

Abstract
Individuals with Down syndrome (DS) have an increased risk of early-onset Alzheimer’s Disease (AD), largely owing to a triplication of the APP gene, located on chromosome 21. In DS and AD, defects in endocytosis and lysosomal function appear at the earliest stages of disease development and progress to widespread failure of intraneuronal waste clearance, neuritic dystrophy and neuronal cell death. The same genetic factors that cause or increase AD risk are also direct causes of endosomal-lysosomal dysfunction, underscoring the essential partnership between this dysfunction and APP metabolites in AD pathogenesis. The appearance of APP-dependent endosome anomalies in DS beginning in infancy and evolving into the full range of AD-related endosomal-lysosomal deficits provides a unique opportunity to characterize the earliest pathobiology of AD preceding the classical neuropathological hallmarks. Facilitating this characterization is the authentic recapitulation of this endosomal pathobiology in peripheral cells from people with DS and in trisomy mouse models. Here, we review current research on endocytic-lysosomal dysfunction in DS and AD, the emerging importance of APP/βCTF in initiating this dysfunction, and the potential roles of additional trisomy 21 genes in accelerating endosomal-lysosomal impairment in DS. Collectively, these studies underscore the growing value of investigating DS to probe the biological origins of AD as well as to understand and ameliorate the developmental disability of DS.
Keywords: Down Syndrome, Alzheimer’s Disease, Endosomes, Lysosomes, Autophagy
Graphical abstract
INTRODUCTION
Arising from an extra copy of chromosome 21 (HSA21), Down syndrome (DS) is characterized by varying degrees of intellectual disability (learning, memory and language), heart defects, craniofacial abnormality and childhood leukemia (Lott and Dierssen, 2010; Wiseman et al., 2009). The average life expectancy of people with DS has increased to 60 years, as medical treatment has advanced and social acceptance has led to decreased institutionalization. Despite these positive developments, however, almost all individuals with DS reaching the age of 40 will develop progressive Alzheimer’s Disease (AD) with characteristic neuropathological features including β-amyloid plaques, neurofibrillary tangles, and neuronal cell loss (Leverenz and Raskind, 1998; Wisniewski et al., 1985). Among DS individuals, 2–5% exhibit symptoms of AD dementia by the age of 40, while the occurrence of AD dementia approaches 100% in DS individuals by the age of 70 years (Lott and Dierssen, 2010). The extra copy of chromosome 21 is the fundamental source of this increased AD risk and can be mainly attributed to the gene encoding for amyloid precursor protein (APP), which is itself linked to familial AD (Tanzi, 2012). The observation that, in rare cases, a trisomy 21 state in which APP gene dosage is 2N rather than 3N, is associated with a DS phenotype but without an AD phenotype, implies that 3N APP is necessary, though not necessarily sufficient, for AD to develop in individuals with DS (Doran et al., 2017; Korbel et al., 2009; Prasher et al., 1998). Beyond APP, many of the triplicate genes on the HSA21 of DS individuals, such as superoxide dismutase 1 (SOD1), DYRK1A, BACE2, Down-Syndrome critical region (DSCR) and S100β have been implicated in AD development (Lott and Dierssen, 2010). Common genetic and pathological factors operating in both AD and DS suggest commonalities in the underlying disease mechanisms. These shared pathological mechanisms are compounded by ageing in both conditions, but accelerated in DS relative to the disease progression observed in AD.
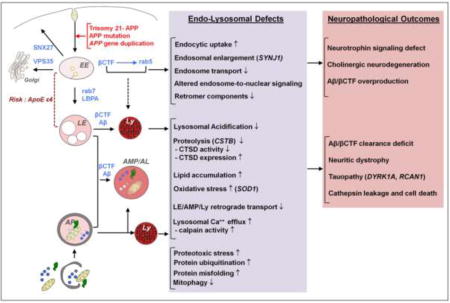
In AD, defects in endocytosis and autophagy have emerged as key drivers of accumulation of cellular waste products, progressive neuronal dysfunction, and neurodegeneration; which we have described previously (Nixon, 2017). Notably, many elements of endocytic and autophagic dysfunction observed in AD are also found in DS brain (as summarized in Figure 1), suggesting common disease-associated pathogenic processes, and a prospect of gaining key insights into these processes by investigating the early onset of AD-related pathophysiological features in DS brain, peripheral tissues, and experimental models of DS. A distinct advantage of the analysis of early AD pathobiology is the authentic recapitulation of the endosomal-lysosomal phenotype and its APP gene dependence in primary skin fibroblasts, other peripheral cell elements, and in induced pluripotent stem (iPSC) neuronal cell lines derived from these cell sources. This has enabled increasingly detailed analyses of the molecular pathways underlying endosomal signaling defects linked to neurodegeneration (Jiang et al., 2010; Park et al., 2008; Weick et al., 2013). In this review, we will address the current state of research on endocytosis and autophagy defects in DS and the relevance of defects in these processes to the development of AD.
Figure 1.
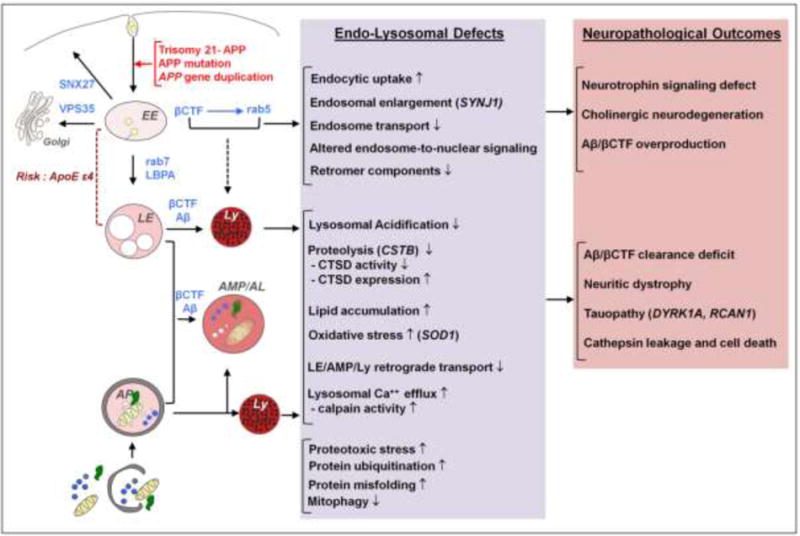
Overview of the shared pathways and pathological mechanisms driven by the APP gene and its cleavage products (βCTF/Aβ) in DS and AD: known and predicted endocytic and lysosomal factors contributing to neurodegeneration. Genetic contributors to neurodegeneration in DS other than APP are shown in parentheses. ApoE ε4, a known genetic risk factor for AD, contributes to endolysosomal dysfunction in AD and potentially in DS. EE, early endosome; LE, late endosome; AP, autophagosome; Ly, Lysosome; AMP/AL, amphisome/autolysosome.
ENDOCYTIC DYSFUNCTION IN DS AND RELATIONSHIPS TO AD
Endosomal pathology in AD and DS and the role of specific APP metabolites
Early endosomes sort internalized and plasma membrane-associated cargoes that will be either recycled to the plasma membrane, transported to and from the Golgi via the retromer complex, or directed to lysosomes for degradation (Di Fiore and von Zastrow, 2014; Huotari and Helenius, 2011). Importantly, in addition, endosomes contain on their surface a signaling platform that transduces external or membrane receptor signals, via activation of intracellular signaling molecules, into multiple vesicular trafficking events controlling cargo fates, receptor occupancy on membranes, and endosome-nuclear translocation of transcriptional regulators like tropomyosin receptor kinases (Trks), receptor tyrosine kinases (RTKs), and cell cycle and inflammatory factors (Cendrowski et al., 2016; Cosker and Segal, 2014; Howe and Mobley, 2004; Kim et al., 2015; Miaczynska, 2013; Pyrzynska et al., 2009). The master regulator of endocytosis is the small Rab GTPase rab5, whose functions, similarly to other rabGTPases, depend on a) its membrane localization, regulated by GDP dissociation inhibitors (GDIs) (Felberbaum-Corti et al., 2005) and GDI displacement factors (GDFs) (Dirac-Svejstrup et al., 1997); and b) cycling between a GTP-bound/active and a GDP-bound/inactive state, via guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (RabGAPs) activities, respectively (Wandinger-Ness and Zerial, 2014).
Upon activation, Rab5 recruits effector proteins important for endosome maturation, trafficking and signaling. These effectors include: Rabaptin-5 (Stenmark et al., 1995), forming a complex with, and regulating the GEF activity of Rabex-5, a specific GEF for rab5 (Horiuchi et al., 1997; Zhang et al., 2014); phosphatidylinositol 3-kinase (PI3K)/Vps34 and its product PI(3)P, required for docking and fusion of incoming rab5 positive early endosomes via recruitment of EEA1 and SNAREs (Christoforidis et al., 1999; Simonsen et al., 1998). Rabenosyn-5, similarly to EEA1, specifically binds to PI(3)P via its FYVE finger motif and is also necessary for endosomal membrane targeting (Sonnichsen et al., 2000; Zerial and McBride, 2001). APPL1 (adaptor protein containing pleckstrin homology domain, phosphotyrosine binding (PTB) domain, and leucine zipper motif) (Miaczynska et al., 2004), enriched on a subpopulation of very early endosomes with distinct spatial distributions and cargo sorting activity (Kalaidzidis et al., 2015) has emerged as another important rab5 regulatory factor highly relevant to emergence of early endosome dysfunction in AD. Owing to its BAR-PH and PTB domains (Li et al., 2007; Zhu et al., 2007), APPL1 binding to GTP-rab5 is key to mediate the signaling of growth factors (EGF, NGF, BDNF) (Fu et al., 2011; Lin et al., 2006; Miaczynska et al., 2004), NF-kB (Hupalowska et al., 2012), adiponectin (Mao et al., 2006), insulin (Ryu et al., 2014) and phosphoinositides (Bohdanowicz et al., 2012).
In AD, acceleration of neuronal endocytosis (as measured directly in DS fibroblasts), is reflected in increased fusion and the swelling of rab5-positive early endosomes (Cataldo et al., 1997; Cataldo et al., 2008) and upregulated expression of rab5 and related rab effectors (Ginsberg et al., 2010a; Ginsberg et al., 2011; Ginsberg et al., 2010b) are the earliest AD neuronal pathology thus far identified. Our previous studies had linked the endosomal abnormality in DS to its extra copy of APP in neurons of the Ts65Dn and Ts2 mouse (Cataldo et al., 2000; Jiang et al., 2016), and also in fibroblasts derived from individuals with DS (Cataldo et al., 2008; Jiang et al., 2010). Notably for AD and DS, endosomes are sites of active APP processing due to the enrichment of β-site APP cleaving enzyme 1 (BACE1) and γ-site APP secretase (Israel et al., 2012; Nixon, 2013). Growing evidence shows that β-site cleavage of APP on endosomes (Kaether et al., 2006) to form APP-βCTF is actively modulated by the varied regulatory influences on BACE1 activity and localization, which include many AD risk factors.
