathology in DS (figure 2).
Recent studies, by Iyer et al. [59], analyzed the alteration of the main components of the mTOR pathway in human hippocampi of DS subjects during prenatal, early postnatal development and in presence of AD pathology. The researchers confirmed the upregulation of the pathway in DS and associated such results with p-tau aggregation in DS during the transition to AD. Indeed, DSAD patients showed a positive correlation between the upregulation of mTORC1 components and downstream signals and total and phosphorylated tau levels [59].
The two major protein quality control mechanisms in CNS cells involved in the clearence of aberrantly phosphorylated and aggregated tau are the ubiquitin-proteasome system (UPS) and autophagy [100]. The hyperactivation of mTOR is deeply involved in the inhibition of autophagy observed in DS individuals and in DS and AD mouse models [15]. The inhibition of autophagy by several different compounds demonstrated delayed tau degradation and enhanced formation of tau aggregates [101, 102]. Moreover, autophagy inducers (e.g., rapamycin) facilitate the degradation of insoluble forms of tau and also protect against its toxicity in tau transgenic and AD mouse models [97, 103]. In agreement, studies from our laboratory demonstrated a tight association between reduced autophagosome formation and increased p-tau levels in DS individuals prior to and after AD development [16, 49]. These results support the crucial role of mTOR-related autophagy impairment in tau hyperphosphorylation and NFTs formation in DS and in AD brains [14]. DS and AD individuals also demonstrated reduced proteasome activity, reduced de-ubiquitinylating activity, and increased protein poly-ubiquitinylation that also supported the contribution of altered ATP/ubiquitin-dependent 26S proteasome in p-tau aggregation and subsequent NFT formation [49, 104]. However, tau degradation can also be mediated by the 20S proteasome in and ATP/ubiquitin independent manner [105], which, interestingly, might be regulated by the mTOR-inhibitor rapamycin [106, 107].
Increased levels of oxidative stress (OS) appears early in DS individuals, from the fetal stage, as a result of triplication of genes involved in redox homeostasis, such as SOD1 or BACH1 [108, 109]. It has been well established that altered redox homeostasis in neurons contribute to the occurrence of the AD hallmarks NFT in DS and healthy population [18, 110, 111]. Moreover, increased OS led to alteration of insulin signaling associated with its mTOR-related branch [112, 113]. Studies from our laboratory showed the oxidative modification of lysosomal V0-ATPase and cathepsin D, which can result in reduced autophagy in DS individuals [49, 114]. Within this context, increased OS might represent a further bridge coupling mTOR/autophagy impairment with tau hyperphosphorylation an NFT deposition [112].
Studies on Ts65Dn mice showed that increased translocation of SET to the cytoplasm led to an increase of tau phosphorylation at Ser-202/Thr-205 as observed in DS and AD patients [96, 115]. Cytoplasmic SET is able to inhibit PP2A, leading to increased activation of mTOR/AMPK signaling pathways, inhibition of autophagy, and subsequently hyperphosphorylated tau.
A further mTOR interacting molecules is SIRT1, which regulates mTOR phosphorylation through TSC1/2, regulates S6K1 acetylation and alters mTORC1-dependent S6K1 phosphorylation. Reduced levels of SIRT1 were observed in DS/AD compared with younger DS cases [101]. Moreover, decreased SIRT1 levels were observed in 12 months-old Ts65Dn mice compared with 6 months-old Tg mice, and between 12 months-old Ts65Dn mice and their age-matched controls [116]. Furthermore, SIRT1 is a substrate of DYRK1A which, can promote tau accumulation by controlling its de-acetylation process [117, 118]. Therefore, SIRT1 alteration might represent an additional link between aberrant mTOR signaling and NFT deposition in DS brain.
Finally, tau phosphorylation is also tightly related to tau O- GlcNAcylation levels, a dynamic protein posttranslational modification, by which O-linked β-N-acetylglucosamine (OGlcNAc) is transferred enzymatically to serine or threonine residues of proteins. O-GlcNAcylation modifies nucleocytoplasmic proteins and is more like protein phosphorylation [119, 120]. OGlcNAcylation and phosphorylation often occur at identical or proximal sites of a protein and thus are reciprocal to each other. The crosstalk between O-GlcNAcylation and phosphorylation has been implicated to be essential for the control of vital cellular processes and for understanding the mechanisms of certain diseases [119, 121]. Intriguingly, O-GlcNAcylation levels serve as a sensor of intracellular glucose metabolism, because the UDP-GlcNAc donor for O-GlcNAcylation is formed from glucose metabolism via the hexosamine biosynthetic pathway [122]. Recent studies demonstrated that O-GlcNAc can antagonize phosphorylation of tau in tissue culture cells, rat brain slices and human AD patients[121-123].
