

Abstract

A BioFocus DPI SoftFocus library of ∼35 000 compounds was screened against Mycobacterium tuberculosis (Mtb) in order to identify novel hits with antitubercular activity. The hits were evaluated in biology triage assays to exclude compounds suggested to function via frequently encountered promiscuous mechanisms of action including inhibition of the QcrB subunit of the cytochrome bc1 complex, disruption of cell–wall homeostasis, and DNA damage. Among the hits that passed this screening cascade, a 6-dialkylaminopyrimidine carboxamide series was prioritized for hit to lead optimization. Compounds from this series were active against clinical Mtb strains, while no cross-resistance to conventional antituberculosis drugs was observed. This suggested a novel mechanism of action, which was confirmed by chemoproteomic analysis leading to the identification of BCG_3193 and BCG_3827 as putative targets of the series with unknown function. Initial structure–activity relationship studies have resulted in compounds with moderate to potent antitubercular activity and improved physicochemical properties.
Introduction
Tuberculosis (TB) is one of the world’s most deadly infectious diseases, leading to 1.4 million deaths in 2015.1 The disease is caused by Mycobacterium tuberculosis (Mtb) and primarily affects lower to middle income countries such as those in sub-Saharan Africa. A standard first-line four-drug regimen of rifampin, isoniazid, pyrazinamide, and ethambutol is used to treat drug-susceptible TB.2 Although these drugs form the core of TB treatment regimens, they have become ineffective against resistant Mtb strains. TB that does not respond to at least isoniazid and rifampin, the two frontline drugs, is defined as multidrug resistant (MDR) TB. MDR TB that develops additional resistance to any fluoroquinolone and at least one of the three injectable second-line drugs (amikacin, kanamycin, or capreomycin) is referred to as extensively drug resistant (XDR) TB and is virtually untreatable.3 One of the main drivers behind the rise of resistance is the 6-month-long treatment required for drug-sensitive disease, resulting in poor patient adherence. Thus, the main goal of many TB drug discovery programs is the identification of compounds that will potentially contribute to treatment shortening, albeit there is no clear evidence as to how this can be accomplished.4 One possible approach is to identify compounds that inhibit novel targets and do not show cross-resistance to currently used anti-TB drugs. Various novel drug combinations can then be evaluated for their potential toward treatment shortening while establishing clinical effectiveness. Identifying compounds that exert their anti-TB effect through novel targets is not a trivial process. This is exemplified by the approval in 2012 of bedaquiline, the first new drug approved for the treatment of TB in more than 40 years with a novel mechanism of action (MOA) that involves inhibition of the mycobacterial ATP synthase.5−7
Two hit-generation approaches that have been traditionally used are target-based and whole-cell screening.8,9 Historically, there has been extremely limited success with the target-based approach owing to the lack of translation from target activity to whole-cell activity. Nevertheless, efforts to discover and validate novel drug targets using various approaches must continue. This is exemplified by the recently reported validation of CoaBC as a bactericidal target using a chemical biology approach.10 On the other hand, a more successful approach has been phenotypic whole-cell high-throughput screening (HTS) of chemical libraries to identify novel chemotypes whose target is then identified retrospectively. There are several compounds in the TB drug pipeline that have been identified through whole-cell HTS. Examples include Q203 (Figure 1), an inhibitor of the QcrB subunit of the cytochrome bc1 complex which is involved in electron transport.11 Another example is SQ109 (Figure 1) whose precise MOA remains to be elucidated but is believed to involve the mycobacterial membrane protein large 3, MmpL3, a mycolic acid transporter involved in cell wall synthesis,.12,13 Similarly, some inhibitors of decaprenylphosphoryl-β-d-ribose 2′-epimerase (DprE1), e.g., pBTZ,14 and azaindoles TBA737115,16 are poised to enter clinical trials (Figure 1). While these compounds show promise as new anti-TB agents, their targets are yet to be clinically validated. This necessitates the need to continuously identify new chemical matter with alternative novel MOAs toward populating the TB drug pipeline.
Figure 1.

New TB drug development candidates identified from whole cell screening.
In line with this, we initiated a HTS campaign using a diverse SoftFocus compound library17 acquired from BioFocus and complemented this with a biology triage process that aimed specifically to exclude compounds acting on targets such as QcrB and the cell wall (e.g., MmpL3 and DprE1), as well as DNA damaging agents. The rationale was to exclude targets known for their promiscuity, with a large proportion of HTS hits having these as part of their MOA.18,19 There is also the question around whether or not compounds targeting cell wall metabolism can contribute to the much sought after treatment shortening.19 Compounds that induce DNA damage were also excluded in order to avoid general cytotoxicity, as well as targets such as DNA gyrase whose clinically used inhibitors are exemplified by the fluoroquinolones. Along with an aminopyrazolo[1,5-a]pyrimidine chemical series,20 a 6-dialkylaminopyrimidine carboxamide scaffold was identified as a hit series with a potentially novel MOA, based on the initial biology triage process (Figure 2). Herein, we describe the synthesis, structure–activity relationship (SAR), in vitro absorption, distribution, metabolism, and excretion (ADME), and in vivo pharmacokinetic (PK) and biological profiles of this chemical series.
Figure 2.

Compound 1 and biology triage data.
Results and Discussion
Phenotypic Whole-Cell HTS and Biology Triage
A screen of a BioFocus DPI SoftFocus library of ∼35 000 compounds against virulent Mtb H37Rv conducted at the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIAID/NIH, U.S.) led to a number of confirmed hits, which included compound 1 with a moderate minimum inhibitory concentration (MIC) value of 20 μM in albumin–dextrose complex (ADC) medium containing ∼0.4% bovine serum albumin (BSA). The compound was slightly more potent in GAST-Fe (glycerol-alanine-salts)20 minimal medium, likely driven by the absence of BSA which allowed for higher free concentration of the compound under the assay conditions. Hence, GAST-Fe was used alongside the 7H9/ADC medium during further SAR exploration of the series. Compound 1 was tested in the three biology triage assays in order to assess activity against promiscuous targets. The compound did not show hypersensitivity against a cytochrome bd oxidase knockout mutant strain (cydKO)21,22 and also retained activity against a QcrB mutant (qcrBA317T), thereby eliminating this as potential target. Compound 1 also did not elicit a positive response in two standard bioluminescence reporter assays: PiniB-LUX,23 which is designed to detect compounds targeting Mtb cell wall biosynthesis, and PrecA-LUX,23 which allows detection of genotoxic compounds (data in Supporting Information). Similar results were obtained with other analogues in the series obtained during SAR studies (data in Supporting Information). These results, along with chemoproteomics and cross-resistance studies (see biology results below), indicate that the 6-dialkylaminopyrimidine carboxamide series of compounds exemplified by compound 1 possess a novel MOA that is not shared with known TB drugs.
SAR Exploration Strategy
Compound 1 was identified as a suitable hit with a moderate minimum inhibitory concentration (MIC99) of 20 μM in 7H9/ADC medium, low toxicity against the mammalian Vero cell line (IC50 of 287 μM), yet poor kinetic solubility (KS, <5 μM). The poor solubility was assumed to be related to the high lipophilicity and flat aromatic character of the molecule. Since there were very few active near neighbors identified from the screen, very limited information was available on further optimization scope with the scaffold to improve anti-TB activity while addressing the poor physicochemical properties and potential safety issues that may be associated with the scaffold. A detailed SAR plan as summarized in Figure 3 was embarked upon. The molecule was divided into three parts for convenience: the central pyrimidine core, right-hand side (RHS) amide, and left-hand side (LHS) hydrophobic N-benzyl moiety (Figure 3). The major SAR exploration focus was on understanding key hydrogen bond (HB) donor–acceptor interactions critical for activity, shape, and size of the central core, and scope for substitution/modification on either the RHS or LHS parts of the molecule to improve potency. The scope for addition of heteroatoms or polar groups into the molecule was also explored with the aim of reducing lipophilicity along with addressing related issues such as poor physicochemical properties and structure alerts such as the presence of potentially AMES positive anilines upon cleavage of the amide group.
Figure 3.

SAR exploration strategy.
Synthesis
Compounds 1–20 were synthesized to explore the SAR around the N-methyl-1-phenylmethanamine motif (SAR on LHS, Table 1). Compounds 21–30 were synthesized to explore the central pyrimidine core of the scaffold (SAR on central core, Table 2), and compounds 31–54 were synthesized to explore the SAR around the amide portion of the scaffold (SAR on RHS, Table 3). In general, target compounds were synthesized using either one of two routes. Scheme 1 was used to synthesize an advanced intermediate with a specific amide in place allowing for the exploration of the LHS of the molecule. The same route was also used to synthesize several of the core modifications. The route involved a straightforward two-step procedure starting from a chloro-heterocyclic-carboxylic acid, where coupling with corresponding amine using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) was first performed, followed by nucleophilic displacement of the relevant chloro group by an appropriate amine. Thus, synthesis of compounds 1–20 and 39 (Table 1) was achieved by coupling of 6-chloropyrimidine-4-carboxylic acid with aniline, 4-methoxyaniline, and 4-aminopyridine to give intermediates 55, 56, and 57, respectively, followed by displacement of 6-chloro group with various amines. Compound 21 was synthesized by coupling of 4-chloropicolinic acid with aniline to form intermediate 58 followed by reaction with 1-(4-fluorophenyl)-N-methylmethanamine.
Table 1. SAR on the LHS.
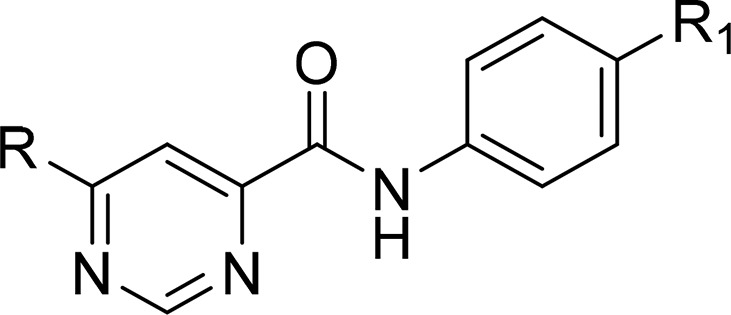
Kinetic solubility at pH 7.4.
IC50 on Vero cell lines.
MIC99 from GFP strain based assay. ND: not determined.
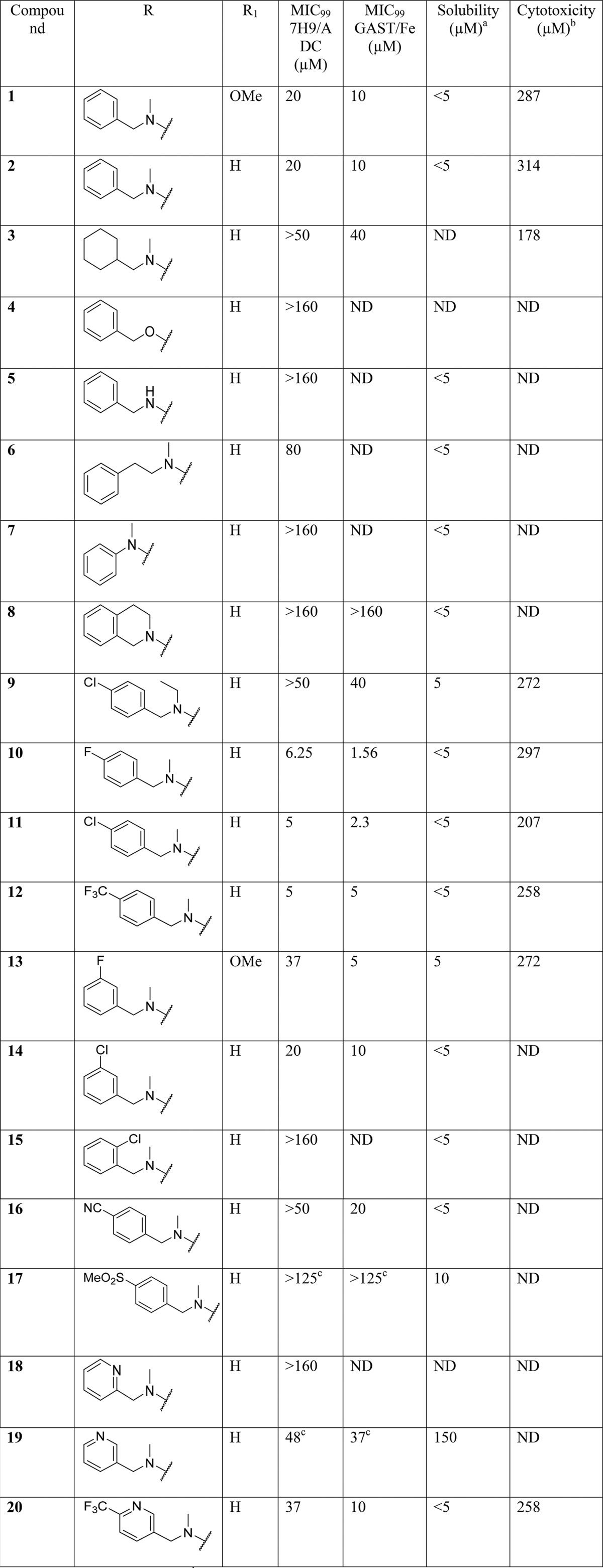
Table 2. SAR on the Central Core.