APP-βCTF is elevated in both AD and DS human brains (Kim et al., 2015; Pera et al., 2013) and gene mutations in presenilin 1 (PSEN1) and APP that cause early-onset forms of AD accelerate APP-βCTF formation and/or accumulation (Cacquevel et al., 2012; Chang and Suh, 2005; De Jonghe et al., 2001; Deyts et al., 2016; van der Kant and Goldstein, 2015). APP-βCTF has been shown recently to pathologically activate rab5 by recruiting to early endosomes the adaptor protein APPL1, which binds both APP-βCTF and GTP-rab5, causing stabilization and further activation of rab5 (Figure 2A). This results in the upregulation of endocytosis, endosomal trafficking defects and abnormal endosome-mediated signaling (Kim et al., 2015). By crossing Ts2 mice with the BACE1+/− line, we generated Ts2.BACE1+/− mice that have reduced APP-βCTF but undetectable change in regional Aβ levels compared to Ts2.BACE1+/+ mice; this modulation rescued rab5-positive endosome size and normalized basal forebrain cholinergic neuron (BFCN) number (Jiang et al., 2016). Combined with findings from others (Choi et al., 2013; Lauritzen et al., 2012; Lauritzen et al., 2016; Salehi et al., 2006; Tamayev et al., 2012) our studies (Jiang et al., 2010; Jiang et al., 2016; Kim et al., 2015) implicate APP-βCTF in the pathogenesis of AD and DS, while also providing support for ongoing clinical study of BACE1 inhibitors as a therapy for AD. Genetic reduction of BACE1 has been shown to reduce amyloid burden, delay the onset BFCN degeneration, and improve cognitive function in several APP transgenic mouse models (McConlogue et al., 2007; Ohno et al., 2007; Singer et al., 2005).
Figure 2.
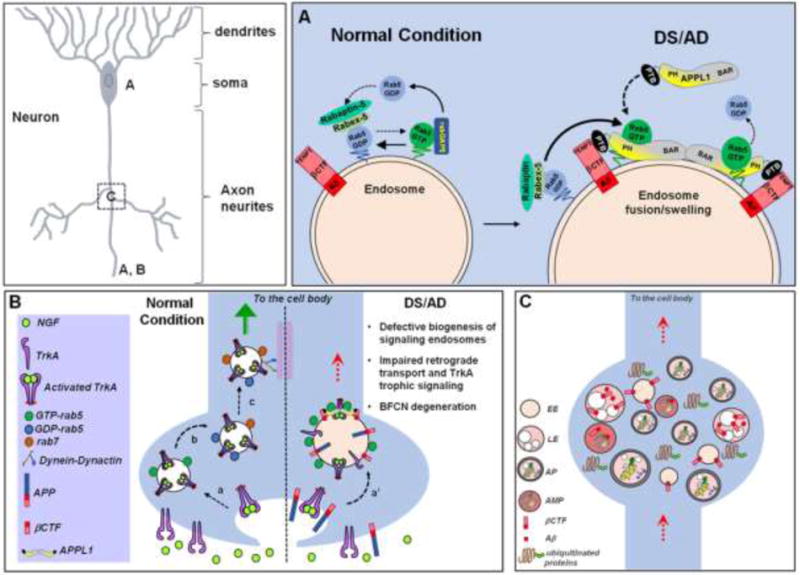
Endosomal pathology and altered endosome trafficking in DS and AD contribute to aberrant neurotrophin transport and signaling and neuritic dystrophy. A) Under normal conditions, the conversion of rab5 from an active to an inactive state occurs rapidly and it is highly regulated by the coordinated action of rab5 effectors rabex-5/rabaptin-5 and rabGAP5, respectively. In DS and AD, APP-βCTF mediates pathological activation of rab5 by recruiting APPL1 to endosomes. APPL1 binds to the “YENPT” motif of βCTF through its phosphotyrosine binding (PTB) domain. Upon dimerization via its BAR domains to facilitate vesicle curvature, APPL1 binds to GTP-rab5 via its PH domain, which stabilizes rab5 in its activated state on endosomes. Over-activation of rab5 causes acceleration of endocytosis and endosome fusion leading to characteristic swelling of endosomes. B) Under normal conditions, biogenesis of long-lived signaling endosomes involves endocytosis of activated TrkA receptors (NGF-bound TrkA) into rab5-positive endosomes (a), followed by deactivation of rab5 and acquisition of rab7 (b), which binds to dynein/dynactin motor complexes regulating long-range retrograde axonal transport (c, green arrow). In DS and AD, prolonged rab5 hyperactivation by APP/APP-βCTF and APPL1 causes enlargement of NGF/TrkA endosomes (a’) and slows retrograde endosome transport (red dotted arrow), thus diminishing TrkA signaling which ultimately leads to the loss of trophic support for basal forebrain cholinergic neurons (BFCNs). C) DS and AD are characterized by the pathological accumulation of ubiquitinated misfolded/aggregated proteins and of enlarged early and late endosomal structures (EE and LE, respectively) containing βCTF/Aβ fragments. Autophagosomes (AP), amphisomes (AMP), and endolysosomal compartments containing incompletely degraded material accumulate within neuritic swellings (“dystrophic neurites”) as a result of the βCTF/Aβ-mediated impairment of retrograde transport and lysosomal function.
In addition to APP, another gene from HSA21 that has been implicated in the observed endosomal dysfunction in DS is the synaptojanin1 (SYN1) gene (Cossec et al., 2012). SYNJ1 encodes for a phosphatase, the activity of which is directed to phosphatidylinositol-4,5-biphosphate PI(4,5)P2, a signaling molecule involved in the regulation of clathrin-mediated endocytosis (Mani et al., 2007). SYNJ1 levels are elevated in the brain of human DS patients (Arai et al., 2002) and even greater in DS with AD, compared to sporadic AD and control brains (Martin et al., 2014). Moreover, elevated expression of SYNJ1 was shown to alter PI(4,5)P2 metabolism and cause cognitive deficits in the T65Dn mouse model of DS (Voronov et al., 2008).
Finally, inheritance of the APOE ε4 allele, which is the strongest genetic risk factor for sporadic AD (Liu et al., 2013), promoted earlier appearance of endosomal enlargement at preclinical stages of AD (Cataldo et al., 2000). Notably, APOE ε4 is a significant factor in the severity of AD pathology in DS. Several studies have shown that DS patients who carry APOE ε4 exhibit increased risk of AD-DS, earlier dementia onset, and greater amyloid load; whereas APOE ε2 has been shown to confer protective effects in DS (Lai et al., 1999; Prasher et al., 2008; Wiseman et al., 2015). Considering the shared genetics between AD and DS, it may be speculated that APOE ε4 accelerates endosomal pathology in DS as well as in AD in the early development of disease.
Endosomal dysfunction in AD and DS: Consequences on downstream endocytic events relevant to AD pathogenesis
Endosome maturation defects
The conversion from rab5- to rab7-positive endosomes underlies the transition of cargos from early to late endosomes (Huotari and Helenius, 2011). Such transition is mediated by GTP-rab5, which recruits the SAND-1/Mon1 and Ccz1 complex (Poteryaev et al., 2007; Wang et al., 2003) and the class C vacuolar protein sorting/homotypic fusion and protein sorting (Class C VPS/HOPS) complex (Rink et al., 2005) for recruitment and activation of rab7 and suppression of rab5. The molecular link between the rab5 and rab7 machineries comes from the observation that expression of GTP hydrolysis-defective rab5 mutant or deletion of Vps39, one of the class C VPS/HOPS complex subunit known to be a GEF for rab7, caused enlargement and colocalization of rab5/EEA1 and rab7/LAMP1-positive vacuoles (Rink et al., 2005). As the endosome moves towards the nucleus, sorting complexes required for transport (ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III) regulate the sequestration of ubiquitinated cargoes (Raiborg and Stenmark, 2009) and initiate the lipid-driven formation of intra-lumenal vesicles (ILVs), which are enriched in cholesterol, sphingolipids and PI(3)P in early phases and lyso-bisphosphatidic acid (LBPA) and phosphoinositol-3,5-bis phosphate (PI(3,5)P2) in later phases (Babst, 2011; Solinger and Spang, 2013).
As a result of the upregulation of endocytosis, DS fibroblasts (Cataldo et al., 2008), like neurons in DS/AD brain, exhibit late endosomal abnormalities, manifesting in increased numbers and size of rab7- and LBPA-positive perinuclear vacuoles. Microarray analysis of mild cognitive impairment (MCI) and AD hippocampal CA1 neurons revealed upregulation of both rab5 and rab7 GTPases (Ginsberg et al., 2010a). Subsequent analysis on rab5 and rab7 protein expression revealed a region-specific increase in their levels in basal forebrain, frontal cortex, hippocampus in MCI and AD (Ginsberg et al., 2011; Ginsberg et al., 2010b). Also, lipidomic analysis showed specific enrichment of LBPA, sphingomyelin, ganglioside GM3, and cholesterol esters in human AD brain entorhinal cortex (Chan et al., 2012). A similar increase in cholesterol esters and GM3 was also observed in PS1-APP mice, altogether suggesting a common collection of pathogenic mechanisms associated with endo-lysosomal pathology in DS and AD (Chan et al., 2012). Disruption of the ESCRT machinery has also been shown to promote neuronal accumulation of ubiquitin-positive aggregates and autophagosomes (APs), as well as neurodegeneration (Kurashige et al., 2013; Lee et al., 2007; Tanikawa et al., 2012; Yamazaki et al., 2010), and causes an early accumulation of large lipid-related autofluorescent intraneuronal aggregates derived from the endolysosomal system (Clayton et al., 2015).
Aberrant endosomal signaling of neurotrophins leading to cholinergic neurodegeneration
The coordinated action of Rab5 and rab7 is key to the regulation of axonal uptake and long-range retrograde transport of NGF-activated TrkA receptor within long-lived signaling endosomes (Howe and Mobley, 2004) (Deinhardt et al., 2006; Philippidou et al., 2011; Saxena et al., 2005) which play roles in the modulation of gene expression promoting neuronal growth, survival and synapse assembly and maintenance (Cosker and Segal, 2014; Lehigh et al., 2017; Sharma et al., 2010).
There are multiple lines of evidence supporting impaired endosomal signaling in AD and DS (Figure 2B). Activity-dependent release of NGF from cortical and hippocampal neurons and NGF signaling are crucial for the survival of BFCNs that are lost in DS and AD, contributing to cognitive decline (Grothe et al., 2012; Iulita and Cuello, 2016). Retrograde transport of NGF is disrupted and the cholinergic phenotype is lost in mouse models of AD and DS (Granholm et al., 2000; Jiang et al., 2016; Salehi et al., 2006) and reversed by administration of NGF (Cooper et al., 2001; Granholm et al., 2000) or by partial reduction of BACE-1 (Jiang et al., 2016). Additionally, microarray analysis of human AD hippocampal neurons and BFCN revealed an early down-regulation of Trk receptors accompanying the upregulation of early endocytic genes (Ginsberg et al., 2010a; Ginsberg et al., 2011).
APP-βCTF-mediated rab5 hyper-activation and endosomal enlargement (Kim et al., 2015; Xu et al., 2016) lead to BFCN atrophy (Xu et al., 2016) and disrupt retrograde transport and signaling causing a net slowing down of vesicle movements, which are prevented by reducing GTP-rab5 levels either by siAPPL1 (Kim et al., 2015) or by expression of dominant negative rab5 mutant (Xu et al., 2016). βCTF-mediated rab5 hyperactivation may also affect the biogenesis of signaling endosomes, which requires inactivation of Rab5 by rabGAP5, and constitutively active rab5 mutant blocks NGF-mediated neurite outgrowth (Haas et al., 2005; Liu et al., 2007). Finally, APPL1 recruitment to βCTF-GTP-rab5 endosomes may prevent APPL1-PTB binding to activated TrkA receptor mediating NGF signaling (Lin et al., 2006; Varsano et al., 2006) and contribute to slow down endosome transport by increasing rab5 activation and reducing TrkA signaling (Nixon, 2017). Aside from its effects on NGF, rab5 hyperactivation promotes activation of signaling pathways leading to cell death. APPL1-mediated nuclear delivery of transcription factors activates nuclear factor-κB (NF-κB) in human DS fibroblasts (Kim et al., 2015). Rab5 interaction with APP-BP1 induces cell cycle re-entry followed by apoptosis in London mutant APP (V642I)-FAD expressing primary rat cortical neurons (Laifenfeld et al., 2007).