Therefore, it is tempting to hypothesize that the early alteration of insulin signaling, through mTOR-related IRS-1 inhibition, might lead to glucose hypometabolism, reduced tau OGlcNAcylation and NFT formation in DS brain.
4. Conclusion
The roles of mTOR in the brain of individuals with DS and how the impairment of mTOR signaling can influence Aβ deposition and the formation of NFT in this syndrome of trisomy of chr 21 is a key to better understanding of the transition from DS to DS with AD, which occurs typically in the 40-50 years of age range, and to AD itself. mTOR activation affects the regulation of Aβ generation/clearance and tau phosphorylation by two main mechanisms: a) inhibition of autophagy; and b) the interaction with several key signaling pathways, including PI3K/Akt, GSK-3β, AMPK and insulin/IGF (Figure 1). We opine that the PI3K/Akt/mTOR axis is a primary candidate to transmit pathophysiological responses from Aβ to tau. In brains of DS subjects the components of mTOR signaling are associated with the pathogenesis and progression of AD [14,15,18]. Moreover, the extra copy of chr 21-encoded genes also appears to have an effect on APP processing and tau phosphorylation. These findings have profound implications for further development of clinical interventions in DS and AD by inhibiting mTOR activation for the treatment and prevention of AD and AD-like dementia in DS. Several reports showed the beneficial effects of mTOR inhibitors in the brain of AD mouse models. Since oxidative stress is extensively present in brain of persons with DS and DS with AD, further studies into the role of oxidative modification to components of the mTOR pathway are warranted. Indeed, though much has been learned, continued basic and clinical research is necessary to further clarify the roles of the mTOR activation/inhibition in DS.
List of Abbreviation
- mTOR
mammalian target of rapamycin
- AD
Alzheimer Disease
- DS
Down Syndrome
- NTF
Neurofibrillary tangles
- Aβ
β-amyloid
- Chr21
Chromosome 21
- AMPK
5′ AMP-activated protein kinase
- GSK-3β
Glycogen synthase kinase 3β
- KIN
Kinase catalytic domain
- FRB
FKBP12 rapamycin-binding domain
- MTORC
Mammalian target of rapamycin complex
- Raptor
Regulatory-associated protein of mTOR
- PRAS40
Proline-rich Akt substrate 40 kDa
- mLST8
mammalian homolog of protein Lethal with Sec Thirteen
- Rictor
Rapamycin-insensitive companion of mTOR
- mSin1
mammalian stress-activated map kinase-interacting protein 1
- PI3K
phosphoinositide 3-kinase
- IGF1
Insulin growth factor 1
- IKK
IkappaB kinase
- 4EBP1
4E binding protein 1
- IRS
Insulin receptor substrate
- CNS
Central nervous system
- APP
Amyloid precursor protein
- DYRK1A
Dual specificity tyrosine phosphorylation regulated kinase 1A
- RCAN1
Regulator of calcineurin 1
- SOD1
Superoxido dismutase 1
- BACE
Beta secretase
- SUMO3
Small ubiquitin-related modifier 3
- MMU16
Mouse Chromosome 16
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- GRP78
Glucose related protein 78
- UCH-L1
Ubiquitin carboxyl-terminal hydrolase isozyme L1
- GFAP
Glial fibrillary acidic protein
- CHO
Chinese hamster ovary
- Ulk
unc-51 like autophagy activating kinase 1
- EGCG
Epigallocatechin gallate
- AICAR
5-Aminoimidazole-4-carboxamide ribonucleotide
- MAPT
Microtubule-associated protein tau
- DSCR
Down Syndrome critical region
- Caln
Calcineurin
- NFAT
Nuclear factor of activated T cells
- CDK5
Cyclin-dependent kinase 5
- PP2A
Protein phosphatase 2A
- SET
PP2A inhibitor 1 and 2
- UPS
Ubiquitin proteasome system
- BACH1
BTB domain and CNC homolog 1
- OS
Oxidative stress
- SIRT1
Sirtuin 1
- TSC1/2
Tuberous Sclerosis 1/2
- GlcNAc
N-acetylglucosamine
References
- 1.O' Neill C. PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer's disease. Exp Gerontol. 2013;48:647–653. doi: 10.1016/j.exger.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 2.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gharibi B, Farzadi S, Ghuman M, Hughes FJ. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells. 2014;32:2256–2266. doi: 10.1002/stem.1709. [DOI] [PubMed] [Google Scholar]