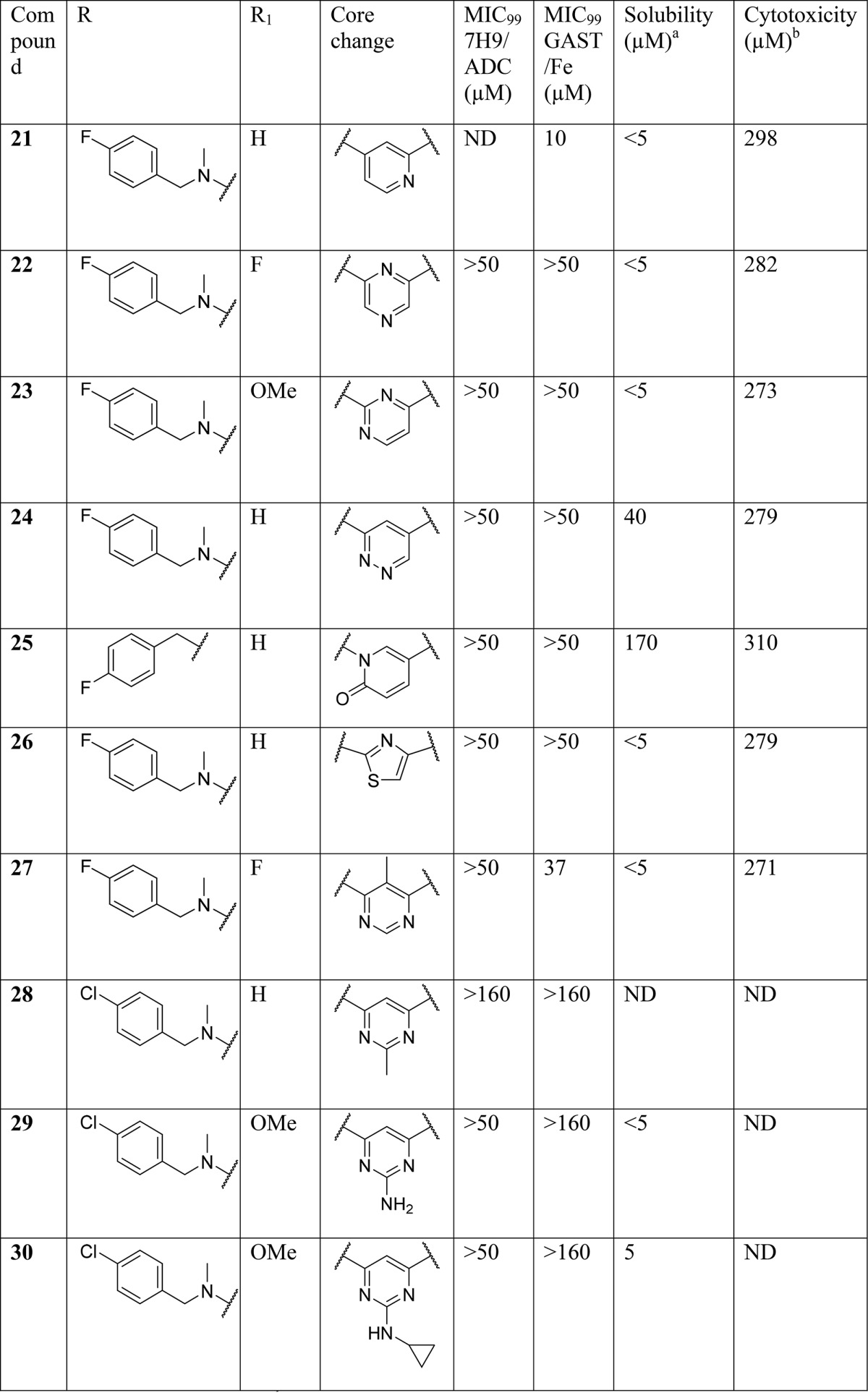
Kinetic solubility at pH 7.4.
IC50 on Vero cell lines. ND: not determined.
Table 3. SAR on the RHS.
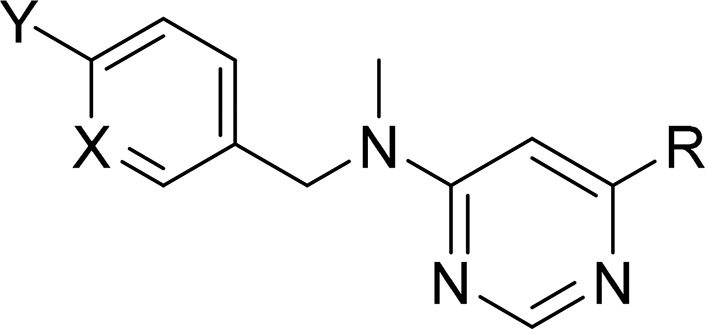

Kinetic solubility at pH 7.4.
IC50 on Vero cell lines.
MIC99 from Mtb-GFP strain based assay. ND: not determined.
Scheme 1. General Route for the Synthesis of 6-Aminopyrimidine-4-carboxamides and Alternative Cores.

Reagents and conditions: (i) amine, DIPEA, HATU, DMF, 25 °C; (ii) amine, i-PrOH, DIPEA, 100 °C.
Synthesis of compounds 22–26 (Table 2) with changes in the central core ring are summarized in Scheme 2. Compound 22 was synthesized by nucleophilic displacement of 59 with 1-(4-fluorophenyl)-N-methylmethanamine followed by carbamoylation according to the procedure described by Ren et al.24 Compound 23 was synthesized in a similar manner except the initial nucleophilic displacement step of 60 resulted in a mixture of the intended 4-chloro-N-(4-fluorobenzyl)-N-methylpyrimidin-2-amine (61) as well as the 2-chloro regioisomer in a 1:2 ratio. The pyridazine core replacement analogue was synthesized by heating 6-oxo-1,6-dihydropyridazine-4-carboxylic acid with POCl3 followed by quenching of the 6-chloropyridazine-4-carbonyl chloride intermediate with aniline to afford 63. Intermediate 63 was subsequently subjected to nucleophilic displacement with 1-(4-fluorophenyl)-N-methylmethanamine under microwave heating to deliver the final compound 24. The pyridone analogue (25) was synthesized from the commercially available precursor, methyl coumalate (64), which was reacted with 4-fluorobenzylamine at room temperature (25 °C) to give the N-substituted pyridinone ester (65). The ester was then hydrolyzed using LiOH followed by an amide coupling to afford 25. The thiazole analogue 26 was synthesized by reductive amination of methyl 2-aminothiazole-4-carboxylate 66 with 4-fluorobenzaldehyde and NaBH4, followed by methylation under basic conditions to afford 67. Intermediate 67 was then subjected to the same hydrolysis and amide coupling conditions as 65 to afford 26.
Scheme 2. Synthetic Routes Used To Access Core Modifications.

Reagents and conditions: (i) 1-(4-fluorophenyl)-N-methylmethanamine, Et3N, dioxane, 60 °C; (ii) aniline, Mo(CO)6, Et3N, TBAC, toluene, tetraglyme, 150 °C; (iii) (a) POCl3, 110 °C; (b) aniline, Et3N; (iv) 4-fluorobenzylamine, Et3N, dioxane, microwave, 125 °C; (v) 4-fluorobenzylamine, H2O/EtOH (1.5:1), 25 °C; (vi) LiOH, MeOH; (vii) aniline, HATU, DMF, Et3N; (viii) 4-fluorobenzaldehyde, NaBH4, EtOH; (ix) MeI, K2CO3, DMF.
The 2- and 5-methylpyrimidine analogues 27 and 28 (Table 2) were synthesized in a similar fashion by condensation of the appropriate 1,4-diethyl ester and imidamide 68 and 69 to form the corresponding dihydropyrimidine carboxylic ester and acid intermediates 70 and 71, respectively (Scheme 3). Intermediate 70 was chlorinated using SOCl2 followed by nucleophilic displacement of the 6-chloro with 1-(4-fluorophenyl)-N-methylmethanamine delivered 72, which was then hydrolyzed under standard basic conditions followed by an amide coupling with HATU to produce 27. Intermediate 71 was subjected to chlorination of both the hydroxyl and carboxylic acid groups using oxalyl chloride. The reaction was then quenched with aniline to afford 73 which was subsequently aminated with 1-(4-chlorophenyl)-N-methylmethanamine to afford 28.
Scheme 3. Synthesis of 27 and 28.

Reagents and conditions: (i) (a) KOH, EtOH; (b) HCl; (ii) SOCl2, DMF; (iii) 1-(4-fluorophenyl)-N-methylmethanamine, K2CO3, DMF; (iv) LiOH, H2O:/THF (2:1), 25 °C; (v) HATU, DIPEA, DMF, 25 °C; (vi) (COCl)2, EtOAc, aniline; (vii) 1-(4-chlorophenyl)-N-methylmethanamine, i-PrOH, DIPEA, 100 °C.
In order to access an advanced intermediate that would allow for exploration of the RHS of the molecule (31, 33–38, 40–52), another relatively straightforward approach (Scheme 4), involving four steps from the commercially available methyl 2,4-dichloropyrimidine-6-carboxylate, was used. Briefly, quantitative amination by selective displacement of the 6-chloro group using an appropriate N-methyl-1-phenylmethanamine at 0 °C in THF followed by hydrogenation with 10% Pd/C and subsequent hydrolysis using LiOH in methanol afforded the carboxylic acid (78–80). Lastly, amide coupling with appropriate amines using HATU gave the desired carboxamide final compound. The intermediate as represented by 74 could be utilized to access 2-amino substiuted analogues such as 29 and 30. Intermediate 74 was hydrolyzed using LiOH in THF/MeOH (1:1) followed by amide coupling with HATU and substitution of the 2-chloro group with an appropriate amine through heating in 2-propanol in the presence of triethylamine in a sealed tube, affording compounds 29 and 30.
Scheme 4. General Route for the Synthesis of 6-Aminopyrimidine-4-carboxamides and Selected Core Modifications.

Reagents and conditions: (i) R-CH2NHCH3, THF, 0 °C; (ii) 10% Pd/C, H2, MeOH; (iii) LiOH, MeOH; (iv) amine, DIPEA, HATU, DMF; (v) LiOH, THF/H2O (1:1); (vi) NH3 or cyclopropylamine, i-PrOH, Et3N, 100 °C.
The reverse amide 32 was synthesized by reacting 6-chloropyrimidin-4-amine 82 with benzoyl chloride in the presence of a base followed by nucleophilic substitution of the 6-chloro with 1-(4-fluorophenyl)-N-methylmethanamine (Scheme 5). The trifluoromethyl ketone, 83, was synthesized from the free acid, 78, via formation of a Weinreb amide followed by trifluoromethylation using TMS-CF3.25 Reductive amination using NaCNBH3 in the presence of TiCl4 then afforded compound 53. Finally, triazolyl compound 54 was synthesized by heating the hydrazide 84 with benzonitrile in refluxing butanol in the presence of potassium carbonate.
Scheme 5. Synthetic Schemes Used To Access Reverse Amide and Amide Isosteres.

Reagents and conditions: (i) BzCl, DIPEA, THF, rt to 50 °C; (ii) 1-(4-fluorophenyl)-N-methylmethanamine, i-PrOH, Et3N, 100 °C; (iii) HATU, Et3N, N,O-dimethylhydroxylamine, DMF, rt; (iv) (a) TMS-CF3, toluene, 20% CsF; (b) TBAF, H2O, 50 °C; (v) 4-fluoroaniline, NaCNBH3, Et3N, TiCl4, MeOH, DCM (50:50), N2; (vi) benzonitrile, K2CO3, BuOH, 120 °C.
SAR on the LHS
After ascertaining that the 4-methoxy substituent of the phenyl carboxamide was not crucial for activity (Table 1; compound 2, R1 = H) the LHS (Table 1) of the scaffold was varied to identify the key pharmacophore needed for activity while keeping the central pyrimidine core unchanged and the RHS constant as 4-methoxyphenyl or phenyl.
Replacement of the aryl ring with a saturated cyclohexyl ring (compound 3) led to considerable loss of activity, suggesting essentiality of an aromatic ring on the LHS. Similarly, replacement of the N-CH3 linker with “O” or “NH” led to inactive compounds 4 and 5, respectively, suggesting the critical importance of a tertiary nitrogen at the linker position or intolerance toward more polar “O” or “NH” groups. Furthermore, linker modifications such as addition of a methylene group (N-methylphenethyl, 6) or removal of CH2 (N-methylaniline, 7), cyclization of the N onto the phenyl ring as tetrahydroisoquinoline moiety (8) or extension of N-methyl to N-ethyl (9) all led to loss or deterioration of MICs. This all indicated stringent shape requirements at this presumably hydrophobic part of the molecule.