Similarly to TrkA receptors (Ascano et al., 2009), also APP (Woodruff et al., 2016) and BACE1 (Buggia-Prevot et al., 2014) have been shown to require rab11-mediated transcytosis for their axonal localization. Following APP endocytosis into in rab5 positive early endosomes, BACE1-mediated cleavage of APP redistributes a portion of full-length APP and βCTF into rab11 recycling endosomes where the γ-secretase-mediated cleavage of βCTF is the “signal” promoting transcytosis (Woodruff et al., 2016). fAD mutations that increase βCTF levels, either by enhancing BACE1 activity or impairing γ-secretase cleavage of APP, disrupt APP transcytosis, causing an accumulation of APP/βCTF and rab11-positive endosomes in the soma and reduced levels in the axon (Woodruff et al., 2016). APP has also been proposed to influence TrkA anterograde transport via direct (Kamal et al., 2001; Stokin et al., 2005) or indirect (Inomata et al., 2003) binding to kinesins that may affect NGF signaling (Zhang et al., 2013). Therefore, decreased anterograde transport of TrkA due to defects in axonal transport of APP caused by overexpression or mutations of APP may also contribute to NGF retrograde transport defects.
Retromer defects
The sequential action of rab5 and rab7 has also been shown to regulate the retrograde transport from the sorting compartment to the trans-Golgi network (TGN), via recruitment of the retromer complex (Rojas et al., 2008), which also regulates the recycling of cargoes from endosomes back to the plasma membrane; a process which is necessary for synaptic remodeling and plasticity (Loo et al., 2014; Temkin et al., 2011; Zhang et al., 2012). Rab5 indirectly promotes the recruitment of the sorting nexin (SNX) subcomplex (different dimeric combinations of SNX1, SNX2, SNX5, and SNX6), likely via PI3K activation (Rojas et al., 2008). On the other hand, rab7, along with SNX3 and TBC1 domain family member 5 (TBC1DF5), also known as the “membrane-recruiting module” (Small and Petsko, 2015), directly interacts with the vacuolar protein sorting (Vps) subcomplex (a heterotrimer composed of Vps26, Vps29, and Vps35). Recently, another SNX, SNX27, was linked to retromer and its function (Steinberg et al., 2013; Temkin et al., 2011). SNX27 functions as an adaptor for binding to PDZ ligand-containing cargos that are directed to the cell surface via the recycling pathway.
Defects in the retromer complex have been linked to neurodegenerative diseases, including AD and DS. While autosomal-dominant mutations in VPS35 link late-onset PD with retromer dysfunction, AD brains are deficient in Vps35 and Vps26 components (Small and Petsko, 2015). Moreover, a recent study provides evidence for SNX27 deficiency in DS brains possibly due to an extra copy of a microRNA encoded by human chromosome 21, which causes decreased SNX27 expression (Wang et al., 2013). This resulted in disrupted glutamate receptor recycling in the hippocampus and led to dendritic dysfunction. Notably, upregulation of SNX27 in the hippocampus of Ts65Dn mice rescued both synaptic and cognitive deficits (Wang et al., 2013). It might be speculated that correcting the retromer-dependent trafficking from endosomes would also have a salutary effect on endosome enlargement and its consequences.
Altered endosome trafficking contributing to autophagic defects and neuritic pathology in AD and DS
Before fusing with lysosomes, late endosomes can fuse with autophagosomes (AP) to form prelysosomal compartments known as amphisomes (Nixon, 2013). Amphisome formation requires some of the components that regulate late endosome/multivesicular body (MVB) formation, such as HOPS and ESCRT protein complexes. Additionally, Rab7 is found on nascent autophagosomes and is required for AP maturation (Gutierrez et al., 2004; Jager et al., 2004). In distal axons, late endosomes commonly fuse with APs and acquire retrograde motility by recruiting dynein-snapin complexes (Cheng et al., 2015). Snapin, originally implicated in vesicle fusion and synaptic transmission (Ilardi et al., 1999), has been shown to play a crucial role in regulating late endosome retrograde transport and maturation in neurons (Cai et al., 2010). Indeed, Snapin deletion causes accumulation of late endosomes and impairs lysosomal function.
Recent evidence indicates that amphisome retrograde transport is impaired in the mossy fiber tracts of the dentate gyrus in young 3×Tg-AD mice, compared to WT, as detected by increased colocalization of LC3-II with the late endosomal marker mannose-6-phosphate receptor (CI-MPR) in distal axons. Additionally, these axons were positive for ubiquitin, p62 and appeared swollen and dystrophic (Tammineni et al., 2017). APs have been shown to be key organelle for the degradation of βCTF via the MVB/lysosomal pathway, and inhibition of AP formation or fusion with the endolysosomal compartment, caused accumulation of βCTF in ILV/MVB (Gonzalez et al., 2017). Disrupted anterograde transport of βCTF-containing endosomes results in their accumulation in axons terminals along with autophagic vacuoles, presumably formed in an effort to facilitate clearance of endosomes, contributing to axonal swelling and neuritic dystrophy similar to that observed at early stages of AD (Stokin et al., 2005).
The majority of dystrophic neurites in human and mouse AD brains are characterized by the presence of autophagic vacuoles (Nixon, 2005). A sustained induction of autophagy and endocytosis paralleled by a progressive decline in lysosomal proteolysis and a slowing of retrogradely transported cargoes within vulnerable neuronal populations likely contributes to the extensive autophagic and neuritic pathologies implicated in AD (as illustrated in Figure 2C) (Bordi et al., 2016). Notably, even in the absence of APP-βCTF/amyloid, defective proteolysis or inhibition of endolysosomal acidification can induce the accumulation of vacuoles filled with incompletely degraded, ubiquitinated material that accumulates, causing neurite swelling and dystrophy (Boland et al., 2008; Lee et al., 2011). In DS fibroblasts, an increase in the number of large compartments with the structural features of early/late endosomal hybrids, MVB, autophagic vacuoles, late endosomes, or lysosomes, has also been reported (Cataldo et al., 2008), further highlighting the relationship between early endosomal dysfunction and downstream endocytic and proteolytic compartments. Extensive accumulation of misfolded and ubiquitinated proteins in the brain (Tramutola et al., 2016) and dystrophic neurites (as illustrated in Figure 2C) (Mattiace et al., 1991) of DS individuals suggests that global defects in protein quality control ultimately disrupt autophagic and lysosomal clearance (see following section), despite the lowered autophagy induction rate due to hyperactivation of the PI3K/Akt/mTOR axis (Perluigi et al., 2014). mTOR activation has also been reported to be elevated in inferior parietal lobule (Tramutola et al., 2015) and medial temporal cortex neurons bearing hyperphosphorylated tau in human AD brains (Li et al., 2005), as well as in non-neural cultured cells transfected with mutant APP (Caccamo et al., 2010) or exposed to Aβ oligomers (Bhaskar et al., 2009). On the other hand, a reduction in mTOR activity has been documented in AD hippocampus (Bordi et al., 2016), as well as murine neuroblastoma cells exposed to Aβ42, PSEN1/APP mouse brain and lymphocytes from AD patients (Lafay-Chebassier et al., 2005). There is also evidence for an early transcriptional upregulation of genes promoting autophagy and downregulation of negative regulators of autophagy flux in human AD entorhinal cortex (Lipinski et al., 2010) and in CA1 pyramidal neurons (Bordi et al., 2016). Taken together, these observations highlight the complexity in defining the net direction of autophagic signaling changes during the course of AD, due to the different experimental models used, the specific brain regions and cell populations examined at a given stage of AD pathology development and the different methods of analyzing mTOR activity itself and downstream effects on its substrates and autophagy.
LYSOSOMAL DYSFUNCTION IN DS AND RELATIONSHIPS TO AD
APP and APP β-C-terminal fragments (βCTF) and lysosomal dysfunction
The triplication of the APP gene in DS is of particular interest in the study of the AD-DS relationship, due to the role of APP mutations in fAD and the broader role of its metabolites in sAD (Selkoe and Hardy, 2016). In the context of lysosomal dysfunction, this attention towards APP is well-supported; APP holoprotein and its cleavage products are implicated in lysosomal defects in DS and AD (Nixon, 2007) (as described in Figure 3). Numerous lines of evidence connect APP and its products with lysosomal dysfunction in AD (Umeda et al., 2011; Xu et al., 2016; Xue et al., 2014; Yang et al., 2008; Yang et al., 2014), and the triplication of the APP gene in DS suggests that APP-mediated lysosomal defects occur in DS in a similar fashion. A recent study on an elderly DS individual lacking APP triplication due to partial trisomy 21 revealed a lack of AD pathology or dementia (Doran et al., 2017), providing human evidence for the role of APP in the development of AD pathology observed in older individuals with DS. This finding is supported by evidence from primary DS fibroblasts which shows that their AD-like endosomal phenotype is APP-dependent (Jiang et al., 2016). The relationship between lysosomal dysfunction and APP is thought to be bi-directional, as lysosomal failure permits formation of misfolded aggregates of APP products and other pathogenic proteins (Ivy et al., 1984; Mangieri et al., 2014), while both APP holoprotein and APP cleavage products (particularly βCTF) negatively impact lysosomal function (Ditaranto et al., 2001; Lauritzen et al., 2012; Lauritzen et al., 2016; Yang et al., 1998; Yang et al., 2011b; Yang et al., 2014). This relationship is observed in studies of the CRND8 APP mutant mouse line, which exhibits age-related amyloid pathology and learning/memory deficits. These mice exhibit markers of lysosomal dysfunction, specifically swollen Cathepsin D (CTSD)-positive compartments, accumulation of amyloid products in the lysosome, and impaired lysosomal lipid processing (Yang et al., 2011b; Yang et al., 2014). Further, the amelioration of learning and memory defects in conjunction with reduced amyloid via rescue of lysosomal function in the CRDN8 model is compelling evidence for the centrality of lysosomal dysfunction in the progression of APP-associated disease phenotypes. Additional strong pathological evidence for autophagic-lysosomal dysfunction in APP mutant animal models has been reported in the 3xTG-AD mouse as well, specifically in regard to the role of βCTF (Lauritzen et al., 2012; Lauritzen et al., 2016).
Figure 3.
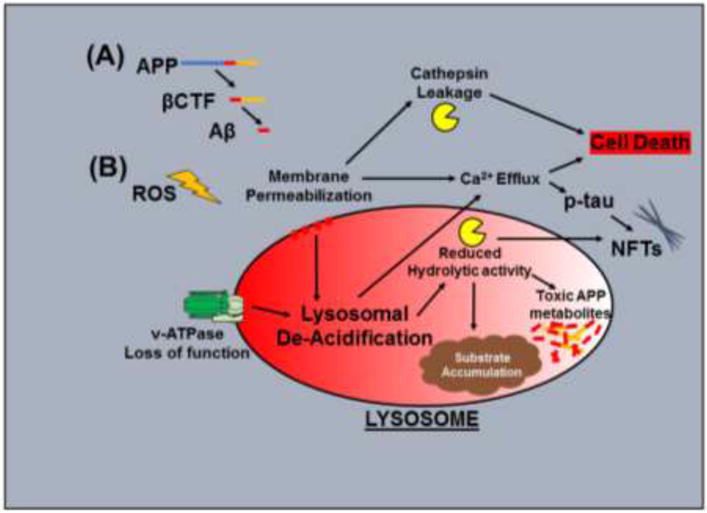
Common mechanisms of lysosomal dysfunction in neurons during AD and DS: (A) APP and its cleavage products, βCTF and Aβ, contribute to lysosomal de-acidification and lysosomal membrane permeabilization, leading to a decline in lysosomal function. (B) Reactive oxygen species (ROS), which are increased in DS and in AD, also contribute to impaired lysosomal function. Congruent elements of lysosomal dysfunction; including lysosomal de-acidification, reduced proteolysis, lysosomal membrane permeabilization leading to loss of membrane integrity, and the release of Ca2+ and cathepsins represent common pathological outcomes in AD and DS, leading to accumulation of intralysosomal waste, toxic APP and tau metabolite accumulation, neurofibrillary tangle (NFT) formation, and cell death.