- 4.Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16:378–386. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Martinez E, Jurado-Lopez R, Cervantes-Escalera P, Cachofeiro V, Miana M. Leptin, a mediator of cardiac damage associated with obesity. Horm Mol Biol Clin Investig. 2014;18:3–14. doi: 10.1515/hmbci-2013-0060. [DOI] [PubMed] [Google Scholar]
- 6.Chong ZZ, Shang YC, Maiese K. Cardiovascular disease and mTOR signaling. Trends Cardiovasc Med. 2011;21:151–155. doi: 10.1016/j.tcm.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, Tampellini D, Klann E, Blitzer RD, Gouras GK. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17:734–742. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Laplante M, Sabatini DM. mTOR Signaling. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a011593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- 12.Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer's disease. J Cell Mol Med. 2008;12:2525–2532. doi: 10.1111/j.1582-4934.2008.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Troca-Marin JA, Alves-Sampaio A, Montesinos ML. Deregulated mTOR-mediated translation in intellectual disability. Prog Neurobiol. 2012;96:268–282. doi: 10.1016/j.pneurobio.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133:739–749. doi: 10.1111/jnc.13037. [DOI] [PubMed] [Google Scholar]
- 15.Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015;84:39–49. doi: 10.1016/j.nbd.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, Head E, Butterfield DA, Di Domenico F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta. 2014;1842:1144–1153. doi: 10.1016/j.bbadis.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fillat C, Dierssen M, de Lagran MM, Altafaj X. Insights from mouse models to understand neurodegeneration in Down syndrome. CNS Neurol Disord Drug Targets. 2010;9:429–438. doi: 10.2174/187152710791556159. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield DA, Di Domenico F, Swomley AM, Head E, Perluigi M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: overlaps in Down's syndrome and Alzheimer's disease brain. Biochem J. 2014;463:177–189. doi: 10.1042/BJ20140772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VL, Fisher EM, Strydom A. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16:564–574. doi: 10.1038/nrn3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheon MS, Dierssen M, Kim SH, Lubec G. Protein expression of BACE1, BACE2 and APP in Down syndrome brains. Amino Acids. 2008;35:339–343. doi: 10.1007/s00726-007-0618-9. [DOI] [PubMed] [Google Scholar]
- 22.Choi JH, Berger JD, Mazzella MJ, Morales-Corraliza J, Cataldo AM, Nixon RA, Ginsberg SD, Levy E, Mathews PM. Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn Down syndrome mouse model. J Neurochem. 2009;110:1818–1827. doi: 10.1111/j.1471-4159.2009.06277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seo H, Isacson O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down's model mice. Exp Neurol. 2005;193:469–480. doi: 10.1016/j.expneurol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 24.De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markesbery WR. Neuropathological criteria for the diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18:S13–19. doi: 10.1016/s0197-4580(97)00064-x. [DOI] [PubMed] [Google Scholar]
- 26.Wolvetang EW, Bradfield OM, Tymms M, Zavarsek S, Hatzistavrou T, Kola I, Hertzog PJ. The chromosome 21 transcription factor ETS2 transactivates the beta-APP promoter: implications for Down syndrome. Biochim Biophys Acta. 2003;1628:105–110. doi: 10.1016/s0167-4781(03)00121-0. [DOI] [PubMed] [Google Scholar]
- 27.Dorval V, Mazzella MJ, Mathews PM, Hay RT, Fraser PE. Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochem J. 2007;404:309–316. doi: 10.1042/BJ20061451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc Natl Acad Sci U S A. 2003;100:259–264. doi: 10.1073/pnas.0235361100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Huang T, Zhao Y, Zheng Q, Thompson RC, Bu G, Zhang YW, Hong W, Xu H. Sorting nexin 27 regulates Abeta production through modulating gamma-secretase activity. Cell Rep. 2014;9:1023–1033. doi: 10.1016/j.celrep.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webb RL, Murphy MP. beta-Secretases, Alzheimer's Disease, and Down Syndrome. Curr Gerontol Geriatr Res. 2012;2012:362839. doi: 10.1155/2012/362839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai Z, Chen G, He W, Xiao M, Yan LJ. Activation of mTOR: a culprit of Alzheimer's disease? Neuropsychiatr Dis Treat. 