Realizing the essential nature of the aryl ring and the linker, we focused efforts on exploring substitution on the phenyl ring to improve potency and introduce polarity. Para-halogenated compounds 10 (4-F), 11 (4-Cl), and 12 (4-CF3) were at least ≥4-fold and ≥2-fold more potent in 7H9/ADC and GAST/Fe, respectively, compared to 1. This was encouraging and the first sign of a SAR trend emerging and leading to the conclusion that a para-halogenated phenyl ring gave the best activity with 4-F phenyl compound 10 showing the most promise. Compounds with 3-F (13) and 3-Cl (14) were less potent compared to corresponding 4-halogenated analogues, whereas the 2-Cl analogue (15) was inactive up to the highest concentration tested, once again suggesting stringent shape requirements on the LHS. Addition of more polar groups such as 4-CN (16) and 4-SO2Me (17) led to deterioration or loss of MICs.
Up to this point, all compounds tested had poor aqueous solubility of ≤5 μM. In an attempt to improve solubility by introducing polarity, the phenyl ring was replaced with a pyridyl ring. Compound 18 with a 2-pyridyl ring was found to be inactive, whereas compound 19 with a 3-pyridyl ring was equipotent to compound 1 albeit with a much improved aqueous solubility of 150 μM at pH 7.4. Further substitution of the 3-pyridyl ring as in compound 20 with a 3-pyridyl-4-trifluoromethyl group retained similar activity but now with loss of solubility, albeit this substitution improved the in vivo pharmacokinetic properties as will be discussed in the DMPK section.
SAR at the Central Core
Exploration of the SAR around the central core of the scaffold was aimed at understanding whether or not the 4,6-disubstituted pyrimidine was essential for activity while retaining a 4-halo group on the LHS phenyl ring and a 4-methoxy, halogen, or hydrogen on the RHS phenyl ring. Initial efforts looked at shuffling the nitrogen atoms around the core ring, including the pyridine (21), pyrazine (22), and pyrimidine (23), with one of the nitrogen atoms shifted between the aminobenzyl and carboxamide groups, and the pyridazine (24) analogues. Only the pyridine retained partial activity at 10 μM in the GAST/Fe medium, which was not sufficient to warrant a scaffold hop. Compounds based on the pyrimidinone isostere (25) and thiazole (26) all lost activity. Lastly 5- and 2-substitutions (27, 28, 29, and 30) on the pyrimidine core were evaluated and found to be similarly inactive. It was thus concluded that the original pyrimidine core was optimal for activity and could not be replaced with other diazine and heteroaromatic rings.
SAR on the RHS
An extensive SAR exploration on the RHS was undertaken in order to evaluate the scope for optimization of potency and physicochemical properties (Table 3). The free amide NH was deemed critical for potency as N-methylation (31) led to a complete loss in activity. Similarly, reversal of the amide functionality led to inactive compound 32. For further exploration of substitution on the aryl ring of the amide, a 4-halophenyl or 4-CF3-3-pyridyl was fixed on the LHS of the scaffold. Replacement of the phenyl ring with an aliphatic chain such as isopropyl (33) was detrimental to activity, while replacement with a saturated cyclohexyl ring (34) resulted in retention of activity in the GAST/Fe medium albeit it was completely inactive in 7H9/ADC. The overall trend suggested a preference for an aryl ring on the RHS. Since the original hit contained a methoxy group, additional ethers were explored in the para position, including isopropoxy (36), 2-methoxyethoxy (37), and a N,N-dimethyl-2-ethoxy ether (38). Encouragingly, compounds 36 and 37 were some of the most active compounds synthesized up to this point but were poorly soluble (solubility pH 7.4 ≤ 5 μM). Compound 38 with the basic dimethylaminoethoxy side chain (predicted pKa of 8.77) was less active (7H9/ADC MIC of 25 μM) even though it showed considerably improved aqueous solubility of 155 μM. Nonetheless, these results suggested scope for substitutions on the aryl ring to modulate physicochemical properties while retaining potency. Replacement of the phenyl ring with a 4-pyridyl ring, as in compound 39, was tolerated indicating a wider scope for SAR exploration at this position with heterocycles and weakly basic substituents. Addition of a less basic N-methylpiperazine ring at the 4-position of the phenyl ring (predicted pKa of 7.85) led to potent compound 40 which was poorly soluble at pH 7.4 but sufficiently soluble at a slightly acidic pH of 6.5 (refer to DMPK section). Solubility could be improved further by combining N-methylpiperazinylphenylamide on the RHS with a 4-CF3-3-pyridyl group on the LHS as in compound 41 with some loss in potency (MIC = 12.5 μM, solubility at pH 7.4 of 165 μM). Moving the N-methylpiperazine ring to the 3-position of the phenyl ring (42) was detrimental to activity with MIC values dropping by 10- to 30-fold. Similarly, 3-pyridyl-4-(N-methylpiperazinyl) analogue 43 was found to be much less potent (7H9/ADC MIC > 50 μM; GAST/Fe MIC = 19 μM). Compounds with neutral substituents at the 4-position of the phenyl ring like N-acetylpiperazinyl (44) or morpholino (compound 45) retained good MICs (<5 μM) but were much less soluble. Interestingly, the 4-morpholino-2-pyridyl analogue (46) retained good MIC activity (2–3 μM) indicating scope for the introduction of a nitrogen into the phenyl ring to address potential reactivity issues of electron-rich anilines during further optimization.
A small set of amides with five-membered heterocycles was synthesized to evaluate the scope for further SAR exploration at this position. Compounds with N-methylpyrazole (47), N-methyltriazolyl (48), and isoxazolyl (49) substituents were either inactive or weakly active. Compound 50 containing a 4-methylthiazolyl group showed a GAST MIC equivalent to its phenyl counterpart (5 μM). However, its MIC in 7H9/ADC medium (37 μM) was weaker and had a poor solubility (<5 μM). Similarly, compounds containing 1,3,4-thiadiazole moieties (51 and 52) showed reasonable MIC in GAST/Fe medium (∼5 μM) but had weaker potency in 7H9/ADC medium (12.5 and 50 μM, respectively). Unfortunately compounds with these moieties were cytotoxic in vitro and were also found to be toxic at higher doses in animal studies. Hence, these modifications were abandoned even though more lead-like DMPK properties were achieved with these compounds.
Lastly, two amide isosteres CF3-amine (53)26 and triazole (54)27 were synthesized in an attempt to explore the scope for variation and in line with metabolite identification studies (Supporting Information Table S1). Metabolite identification, performed on 12 and 51, indicated cleavage of the amide bound on the RHS as a primary route of metabolism compared to N-demethylation on the LHS. Even though both these modifications resulted in compounds showing some activity, there was no improvement in other properties like solubility and hence were not explored further. Overall, as summarized in Figure 4, the SAR around the LHS and the central core was narrow with fewer modifications tolerated; SAR around the RHS was more dynamic, allowing scope for further optimization. From this, several compounds were identified for further profiling both in vitro and in vivo.
Figure 4.

Summary of SAR.
Biology Profiling
A spontaneous resistant mutant of Mtb H37Rv was successfully raised against compound 13. Most of the analogues from the series were cross-resistant against this mutant except the thiadiazole analogues 50 and 51, indicating additional targets and/or a different MOA (Table 4). Additionally, these mutants were not cross-resistant to any known TB drugs, suggesting a potential novel MOA for the series. Unfortunately, whole genome sequencing of the mutant did not reveal any genetic polymorphism(s) suggestive of the target or MOA. Hence, further studies using different target identification approaches were explored in order to identify the molecular target of these compounds. Representative compounds were screened against drug-sensitive clinical isolates and were found to retain MICs similar to those in H37Rv (Table 5).
Table 4. Cross-Screening against 13-Resistant Mutanta.
| 14 day GAST/Fe MIC99 (μM) |
||
|---|---|---|
| against WT H37Rv | against 13-resistant mutant | |
| 1 | 20 | >160 |
| 10 | 10 | >160 |
| 13 | 2.5–5 | >160 |
| 20 | 10 | >160 |
| 36 | 1.25–5 | >160 |
| 51 | 5 | 10 |
| Rif | 0.012 | 0.012 |
| INH | 0.18 | 0.36 |
| STREP | 1.62 | 1.62 |
| ETB | 2.3 | 2.3 |
| LEVO | 0.64 | 1.3 |
| KAN | 0.32 | 0.32 |
Rif, rifampicin; INH, isoniazid; STREP, streptomycin; ETB, ethambutol; LEVO, levofloxacin; KAN, kanamycin.
Table 5. Activity against Clinical Isolates.
| MIC on ref strain (μM)a | MIC against clinical isolates (μM)a |
||||||
|---|---|---|---|---|---|---|---|
| compd | H37Rv | SAWC1125 | SAWC3933 | SAWC3385 | SAWC3200 | SAWC3388 | SAWC3906 |
| 36 | 1.25 | 2.5 | 2.5 | 0.31 | <0.08 | 0.31 | 0.31 |
| 40 | 1.25–2.5 | 2.5–5 | 1.25–2.5 | 1.25 | 1.25 | 1.25–2.5 | 0.31–0.63 |
| 51 | 2.5 | <0.08 | 0.625 | <0.08 | <0.08 | 0.15 | <0.08 |
| 52 | 50 | 2.5 | 10–20 | 10 | 2.5–5 | 10 | 2.5–5 |
MIC90 readout from MABA assay in Middlebrook 7H9 medium; SAWC 1125, typical Beijing (lng 2.2); SAWC 3933, T1 (lng 4.2); SAWC 3385, Cas 1/Delhi (lng 3); SAWC 3200, X1 (lng 4.9); SAWC 3388, T1, Tuscany, T5/Rus (lng4.3); SAWC 3906, atypic Beijing (lng 2).
Chemoproteomics
In order to supplement our MOA identification efforts, we used a chemoproteomics approach to identify the potential target proteins of the 6-dialkylaminopyrimidine carboxamides. This affinity-based approach was recently successfully employed for the direct target identification of antitubercular compounds.28−30 This approach is based on the modification of the compound 40 with a linker for covalent immobilization on sepharose-beads by N-hydroxysuccinamide (NHS) coupling. The modified compound 85 (Figure 5, synthesis Scheme S1) retained antimycobacterial activity (MIC90 = 18.6 μM as determined against M. bovis BCG) suggesting that the derivatization with the linker moiety did not interfere with target binding. The 85 beads were incubated with Mycobacterium bovis BCG extract, and proteins captured by the beads were digested with trypsin, labeled with isobaric mass tags (TMT 10plex), and quantitatively identified by liquid chromatography–tandem mass spectrometry (LC–MS/MS). In order to distinguish true targets from nonspecific background binding, the bacterial extracts were incubated prior to the bead-incubation step with the active test compound 40, the inactive test compound 25, or a vehicle (DMSO only) control. Only the active compound should bind to its target proteins in the lysate and thus reduce the binding of these proteins to the beads, but not the inactive compound. Indeed, two proteins, BCG_3193 (Rv3169) and BCG_3827 (Rv3768), showed reduced binding to the compound beads in the presence of competing “free” 40 (Figure 5A) but not with inactive 25, which suggests that these proteins are the potential targets of 40. In order to estimate inhibitor potency, we performed the affinity step in the presence of different concentrations of active compound 40 in the protein extract, which allowed the determination of an apparent dissociation constant Kdapp (Figure 5C). The Kdapp takes into consideration the affinity of the target for the bead-immobilized ligand. The latter effect can be deduced by measuring the depletion of the target protein by the beads.31 The apparent dissociation constant of compound 40 for BCG_3193 was determined in two separate experiments as 0.61 μM and 0.69 μM and for BCG_3827 as 2.1 μM and 3.8 μM (Figure 5C). Both potential novel target proteins, BCG_3193/Rv3169 and BCG_3827/Rv3768 (contains a polyketide cyclase SnoaL-like domain, IPR009959), are conserved hypothetical proteins of unknown function. Both proteins are nonessential but notably found using Triton X-114 detergent phase separation for extraction of lipophilic proteins, suggesting they are associated with the cell envelope despite lacking detectable transmembrane helices.32
Figure 5.

Affinity chemoproteomics revealed BCG_3193 and BCG_3827, novel TB targets with unknown function, as targets of the 6-dialkylaminopyrimidine carboxamides. (A) BCG_3193 and BCG_3827 show reduced binding to 6-dialkylaminopyrimidine carboxamide analogue beads due to compound 40 binding, but no effect on these proteins was seen by the inactive compound. Shown are relative protein amounts captured by the beads in a log 2 scale. (B) Compound structures used for the affinity chemoproteomics experiments. (C) The apparent dissociation constant of compound 40 for BCG_3193 was determined in two different experiments as 0.61 μM and 0.69 μM and for BCG_3827 as 2.1 μM and 3.8 μM.