While a great deal of research in AD has focused on Aβ1–40/42 and the products of their aggregation, current studies indicate that the APP cleavage product βCTF exert significant pathological effects on lysosomal function, in addition to the demonstrated roles of βCTF in endosomal dysfunction (described above). βCTF was found to promote neurodegeneration and formation of lesions in vivo following viral induction of βCTF expression (Lauritzen et al., 2012), and these pathological signs appear to be driven by impairments of lysosomal function. βCTF contributes to morphological changes in lysosomes, reduced lysosomal proteolysis, and accumulation of intracellular waste products (Lauritzen et al., 2016). Further studies of this phenomenon demonstrate that gamma-secretase inhibition, which blocks Aβ formation while increasing βCTF levels, significantly exacerbates the lysosomal dysfunction. These detrimental effects on endolysosomal function also occur independently of Aβ in models of DS (Jiang et al., 2010; Jiang et al., 2016). It is also shown that βCTF fragments can be found in the membranes of lysosomes and autophagic compartments, where they may impair the integrity of the membrane. This could impair acidification of lysosomes via loss of membrane stability and/or through the potential interactions of βCTF with any of the various membrane proteins of the lysosome which are essential to proper acidification. βCTF-induced membrane permeability could also promote cell death via release of lysosomal calcium and pro-apoptotic functions of leaked lysosomal enzymes (Guicciardi et al., 2004)(as described in Figure 3).
Amyloid-β
Additionally, there is also evidence that forms of Aβ can negatively impact lysosomes en route to promoting neuronal death. In vitro studies suggest that various forms of Aβ can permeabilize lysosomal membranes (Ditaranto et al., 2001; Kosenko et al., 2011; Song et al., 2011; Umeda et al., 2011; Yang et al., 1998), which is detrimental to the stability of the lysosome, permitting release of lysosomal enzymes and calcium into the cytoplasm, promoting apoptosis (Guicciardi et al., 2004; Nixon and Cataldo, 1993). Many of these studies tested the effects of intraneuronal amyloid peptides, which accumulate early in DS pathogenesis (Mori et al., 2002). Notably, in animal models of DS and AD, the identity of APP cleavage products is heterogeneous; βCTF and various forms of Aβ are generated, and Aβ peptides can be present as monomers or various stages of oligomerization. In the case of DS, increased expression of full-length APP is likely to increase the autophagic burden of clearing all types of APP cleavage products, but positive effects of BACE1 inhibition (Jiang et al., 2016) suggest potential benefit in targeting APP cleavage by BACE1 to reduce both βCTF and Aβ.
Cathepsin D (CTSD) defects and lysosomal pathology in DS and AD
Lysosomal CTSD-mediated proteolysis plays a very important role in maintaining neuronal cell homeostasis by degrading unwanted material delivered to lysosomes via autophagy or endocytosis. Many disease-associated aggregated proteins: Aβ/βCTF, α-synuclein, tau and huntingtin, abnormally accumulate in autophagic compartments when they are not efficiently degraded by CTSD and other lysosomal enzymes (Nixon, 2013). Numerous studies have shown that mice lacking CTSD have neurodevelopmental deficits and exhibit progressive neurodegeneration (Koike et al., 2003; Shacka et al., 2007; Tyynela et al., 2000; Yamasaki et al., 2007), demonstrating the essential functions of CTSD in efficient lysosomal function and subsequently, neuronal health. In human disease, a subtype of neuronal ceroid lipofuscinosis (NCL) is a lysosomal storage disease directly caused by mutations in CTSD (Ketterer et al., 2017; Stoka et al., 2016). Also, a rare form of Gaucher disease (another lysosomal storage disease) is the result of lowered activity of lysosomal CTSD, caused by a pro-saposin gene mutation (Tatti et al., 2013). Abnormal CTSD protein levels and lowered specific enzyme activity, reflecting a accumulation of inactive or less active CTSD, are seen in the brains of individuals with either sporadic AD or PSEN-fAD (Cataldo et al., 2004). Moreover, also seen in AD is a) an elevated level of CTSD transcript in CA1 neurons of AD brains (Ginsberg et al., 2010a); b) increases in both mRNA expression and cellular content of CTSD in neocortical pyramidal neurons of AD brain (Cataldo et al., 1995); c) presence of CTSD immunoreactivity and active forms of other cathepsins and lysosomal enzymes within amyloid plaques of both DS and AD postmortem brains (Cataldo et al., 1996), reflecting the robust autophagy-lysosomal pathway pathology in neuritic plaques; and d) elevated level of CTSD of abnormally lowered specific enzyme activity in the cerebrospinal fluid of AD patients (Schwagerl et al., 1995). In addition, altered expression and subcellular localization of CTSD were found in both neurons and glia in aged brains (Nakanishi, 2003). CTSD has been associated with the clearance of Aβ, tau, and APOE, all of which are involved in the pathogenesis of AD (Kenessey et al., 1997; Zhou et al., 2006). Our recent studies in DS human fibroblasts show decreased lysosomal CTSD specific and in situ activities, despite an elevation of total CTSD activity (Nixon et al., 2017). Further research on CTSD dysfunction and related pathologic effects on DS and AD are needed to fully understand the mechanism underlying these changes, although impaired lysosomal acidification is a significant contributing factor (Nixon et al., 2017).
Lysosomal acidification defects in autophagic dysfunction during AD and DS
Lysosomal acidification is crucial to many functions of the lysosome, most notably proteolysis; as activities and maturation of multiple lysosomal hydrolases, including CTSD, are pH-dependent. But acidification is also critical for axonal transport of acidified vesicular compartments (Lee et al., 2011), fusion with AVs, lysosomal signaling functions via mTOR (Perera and Zoncu, 2016), lysosomal calcium release, and exocytosis (Wolfe et al., 2013). Acidification of lysosomes is fundamental to lysosomal function and autophagy in general throughout cell types, but is particularly important in neurons which are post-mitotic and need to exert quality control over large expanses of cytoplasm. The rapid constitutive rate of autophagy necessitates also having a highly efficient lysosomal processing of substrates (Boland et al., 2008). Lysosomal acidification defects have been implicated in a number of late-onset neurodegenerative diseases, including AD (Lee et al., 2010; Lee et al., 2015), and PD (Dehay et al., 2013), but also in a group of childhood genetic diseases affecting the CNS (Colacurcio and Nixon, 2016). These acidification-related diseases are often linked to mutation of a component of the vacuolar ATPase (v-ATPase) complex, the proton pump that is the primary driver of lysosomal acidification (McGuire et al., 2017). In these genetic disease contexts, a mutation of the v-ATPase or a v-ATPase associated protein leads to impaired lysosomal acidification, which can drive aggressive (i.e. childhood-onset) CNS pathology via progressive failure of lysosomal function in neurons.
While we have previously addressed the genetic and pathological overlap between AD and DS, it is noteworthy that lysosomal de-acidification causes another form of mental retardation in the case of Mental Retardation Hedera Type, during which a chaperone (ATP6AP2) of the lysosomal v-ATPase is mutated (Hedera et al., 2002; Kinouchi et al., 2013; Korvatska et al., 2013). Combined with the observations of lysosomal de-acidification in AD and AD models (Avrahami et al., 2013; Lee et al., 2015), florid lysosomal pathology of APP mutant mice (Lauritzen et al., 2016; Yang et al., 2011a; Yang et al., 2014), and the overlapping role of APP in AD and DS, lysosomal de-acidification may contribute substantially to the lysosomal dysfunction in DS. Lysosomal de-acidification may also explain how the activities of CTSD and other lysosomal hydrolases become altered and why inactive enzyme forms accumulate. In this regard, our recent data demonstrate de-acidification of lysosomes, driven by loss of v-ATPase function in primary human DS fibroblasts relative to control cells (Nixon et al., 2017).
Also, recent proteomic studies have identified accumulation of ubiquitinated proteins in DS (Tramutola et al., 2016), indicative of aforementioned failures of both proteasomal function and autophagic clearance of proteins (Dephoure et al., 2014). In addition to a global increase in ubiquitinated proteins and loss of proteasomal function, this study (Tramutola et al., 2016) demonstrated progressive ubiquitination of certain proteins between brains of young DS individuals and older DS individuals with AD pathology compared to non-DS controls. For example, V1B2 subunits of the v-ATPase were increasingly ubiquitinated in the brains of DS individuals and further increased in DS individuals with AD pathology. Moreover, this ubiquitination increased with age in non-DS individuals. These changes in the v-ATPase could possibly impair the acidification of lysosomes. The correlation of v-ATPase ubiquitination with DS, DS-associated AD pathology, and ageing therefore further adds to previous evidence (Colacurcio and Nixon, 2016) that v-ATPase dysfunction and lysosomal de-acidification are significant contributors to a common theme of lysosomal dysfunction throughout neurodegenerative disease.
Downstream of lysosomal acidification defects, a number of pathological events can occur progressively throughout the lysosomal network, and ultimately impact the overall health of the cell as this progressive lysosomal corruption worsens (Guicciardi et al., 2004). De-acidification of lysosomes impairs maturation of cathepsins and their activity, leading to oxidative stress in conjunction with impaired clearance of oxidatively damaged proteins (Wolfe et al., 2013). Beyond this hydrolytic dysfunction, abnormal rise in lysosomal pH interferes with axonal trafficking of autophagic compartments (Lee et al., 2011), stalls clearance of defective mitochondria (mitophagy), causes release of lysosomal calcium (McBrayer and Nixon, 2013) leading to harmful downstream effects of calpain activation (Rao et al., 2014) and kinase activation. Many of these impairments overlap and synergize, such as mitophagy failure as an additional source of oxidative stress. The accumulated effects destabilize lysosomal membrane integrity, sending neurons into a “death spiral” (Malkus et al., 2009).
APP-independent lysosomal defects in DS and AD
While the aforementioned evidence from individuals with DS (Doran et al., 2017) and DS cells (Jiang et al., 2010; Jiang et al., 2016) implicates APP gene dosage with AD-like phenotypes in DS, a number of cellular abnormalities associated with DS may also contribute to lysosomal dysfunction. Without regard for specific Ts21 genes, aneuploidy alone has been shown to exert stress on the proteostatic network (Dephoure et al., 2014; Oromendia et al., 2012; Torres et al., 2007). The abnormally high mRNA expression in trisomy 21 leads to proteotoxic stress, ER stress, activation of the unfolded protein response (UPR), oxidative stress, elevated ubiquitinated proteins, and decreased proteasomal degradation. This general disruption of the entire proteostatic network represents an increased burden on lysosomal clearance, especially considering the decrease in proteasomal function. Extensive accumulation of ubiquitinated proteins has also been observed in DS brain tissue (Di Domenico et al., 2013; Tramutola et al., 2016); further suggesting systemic defects in protein quality control and failed waste clearance by both proteasomal and lysosomal systems.