2015;11:1015–1030. doi: 10.2147/NDT.S75717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 34.Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer's disease. Alzheimers Res Ther. 2013;5:53. doi: 10.1186/alzrt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010;21:691–698. doi: 10.1016/j.semcdb.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 37.Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease--locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15:217–224. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 40.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nilsson P, Saido TC. Dual roles for autophagy: degradation and secretion of Alzheimer's disease Abeta peptide. Bioessays. 2014;36:570–578. doi: 10.1002/bies.201400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal abetaamyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001;125:489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- 43.Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- 44.Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 45.Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- 46.Holtzman DM, Santucci D, Kilbridge J, Chua-Couzens J, Fontana DJ, Daniels SE, Johnson RM, Chen K, Sun Y, Carlson E, Alleva E, Epstein CJ, Mobley WC. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc Natl Acad Sci U S A. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 49.Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, Perluigi M. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer's disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta. 2013;1832:1249–1259. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tramutola A, Lanzillotta C, Arena A, Barone E, Perluigi M, Di Domenico F. Increased Mammalian Target of Rapamycin Signaling Contributes to the Accumulation of Protein Oxidative Damage in a Mouse Model of Down's Syndrome. Neurodegener Dis. 2016;16:62–68. doi: 10.1159/000441419. [DOI] [PubMed] [Google Scholar]
- 51.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 52.Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78:1000–1008. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 53.Zhang F, Beharry ZM, Harris TE, Lilly MB, Smith CD, Mahajan S, Kraft AS. PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol Ther. 2009;8:846–853. doi: 10.4161/cbt.8.9.8210. [DOI] [PubMed] [Google Scholar]
- 54.Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-AktmTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. doi: 10.1186/1750-1326-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, Pradier L, Hugon J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94:215–225. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- 56.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- 57.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 58.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 59.Iyer AM, van Scheppingen J, Milenkovic I, Anink JJ, Adle-Biassette H, Kovacs GG, Aronica E. mTOR Hyperactivation in down syndrome hippocampus appears early during development. J Neuropathol Exp Neurol. 2014;73:671–683. doi: 10.1097/NEN.0000000000000083. [DOI] [PubMed] [Google Scholar]
- 60.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer's disease. Neuromolecular Med. 2012;14:1–14. doi: 10.1007/s12017-012-8173-2. [DOI] [PubMed] [Google Scholar]
- 62.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valenti D, de Bari L, de Rasmo D, Signorile A, Henrion-Caude A, Contestabile A, Vacca RA. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta. 2016;1862:1093–1104. doi: 10.1016/j.bbadis.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 65.Head E, Lott IT, Hof PR, Bouras C, Su JH, Kim R, Haier R, Cotman CW. Parallel compensatory and pathological events associated with tau pathology in middle aged individuals with Down syndrome. J Neuropathol Exp Neurol. 2003;62:917–926. doi: 10.1093/jnen/62.9.917. [DOI] [PubMed] [Google Scholar]