Physicochemical, DMPK, and hERG Profiles
Representative compounds were profiled for physicochemical properties and in vitro DMPK properties which included kinetic solubility, human plasma protein binding (PPB), microsomal metabolic stability (MS), and permeability, as shown in Table 6.
Table 6. In Vitro ADME Properties, hERG Activity, and Caseum Distribution.
| compound |
||||||
|---|---|---|---|---|---|---|
| 12 | 20 | 36 | 40 | 51 | 52 | |
| cLogPa | 5.0 | 3.7 | 5.0 | 5.0 | 2.0 | 1.4 |
| FassiF solubility at pH 6.5 (μM)b | 5 | <5 | <5 | 150 | <5 | 195 |
| solubility at pH 7.4 (μM)c | <5 | <5 | 5 | <5 | 128 | 191 |
| PPB human fud | 0.11 | 0.05 | 0.04 | 0.14 | 0.01 | 0.32 |
| PAMPA log Pappe | –4.9 | –3.8 | ND | –4.5 | ND | –3.9 |
| MS H/R/M % remaining after 30 minf | 55/87/60 | 85/72/87 | 64/43/37 | 57/37/21 | 67/65/74 | 100/88/78 |
| hERG IC50 (μM)g | 3.3 | 5.6 | 4.1 | 0.9 | 4.2 | 14 |
| caseum distribution unbound (%)h | 0.1 | ND | ND | 0.1 | 0.5 | 3.6 |
Calculated log P (StarDrop).
Fasted-state simulated intestinal fluid, pH 6.5.
Kinetic solubility.
Plasma proteing binding fu - fraction unbound.
Parallel artificial membrane permeability assay.
Metabolic stability: H, human; R, rat; M, mice.
Tested at Essen BioScience using IonWorks patch clamp electrophysiology.
Assay using surrogate caseum, ND: not determined.
As discussed previously, most of the compounds based on phenyl amides had poor to moderate solubility at pH 7.4 except those with basic solubilizing side chains (e.g., compounds 38 and 41). These compounds showed considerably improved solubility in slightly acidic Fassif medium (fasted-state simulated intestinal fluid, pH 6.5),33 e.g., compound 40 (kinetic solubility at pH 7.4 of <5 μM; kinetic solubility in Fassif of 150 μM). This may be relevant for oral absorption of these compounds. Compounds containing heterocyclic amides like thiadiazole compounds 51 and 52 had good solubility of >100 μM in both media. In general, compounds in the series had 5–10% free fraction in human plasma as measured in a PPB assay while some more polar compounds like 52 had up to 30% free fraction in human plasma. The ability of compounds to cross biological membranes by passive diffusion was assessed using the PAMPA assay.34 All compounds tested showed good to moderate permeability with values ranging between −3.8 and −4.9 log Papp presumably due to their lipophilic character. Caseum binding was also assessed for certain compounds in order to give some insight into the ability of the compounds to permeate the caseous necrotic regions of granulomas to access the bacteria.35 Compound 52 had the highest free fraction (3.6% unbound) from the compounds assessed with phenyl compounds generally being highly bound (0.06–0.1% unbound). Further safety profiling was conducted against the human ether-a-go-go-related gene (hERG) potassium channel, and compounds were found to be low micromolar inhibitors. While hERG activity is a well-established safety concern,36 compounds were not optimized to remove hERG activity due to the initial focus being on increasing potency and solubility.
In general, compounds showed moderate metabolism in human, mouse, and rat liver microsomes except compound 40, which showed rapid in vitro clearance. The compounds were also tested in male C57/BL6 mice for pharmacokinetic parameters at an intravenous dose of 2 mg/kg and an oral dose of 20 mg/kg (Table 7). Most of the compounds showed low in vivo clearance in mice in accordance with their in vitro microsomal metabolic stability. Compound 40 showed low clearance iv clearance despite its poor microsomal stability, probably due to extensive tissue distribution as indicated by high volume of distribution (Vd = 21.8 L/kg). Most of the compounds had moderate to good oral bioavailability. Moderate permeability was sufficient to give good oral absorption as well as moderate bioavailability for compounds with low solubility (12 and 20). Compound, 52 with a thiadiazole group at LHS showed improved physicochemical and ADME parameters such as good solubility, improved microsomal stability leading to higher oral absorption, higher AUC and better bioavailability of 67%. Unfortunately, this compound was found to be toxic to mice when dosed orally above 50 mg/kg and hence was not progressed any further.
Table 7. Pharmacokinetic Parameters in Male C57/BL6 Mouse Blooda.
|
12 |
20 |
40 |
51 |
52 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| dose (mg/kg) | oral 20 | iv 5 | oral 20 | iv 5 | oral 20 | iv 5 | oral 20 | iv 5 | oral 20 | iv 5 |
| apparent t1/2 (h) | 8.7 | 4.6 | 7.2 | 0.7 | 15.8 | 10.3 | 4.4 | 5.1 | 3.0 | 3.0 |
| CLtotal (mL min–1 kg–1) | 11.3 | 11.2 | 26.3 | 11.1 | 6.6 | |||||
| Vd (L/kg) | 4.3 | 0.7 | 21.8 | 4.8 | 1.7 | |||||
| Cmax (μM) | 2.2 | 3.4 | 0.42 | 2.58 | 10.8 | |||||
| Tmax (h) | 1 | 4 | 5 | 1 | 1 | |||||
| AUC0–∞ (min·μM) | 1334 | 1224 | 1999 | 1179 | 564 | 540 | 1022 | 1329 | 5614 | 2106 |
| oral bioavailability (%) | 28 | 42 | 26 | — | 19 | 67 | ||||
iv, intravenous; t1/2, elimination half-life; CL, plasma clearance; Vd, volume of distribution; Cmax, maximum (peak) plasma concentration following oral administration; AUC, area under the curve.
Conclusion
Screening of a Biofocus library led to the identification of 6-dialkylaminopyrimidine carboxamides with potent antitubercular properties and a novel mode of action. A biology triage cascade allowed deprioritization of compounds potentially acting through inhibition of QcrB, cell-wall targets and DNA damage. Cross-resistance studies of TB drugs against the resistant mutant raised against this series indicate a potentially novel MOA for the series, and this was reinforced by chemoproteomic studies albeit the exact target (and MOA) remains unknown. Detailed SAR studies have demonstrated limitations as well as scope for further optimization of these series to deliver lead-like compounds with potent anti-TB properties and oral bioavailability.
Experimental Section
Biology
Biology triage assays were carried out as described in ref (23).
DMPK
All protocols for in vitro DMPK studies and mouse PK studies are available in the supplementary document. Animal studies were conducted following guidelines and policies as stipulated in the UCT Research Ethics Code for Use of Animals in Research and Teaching after review and approval of the experimental protocol by the UCT Senate Animal Ethics Committee (Protocol FHS-AEC 013/032)
Chemoproteomics
Chemoproteomics experiments were performed as previously described.31 Briefly, sepharose beads were derivatized with compound 85 at a concentration of 0.04 mM and subsequently washed and equilibrated in lysis buffer (50 mM Tris-HCl, pH 7.4, 0.4% Igepal-CA630, 1.5 mM MgCl2, 5% glycerol, 150 mM NaCl, 25 mM NaF, 1 mM Na3VO4, 1 mM DTT, and one Complete EDTA-free protease inhibitor tablet (Roche) per 25 mL). The 85 beads were incubated at 4 °C for 1 h with 0.1 mL (0.25 mg) M. bovis BCG extract, which was preincubated with compound or DMSO (vehicle control). Our experimental design employed isobaric tandem mass tags (TMTs)37 which allowed us to analyze 10 samples together by LC–MS/MS enabling the relative quantification of proteins across these samples. The experiments were configured to generate values for the affinity of the beads to the bound proteins (“depletion” values, four samples) and to allow determination of IC50 values (six samples) in a single experiment. Samples 1 and 2 were vehicle control duplicates, samples 3 and 4 were duplicates of the “rebinding” experiment, and samples 5–10 served to generate IC50 values by adding compound over a range of 5 concentrations (highest concentration of 30 μM, then four dilution steps of 1:4 each). In the “rebinding” experiment, the nonbound fraction from the first bead incubation step was incubated again with “fresh” beads, allowing the determination of target depletion by the beads. Apparent dissociation constants were determined by taking into account the protein depletion by the beads.31 A repeat experiment was performed with vehicle control versus 20 μM and 5 μM compound. The beads were transferred to filter plates (Durapore PVDF membrane, Merck Millipore), washed extensively with lysis buffer, and eluted with SDS sample buffer. Proteins were alkylated, separated on 4–12% Bis-Tris NuPAGE (Life Technologies), and stained with colloidal Coomassie. Gel lanes were cut into three slices and subjected to in-gel digest using LysC for 2 h and trypsin overnight. Digestion, labeling with TMT isobaric mass tags, peptide fractionation, and mass spectrometric analyses were performed.31 Proteins were quantified by isobaric mass tagging and LC–MS/MS. The proteins.fasta file for M. bovis BCG was downloaded on May 11, 2011, and supplemented with the sequences of bovine serum albumin, porcine trypsin, and mouse, rat, sheep, and dog keratins. Decoy versions of all proteins were created and added. The search database contained a total of 11 492 protein sequences, 50% forward and 50% reverse. Protein identification and quantification were performed.37 Proteins identified with >1 unique peptide matches were considered for further data analysis.
All commercial reagents were purchased from Sigma-Aldrich, Combi-blocks, Waterstone, or Fluorochem and were used without further purification. Solvents were used as received unless otherwise stated. Analytical thin-layer chromatography (TLC) was performed on SiO2 plates on aluminum backing. Visualization was accomplished by UV irradiation at 254 and 220 nm. Flash column chromatography was performed using a Teledyne ISCO flash purification system with SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh). Purity of all final derivatives for biological testing was confirmed to be >95% as determined using an Agilent 1260 Infinity binary pump, Agilent 1260 Infinity diode array detector (DAD), Agilent 1290 Infinity column compartment, Agilent 1260 Infinity standard autosampler, and a Agilent 6120 quadrupole (single) mass spectrometer, equipped with APCI and ESI multimode ionization source. Using a Kinetex Core C18 2.6 μm column (50 mm × 3 mm); mobile phase B of 0.4% acetic acid, 10 mM ammonium acetate in a 9:1 ratio of HPLC grade methanol and type 1 water, mobile phase A of 0.4% acetic acid in 10 mM ammonium acetate in HPLC grade (type 1) water, with flow rate of 0.9 mL/min, detector diode array (DAD). Or an Agilent UPLC–MS was used: Agilent Technologies 6150 quadrupole, ES ionization, coupled with an Agilent Technologies 1290 Infinity II series UPLC system Agilent 1290 series HPLC at two wavelengths 254 and 290 nm using the following conditions: Kinetex 1.7 μm Evo C18 100A, LC column 50 mm × 2.1 mm, solvent A of 0.1% (formic acid) water, solvent B of 0.1% (formic acid) acetonitrile. The structures of the intermediates and end products were confirmed by 1H NMR and mass spectrometry. Proton magnetic resonance spectra were determined in an appropriate deuterated solvent on a Varian Mercury spectrometer at 300 MHz or a Varian Unity spectrometer at 400 MHz.
General Protocol for Synthesis of Compounds 1–21 and 28
The appropriate 6-chloro-N-phenylpyrimidine-4-carboxamide (1 equiv), N-methylbenzylamine (1.2 equiv), and DIPEA (2 equiv) were added to a 50 mL round-bottomed flask containing i-PrOH (10 mL) and heated to reflux overnight. Compounds that precipitated upon cooling were filtered and washed with cold EtOH. For those that did not precipitate, the i-PrOH was removed under vacuum and the residue redissolved in EtOAc (20 mL) and washed with a saturated aqueous solution of NaHCO3, the organic layer dried and removed under reduced pressure. The resulting solid or residue was either recrystallized from EtOH or purified by flash column chromatography to afford the appropriate 6-(benzyl(methyl)amino)-N-phenylpyrimidine-4-carboxamide.
6-(Benzyl(methyl)amino)-N-(4-methoxyphenyl)pyrimidine-4-carboxamide (1)
Yield 52%. 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.67 (s, 1H) 7.91–7.71 (m, 2H), 7.46–7.17 (m, 6H), 7.05–6.80 (m, 2H), 4.92 (s, 2H), 3.75 (s, 3H), 3.17 (s, 3H). LC–MS (ESI): m/z 349.2 [M + H]+. HPLC purity 100%.
6-(Benzyl(methyl)amino)-N-phenylpyrimidine-4-carboxamide (2)
Yield 98%. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.66 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.36–7.21 (m, 8H), 7.11 (t, J = 7.2 Hz, 1H), 4.90 (s, 2H) and 3.15 (s, 3H). LC–MS (ESI): m/z 319.2 [M + H]+. HPLC purity 99%.