Beyond global defects in protein quality control, a number of individual genes triplicated in DS are linked to lysosomal dysfunction. Notably, the triplication of SOD1 contributes to an oxidative stress burden in DS, which has been shown in DS primary cells and in amniotic fluid samples from DS individuals (Butterfield et al., 2014; Lott and Dierssen, 2010; Pagano and Castello, 2012; Perluigi et al., 2011; Zana et al., 2007; Zana et al., 2006). The increased oxidative stress in DS drives accelerated senescence in DS cells (Rodriguez-Sureda et al., 2015). Oxidative stress is also shown pathologically in brain tissue from DS patients and correlates with amyloid pathology (Cenini et al., 2012). A proteomic study of oxidatively modified proteins in DS brain tissue showed that a number of lysosomal proteins (including v-ATPase, CTSD, GRP78, UCH-L1, and GFAP) become modified, in a manner that couples with decreased activity of the proteasome and AP formation (Di Domenico et al., 2013). The v-ATPase, which is carbonylated on the V1B2 subunit, may be impaired functionally by such changes, contributing to de-acidification (Porter et al., 2013; Wang and Floor, 1998). Oxidative modifications to other lysosomal proteins can also hamper the functions of the lysosome, as evidenced in models of acute oxidative stress (Pal et al., 2016; Zhang et al., 2017). The role of oxidative stress in DS is an intriguing area of research, and is addressed further in other reviews in this monograph.
Cystatin B functions as an endogenous lysosomal protease inhibitor, specifically found to inhibit the functions of cathepsins. Previous research from our group (Yang et al., 2011a; Yang et al., 2014) demonstrates in vivo that deletion of cystatin B stimulates lysosomal enzyme function, leading to improvement of lysosomal proteolysis. CRND8 APP mutant mice, when crossed with cystatin B-deficient mice, exhibit improved learning/memory function and amelioration of amyloid pathology compared to the CRND8 mice. These findings are highly relevant to the discussion of lysosomal dysfunction in AD and DS because cystatin B is located on chromosome 21 (Pennacchio and Myers, 1996), and the increased gene dosage (due to trisomy of chromosome 21) of Cystatin B in DS could impair cathepsin function, contributing to lysosomal dysfunction in DS.
As in AD and numerous other neurodegenerative diseases (Guo et al., 2017), neurofibrillary pathology is observed in DS (Head et al., 2016). Neurofibrillary pathology in DS does not appear until age 30–40, manifesting in a similar neuroanatomical pattern as in AD (Hof et al., 1995). Despite the timing of neurofibrillary pathology in relation to amyloid deposition, tau pathogenesis in DS may occur due to both APP-dependent and APP-independent causes. Conversely to this time frame, there is evidence for changes in tau phosphoryation in DS even at a prenatal stage, so initial intracellular changes in tau phosphorylation and proteolysis could occur long before tangle pathology is observed (Milenkovic et al., 2017). Rather than changes in the MAPT gene, the triplication of the Dyrk1A gene in DS (Kay et al., 2016; Yin et al., 2017) is shown to increase the 3R forms of tau, which is associated with tau pathology (Cardenas et al., 2012; Wegiel et al., 2011). Concurrently, the neurofibrillary tangles in DS brain show increased amounts of 3R tau isoforms and Dyrk1A-mediated phosphorylation relative to AD, suggesting a specific role for Drk1A in DS tau pathology. Tau itself, particularly in aggregated form, is also a substrate of lysosomal degradation (Vinicia et al., 2014), and so impairments in lysosomal function could favor the accumulation of Drk1A-phosphorylated and/or misfolded/aggregated tau. RCAN1 (Cardenas et al., 2012) is also implicated in tau pathology in DS, via the stimulation of the tau kinase GSK3β and inhibition of calcineurin. The progression of tau pathology in DS has been reported to occur subsequently to early-life accumulation of intracellular APP metabolites (Mori et al., 2002) but on a more rapid time frame than in AD possibly due to excessive Drk1A tau phosphorylation and RCAN1 gain of function as contributing genetic factors. Additionally, progression of tauopathy in both DS and AD is potentially accelerated by lysosomal dysfunction via release of lysosomal calcium, which promotes tau pathology through activation of calpains and tau kinases (Rao et al., 2014) (as described in Figure 3).
DS CELL LINES AND TRISOMIC MICE: WINDOWS INTO THE EARLIEST STAGES OF AD AND THE MULTIFACTORIAL ROLES OF APP IN PATHOGENESIS
Beside human postmortem fixed or frozen brain tissue from control or DS individuals, both primary cell lines obtained from human and various trisomy 21 mice are widely used in studies of DS (Choong et al., 2015; Prandini et al., 2007). Human DS and age-matched control fibroblast cell lines and Epstein Barr virus (EBV)-transformed human lymphoblastoid cell lines (LCLs) are widely available. In addition, iPSC cells derived from fibroblast of multiple DS patients (Park et al., 2008; Weick et al., 2013), including fetal fibroblasts of monozygotic twins (Hibaoui et al., 2014), have been differentiated into neurons and astroglia (Chen et al., 2014) for further functional studies.
The most widely used DS mouse models is Ts65Dn [B6EiC3Sn a/A-Ts(1716)65Dn/J], which has triplicate App and other HSA21 analogous genes on mouse chromosome 16 (MMU16) (Davisson et al., 1993). Ts65Dn and other mouse models with various lengths of triplicate genes comparable to human HSA21, with or without App have been reviewed in detail (Choong et al., 2015). Therefore, we would like to focus on the Ts2 mouse (Ts[Rb(12.1716)]2Cje), a model of DS generated by a spontaneous Robertsonian fusion, was characterized by Villar and colleagues (Villar et al., 2005). Ts2 mice possess higher transmission rate of MMU16, and male fertility, resulting in a ~3-fold higher viable offspring compared to Ts65Dn mice (Davisson et al., 1993). Our study (Jiang 2016) showed that Ts2 mice develop a similar regional pattern of APP-dependent neuronal endosomal abnormality (Cataldo et al., 2003), and a similar loss of basal forebrain cholinergic neurons (BFCN) as seen in Ts65Dn mice at the similar age (Davisson and Costa, 1999; Davisson et al., 1990; Granholm et al., 2000; Holtzman et al., 1996). The evidence of endosomal pathology prior to the onset of substantial BFCN loss in Ts2 mice indicates that early endosomal alterations are an early manifestation of pathology and precede BFCN neuron loss. Ts2 and Ts65Dn mice also show similar hippocampal glutamatergic neuronal deficits (Kaur et al., 2014) and similar MRI anomalies (Chen et al., 2009). The breeding advantages of Ts2 mouse over the more extensively used Ts65Dn model recommend it strongly for future studies on the role of APP and additional relevant genes in recapitulating the earliest pathobiology in AD.
Compared to AD mouse models, the triplication of APP in trisomy models of DS is a relatively precise genetic model of DS, as the key features of early pathology mirror the human disease authentically, and have been cross validated in primary cells from DS individuals (Jiang et al., 2016). The utility of DS model mouse systems makes them a valuable resource in addition to existing AD mouse models, which are typically based on the induced expression of mutant forms of APP, PSEN1 or MAPT, but are unfortunately narrow in their recapitulation of a full human AD phenotype. Models based on familial AD-associated genetic mutations are limited in their applicability to sporadic AD, and current APP overexpression models of AD may cause a range of artifactual responses in neurons that obscure the disease effects exerted at endogenous levels of a pathogenic protein (Nilsson et al., 2014). A frequent criticism of common AD mouse models is that high expression of mutant APP induces an accelerated amyloid phenotype which doesn’t fully model the gradual onset of human AD in relationship with ageing, and can mask the detection of earlier changes within the endocytic and lysosomal systems in human AD (Cataldo et al., 2000) due to excessive amyloid deposition and widespread neuronal death early in life. Considering the relevance of endocytic and lysosomal dysfunction in the early stages of AD, and the commonality of these defects in the genetics and pathology of DS and AD, we suggest DS models as useful tools in the study of endocytic-lysosomal defects fundamental to AD pathogenesis.
CONCLUSIONS
Combined with the genetic and neuropathological similarities between AD and Down syndrome, emerging findings indicate that disruption of endocytosis and autophagy are fundamental factors in the pathogenesis of both AD and DS (as summarized in Figure 1). The same endosomal-lysosomal disruptions that occur at a rapid pace during DS (and familial early onset forms of AD) develop at a slower pace during sporadic AD when neuronal ageing becomes a key additional factor in the progressive corruption of endocytosis and autophagy. The ability to characterize these earliest endosomal anomalies in tractable models of the authentic disease (DS fibroblasts and trisomic mice) has greatly facilitated the elucidation of pathogenic APP-dependent mechanisms in the absence of massive amyloid deposition and secondary reactions to it. The continued use of DS cellular and animal models represents an appealing complement to the limitations of models often used in AD research.
Given that effective therapies for AD and DS remain elusive, expansion of drug development pursuits to include potential targets in the endosomal-lysosomal and autophagy pathways may lead to improved therapies for both DS and AD. For example, preclinical research has shown that stimulation of lysosomal CTSD function (via genetic deletion of cystatin B) in mouse models of AD reverses autophagic-lysosomal pathology and improves learning and memory (Yang et al., 2011a). Our group and others also demonstrated improvement of lysosomal function in in vitro AD models using acidic nanoparticles to restore lysosomal acidification (Bourdenx et al., 2016; Lee et al., 2015; Xue et al., 2014). These studies, in conjunction with others (Menzies et al., 2017), demonstrate the potential of targeting endocytic-lysosomal function as a therapeutic avenue in neurodegenerative disease.
Additionally, modulation of βCTF generation has previously been ignored as a therapeutic target in AD and DS, even when clinical trials have been carried out with BACE1 inhibitors developed with the goal of lowering Aβ levels. The complex regulation of BACE1 compared with gamma-secretase, the worsening of cognition seen in clinical trials of gamma-secretase inhibitors that elevate βCTF levels as they lower Aβ levels, and the existence of a “protective mutation” of APP (Hashimoto and Matsuoka, 2014) that acts to reduce βCTF (not only Aβ), and the effects of mutation of PSEN1 and APP which alter βCTF as well as Aβ, are all consistent with the mounting data showing that βCTF is a regulator of endosomal signaling that becomes aberrant at the earliest stages of AD and in DS and is linked to early cholinergic neurodegeneration.
Acknowledgments
The laboratory of R.A.N. is funded by the U.S. National Institutes of Health (NIH); National Institute on Aging (NIA) (P01AG017617-16).