- 66.Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, Ramakrishna N, Gong CX. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22:3224–3233. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shukkur EA, Shimohata A, Akagi T, Yu W, Yamaguchi M, Murayama M, Chui D, Takeuchi T, Amano K, Subramhanya KH, Hashikawa T, Sago H, Epstein CJ, Takashima A, Yamakawa K. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum Mol Genet. 2006;15:2752–2762. [Google Scholar]
- 68.Jones EL, Margallo-Lana M, Prasher VP, Ballard CG. The extended tau haplotype and the age of onset of dementia in Down syndrome. Dement Geriatr Cogn Disord. 2008;26:199–202. doi: 10.1159/000152044. [DOI] [PubMed] [Google Scholar]
- 69.Flament S, Delacourte A, Mann DM. Phosphorylation of Tau proteins: a major event during the process of neurofibrillary degeneration. A comparative study between Alzheimer's disease and Down's syndrome. Brain Res. 1990;516:15–19. doi: 10.1016/0006-8993(90)90891-e. [DOI] [PubMed] [Google Scholar]
- 70.Cardenas AM, Ardiles AO, Barraza N, Baez-Matus X, Caviedes P. Role of tau protein in neuronal damage in Alzheimer's disease and Down syndrome. Arch Med Res. 2012;43:645–654. doi: 10.1016/j.arcmed.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 71.Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dowjat WK, Adayev T, Kuchna I, Nowicki K, Palminiello S, Hwang YW, Wegiel J. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci Lett. 2007;413:77–81. doi: 10.1016/j.neulet.2006.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 74.Wegiel J, Dowjat K, Kaczmarski W, Kuchna I, Nowicki K, Frackowiak J, Mazur Kolecka B, Wegiel J, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Gong CX, Liu F, Adayev T, Chen-Hwang MC, Hwang YW. The role of overexpressed DYRK1A protein in the early onset of neurofibrillary degeneration in Down syndrome. Acta Neuropathol. 2008;116:391–407. doi: 10.1007/s00401-008-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marti E, Altafaj X, Dierssen M, de la Luna S, Fotaki V, Alvarez M, Perez-Riba M, Ferrer I, Estivill X. Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res. 2003;964:250–263. doi: 10.1016/s0006-8993(02)04069-6. [DOI] [PubMed] [Google Scholar]
- 76.Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J. 2001;355:609–615. doi: 10.1042/bj3550609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryoo SR, Jeong HK, Radnaabazar C, Yoo JJ, Cho HJ, Lee HW, Kim IS, Cheon YH, Ahn YS, Chung SH, Song WJ. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;282:34850–34857. doi: 10.1074/jbc.M707358200. [DOI] [PubMed] [Google Scholar]
- 78.Sheppard O, Plattner F, Rubin A, Slender A, Linehan JM, Brandner S, Tybulewicz VL, Fisher EM, Wiseman FK. Altered regulation of tau phosphorylation in a mouse model of down syndrome aging. Neurobiol Aging. 2012;33:828 e831–844. doi: 10.1016/j.neurobiolaging.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, Ihara Y. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–371. doi: 10.1016/0197-4580(95)00027-c. discussion 371-380. [DOI] [PubMed] [Google Scholar]
- 80.Yin X, Jin N, Shi J, Zhang Y, Wu Y, Gong CX, Iqbal K, Liu F. Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci Rep. 2017;7:619. doi: 10.1038/s41598-017-00682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jung MS, Park JH, Ryu YS, Choi SH, Yoon SH, Kwen MY, Oh JY, Song WJ, Chung SH. Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J Biol Chem. 2011;286:40401–40412. doi: 10.1074/jbc.M111.253971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davies KJ, Ermak G, Rothermel BA, Pritchard M, Heitman J, Ahnn J, Henrique-Silva F, Crawford D, Canaider S, Strippoli P, Carinci P, Min KT, Fox DS, Cunningham KW, Bassel-Duby R, Olson EN, Zhang Z, Williams RS, Gerber HP, Perez-Riba M, Seo H, Cao X, Klee CB, Redondo JM, Maltais LJ, Bruford EA, Povey S, Molkentin JD, McKeon FD, Duh EJ, Crabtree GR, Cyert MS, de la Luna S, Estivill X. Renaming the DSCR1/Adapt78 gene family as RCAN: regulators of calcineurin. FASEB J. 2007;21:3023–3028. doi: 10.1096/fj.06-7246com. [DOI] [PubMed] [Google Scholar]
- 83.Abdul HM, Furman JL, Sama MA, Mathis DM, Norris CM. NFATs and Alzheimer's Disease. Mol Cell Pharmacol. 2010;2:7–14. [PMC free article] [PubMed] [Google Scholar]
- 84.Garver TD, Kincaid RL, Conn RA, Billingsley ML. Reduction of calcineurin activity in brain by antisense oligonucleotides leads to persistent phosphorylation of tau protein at Thr181 and Thr231. Mol Pharmacol. 1999;55:632–641. [PubMed] [Google Scholar]