6-((Cyclohexylmethyl)(methyl)amino)-N-phenylpyrimidine-4-carboxamide (3)
Yield 12.5%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.68 (s, 1H), 7.93 (d, J = 8.0 Hz, 2H), 7.40 (m, 2H), 7.27 (s, 1H), 7.16 (t, J = 8.0 Hz, 1H), 3.54 (s, 2H), 3.13 (s, 3H), 1.86–1.62 (m, 6H), 1.25–0.96 (m, 5H). LC–MS (ESI): m/z 325.2 [M + H]+. HPLC purity 99%.
6-(Benzyloxy)-N-phenylpyrimidine-4-carboxamide (4)
Yield 86%. 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 8.99 (s, 1H), 7.87 (d, J = 8.0 Hz, 2H), 7.51–7.47 (m, 3H), 7.41–7.33 (m, 4H), 7.21 (m, 1H), 7.13 (t, J = 8.0 Hz, 1H) and 5.52 (s, 2H). LC–MS (ESI): m/z 306.1 [M + H]+. HPLC purity 98%.
6-(Benzylamino)-N-phenylpyrimidine-4-carboxamide (5)
Yield 85%. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.57 (d, J = 0.8 Hz, 1H), 8.39 (t, J = 8.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.36–7.24 (m, 8H), 7.11 (t, J = 7.2 Hz, 1H), 4.61 (s, 2H). LC–MS (ESI): m/z 305.2 [M + H]+. HPLC purity 99%.
6-(Methyl(phenethyl)amino)-N-phenylpyrimidine-4-carboxamide (6)
Yield 97%. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.62 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.39–7.10 (m, 9H), 3.84 (m, 2H), 3.05 (s, 3H) and 2.87 (t, J = 7.2 Hz, 2H). LC–MS (ESI): m/z 333.2 [M + H]+. HPLC purity 99%.
6-(Methyl(phenyl)amino)-N-phenylpyrimidine-4-carboxamide (7)
Yield 84%. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.75 (d, J = 1.2 Hz, 1H), 7.81 (dd, J = 8.4, 1.2 Hz, 2H), 7.57–7.30 (m, 7H), 7.10 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 1.2 Hz, 1H), 3.49 (s, 3H). LC–MS (ESI): m/z 305.2 [M + H]+. HPLC purity 99%.
6-(3,4-Dihydroisoquinolin-2(1H)-yl)-N-phenylpyrimidine-4-carboxamide (8)
Yield 40%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.69 (s, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.45 (s, 1H) 7.38–7.20 (m, 6H), 7.12 (t, J = 8.0 Hz, 1H), 4.84 (s, 2H), 3.92 (m, 2H) and 2.93 (t, J = 8.0 Hz, 2H). LC–MS (ESI): m/z 331.2 [M + H]+. HPLC purity 99%.
6-((4-Chlorobenzyl)(ethyl)amino)-N-phenylpyrimidine-4-carboxamide (9)
Yield 15%. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 8.66 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.39–7.24 (m, 7H), 7.11 (t, J = 8.0 Hz, 1H), 4.84 (s, 2H), 3.62 (s, 2H) and 1.12 (t, J = 8.0 Hz, 3H). LC–MS (ESI): m/z 367.1 [M + H]+. HPLC purity 99%.
6-((4-Fluorobenzyl)(methyl)amino)-N-phenylpyrimidine-4-carboxamide (10)
Yield 48%. 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1H), 8.70 (s, 1H), 7.90 (d, J = 8.0 Hz, 2H), 7.40–7.12 (m, 8H), 4.93 (s, 2H) and 3.21 (s, 3H). LC–MS (ESI): m/z 337.2 [M + H]+. HPLC purity 99%.
6-((4-Chlorobenzyl)(methyl)amino)-N-phenylpyrimidine-4-carboxamide (11)
Yield 81%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.66 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.39–7.24 (m, 7H), 7.11 (t, 8.0 = Hz, 1H), 4.89 (s, 2H) and 3.15 (s, 3H). LC–MS (ESI): m/z 353.1 [M + H]+. HPLC purity 99%.
6-(Methyl(4-(trifluoromethyl)benzyl)amino)-N-phenylpyrimidine-4-carboxamide (12)
Yield 92%. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.55 (s, 1H), 7.85 (m, 3H), 7.34 (t, J = 8.0 Hz, 2H), 7.17 (s, 1H), 7.11 (t, J = 8.0 Hz, 1H), 4.10 (m, 1H), 3.86 (m, 2H), 3.41 (m, 2H), 1.87 (m, 2H) and 1.46 (m, 2H). LC–MS (ESI): m/z 387.1 [M + H]+. HPLC purity 99%.
6-((3-Fluorobenzyl)(methyl)amino)-N-(4-methoxyphenyl)pyrimidine-4-carboxamide (13)
Yield 58%. 1H NMR (300 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.64 (s, 1H), 7.77 (d, J = 9.0 Hz, 2H), 7.38 (m, 1H), 7.28 (s, 1H), 7.10–7.04 (m, 3H), 6.92 (d, J = 9.0 Hz, 2H), 4.91 (s, 2H), 3.73 (s, 3H) and 3.16 (s, 3H). LC–MS (ESI): m/z 367.2 [M + H]+. HPLC purity 99%.
6-((3-Chlorobenzyl)(methyl)amino)-N-phenylpyrimidine-4-carboxamide (14)
Yield 91%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.67 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.36–7.12 (m, 8H), 4.91 (s, 2H), and 3.16 (s, 3H). LC–MS (ESI): m/z 353.1 [M + H]+. HPLC purity 98%
6-((2-Chlorobenzyl)(methyl)amino)-N-phenylpyrimidine-4-carboxamide (15)
Yield 91%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.65 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.50–7.07 (m, 8H), 4.95 (s, 2H), and 3.20 (s, 3H). LC–MS (ESI): m/z 353.1 [M + H]+. HPLC purity 96%.
6-((4-Cyanobenzyl)(methyl)amino)-N-phenylpyrimidine-4-carboxamide (16)
Yield 71%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.65 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H) 7.42–7.31 (m, 5H), 7.11 (t, J = 8.0 Hz, 1H), 4.99 (s, 2H) and 3.19 (s, 3H). LC–MS (ESI): m/z 344.2 [M + H]+. HPLC purity 99%.
6-(Methyl(4-(methylsulfonyl)benzyl)amino)-N-phenylpyrimidine-4-carboxamide (17)
Yield 48%. 1H NMR (300 MHz, DMSO-d6) δ 10.54 (s, 1H), 8.69 (s, 1H), 7.89 (dd, J = 8.6, 7.0 Hz, 4H), 7.61–7.44 (m, 2H), 7.44–7.25 (m, 3H), 7.25–7.06 (m, 1H), 5.05 (s, 2H), 3.21 (d, J = 7.0 Hz, 6H). LC–MS (ESI): m/z 397.1 [M + H]+. HPLC purity 100%.
6-(Methyl(pyridin-2-ylmethyl)amino)-N-phenylpyrimidine-4-carboxamide (18)
Yield 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 8.63 (s, 1H), 8.51 (d, J = 4 Hz, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.74 (t, J = 8.0 Hz, 1H), 7.37–7.23 (m, 5H), 7.11 (t, J = 8.0 Hz, 1H), 4.97 (s, 2H) and 3.24 (s, 3H). LC–MS (ESI): m/z 320.2 [M + H]+. HPLC purity 97%.
6-(Methyl(pyridin-3-ylmethyl)amino)-N-phenylpyrimidine-4-carboxamide (19)
Yield 84%. 1H NMR (300 MHz, DMSO-d6) δ 10.53 (s, 1H), 8.69 (d, J = 1.1 Hz, 1H), 8.59–8.38 (m, 2H), 7.99–7.75 (m, 2H), 7.66 (dt, J = 7.9, 2.0 Hz, 1H), 7.50–7.25 (m, 4H), 7.25–6.97 (m, 1H), 4.96 (s, 2H), 3.19 (s, 3H). LC–MS (ESI): m/z 320.2 [M + H]+. HPLC purity 100%.
6-(Methyl((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)-N-phenylpyrimidine-4-carboxamide (20)
Yield 35%. 1H NMR (300 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.69 (m, 2H), 7.86 (m, 4H), 7.35 (m, 3H), 7.13 (m, 1H), 5.05 (s, 2H) and 3.22 (s, 3H). LC–MS (ESI): m/z 388.2 [M + H]+. HPLC purity 98%.
4-((4-Fluorobenzyl)(methyl)amino)-N-phenylpicolinamide (21)
Yield 26%. 1H NMR (300 MHz, CDCl3): 10.40 (br s, 1H), 8.82 (d, J = 6 Hz, 1H), 7.82 (d, J = 6 Hz, 2H), 7.72 (s, 1H), 7.39 (d, J = 9 Hz, 2H), 7.15–7.25 (m, 3H), 7.1 (t, J = 9 Hz, 2H), 6.65–6.75 (m, 1H), 4.68 (s, 2H), δ 3.19 (s, 3H) . LC–MS (ESI): m/z 336.1 [M + H]+. HPLC purity 98%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-fluorophenyl)pyrazine-2-carboxamide (22)
Yield 30%. A mixture of 95 mg (0.37 mmol) of 6-chloro-N-(4-fluorobenzyl)-N-methylpyrazin-2-amine, 100 mg (0.90 mmol) of 4-fluoroaniline, 100 mg (0.38 mmol) of molybdenum hexacarbonyl, 100 mg of tetrabutylammonium chloride, and 500 mg of triethylamine in 3 mL of toluene and 2 mL of tetraethylene glycol dimethyl ether was heated under nitrogen in a pressure tube to 150 °C for 4 h. The cooled mixture was evaporated onto silica gel. Silica flash chromatography (EtOAc/Hex 30/70 to 50/50) yielded 40 mg as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.24 (s, 1H), 8.65 (s, 1H), 8.22 (s, 1H), 7.55–7.50 (m, 2H), 7.19–7.13 (m, 2H), 7.02–6.96 (m, 4H), 4.75 (s, 2H), 3.20 (s, 3H). LC–MS (ESI): m/z 355.1 [M + H]+. HPLC purity 99%.
2-((4-Fluorobenzyl)(methyl)amino)-N-(4-methoxyphenyl)pyrimidine-4-carboxamide (23)
Yield 93%. A mixture of 50 mg (0.20 mmol) of 4-chloro-N-(4-fluorobenzyl)-N-methylpyrimidin-2-amine, 100 mg (0.81 mmol) of 4-methoxyaniline, 100 mg (0.38 mmol) if molybdenum hexacarbonyl, 100 mg of tetrabutylammonium chloridem and 500 mg of triethylamine in 3 mL of toluene and 2 mL of tetraethylene glycol dimethyl ether was heated under nitrogen in a pressure tube to 150 °C for 4 h. The cooled mix was diluted with 60 mL of water and extracted with EtOAc (2 × 50 mL), dried over Na2SO4 and the solvent evaporated. Flash chromatography (EtOAc/Hex 20/80 to 40/60) yielded 67 mg (93%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.51 (s, 1H), 8.63 (d, J = 6.0 Hz, 1H), 7.59 (d, J = 9.0 Hz, 2H), 7.44 (d, J = 6.0 Hz, 1H), 7.31–7.26 (m, 2H), 7.09–7.03 (m, 2H), 6.94 (d, J = 9.0 Hz, 2H), 4.95 (s, 2H), 3.84 (s, 3H), 3.30 (s, 3H). LC–MS (ESI): m/z 367.1 [M + H]+. HPLC purity 99%.
6-((4-Fluorobenzyl)(methyl)amino)-N-phenylpyridazine-4-carboxamide (24)
Yield 28%. In a 10 mL CEM microwave tube, 6-chloro-N-phenylpyridazine-4-carboxamide (50 mg, 0.214 mmol), 1-(4-fluorophenyl)-N-methylmethanamine (32.8 mg, 0.235 mmol), and Et3N (0.060 mL, 0.428 mmol) were mixed in 1,4-dioxane (0.5 mL). The mixture was microwaved at 125 °C and 200 W in CEM microwave for 6 h. Solvent was evaporated and the residue was purified by prep-HPLC to give a buff colored powder. 1H NMR (300 MHz, DMSO-d6): δ 10.50 (br s, 1H), 8.91 (d, J = 3 Hz, 1H), 7.72–7.75 (m, 2H), 7.46 (d, J = 3 Hz, 1H), 7.30–7.42 (m, 4H), 7.14–7.20 (m,3H), 4.95 (s, 2H),3.18 (s, 3H). LC–MS (ESI): m/z 337.1 [M + H]+. HPLC purity 99%.
1-(4-Fluorobenzyl)-6-oxo-N-phenyl-1,6-dihydropyridine-3-carboxamide (25)
Yield 44% over 2 steps.