Abbreviations
- AD
Alzheimer Disease
- DS
Down Syndrome
- Aβ
amyloid-β
- HSA21
Chromosome 21
- APP
Amyloid precursor protein
- BACE1/2
β-site APP cleaving enzyme ½
- βCTF
β carboxyl-terminal fragment
- PSEN1
presenilin 1
- APPL1
adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain, and leucine zipper motif
- GEF
guanine nucleotide exchange factor
- GAP
GTPase-activating protein
- GDI
GDP dissociation inhibitor
- GDF
GDI displacement factor
- PI3K/vps34
phosphoinositide 3-kinase
- SYN1
synaptojanin1
- Class C VPS/HOPS
class C vacuolar protein sorting/homotypic fusion and protein sorting
- ESCRT
sorting complexes required for transport
- LBPA
lyso-bisphosphatidic acid
- BFCN
basal forebrain cholinergic neurons
- NGF
nerve growth factor
- Trk
tropomyosin receptor kinase
- ILV
intra-lumenal vesicles
- MVB
multivesicular body
- SNX
sorting nexin
- Vps
vacuolar protein sorting
- AP
autophagosome
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- mTOR
mammalian target of rapamycin
- CTSD
Cathepsin D
- v-ATPase
vacuolar ATPase
- UPR
unfolded protein response
- DYRK1A
Dual specificity tyrosine phosphorylation regulated kinase 1A
- RCAN1
Regulator of calcineurin 1
- SOD1
Superoxide dismutase 1
- CSTB
Cystatin B
- MAPT
Microtubule-associated protein tau
- GSK-3β
Glycogen synthase kinase 3β
- cdk5
cyclin-dependent kinase 5
- LCLs
lymphoblastoid cell lines
- iPSCs
induced pluripotent stem cells
- MMU16
mouse chromosome 16
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arai Y, Ijuin T, Takenawa T, Becker LE, Takashima S. Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev. 2002;24:67–72. doi: 10.1016/s0387-7604(01)00405-3. [DOI] [PubMed] [Google Scholar]
- Ascano M, Richmond A, Borden P, Kuruvilla R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J Neurosci. 2009;29:11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr Opin Cell Biol. 2011;23:452–457. doi: 10.1016/j.ceb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. doi: 10.1186/1750-1326-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohdanowicz M, Balkin DM, De Camilli P, Grinstein S. Recruitment of OCRL and Inpp5B to phagosomes by Rab5 and APPL1 depletes phosphoinositides and attenuates Akt signaling. Mol Biol Cell. 2012;23:176–187. doi: 10.1091/mbc.E11-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, Ginsberg SD, Nixon RA. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–2483. doi: 10.1080/15548627.2016.1239003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdenx M, Daniel J, Genin E, Soria FN, Blanchard-Desce M, Bezard E, Dehay B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy. 2016;12:472–483. doi: 10.1080/15548627.2015.1136769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggia-Prevot V, Fernandez CG, Riordan S, Vetrivel KS, Roseman J, Waters J, Bindokas VP, Vassar R, Thinakaran G. Axonal BACE1 dynamics and targeting in hippocampal neurons: a role for Rab11 GTPase. Mol Neurodegener. 2014;9:1. doi: 10.1186/1750-1326-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Di Domenico F, Swomley AM, Head E, Perluigi M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: overlaps in Down’s syndrome and Alzheimer’s disease brain. Biochem J. 2014;463:177–189. doi: 10.1042/BJ20140772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacquevel M, Aeschbach L, Houacine J, Fraering PC. Alzheimer’s disease-linked mutations in presenilin-1 result in a drastic loss of activity in purified gamma-secretase complexes. PLoS One. 2012;7:e35133. doi: 10.1371/journal.pone.0035133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68:73–86. doi: 10.1016/j.neuron.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas AM, Ardiles AO, Barraza N, Baez-Matus X, Caviedes P. Role of tau protein in neuronal damage in Alzheimer’s disease and Down syndrome. Arch Med Res. 2012;43:645–654. doi: 10.1016/j.arcmed.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Mann DM, Nixon RA. Colocalization of lysosomal hydrolase and beta-amyloid in diffuse plaques of the cerebellum and striatum in Alzheimer’s disease and Down’s syndrome. Journal of neuropathology and experimental neurology. 1996;55:704–715. doi: 10.1097/00005072-199606000-00004. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Mathews PM, Boiteau AB, Hassinger LC, Peterhoff CM, Jiang Y, Mullaney K, Neve RL, Gruenberg J, Nixon RA. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008;173:370–384. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, Nixon RA. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. JNeurosci. 2003;23:6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, Mathews PM, Nixon RA. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. Journal of neuropathology and experimental neurology. 2004;63:821–830. doi: 10.1093/jnen/63.8.821. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cendrowski J, Maminska A, Miaczynska M. Endocytic regulation of cytokine receptor signaling. Cytokine Growth Factor Rev. 2016;32:63–73. doi: 10.1016/j.cytogfr.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Cenini G, Dowling AL, Beckett TL, Barone E, Mancuso C, Murphy MP, Levine H, 3rd, Lott IT, Schmitt FA, Butterfield DA, et al. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta. 2012;1822:130–138. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KA, Suh YH. Pathophysiological roles of amyloidogenic carboxy-terminal fragments of the beta-amyloid precursor protein in Alzheimer’s disease. J Pharmacol Sci. 2005;97:461–471. doi: 10.1254/jphs.cr0050014. [DOI] [PubMed] [Google Scholar]
- Chen C, Jiang P, Xue H, Peterson SE, Tran HT, McCann AE, Parast MM, Li S, Pleasure DE, Laurent LC, et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat Commun. 2014;5:4430. doi: 10.1038/ncomms5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dyakin VV, Branch CA, Ardekani B, Yang D, Guilfoyle DN, Peterson J, Peterhoff C, Ginsberg SD, Cataldo AM, et al. In vivo MRI identifies cholinergic circuitry deficits in a Down syndrome model. Neurobiol Aging. 2009;30:1453–1465. doi: 10.1016/j.neurobiolaging.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209:377–386. doi: 10.1083/jcb.201412046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Kaur G, Mazzella MJ, Morales-Corraliza J, Levy E, Mathews PM. Early endosomal abnormalities and cholinergic neuron degeneration in amyloid-beta protein precursor transgenic mice. J Alzheimers Dis. 2013;34:691–700. doi: 10.3233/JAD-122143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choong XY, Tosh JL, Pulford LJ, Fisher EM. Dissecting Alzheimer disease in Down syndrome using mouse models. Front Behav Neurosci. 2015;9:268. doi: 10.3389/fnbeh.2015.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, Robbins M, Damirji H, Holm IE, Johannsen P, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 2015;130:511–523. doi: 10.1007/s00401-015-1475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev. 2016 doi: 10.1016/j.arr.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JD, Salehi A, Delcroix JD, Howe CL, Belichenko PV, Chua-Couzens J, Kilbridge JF, Carlson EJ, Epstein CJ, Mobley WC. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci U S A. 2001;98:10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosker KE, Segal RA. Neuronal signaling through endocytosis. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, Ripoll C, Mircher C, Grattau Y, Olivomarin JC, et al. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum Mol Genet. 2012;21:3156–3172. doi: 10.1093/hmg/dds142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson MT, Costa AC. Mouse models of Down Syndrome. In: Popko B, editor. Mouse models of human genetic neurological diseases. New York: Plenum; 1999. pp. 297–327. [Google Scholar]
- Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog Clin Biol Res. 1990;360:263–280. [PubMed] [Google Scholar]
- Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT. Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res. 1993;384:117–133. [PubMed] [Google Scholar]
- De Jonghe C, Esselens C, Kumar-Singh S, Craessaerts K, Serneels S, Checler F, Annaert W, Van Broeckhoven C, De Strooper B. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum Mol Genet. 2001;10:1665–1671. doi: 10.1093/hmg/10.16.1665. [DOI] [PubMed] [Google Scholar]
- Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E. Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013;28:725–732. doi: 10.1002/mds.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O’Sullivan C, Dodgson SE, Gygi SP, Amon A, Torres EM. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife. 2014;3:e03023. doi: 10.7554/eLife.03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyts C, Clutter M, Herrera S, Jovanovic N, Goddi A, Parent AT. Loss of presenilin function is associated with a selective gain of APP function. Elife. 2016;5 doi: 10.7554/eLife.15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta. 2013;1832:1249–1259. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore PP, von Zastrow M. Endocytosis, signaling, and beyond. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirac-Svejstrup AB, Sumizawa T, Pfeffer SR. Identification of a GDI displacement factor that releases endosomal Rab GTPases from Rab-GDI. EMBO J. 1997;16:465–472. doi: 10.1093/emboj/16.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditaranto K, Tekirian TL, Yang AJ. Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiology of disease. 2001;8:19–31. doi: 10.1006/nbdi.2000.0364. [DOI] [PubMed] [Google Scholar]
- Doran E, Keator D, Head E, Phelan MJ, Kim R, Totoiu M, Barrio JR, Small GW, Potkin SG, Lott IT. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J Alzheimers Dis. 2017;56:459–470. doi: 10.3233/JAD-160836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felberbaum-Corti M, Cavalli V, Gruenberg J. Capture of the small GTPase Rab5 by GDI: regulation by p38 MAP kinase. Methods Enzymol. 2005;403:367–381. doi: 10.1016/S0076-6879(05)03032-6. [DOI] [PubMed] [Google Scholar]
- Fu X, Yang Y, Xu C, Niu Y, Chen T, Zhou Q, Liu JJ. Retrolinkin cooperates with endophilin A1 to mediate BDNF-TrkB early endocytic trafficking and signaling from early endosomes. Mol Biol Cell. 2011;22:3684–3698. doi: 10.1091/mbc.E11-04-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010a;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, Che S. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Chem Neuroanat. 2011;42:102–110. doi: 10.1016/j.jchemneu.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Mufson EJ, Counts SE, Wuu J, Alldred MJ, Nixon RA, Che S. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2010b;22:631–639. doi: 10.3233/JAD-2010-101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez AE, Munoz VC, Cavieres VA, Bustamante HA, Cornejo VH, Januario YC, Gonzalez I, Hetz C, daSilva LL, Rojas-Fernandez A, et al. Autophagosomes cooperate in the degradation of intracellular C-terminal fragments of the amyloid precursor protein via the MVB/lysosomal pathway. FASEB J. 2017;31:2446–2459. doi: 10.1096/fj.201600713R. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Sanders LA, Crnic LS. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. ExpNeurol. 2000;161:647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol Psychiatry. 2012;71:805–813. doi: 10.1016/j.biopsych.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704. doi: 10.1007/s00401-017-1707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- Haas AK, Fuchs E, Kopajtich R, Barr FA. A GTPase-activating protein controls Rab5 function in endocytic trafficking. Nat Cell Biol. 2005;7:887–893. doi: 10.1038/ncb1290. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Matsuoka M. A mutation protective against Alzheimer’s disease renders amyloid beta precursor protein incapable of mediating neurotoxicity. J Neurochem. 2014;130:291–300. doi: 10.1111/jnc.12717. [DOI] [PubMed] [Google Scholar]
- Head E, Lott IT, Wilcock DM, Lemere CA. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr Alzheimer Res. 2016;13:18–29. doi: 10.2174/1567205012666151020114607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedera P, Alvarado D, Beydoun A, Fink JK. Novel mental retardation-epilepsy syndrome linked to Xp21.1-p11.4. Ann Neurol. 2002;51:45–50. doi: 10.1002/ana.10051. [DOI] [PubMed] [Google Scholar]
- Hibaoui Y, Grad I, Letourneau A, Sailani MR, Dahoun S, Santoni FA, Gimelli S, Guipponi M, Pelte MF, Bena F, et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol Med. 2014;6:259–277. doi: 10.1002/emmm.201302848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Bouras C, Perl DP, Sparks DL, Mehta N, Morrison JH. Age-related distribution of neuropathologic changes in the cerebral cortex of patients with Down’s syndrome. Quantitative regional analysis and comparison with Alzheimer’s disease. Arch Neurol. 1995;52:379–391. doi: 10.1001/archneur.1995.00540280065020. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Santucci D, Kilbridge J, Chua-Couzens J, Fontana DJ, Daniels SE, Johnson RM, Chen K, Sun Y, Carlson E, et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc Natl Acad Sci U S A. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi H, Lippe R, McBride HM, Rubino M, Woodman P, Stenmark H, Rybin V, Wilm M, Ashman K, Mann M, et al. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 1997;90:1149–1159. doi: 10.1016/s0092-8674(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Howe CL, Mobley WC. Signaling endosome hypothesis: A cellular mechanism for long distance communication. J Neurobiol. 2004;58:207–216. doi: 10.1002/neu.10323. [DOI] [PubMed] [Google Scholar]
- Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupalowska A, Pyrzynska B, Miaczynska M. APPL1 regulates basal NF-kappaB activity by stabilizing NIK. J Cell Sci. 2012;125:4090–4102. doi: 10.1242/jcs.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilardi JM, Mochida S, Sheng ZH. Snapin: a SNARE-associated protein implicated in synaptic transmission. Nat Neurosci. 1999;2:119–124. doi: 10.1038/5673. [DOI] [PubMed] [Google Scholar]
- Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iulita MF, Cuello AC. The NGF Metabolic Pathway in the CNS and its Dysregulation in Down Syndrome and Alzheimer’s Disease. Curr Alzheimer Res. 2016;13:53–67. doi: 10.2174/1567205012666150921100030. [DOI] [PubMed] [Google Scholar]
- Ivy GO, Schottler F, Wenzel J, Baudry M, Lynch G. Inhibitors of lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain. Science. 1984;226:985–987. doi: 10.1126/science.6505679. [DOI] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C, Stavrides P, Smiley JF, Ginsberg SD, Mathews PM, et al. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol Aging. 2016;39:90–98. doi: 10.1016/j.neurobiolaging.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaether C, Haass C, Steiner H. Assembly, trafficking and function of gamma-secretase. Neurodegener Dis. 2006;3:275–283. doi: 10.1159/000095267. [DOI] [PubMed] [Google Scholar]
- Kalaidzidis I, Miaczynska M, Brewinska-Olchowik M, Hupalowska A, Ferguson C, Parton RG, Kalaidzidis Y, Zerial M. APPL endosomes are not obligatory endocytic intermediates but act as stable cargo-sorting compartments. J Cell Biol. 2015;211:123–144. doi: 10.1083/jcb.201311117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kaur G, Sharma A, Xu W, Gerum S, Alldred MJ, Subbanna S, Basavarajappa BS, Pawlik M, Ohno M, Ginsberg SD, et al. Glutamatergic transmission aberration: a major cause of behavioral deficits in a murine model of Down’s syndrome. JNeurosci. 2014;34:5099–5106. doi: 10.1523/JNEUROSCI.5338-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay LJ, Smulders-Srinivasan TK, Soundararajan M. Understanding the Multifaceted Role of Human Down Syndrome Kinase DYRK1A. Adv Protein Chem Struct Biol. 2016;105:127–171. doi: 10.1016/bs.apcsb.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Kenessey A, Nacharaju P, Ko LW, Yen SH. Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J Neurochem. 1997;69:2026–2038. doi: 10.1046/j.1471-4159.1997.69052026.x. [DOI] [PubMed] [Google Scholar]
- Ketterer S, Gomez-Auli A, Hillebrand LE, Petrera A, Ketscher A, Reinheckel T. Inherited diseases caused by mutations in cathepsin protease genes. FEBS J. 2017;284:1437–1454. doi: 10.1111/febs.13980. [DOI] [PubMed] [Google Scholar]
- Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, Jiang Y, Nixon RA. Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol Psychiatry. 2015 doi: 10.1038/mp.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Ochi H, Fukuda T, Bokuda K, Kurosawa H, Yoshida N, et al. The role of individual domains and the significance of shedding of ATP6AP2/(pro)renin receptor in vacuolar H(+)-ATPase biogenesis. PLoS One. 2013;8:e78603. doi: 10.1371/journal.pone.0078603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Ohsawa Y, Nakanishi H, Koga T, Kametaka S, Waguri S, Momoi T, Kominami E, Peters C, et al. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol Cell Neurosci. 2003;22:146–161. doi: 10.1016/s1044-7431(03)00035-6. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106:12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korvatska O, Strand NS, Berndt JD, Strovas T, Chen DH, Leverenz JB, Kiianitsa K, Mata IF, Karakoc E, Greenup JL, et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS) Hum Mol Genet. 2013;22:3259–3268. doi: 10.1093/hmg/ddt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko E, Poghosyan A, Kaminsky Y. Subcellular compartmentalization of proteolytic enzymes in brain regions and the effects of chronic beta-amyloid treatment. Brain Res. 2011;1369:184–193. doi: 10.1016/j.brainres.2010.10.078. [DOI] [PubMed] [Google Scholar]
- Kurashige T, Takahashi T, Yamazaki Y, Hiji M, Izumi Y, Yamawaki T, Matsumoto M. Localization of CHMP2B-immunoreactivity in the brainstem of Lewy body disease. Neuropathology. 2013;33:237–245. doi: 10.1111/j.1440-1789.2012.01346.x. [DOI] [PubMed] [Google Scholar]
- Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, Pradier L, Hugon J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J Neurochem. 2005;94:215–225. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- Lai F, Kammann E, Rebeck GW, Anderson A, Chen Y, Nixon RA. APOE genotype and gender effects on Alzheimer disease in 100 adults with Down syndrome. Neurology. 1999;53:331–336. doi: 10.1212/wnl.53.2.331. [DOI] [PubMed] [Google Scholar]
- Laifenfeld D, Patzek LJ, McPhie DL, Chen Y, Levites Y, Cataldo AM, Neve RL. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci. 2007;27:7141–7153. doi: 10.1523/JNEUROSCI.4599-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, Fraser P, St-George-Hyslop P, Le Thuc O, Espin V, et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci. 2012;32:16243–16255a. doi: 10.1523/JNEUROSCI.2775-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, Checler F. Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016;132:257–276. doi: 10.1007/s00401-016-1577-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JHH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015;12:1430–1444. doi: 10.1016/j.celrep.2015.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehigh KM, West KM, Ginty DD. Retrogradely Transported TrkA Endosomes Signal Locally within Dendrites to Maintain Sympathetic Neuron Synapses. Cell Rep. 2017;19:86–100. doi: 10.1016/j.celrep.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverenz JB, Raskind MA. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol. 1998;150:296–304. doi: 10.1006/exnr.1997.6777. [DOI] [PubMed] [Google Scholar]
- Li J, Mao X, Dong LQ, Liu F, Tong L. Crystal structures of the BAR-PH and PTB domains of human APPL1. Structure. 2007;15:525–533. doi: 10.1016/j.str.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272:4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- Lin DC, Quevedo C, Brewer NE, Bell A, Testa JR, Grimes ML, Miller FD, Kaplan DR. APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol Cell Biol. 2006;26:8928–8941. doi: 10.1128/MCB.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:14164–14169. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lamb D, Chou MM, Liu YJ, Li G. Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol Biol Cell. 2007;18:1375–1384. doi: 10.1091/mbc.E06-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo LS, Tang N, Al-Haddawi M, Dawe GS, Hong W. A role for sorting nexin 27 in AMPA receptor trafficking. Nat Commun. 2014;5:3176. doi: 10.1038/ncomms4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9:623–633. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- Malkus KA, Tsika E, Ischiropoulos H. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: how neurons are lost in the Bermuda triangle. Mol Neurodegener. 2009;4:24. doi: 10.1186/1750-1326-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangieri LR, Mader BJ, Thomas CE, Taylor CA, Luker AM, Tse TE, Huisingh C, Shacka JJ. ATP6V0C knockdown in neuroblastoma cells alters autophagy-lysosome pathway function and metabolism of proteins that accumulate in neurodegenerative disease. PLoS One. 2014;9:e93257. doi: 10.1371/journal.pone.0093257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, Ryan TA. The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron. 2007;56:1004–1018. doi: 10.1016/j.neuron.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- Martin SB, Dowling AL, Lianekhammy J, Lott IT, Doran E, Murphy MP, Beckett TL, Schmitt FA, Head E. Synaptophysin and synaptojanin-1 in Down syndrome are differentially affected by Alzheimer’s disease. J Alzheimers Dis. 2014;42:767–775. doi: 10.3233/JAD-140795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiace LA, Kress Y, Davies P, Ksiezak-Reding H, Yen SH, Dickson DW. Ubiquitin-immunoreactive dystrophic neurites in Down’s syndrome brains. J Neuropathol Exp Neurol. 1991;50:547–559. doi: 10.1097/00005072-199109000-00003. [DOI] [PubMed] [Google Scholar]
- McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer’s disease: partners in crime. Biochem Soc Trans. 2013;41:1495–1502. doi: 10.1042/BST20130201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP Transgenic Mice. J Biol Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- McGuire C, Stransky L, Cotter K, Forgac M. Regulation of V-ATPase activity. Front Biosci (Landmark Ed) 2017;22:609–622. doi: 10.2741/4506. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 2017;93:1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- Miaczynska M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2013;5:a009035. doi: 10.1101/cshperspect.a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Milenkovic I, Jarc J, Dassler E, Aronica E, Iyer A, Adle-Biassette H, Scharrer A, Reischer T, Hainfellner JA, Kovacs GG. The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down syndrome. Neuropathol Appl Neurobiol. 2017 doi: 10.1111/nan.12406. [DOI] [PubMed] [Google Scholar]
- Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- Nakanishi H. Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res Rev. 2003;2:367–381. doi: 10.1016/s1568-1637(03)00027-8. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Saito T, Saido TC. New mouse model of Alzheimer’s. ACS Chem Neurosci. 2014;5:499–502. doi: 10.1021/cn500105p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiology of aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. Journal of cell science. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J. 2017;31:2729–2743. doi: 10.1096/fj.201700359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM. The lysosomal system in neuronal cell death: a review. Annals of the New York Academy of Sciences. 1993;679:87–109. doi: 10.1111/j.1749-6632.1993.tb18291.x. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Jiang Y, Sato Y, Im E, Bordi M, Colacurcio DJ, Pensalfini A, Rigoglioso A, Mohan P. APP-beta CTF-mediated pathological endosome signaling also disrupts lysosomal function in Down syndrome (DS) Paper presented at: 13th International Congress on Alzheimers and Parkinsons Disease 2017 [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiology of Disease. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012;26:2696–2708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano G, Castello G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol. 2012;724:291–299. doi: 10.1007/978-1-4614-0653-2_22. [DOI] [PubMed] [Google Scholar]
- Pal R, Bajaj L, Sharma J, Palmieri M, Di Ronza A, Lotfi P, Chaudhury A, Neilson J, Sardiello M, Rodney GG. NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson’s disease. Sci Rep. 2016;6:22866. doi: 10.1038/srep22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Myers RM. Isolation and characterization of the mouse cystatin B gene. Genome Res. 1996;6:1103–1109. doi: 10.1101/gr.6.11.1103. [DOI] [PubMed] [Google Scholar]
- Pera M, Alcolea D, Sanchez-Valle R, Guardia-Laguarta C, Colom-Cadena M, Badiola N, Suarez-Calvet M, Llado A, Barrera-Ocampo AA, Sepulveda-Falla D, et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013;125:201–213. doi: 10.1007/s00401-012-1062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol. 2016;32:223–253. doi: 10.1146/annurev-cellbio-111315-125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M, di Domenico F, Fiorini A, Cocciolo A, Giorgi A, Foppoli C, Butterfield DA, Giorlandino M, Giorlandino C, Schinina ME, et al. Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteomics Clin Appl. 2011;5:167–178. doi: 10.1002/prca.201000121. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, Head E, Butterfield DA, Di Domenico F. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta. 2014;1842:1144–1153. doi: 10.1016/j.bbadis.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippidou P, Valdez G, Akmentin W, Bowers WJ, Federoff HJ, Halegoua S. Trk retrograde signaling requires persistent, Pincher-directed endosomes. Proc Natl Acad Sci U S A. 2011;108:852–857. doi: 10.1073/pnas.1015981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter K, Nallathambi J, Lin Y, Liton PB. Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells: implications for glaucoma pathogenesis. Autophagy. 2013;9:581–594. doi: 10.4161/auto.23568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteryaev D, Fares H, Bowerman B, Spang A. Caenorhabditis elegans SAND-1 is essential for RAB-7 function in endosomal traffic. EMBO J. 2007;26:301–312. doi: 10.1038/sj.emboj.7601498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandini P, Deutsch S, Lyle R, Gagnebin M, Delucinge Vivier C, Delorenzi M, Gehrig C, Descombes P, Sherman S, Dagna Bricarelli F, et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet. 2007;81:252–263. doi: 10.1086/519248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, Butler AC. Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann Neurol. 1998;43:380–383. doi: 10.1002/ana.410430316. [DOI] [PubMed] [Google Scholar]