- 85.Ermak G, Morgan TE, Davies KJ. Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J Biol Chem. 2001;276:38787–38794. doi: 10.1074/jbc.M102829200. [DOI] [PubMed] [Google Scholar]
- 86.Ermak G, Harris CD, Davies KJ. The DSCR1 (Adapt78) isoform1 protein calcipressin 1 inhibits calcineurin and protects against acute calcium-mediated stress damage, including transient oxidative stress. FASEB J. 2002;16:814–824. doi: 10.1096/fj.01-0846com. [DOI] [PubMed] [Google Scholar]
- 87.Lin HY, Michtalik HJ, Zhang S, Andersen TT, Van Riper DA, Davies KK, Ermak G, Petti LM, Nachod S, Narayan AV, Bhatt N, Crawford DR. Oxidative and calcium stress regulate DSCR1 (Adapt78/MCIP1) protein. Free Radic Biol Med. 2003;35:528–539. doi: 10.1016/s0891-5849(03)00358-7. [DOI] [PubMed] [Google Scholar]
- 88.Lloret A, Badia MC, Giraldo E, Ermak G, Alonso MD, Pallardo FV, Davies KJ, Vina J. Amyloid-beta toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer's disease. J Alzheimers Dis. 2011;27:701–709. doi: 10.3233/JAD-2011-110890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chang KT, Shi YJ, Min KT. The Drosophila homolog of Down's syndrome critical region 1 gene regulates learning: implications for mental retardation. Proc Natl Acad Sci U S A. 2003;100:15794–15799. doi: 10.1073/pnas.2536696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 91.Pollonini G, Gao V, Rabe A, Palminiello S, Albertini G, Alberini CM. Abnormal expression of synaptic proteins and neurotrophin-3 in the Down syndrome mouse model Ts65Dn. Neuroscience. 2008;156:99–106. doi: 10.1016/j.neuroscience.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liang Z, Liu F, Iqbal K, Grundke-Iqbal I, Wegiel J, Gong CX. Decrease of protein phosphatase 2A and its association with accumulation and hyperphosphorylation of tau in Down syndrome. J Alzheimers Dis. 2008;13:295–302. doi: 10.3233/jad-2008-13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eckert A, Schulz KL, Rhein V, Gotz J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010;41:107–114. doi: 10.1007/s12035-010-8109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Selenica ML, Brownlow M, Jimenez JP, Lee DC, Pena G, Dickey CA, Gordon MN, Morgan D. Amyloid oligomers exacerbate tau pathology in a mouse model of tauopathy. Neurodegener Dis. 2013;11:165–181. doi: 10.1159/000337230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, Chen PP, Hudspeth B, Chen C, Zhao Y, Vinters HV, Frautschy SA, Cole GM. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dorard E, Gorisse-Hussonnois L, Guihenneuc-Jouyaux C, Albac C, Potier MC, Allinquant B. Increases of SET level and translocation are correlated with tau hyperphosphorylation at ser202/thr205 in CA1 of Ts65Dn mice. Neurobiol Aging. 2016;46:43–48. doi: 10.1016/j.neurobiolaging.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 97.Tramutola A, Lanzillotta C, Di Domenico F. Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies. Expert Rev Neurother. 2017;17:33–45. doi: 10.1080/14737175.2017.1244482. [DOI] [PubMed] [Google Scholar]
- 98.Ermak G, Harris CD, Battocchio D, Davies KJ. RCAN1 (DSCR1 or Adapt78) stimulates expression of GSK-3beta. FEBS J. 2006;273:2100–2109. doi: 10.1111/j.1742-4658.2006.05217.x. [DOI] [PubMed] [Google Scholar]
- 99.Ermak G, Sojitra S, Yin F, Cadenas E, Cuervo AM, Davies KJ. Chronic expression of RCAN1-1L protein induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis in neuronal cells. J Biol Chem. 2012;287:14088–14098. doi: 10.1074/jbc.M111.305342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 101.Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, Deture M, Ko LW. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- 102.Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Richardson A, Galvan V, Lin AL, Oddo S. How longevity research can lead to therapies for Alzheimer's disease: The rapamycin story. Exp Gerontol. 2015;68:51–58. doi: 10.1016/j.exger.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tramutola A, Di Domenico F, Barone E, Arena A, Giorgi A, di Francesco L, Schinina ME, Coccia R, Head E, Butterfield DA, Perluigi M. Polyubiquitinylation Profile in Down Syndrome Brain Before and After the Development of Alzheimer Neuropathology. Antioxid Redox Signal. 2017;26:280–298. doi: 10.1089/ars.2016.6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grune T, Botzen D, Engels M, Voss P, Kaiser B, Jung T, Grimm S, Ermak G, Davies KJ. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch Biochem Biophys. 2010;500:181–188. doi: 10.1016/j.abb.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Osmulski PA, Gaczynska M. Rapamycin allosterically inhibits the proteasome. Mol Pharmacol. 2013;84:104–113. doi: 10.1124/mol.112.083873. [DOI] [PubMed] [Google Scholar]