Step 1
In a 7 mL reaction vial, methyl 1-(4-fluorobenzyl)-6-oxo-1,6-dihydropyridine-3-carboxylate (100 mg, 0.383 mmol) was dissolved in MeOH (4 mL), and LiOH (92 mg, 3.83 mmol) was added to the solution and stirred at rt overnight. Methanol was removed under vacuum, and the residue was dissolved in water. Aqueous layer was extracted with ethyl acetate. Aqueous layer was then cooled in ice-bath and acidified with concentrated HCl. Precipitated solid was extracted with ethyl acetate. Ethyl acetate layer was then dried on anhydrous MgSO4 and concentrated under vacuum to give an off-white solid, 1-(4-fluorobenzyl)-6-oxo-1,6-dihydropyridine-3-carboxylic acid. Yield 95%. 1H NMR (300 MHz, DMSO-d6) δ 12.33 (br s, 1H), 8.60 (d, J= 3 Hz, 1H), 7.80 (dd, J = 9, 3 Hz, 1H), 7.38–7.43 (m, 2H), 7.15–7.21 (m, 2H), 6.45 (d, J = 9 Hz, 1H), 5.18 (s, 2H). LC–MS (ESI): m/z 248.1 [M – H]−. HPLC purity 94%.
Step 2
In a 7 mL reaction vial, 1-(4-fluorobenzyl)-6-oxo-1,6-dihydropyridine-3-carboxylic acid (50 mg, 0.202 mmol) was dissolved in DMF (1 mL), and to it were added Et3N (0.056 mL, 0.404 mmol) and HATU (115 mg, 0.303 mmol). The mixture was stirred for 30 min at 25 °C. Aniline (0.022 mL, 0.243 mmol) was then added and the solution stirred at 25 °C for 24 h. Reaction mixture was diluted with ethyl acetate and washed with 10% LiCl solution (2 × 15 mL), saturated aqueous NaHCO3 solution, and brine. The EtOAc layer was then dried and evaporated under vacuum. The residue was chromatographed on Biotage using 10 g prepacked silica gel column and 0–90% EtOAc in hexane as eluent. Pure fractions were combined and evaporated under vacuum. The syrupy residue was triturated with a 3:1 DCM/hexane mixture, and precipitated solid was removed by filtration. Filtrate was evaporated and the residue was crystallized from ethanol to afford 25 as colorless crystalline needles. 1H NMR (300 MHz, DMSO-d6) δ 9.98 (br s, 1H), 8.62 (d, J = 3 Hz, 1H), 8.01 (dd, J = 9, 3 Hz, 1H), 7.68 (d, J = 6 Hz, 2H), 7.40–7.45 (m, 2H), 7.35 (t, J = 9H, 2H), 7.17–23 (m, 2H), 7.10 (t, J = 9 Hz, 1H), 6.52 (d, J = 12 Hz, 1H),5.18 (s, 2H) . LC–MS (ESI): m/z 323.1 [M + H]+. HPLC purity 95%.
2-((4-Fluorobenzyl)(methyl)amino)-N-phenylthiazole-4-carboxamide (26)
50% over 2-steps.
Step 1. 2-((4-Fluorobenzyl)(methyl)amino)thiazole-4-carboxylic Acid
Yield 81%. In a 7 mL glass vial, methyl 2-((4-fluorobenzyl)(methyl)amino)thiazole-4-carboxylate (150 mg, 0.535 mmol) was dissolved in methanol (2 mL). LiOH (128 mg, 5.35 mmol) was added to the solution, and the mixture was stirred at rt overnight. LC–MS indicated complete hydrolysis. Methanol was removed under reduced pressue. Residue was taken up in water and washed with ethyl acetate. Aqueous layer was cooled and acidified with dilute HCl. Precipitated acid was extracted with dichloromethane. Dichloromethane layer was washed with brine, dried on anhydrous MgSO4, and concentrated under vacuum to give a buff colored powder, 2-((4-fluorobenzyl)(methyl)amino)thiazole-4-carboxylic acid. Yield 81%. 1H NMR (300 MHz, DMSO-d6) δ 7.52 (s, 1H), 7.25–7.29 (m, 2H), 7.07 (t, J = 9 Hz, 2H), 4.71 (s, 2H), 3.09 (s, 3H). LC–MS (ESI): m/z 267.1 [M + H]+. HPLC purity 88%. Product was used without further purification in the next step.
Step 2
In a 7 mL glass vial, 2-((4-fluorobenzyl)(methyl)amino)thiazole-4-carboxylic acid (50 mg, 0.188 mmol) was dissolved in dry DMF (2 mL). HATU (107 mg, 0.282 mmol) and Et3N (0.052 mL, 0.376 mmol) were then added to the mixture and stirred at 25 °C for 30 min. Aniline was then added (0.021 mL, 0.225 mmol) and stirred at 25 °C overnight. LCMS indicated complete reaction. Reaction mixture was diluted with ethyl acetate and washed with successively with dilute HCl, water, saturated NaHCO3 solution, and brine. The sample was dried on anhydrous MgSO4 and concentrated under vacuum. Residue was purified on Biotage using 10 g silica gel column and 0–50% ethyl acetate in hexane as eluent. Pure fractions were combined and concentrated to give 26 as a light yellow colored solid. Yield 62%. 1H NMR (300 MHz, DMSO-d6) δ 9.73 (br s, 1H), 7.77 (dd, J = 9 Hz, 1 Hz, 2H), 7.52 (s, 1H), 7.33–7.44 (m, 4H), 7.17–7.23 (m. 2H), 7.08–7.13 (m, 1H), 4.83 (s, 2H), 3.08 (s, 3H). LC–MS (ESI): m/z 342.1 [M + H]+. HPLC purity 95%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-fluorophenyl)-5-methylpyrimidine-4-carboxamide (27)
Yield 17% over 2 steps.
Step 1
To ethyl 6-((4-fluorobenzyl)(methyl)amino)-5-methylpyrimidine-4-carboxylate 4 (0.25 g, 0.82 mmol) in THF (3 mL) were added lithium hydroxide monohydrate (0.138 g, 3.3 mmol) and water (1.5 mL). The reaction mixture was stirred at room temperature for 16 h. The reaction mixture was concentrated under vacuum, cooled to 0 °C, and acidified with citric acid solution. The acidified mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to yield the product, 6-((4-fluorobenzyl)(methyl)amino)-5-methylpyrimidine-4-carboxylic acid. Yield 88.4%. 1H NMR (400 MHz, DMSO-d6): δ 12.48 (s, 1H), 8.45 (s, 1H), 7.32 (d, J = 5.60 Hz, 2H), 7.17 (d, J = 9.20 Hz, 2H), 4.71 (s, 2H), 3.17 (s, 3H). LC–MS (APCI): m/z 276.0 [M + H]+.
Step 2
To a solution of 6-((4-fluorobenzyl)(methyl)amino)-5-methylpyrimidine-4-carboxylic acid 5 (0.20 g, 0.72 mmol) in DMF (3 mL) was added diisopropylethylamine (0.232 g, 1.8 mmol), HATU (0.41 g, 1.08 mmol), and 4-fluoroanililne (0.08 g, 0.7 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 16 h. Water was added to the reaction mixture and extracted with ethyl acetate. The organic layer was washed with water, dried over anhydrous sodium sulfate, concentrated under vacuum to get a crude product. It was purified by column chromatography on 60–120 mesh silica gel with 10% ethyl acetate in hexanes as eluent to yield 27 (0.055 g). Yield 20%. 1H NMR (400 MHz, CDCl3): δ 10.20 (s, 1H), 8.56 (s, 1H), 7.73–7.70 (m, 2H), 7.30–7.26 (m, 2H), 7.10–7.04 (m, 4H), 4.69 (s, 2H), 3.01 (s, 3H), 2.63 (s, 3H). LC–MS (APCI): m/z 369.2 [M + H]+. HPLC purity 100%.
6-((4-Chlorobenzyl)(methyl)amino)-2-methyl-N-phenylpyrimidine-4-carboxamide (28)
Reaction was according to general synthesis final step of compounds 1–21. Yield 95%. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.36 (m, 4H), 7.25 (d, J = 8.0 Hz, 2H), 7.12 (m, 2H), 4.87 (s, 2H), 3.11 (s, 3H) and 2.52 (s, 3H). LC–MS (ESI): 367.1 m/z [M + H]+. HPLC purity 98%.
Compounds 29 and 30 were both synthesized according to the general synthetic procedure below. Ethyl 2-chloro-6-((4-chlorobenzyl)(methyl)amino)pyrimidine-4-carboxylate (1 equiv), DIPEA (2 equiv), and the appropriate amine (1.2 equiv) were added to a pressure tube containing i-PrOH, and the tube was sealed and heated to 120 °C overnight. The solution was then cooled and the resulting precipitate filtered off to afford the intended product as a white solid.
2-Amino-6-((4-chlorobenzyl)(methyl)amino)-N-(4-methoxyphenyl)pyrimidine-4-carboxamide (29)
Yield 68%. 1H NMR (400 MHz, CDCl3) δ 9.64 (s, 1H), 7.66–7.49 (m, 2H), 7.29–7.14 (m, 2H), 7.13–7.00 (m, 2H), 6.91–6.73 (m, 3H), 5.11–4.49 (m, 4H), 3.74 (s, 3H), 2.98 (s, 3H).. LC–MS (ESI): m/z 398.1 [M + H]+. HPLC purity 96%.
6-((4-Chlorobenzyl)(methyl)amino)-2-(cyclopropylamino)-N-(4-methoxyphenyl)pyrimidine-4-carboxamide (30)
Yield 71%. 1H NMR (400 MHz, CDCl3) δ 9.81 (s, 1H), 7.67–7.45 (m, 2H), 7.26–7.17 (m, 2H), 7.10 (d, J = 8.1 Hz, 2H), 6.88–6.75 (m, 3H), 5.06 (s, 1H), 4.74 (s, 2H), 3.74 (d, J = 0.9 Hz, 3H), 3.03 (s, 3H), 2.69 (td, J = 5.8, 4.8, 2.6 Hz, 1H), 0.74–0.63 (m, 2H), 0.49 (m, 2H). LC–MS (ESI): m/z 438.2 [M + H]+. HPLC purity 95%.
General synthetic protocol for compounds 31 and 33–52 is as follows: The appropriate 6-(benzyl(methyl)amino)pyrimidine-4-carboxylic acid (1 equiv), DIPEA (3 equiv), and HATU (1.5 equiv) were dissolved in DMF and stirred for 10 min. To the solution was then added the appropriate amine, and the mixture was stirred at rt overnight. Final compounds were either isolated by precipitation with water and recrystallization with EtOH or extracted with EtOAc and washed successively with 10% LiCl, saturated aqueous NaHCO3 and brine and then purified by silica flash chromatography.
6-(Benzyl(methyl)amino)-N-methyl-N-phenylpyrimidine-4-carboxamide (31)
Yield 67%. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (s, 1H), 7.34–7.06 (m, 10H), 6.68 (m, 1H), 4.72 (s, 2H), 2.97 (s, 3H) and 2.48 (s, 3H). LC–MS (ESI): m/z 333.2 [M + H]+. HPLC purity 99%.
N-(6-((4-Fluorobenzyl)(methyl)amino)pyrimidin-4-yl)benzamide (32)
Yield 20%. To a stirred solution of 6-chloropyrimidin-4-amine (500 mg, 3.86 mmol) and DIPEA (1.348 mL, 7.72 mmol) in dry THF (20 mL) was added dropwise benzoyl chloride (0.672 mL, 5.79 mmol). The solution was heated at 50 °C for 12 h. The crude reaction mixture was adsorbed onto silica and purification attempted on an ISCO flash purification system; solvent system gradient from 0% to 70% EtOAc in Hex over 15 min. Single peak isolated contained both product and impurity. LC–MS (ESI): m/z 234.1 [M + H]+. Purity 64%. Crude material from previous step was dissolved in i-PrOH and N-methyl-1-phenylmethanamine (349 mg, 2.88 mmol) and DIPEA (0.837 mL, 4.79 mmol) added and heated at 100 °C for 4 h. The resulting solution was adsorbed directly onto silica gel and purified on ISCO purification system using a 4 g silica prepacked cartridge and solvent system from 0% to 100% EtOAc in hexane over 15 min to afford a pale cream solid. 1H NMR (300 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.36 (d, J = 1.1 Hz, 1H), 8.05–7.95 (m, 2H), 7.65–7.57 (m, 1H), 7.57–7.46 (m, 3H), 7.40–7.30 (m, 2H), 7.30–7.19 (m, 2H), 4.85 (s, 2H), 3.07 (s, 3H). LC–MS (ESI): m/z 337.2 [M + H]+. HPLC purity 99%.
N-Isopropyl-6-(methyl(4-(trifluoromethyl)benzyl)amino)pyrimidine-4-carboxamide (33)
Yield 20%. 1H NMR (300 MHz, DMSO-d6) δ 8.58 (s, 1H), 8.41 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.21 (s, 1H), 4.99 (s, 2H), 4.08 (h, J = 6.5 Hz, 1H), 3.18 (d, J = 4.3 Hz, 3H) and 1.18 (d, J = 6.7 Hz, 6H). LC–MS (ESI): m/z 353.2 [M + H]+. HPLC purity 98%.