- Prasher VP, Sajith SG, Rees SD, Patel A, Tewari S, Schupf N, Zigman WB. Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer’s disease and mortality in persons with Down syndrome. Int J Geriatr Psychiatry. 2008;23:1134–1140. doi: 10.1002/gps.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrzynska B, Pilecka I, Miaczynska M. Endocytic proteins in the regulation of nuclear signaling, transcription and tumorigenesis. Mol Oncol. 2009;3:321–338. doi: 10.1016/j.molonc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- Rao MV, McBrayer MK, Campbell J, Kumar A, Hashim A, Sershen H, Stavrides PH, Ohno M, Hutton M, Nixon RA. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:9222–9234. doi: 10.1523/JNEUROSCI.1132-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Sureda V, Vilches A, Sanchez O, Audi L, Dominguez C. Intracellular oxidant activity, antioxidant enzyme defense system, and cell senescence in fibroblasts with trisomy 21. Oxid Med Cell Longev. 2015;2015:509241. doi: 10.1155/2015/509241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P, Bonifacino JS. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J Cell Biol. 2008;183:513–526. doi: 10.1083/jcb.200804048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Galan AK, Xin X, Dong F, Abdul-Ghani MA, Zhou L, Wang C, Li C, Holmes BM, Sloane LB, et al. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep. 2014;7:1227–1238. doi: 10.1016/j.celrep.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Saxena S, Bucci C, Weis J, Kruttgen A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J Neurosci. 2005;25:10930–10940. doi: 10.1523/JNEUROSCI.2029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwagerl AL, Mohan PS, Cataldo AM, Vonsattel JP, Kowall NW, Nixon RA. Elevated levels of the endosomal-lysosomal proteinase cathepsin D in cerebrospinal fluid in Alzheimer disease. Journal of neurochemistry. 1995;64:443–446. doi: 10.1046/j.1471-4159.1995.64010443.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Klocke BJ, Young C, Shibata M, Olney JW, Uchiyama Y, Saftig P, Roth KA. Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J Neurosci. 2007;27:2081–2090. doi: 10.1523/JNEUROSCI.5577-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Deppmann CD, Harrington AW, St Hillaire C, Chen ZY, Lee FS, Ginty DD. Long-distance control of synapse assembly by target-derived NGF. Neuron. 2010;67:422–434. doi: 10.1016/j.neuron.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci. 2015;16:126–132. doi: 10.1038/nrn3896. [DOI] [PubMed] [Google Scholar]
- Solinger JA, Spang A. Tethering complexes in the endocytic pathway: CORVET and HOPS. FEBS J. 2013;280:2743–2757. doi: 10.1111/febs.12151. [DOI] [PubMed] [Google Scholar]
- Song MS, Baker GB, Todd KG, Kar S. Inhibition of beta-amyloid1-42 internalization attenuates neuronal death by stabilizing the endosomal-lysosomal system in rat cortical cultured neurons. Neuroscience. 2011;178:181–188. doi: 10.1016/j.neuroscience.2010.12.055. [DOI] [PubMed] [Google Scholar]
- Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg F, Gallon M, Winfield M, Thomas EC, Bell AJ, Heesom KJ, Tavare JM, Cullen PJ. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 2013;15:461–471. doi: 10.1038/ncb2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Vitale G, Ullrich O, Zerial M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 1995;83:423–432. doi: 10.1016/0092-8674(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. 2016;32:22–37. doi: 10.1016/j.arr.2016.04.010. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Tamayev R, Matsuda S, Arancio O, D’Adamio L. beta- but not gamma-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med. 2012;4:171–179. doi: 10.1002/emmm.201100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammineni P, Ye X, Feng T, Aikal D, Cai Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife. 2017;6 doi: 10.7554/eLife.21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanikawa S, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K. Endosomal sorting related protein CHMP2B is localized in Lewy bodies and glial cytoplasmic inclusions in alpha-synucleinopathy. Neurosci Lett. 2012;527:16–21. doi: 10.1016/j.neulet.2012.08.035. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatti M, Motta M, Di Bartolomeo S, Cianfanelli V, Salvioli R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy. 2013;9:241–243. doi: 10.4161/auto.22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin P, Lauffer B, Jager S, Cimermancic P, Krogan NJ, von Zastrow M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. 2011;13:715–721. doi: 10.1038/ncb2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Tramutola A, Di Domenico F, Barone E, Giorgi A, Di Francesco L, Schinina E, Coccia R, Arena A, Head E, Butterfield DA, et al. Poly-Ubiquitinylation Profile in Down Syndrome Brain before and after the Development of Alzheimer Neuropathology. Antioxid Redox Signal. 2016 doi: 10.1089/ars.2016.6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133:739–749. doi: 10.1111/jnc.13037. [DOI] [PubMed] [Google Scholar]
- Tyynela J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, Baumann M, Haltia M, Lobel P. A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J. 2000;19:2786–2792. doi: 10.1093/emboj/19.12.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011;89:1031–1042. doi: 10.1002/jnr.22640. [DOI] [PubMed] [Google Scholar]
- van der Kant R, Goldstein LS. Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell. 2015;32:502–515. doi: 10.1016/j.devcel.2015.01.022. [DOI] [PubMed] [Google Scholar]
- Varsano T, Dong MQ, Niesman I, Gacula H, Lou X, Ma T, Testa JR, Yates JR, 3rd, Farquhar MG. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol. 2006;26:8942–8952. doi: 10.1128/MCB.00305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ. Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(171)65Dn and mouse chromosome 12. Mamm Genome. 2005;16:79–90. doi: 10.1007/s00335-004-2428-7. [DOI] [PubMed] [Google Scholar]
- Vinicia AP, Hongmei L, Heidi MS, Baiping W, Li Y, Yin X, Daniel BS, Michela P, Alberto R, Virginia MYL, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Molecular Medicine. 2014;6:1142–1160. doi: 10.15252/emmm.201303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, et al. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc Natl Acad Sci U S A. 2008;105:9415–9420. doi: 10.1073/pnas.0803756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandinger-Ness A, Zerial M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol. 2014;6:a022616. doi: 10.1101/cshperspect.a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CW, Stromhaug PE, Kauffman EJ, Weisman LS, Klionsky DJ. Yeast homotypic vacuole fusion requires the Ccz1-Mon1 complex during the tethering/docking stage. J Cell Biol. 2003;163:973–985. doi: 10.1083/jcb.200308071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, Loo LS, Cai L, Thompson RC, Yang B, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Floor E. Hydrogen peroxide inhibits the vacuolar H+-ATPase in brain synaptic vesicles at micromolar concentrations. J Neurochem. 1998;70:646–652. doi: 10.1046/j.1471-4159.1998.70020646.x. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Kaczmarski W, Barua M, Kuchna I, Nowicki K, Wang KC, Wegiel J, Yang SM, Frackowiak J, Mazur-Kolecka B, et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J Neuropathol Exp Neurol. 2011;70:36–50. doi: 10.1097/NEN.0b013e318202bfa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP, Held DL, Bonadurer GF, 3rd, Doers ME, Liu Y, Maguire C, Clark A, Knackert JA, Molinarolo K, Musser M, et al. Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci U S A. 2013;110:9962–9967. doi: 10.1073/pnas.1216575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VL, Fisher EM, Strydom A. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16:564–574. doi: 10.1038/nrn3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman FK, Alford KA, Tybulewicz VL, Fisher EM. Down syndrome–recent progress and future prospects. Hum Mol Genet. 2009;18:R75–83. doi: 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology. 1985;35:957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- Wolfe DM, Lee JHH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. The European journal of neuroscience. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G, Reyna SM, Dunlap M, Van Der Kant R, Callender JA, Young JE, Roberts EA, Goldstein LS. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016;17:759–773. doi: 10.1016/j.celrep.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Weissmiller AM, White JA, 2nd, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016;126:1815–1833. doi: 10.1172/JCI82409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Wang LRR, Sato Y, Jiang Y, Berg M, Yang DSS, Nixon RA, Liang XJJ. Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer’s disease. Nano letters. 2014;14:5110–5117. doi: 10.1021/nl501839q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Zhang J, Koshiishi I, Sastradipura Suniarti DF, Wu Z, Peters C, Schwake M, Uchiyama Y, Kira J, Saftig P, et al. Involvement of lysosomal storage-induced p38 MAP kinase activation in the overproduction of nitric oxide by microglia in cathepsin D-deficient mice. Mol Cell Neurosci. 2007;35:573–584. doi: 10.1016/j.mcn.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Takahashi T, Hiji M, Kurashige T, Izumi Y, Yamawaki T, Matsumoto M. Immunopositivity for ESCRT-III subunit CHMP2B in granulovacuolar degeneration of neurons in the Alzheimer’s disease hippocampus. Neurosci Lett. 2010;477:86–90. doi: 10.1016/j.neulet.2010.04.038. [DOI] [PubMed] [Google Scholar]
- Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Aβ1-42 pathogenesis. Journal of Neuroscience Research. 1998 doi: 10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Yang DSS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, Nixon RA. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. The American journal of pathology. 2008;173:665–681. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DSS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain: a journal of neurology. 2011a;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DSS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson DW, Bandyopadhyay U, Jiang Y, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011b;7:788–789. doi: 10.4161/auto.7.7.15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DSS, Stavrides P, Saito M, Kumar A, Rodriguez-Navarro JA, Pawlik M, Huo C, Walkley SU, Saito M, Cuervo AM, et al. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain: a journal of neurology. 2014;137:3300–3318. doi: 10.1093/brain/awu278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Jin N, Shi J, Zhang Y, Wu Y, Gong CX, Iqbal K, Liu F. Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci Rep. 2017;7:619. doi: 10.1038/s41598-017-00682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zana M, Janka Z, Kalman J. Oxidative stress: a bridge between Down’s syndrome and Alzheimer’s disease. Neurobiol Aging. 2007;28:648–676. doi: 10.1016/j.neurobiolaging.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Zana M, Szecsenyi A, Czibula A, Bjelik A, Juhasz A, Rimanoczy A, Szabo K, Vetro A, Szucs P, Varkonyi A, et al. Age-dependent oxidative stress-induced DNA damage in Down’s lymphocytes. Biochem Biophys Res Commun. 2006;345:726–733. doi: 10.1016/j.bbrc.2006.04.167. [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- Zhang D, Isack NR, Glodowski DR, Liu J, Chen CC, Xu XZ, Grant BD, Rongo C. RAB-6.2 and the retromer regulate glutamate receptor recycling through a retrograde pathway. J Cell Biol. 2012;196:85–101. doi: 10.1083/jcb.201104141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Eitan E, Mattson MP. Early Involvement of Lysosome Dysfunction in the Degeneration of Cerebral Cortical Neurons Caused by the Lipid Peroxidation Product 4-Hydroxynonenal. J Neurochem. 2017 doi: 10.1111/jnc.13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Chen Y, Liu Y, Zhao Y, Liao FF, Xu H. APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS One. 2013;8:e80571. doi: 10.1371/journal.pone.0080571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang T, Wang S, Gong Z, Tang C, Chen J, Ding J. Molecular mechanism for Rabex-5 GEF activation by Rabaptin-5. Elife. 2014;3 doi: 10.7554/eLife.02687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Scott SA, Shelton SB, Crutcher KA. Cathepsin D-mediated proteolysis of apolipoprotein E: possible role in Alzheimer’s disease. Neuroscience. 2006;143:689–701. doi: 10.1016/j.neuroscience.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Zhu G, Chen J, Liu J, Brunzelle JS, Huang B, Wakeham N, Terzyan S, Li X, Rao Z, Li G, et al. Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5. EMBO J. 2007;26:3484–3493. doi: 10.1038/sj.emboj.7601771. [DOI] [PMC free article] [PubMed] [Google Scholar]