- 107.Zhao J, Garcia GA, Goldberg AL. Control of proteasomal proteolysis by mTOR. Nature. 2016;529:E1–2. doi: 10.1038/nature16472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Di Domenico F, Pupo G, Mancuso C, Barone E, Paolini F, Arena A, Blarzino C, Schmitt FA, Head E, Butterfield DA, Perluigi M. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: insights for transition to Alzheimer's disease. J Alzheimers Dis. 2015;44:1107–1120. doi: 10.3233/JAD-141254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Di Domenico F, Pupo G, Tramutola A, Giorgi A, Schinina ME, Coccia R, Head E, Butterfield DA, Perluigi M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: clues for understanding the development of Alzheimer disease. Free Radic Biol Med. 2014;71:270–280. doi: 10.1016/j.freeradbiomed.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perry G, Cash AD, Smith MA. Alzheimer Disease and Oxidative Stress. J Biomed Biotechnol. 2002;2:120–123. doi: 10.1155/S1110724302203010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Head E, Powell D, Gold BT, Schmitt FA. Alzheimer's Disease in Down Syndrome. Eur J Neurodegener Dis. 2012;1:353–364. [PMC free article] [PubMed] [Google Scholar]
- 112.Di Domenico F, Barone E, Perluigi M, Butterfield DA. The Triangle of Death in Alzheimer's Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid Redox Signal. 2017;26:364–387. doi: 10.1089/ars.2016.6759. [DOI] [PubMed] [Google Scholar]
- 113.Butterfield DA, Di Domenico F, Barone E. Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochim Biophys Acta. 2014;1842:1693–1706. doi: 10.1016/j.bbadis.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Di Domenico F, Tramutola A, Perluigi M. Cathepsin D as a therapeutic target in Alzheimer's disease. Expert Opin Ther Targets. 2016;20:1393–1395. doi: 10.1080/14728222.2016.1252334. [DOI] [PubMed] [Google Scholar]
- 115.Facchinetti P, Dorard E, Contremoulins V, Gaillard MC, Deglon N, Sazdovitch V, Guihenneuc-Jouyaux C, Brouillet E, Duyckaerts C, Allinquant B. SET translocation is associated with increase in caspase cleaved amyloid precursor protein in CA1 of Alzheimer and Down syndrome patients. Neurobiol Aging. 2014;35:958–968. doi: 10.1016/j.neurobiolaging.2013.08.039. [DOI] [PubMed] [Google Scholar]
- 116.Tramutola A, Pupo G, Di Domenico F, Barone E, Arena A, Lanzillotta C, Broekaart D, Blarzino C, Head E, Butterfield DA, Perluigi M. Activation of p53 in Down Syndrome and in the Ts65Dn Mouse Brain is Associated with a Pro-Apoptotic Phenotype. J Alzheimers Dis. 2016;52:359–371. doi: 10.3233/JAD-151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yuzwa SA, Vocadlo DJ. O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer's disease and beyond. Chem Soc Rev. 2014;43:6839–6858. doi: 10.1039/c4cs00038b. [DOI] [PubMed] [Google Scholar]
- 120.Di Domenico F, Owen JB, Sultana R, Sowell RA, Perluigi M, Cini C, Cai J, Pierce WM, Butterfield DA. The wheat germ agglutinin-fractionated proteome of subjects with Alzheimer's disease and mild cognitive impairment hippocampus and inferior parietal lobule: Implications for disease pathogenesis and progression. J Neurosci Res. 2010;88:3566–3577. doi: 10.1002/jnr.22528. [DOI] [PubMed] [Google Scholar]
- 121.Gong CX, Liu F, Iqbal K. O-GlcNAcylation: A regulator of tau pathology and neurodegeneration. Alzheimers Dement. 2016;12:1078–1089. doi: 10.1016/j.jalz.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 122.Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem. 2014;289:34472–34481. doi: 10.1074/jbc.R114.601351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]