N-Cyclohexyl-6-(methyl(4-(trifluoromethyl)benzyl)amino)pyrimidine-4-carboxamide (34)
Yield 60%. 1H NMR (300 MHz, DMSO-d6) δ 8.58 (s, 1H), 8.38 (d, J = 8.5 Hz, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.20 (s, 1H), 4.99 (s, 2H), 3.93–3.53 (m, 1H), 3.17 (s, 3H), 1.90–1.51 (m, 5H), 1.51–1.00 (m, 5H). LC–MS (ESI): m/z 393.2 [M + H]+. HPLC purity 100%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-methoxyphenyl)pyrimidine-4-carboxamide (35)
Yield 68%. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.65 (d, J = 1.2 Hz, 1H), 7.77 (d, J = 9.0 Hz, 2H), 7.32–7.24 (m, 3H), 7.15 (t, J = 8.8 Hz, 2H), 6.91 (d, J = 9.1 Hz, 2H), 4.88 (s, 2H), 3.73 (s, 3H), 3.13 (s, 3H). LC–MS (ESI): m/z 367.2 [M + H]+. HPLC purity 97%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-isopropoxyphenyl)pyrimidine-4-carboxamide (36)
Yield 63%. 1H NMR (400 MHz, CDCl3) δ 9.92–9.74 (m, 1H), 8.61 (s, 1H), 7.63 (d, J = 8.9 Hz, 2H), 7.40 (dd, J = 1.2, 0.5 Hz, 1H), 7.18 (dd, J = 8.5, 5.4 Hz, 2H), 7.00 (t, J = 8.6 Hz, 2H), 6.89 (d, J = 8.9 Hz, 2H), 4.86 (s, 2H), 4.51 (hept, J = 6.1 Hz, 1H), 3.11 (s, 3H), 1.40–1.29 (m, 6H). LC–MS (ESI): m/z 395.2 [M + H]+. HPLC purity 99%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-(2-methoxyethoxy)phenyl)pyrimidine-4-carboxamide (37)
Yield 45%. 1H NMR (300 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.67 (d, J = 1.1 Hz, 1H), 7.78 (d, J = 9.0 Hz, 2H), 7.36–7.24 (m, 3H), 7.17 (t, J = 8.8 Hz, 2H), 6.94 (d, J = 9.1 Hz, 2H), 4.90 (s, 2H), 4.25–3.96 (m, 2H), 3.73–3.53 (m, 2H), 3.30 (s, 3H), 3.16 (s, 3H). LC–MS (APCI): m/z 411.2 [M + H]+. HPLC purity 95%.
N-(4-(2-(Dimethylamino)ethoxy)phenyl)-6-((4-fluorobenzyl)(methyl)amino)pyrimidine-4-carboxamide (38)
Yield 25%. 1H NMR (300 MHz, DMSO-d6) δ 10.45 (s, 1H), 8.68 (s, 1H), 7.79 (d, J = 8.6 Hz, 2H), 7.38–7.24 (m, 3H), 7.17 (t, J = 8.7 Hz, 2H), 6.96 (d, J = 8.6 Hz, 2H), 4.90 (s, 2H), 4.10 (t, J = 5.7 Hz, 2H), 3.16 (s, 3H), 2.81 (t, J = 5.7 Hz, 2H), 2.36 (s, 6H). LC–MS (APCI): m/z 424.2.2 [M + H]+. HPLC purity 95%.
6-((4-Chlorobenzyl)(methyl)amino)-N-(pyridin-4-yl)pyrimidine-4-carboxamide (39)
Yield 35%. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.68 (s, 1H), 8.48 (d, J = 5.6 Hz, 2H), 7.89 (d, J = 5.6 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.33–7.22 (m, 3H), 4.90 (s, 2H), 3.15 (s, 2H). LC–MS (ESI): m/z 354.1 [M + H]+. HPLC purity 100%.
6-(Methyl(4-(trifluoromethyl)benzyl)amino)-N-(4-(4-methylpiperazin-1-yl)phenyl)pyrimidine-4-carboxamide (40)
Yield 74%. 1H NMR (300 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.74 (s, 1H), 7.79 (d, J = 9 Hz, 2H), 7.76 (s, 1H), 7.73 (d, J = 9 Hz, 2H), 7.50 (d, J = 9 Hz, 2H), 7.03 (d, J = 6 Hz, 2H), 5.11 (s, 2H), 3.80 (d, J = 9H, 2H), 3.29 (s, 3H), 3.04–3.21 (m, 5H), 2.82 (m, 3H). LC–MS (ESI): m/z 485.1 [M + H]+. HPLC purity 98%.
6-(Methyl((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)-N-(4-(4-methylpiperazin-1-yl)phenyl)pyrimidine-4-carboxamide (41)
Yield 45%. 1H NMR (300 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.71 (d, J = 6 Hz, 2H), 7.94 (d, J = 9 Hz, 1H), 7.87 (d, J = 9 Hz, 1H), 7.77 (d, J = 9H, 2H), 7.47 (s, 1H), 7.02 (d, J = 9 Hz, 2H), 5.09 (s, 2H), 3.80 (d, J = 15 Hz, 2H), 3.26 (s, 3H), 3.00–3.21 (m, 6H), 2.83 (d, J = 6 Hz, 3H). LC–MS (ESI): m/z 486.2 [M + H]+. HPLC purity 98%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(3-(4-methylpiperazin-1-yl)phenyl)pyrimidine-4-carboxamide (42)
Yield 12%. 1H NMR (300 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.68 (s, 1H), 7.51 (t, J = 2.2 Hz, 1H), 7.40–7.23 (m, 4H), 7.24–7.10 (m, 3H), 6.73 (dd, J = 8.2, 2.4 Hz, 1H), 4.90 (s, 2H), 3.38 (s, 4H), 3.16 (s, 7H), 2.26 (s, 3H). LC–MS (APCI): m/z 435.2 [M + H]+. HPLC purity 97%.
6-((4-Fluorobenzyl)(methyl)amino)-N-(6-(4-methylpiperazin-1-yl)pyridin-3-yl)pyrimidine-4-carboxamide (43)
Yield 29%. 1H NMR (300 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.55 (s, 1H), 7.91–7.65 (m, 2H), 7.45–7.26 (m, 2H), 7.28–7.03 (m, 4H), 4.72 (s, 2H), 3.32 (s, 8H) 3.02 (s, 3H), 2.35 (s, 3H). LC–MS (APCI): m/z 436.2 [M + H]+. HPLC purity 97%.
N-(4-(4-Acetylpiperazin-1-yl)phenyl)-6-(methyl((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)pyrimidine-4-carboxamide (44)
Yield 38%. 1H NMR (300 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.71 (s, 1H), 8.67 (s, 1H), 7.96–7.86 (m, 2H), 7.75 (d, J = 9.1 Hz, 2H), 7.34 (s, 1H), 6.97 (d, J = 9.1 Hz, 2H), 5.07 (s, 2H), 3.62–3.50 (m, 4H), 3.23 (s, 3H), 3.18–3.04 (m, 4H), 2.05 (s, 3H). LC–MS (ESI): m/z 514.1 [M + H]+. HPLC purity 87.75%
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-morpholinophenyl)pyrimidine-4-carboxamide (45)
Yield 30%. 1H NMR (300 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.66 (s, 1H), 7.74 (d, J = 9.0 Hz, 2H), 7.35–7.10 (m, 5H), 6.94 (d, J = 9.1 Hz, 2H), 4.90 (s, 2H), 3.83–3.68 (m, 4H), 3.16 (s, 3H), 3.12–3.03 (m, 4H). LC–MS (ESI): m/z 422.1 [M + H]+. HPLC purity 100%
6-(Methyl((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)-N-(5-morpholinopyridin-2-yl)pyrimidine-4-carboxamide (46)
Yield 23%. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.72 (d, J = 2.0 Hz, 1H), 8.68 (s, 1H), 8.12 (m, 2H), 7.93 (d, J = 8.1 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.52 (dd, J = 9.1, 3.1 Hz, 1H), 7.39 (d, J = 1.2 Hz, 1H), 5.08 (s, 2H), 3.76 (m, 4H), 3.25 (s, 3H), 3.17 (m, 4H). LC–MS (ESI): m/z 474.2 [M + H]+. HPLC purity 97%
6-((4-Fluorobenzyl)(methyl)amino)-N-(1-methyl-1H-pyrazol-3-yl)pyrimidine-4-carboxamide (47)
Yield 54%. 1H NMR (400 MHz, CDCl3) δ 10.20 (s, 1H), 8.53 (d, J = 1.2 Hz, 1H), 7.30 (d, J = 1.2 Hz, 1H), 7.21 (d, J = 2.3 Hz, 1H), 7.15–7.08 (m, 2H), 6.93 (t, J = 8.7 Hz, 2H), 6.73 (d, J = 2.3 Hz, 1H), 4.78 (s, 2H), 3.76 (s, 3H), 3.03 (s, 3H). LC–MS (ESI): m/z 341.2 [M + H]+. HPLC purity 100%
6-(Methyl(4-(trifluoromethyl)benzyl)amino)-N-(1-methyl-1H-1,2,4-triazol-3-yl)pyrimidine-4-carboxamide (48)
Yield 49%. 1H NMR (300 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.66 (s, 1H), 8.39 (s, 1H), 7.71 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.28 (s, 1H), 5.02 (s, 2H), 3.85 (s, 3H), 3.20 (s, 3H). LC–MS (ESI): m/z 392.2 [M + H]+. HPLC purity 99%
6-((4-Fluorobenzyl)(methyl)amino)-N-(isoxazol-3-yl)pyrimidine-4-carboxamide (49)
Yield 6%. 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 8.86 (d, J = 1.8 Hz, 1H), 8.67 (d, J = 1.2 Hz, 1H), 7.33–7.24 (m, 3H), 7.16 (t, J = 8.8 Hz, 2H), 6.98 (d, J = 1.7 Hz, 1H), 4.89 (s, 2H), 3.30 (s, 3H). LC–MS (ESI): m/z 328.0 [M + H]+. HPLC purity 99%
6-((4-Fluorobenzyl)(methyl)amino)-N-(4-methylthiazol-2-yl)pyrimidine-4-carboxamide (50)
Yield 6%. 1H NMR (300 MHz, DMSO-d6) δ 11.75 (s, 1H), 8.67 (d, J = 1.1 Hz, 1H), 7.36–7.23 (m, 3H), 7.24–7.09 (m, 2H), 6.91 (q, J = 0.9 Hz, 1H), 4.90 (s, 2H), 3.16 (s, 3H), 2.31 (d, J = 1.1 Hz, 3H). LC–MS (ESI): m/z 358.0 [M + H]+. HPLC purity 99%
6-((4-Fluorobenzyl)(methyl)amino)-N-(1,3,4-thiadiazol-2-yl)pyrimidine-4-carboxamide (51)
Yield 47%. 1H NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 9.26 (d, J = 1.6 Hz, 1H), 8.68 (s, 1H), 7.33 (s, 1H), 7.28 (t, J = 6.2 Hz, 2H), 7.15 (t, J = 8.1 Hz, 2H), 4.88 (s, 2H), 3.13 (s, 3H). LC–MS (ESI): m/z 345.1 [M + H]+. HPLC purity 100%
6-(Methyl((6-(trifluoromethyl)pyridin-3-yl)methyl)amino)-N-(1,3,4-thiadiazol-2-yl)pyrimidine-4-carboxamide (52)
Yield 30%. 1H NMR (400 MHz, DMSO-d6) δ 12.61 (s, 1H), 9.29 (s, 1H), 8.72 (s, 2H), 7.95–7.86 (m, 2H), 7.42 (s, 1H), 5.08 (s, 2H) and 3.24 (s, 3H). LC–MS (ESI): m/z 396.1 [M + H]+. HPLC purity 96%
N-(4-Fluorobenzyl)-N-methyl-6-(2,2,2-trifluoro-1-((4-fluorophenyl)amino)ethyl)pyrimidin-4-amine (53)
A solution of 4-fluoroaniline (42.6 mg, 0.383 mmol), Et3N (0.133 mL, 0.958 mmol), titanium tetrachloride (0.035 mL, 0.319 mmol), and 2,2,2-trifluoro-1-(6-((4-fluorobenzyl)(methyl)amino)pyrimidin-4-yl)ethan-1-one (100 mg, 0.319 mmol) in dry DCM (10 mL) under nitrogen was stirred at rt for 18 h. Sodium cyanoborohydride (80 mg, 1.277 mmol) was then added portionwise and the reaction stirred for a further 30 min. The reaction was then quenched with water (10 mL) and washed with brine (10 mL). The organic layer was separated, dried, and adsorbed onto silica and purified by silica flash chromatography to afford a clear oil. Yield 37%. 1H NMR (300 MHz, CDCl3) δ 8.68 (d, J = 1.1 Hz, 1H), 7.25–7.13 (m, 2H), 7.03 (t, J = 8.6 Hz, 2H), 6.92 (t, J = 8.7 Hz, 2H), 6.81–6.66 (m, 2H), 6.51 (s, 1H), 5.40 (s, 1H), 5.06–4.55 (m, 3H), 3.08 (s, 3H). LC–MS (ESI): m/z 409.1 [M + H]+. HPLC purity 97%
N-(4-Fluorobenzyl)-N-methyl-6-(5-phenyl-4H-1,2,4-triazol-3-yl)pyrimidin-4-amine (54)
A solution of potassium carbonate, benzonitrile, and 6-((4-fluorobenzyl)(methyl)amino)pyrimidine-4-carbohydrazide in n-butanol (4 mL) was heated in a sealed tube overnight. The resulting mixture was concentrated onto silica and purified by silica flash column chromatography, EtOAc/Hex (1:1), to afford an off white solid. Yield 61%. 1H NMR (300 MHz, DMSO-d6) δ 8.66 (d, J = 1.1 Hz, 1H), 8.14–8.00 (m, 2H), 7.60–7.41 (m, 3H), 7.41–7.25 (m, 3H), 7.18 (t, J = 8.9 Hz, 2H), 4.92 (s, 2H), 3.16 (s, 3H). LC–MS (ESI): m/z 361.2 [M + H]+. HPLC 97%.
Acknowledgments
The authors acknowledge the following: Dr. Sridevi Bashayam, Syngene, India, and her team for profound chemistry support; Marianna de Kock and Claudia Spies, DST/NRF Centre of Excellence for Biomedical Tuberculosis Research/SAMRC Centre for Tuberculosis Research, Division of Molecular Biology and Human Genetics, Faculty of Health Sciences, Stellenbosch University, Tygerberg, South Africa, for excellent technical assistance for excellent technical support in generating MICs against clinical TB isolates; Dr. Michael Whitty and Dr. David Waterson for insightful scientific consultation during the course of the project. The project was funded through a Global Health Grant (Grant OPP1066878) received from the Bill and Melinda Gates Foundation, the Division of Intramural Research of the NIAID/NIH and the Strategic Health Innovation Partnerships (SHIP) unit of the South African Medical Research Council (SAMRC). The University of Cape Town, SAMRC, and South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation, are gratefully acknowledged for support (K.C.).
Glossary
Abbreviations Used
- TB
tuberculosis
- Mtb
Mycobacterium tuberculosis
- MIC
minimum inhibitory concentration
- GFP
green fluorescent protein
- SAR
structure–activity relationship
- AUC
area under the curve
- fu
fraction unbound
- CFU
colony-forming unit
- rt
room temperature
- EtOH
ethanol
- EtOAc
ethyl acetate
- DIPEA
diisopropylethylamine
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- BuOH
butanol
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- TBAC
tetrabutylammonium chloride
- POCl3
phosphorus(V) oxychloride
- SOCl2
thionyl chloride
- NaBH4
sodium borohydride
- NaCNBH3
sodium cyanoborohydride
- TiCl4
titanium(IV) chloride
- TMT
tandem mass tag
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01347.
Author Contributions
K.C. and R.G. came up with the original idea of screening the SoftFocus library against Mycobacterium tuberculosis to identify hits for medicinal chemistry progression. H.I.M.B. and C.E.B facilitated the screening of the SoftFocus library and analyzed the data. Chemical synthesis for hit evaluation and SAR expansion were conducted by R.K.G., C.R.W., C.S.d.M., S.B.S., A.N., R.M., and S.R.G. SAR analysis and compound design were conducted by L.S., S.R.G., C.R.W., R.K.G., R.M. C.J.H., and K. C. Biology triage and MIC assays were performed by R.S. D.F.W. and V.M. were involved in the analysis of biology triage results and provided intellectual input into the overall biology of the project. A.M. was responsible for mutant generation and cross-screening of compounds against the mutants. F.A.S. and P.v.H. conducted and analyzed screening data against the clinical isolates. E.A., D.T., M.N., C.B., and N.L. performed and analyzed DMPK experiments. B.U., C.S., and A.A. performed the synthesis of derivatives for chemoproteomics experiments. O.S., G.S., M.J.R.-L., M.J.L.-M., H.P., S.G.-D., M.B., and G.D. performed the chemoproteomics and related biology experiments. J.L. and L.B. provided the leadership and intellectual inputs into the chemoproteomics experiments. C.R.W. and S.R.G. wrote the manuscript which was further edited by L.S., H.I.M.B, C.E.B., F.A.S., D.F.W., and C.B. and was finally reviewed and edited by K.C. The chemoproteomics section of the manuscript was written by the personnel involved in performing the experiments.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organisation. Global Tuberculosis Report. http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1 (accessed Dec 12, 2016).
- Dover L. G.; Coxon G. D. Current Status and Research Strategies in Tuberculosis Drug Development Miniperspective. J. Med. Chem. 2011, 54, 6157–6165. 10.1021/jm200305q. [DOI] [PubMed] [Google Scholar]
- Olaru I. D.; Lange C.; Heyckendorf J. Personalized Medicine for Patients with MDR-TB. J. Antimicrob. Chemother. 2016, 71, 852–855. 10.1093/jac/dkv354. [DOI] [PubMed] [Google Scholar]
- Warner D. F.; Mizrahi V. Shortening Treatment for Tuberculosis - Back to Basics. N. Engl. J. Med. 2014, 371, 1642–1643. 10.1056/NEJMe1410977. [DOI] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Gohlmann H. W. H.; Neefs J.-M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium Tuberculosis. Science 2005, 307, 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Horsburgh C. R. Jr; Barry C. E. III; Lange C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- Fox G. J.; Menzies D. A Review of the Evidence for Using Bedaquiline (TMC207) to Treat Multi-Drug Resistant Tuberculosis. Infect. Dis. Ther. 2013, 2, 123–144. 10.1007/s40121-013-0009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U. H.; Smith P. W. Perspective: Challenges and Opportunities in TB Drug Discovery from Phenotypic Screening. Bioorg. Med. Chem. 2015, 23, 5087–5097. 10.1016/j.bmc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Kana B. D.; Karakousis P. C.; Parish T.; Dick T. Future Target-Based Drug Discovery for Tuberculosis?. Tuberculosis 2014, 94, 551–556. 10.1016/j.tube.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. C.; Trujillo C.; Wang Z.; Eoh H.; Ehrt S.; Schnappinger D.; Boshoff H. I. M.; Rhee K. Y.; Barry C. E. III; Mizrahi V. Validation of CoaBC as a Bactericidal Target in the Coenzyme A Pathway of Mycobacterium Tuberculosis. ACS Infect. Dis. 2016, 2, 958–968. 10.1021/acsinfecdis.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; Christophe T.; Lee H.; Kempf M.; Jackson M.; Lenaerts A. J.; Pham H.; Jones V.; Seo M. J.; Kim Y. M.; Seo M.; Seo J. J.; Park D.; Ko Y.; Choi I.; Kim R.; Kim S. Y.; Lim S.; Yim S.-A.; Nam J.; Kang H.; Kwon H.; Oh C.-T.; Cho Y.; Jang Y.; Kim J.; Chua A.; Tan B. H.; Nanjundappa M. B.; Rao S. P. S.; Barnes W. S.; Wintjens R.; Walker J. R.; Alonso S.; Lee S.; Kim J.; Oh S.; Oh T.; Nehrbass U.; Han S.-J.; No Z.; Lee J.; Brodin P.; Cho S.-N.; Nam K.; Kim J. Discovery of Q203, a Potent Clinical Candidate for the Treatment of Tuberculosis. Nat. Med. 2013, 19, 1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Sacksteder K. A.; Protopopova M.; Barry C. E. III; Andries K.; Nacy C. A. Discovery and Development of SQ109: A New Antitubercular Drug with a Novel Mechanism of Action. Future Microbiol. 2012, 7, 823–837. 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Meshcheryakov V. A.; Poce G.; Chng S.-S. MmpL3 Is the Flippase for Mycolic Acids in Mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7993–7998. 10.1073/pnas.1700062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a New Combination Therapy for Tuberculosis with next Generation Benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R.; Sadler C.; Naik M.; Hosagrahara V.; Hameed S.; Shinde V.; Bathula C.; Humnabadkar V.; Kumar N.; Reddy J.; Panduga V.; Sharma S.; Ambady A.; Hegde N.; Whiteaker J.; McLaughlin R. E.; Gardner H.; Madhavapeddi P.; Ramachandran V.; Kaur P.; Narayan A.; Guptha S.; Awasthy D.; Narayan C.; Mahadevaswamy J.; Vishwas K.; Ahuja V.; Srivastava A.; Prabhakar K.; Bharath S.; Kale R.; Ramaiah M.; Choudhury N. R.; Sambandamurthy V. K.; Solapure S.; Iyer P. S.; Narayanan S.; Chatterji M. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium Tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56, 9701–9708. 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- Chatterji M.; Shandil R.; Manjunatha M. R.; Solapure S.; Ramachandran V.; Kumar N.; Saralaya R.; Panduga V.; Reddy J.; KR P.; Sharma S.; Sadler C.; Cooper C. B.; Mdluli K.; Iyer P. S.; Narayanan S.; Shirude P. S. 1,4-Azaindole, A Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. 10.1128/AAC.03233-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. C.; Hill R. D.; Sheppard D. W.; Slater M. J.; Stouten P. F. W. The Design and Application of Target-Focused Compound Libraries. Comb. Chem. High Throughput Screening 2011, 14, 521–531. 10.2174/138620711795767802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechartier B.; Rybniker J.; Zumla A.; Cole S. T. Tuberculosis Drug Discovery in the Post-Post-Genomic Era. EMBO Mol. Med. 2014, 6, 158–168. 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoagland D. T.; Liu J.; Lee R. B.; Lee R. E. New Agents for the Treatment of Drug-Resistant Mycobacterium Tuberculosis. Adv. Drug Delivery Rev. 2016, 102, 55–72. 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares de Melo C.; Feng T.-S.; van Der Westhuyzen R.; Gessner R. K.; Street L. J.; Morgans G. L.; Warner D. F.; Moosa A.; Naran K.; Lawrence N.; Boshoff H. I. M.; Barry C. E. III; Harris C. J.; Gordon R.; Chibale K. Aminopyrazolo[1,5-A]pyrimidines as Potential Inhibitors of Mycobacterium Tuberculosis: Structure Activity Relationships and ADME Characterization. Bioorg. Med. Chem. 2015, 23, 7240–7250. 10.1016/j.bmc.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Arora K.; Ochoa-Montano B.; Tsang P. S.; Blundell T. L.; Dawes S. S.; Mizrahi V.; Bayliss T.; Mackenzie C. J.; Cleghorn L. A. T.; Ray P. C.; Wyatt P. G.; Uh E.; Lee J.; Barry C. E. III; Boshoff H. I. Respiratory Flexibility in Response to Inhibition of Cytochrome C Oxidase in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6962–6965. 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosa A.; Lamprecht D. A.; Arora K.; Barry C. E. III; Boshoff H. I. M.; Ioerger T. R.; Steyn A. J. C.; Mizrahi V.; Warner D. F. Susceptibility of Mycobacterium Tuberculosis Cytochrome Bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrob. Agents Chemother. 2017, 61, e01338-17. 10.1128/AAC.01338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naran K.; Moosa A.; Barry C. E. III; Boshoff H. I. M.; Mizrahi V.; Warner D. F. Bioluminescent Reporters for Rapid Mechanism of Action Assessment in Tuberculosis Drug Discovery. Antimicrob. Agents Chemother. 2016, 60, 6748–6757. 10.1128/AAC.01178-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W.; Yamane M. Mo(CO)6-Mediated Carbamoylation of Aryl Halides. J. Org. Chem. 2010, 75, 8410–8415. 10.1021/jo101611g. [DOI] [PubMed] [Google Scholar]
- Rudzinski D. M.; Kelly C. B.; Leadbeater N. E. A Weinreb Amide Approach to the Synthesis of Trifluoromethylketones. Chem. Commun. 2012, 48, 9610–9612. 10.1039/c2cc35037h. [DOI] [PubMed] [Google Scholar]
- Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong L. T.; Falgueyret J. P.; Kimmel D. B.; Lamontagne S.; Léger S.; LeRiche T.; Li C. S.; Massé F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa R. M.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Thérien M.; Truong V. L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. The Discovery of Odanacatib (MK-0822), a Selective Inhibitor of Cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Bock V. D.; Speijer D.; Hiemstra H.; van Maarseveen J. H. 1,2,3-Triazoles as Peptide Bond Isosteres: Synthesis and Biological Evaluation of Cyclotetrapeptide Mimics. Org. Biomol. Chem. 2007, 5, 971–975. 10.1039/b616751a. [DOI] [PubMed] [Google Scholar]
- Cox J. A. G.; Abrahams K. A.; Alemparte C.; Ghidelli-Disse S.; Rullas J.; Angulo-Barturen I.; Singh A.; Gurcha S. S.; Nataraj V.; Bethell S.; Remuiñán M. J.; Encinas L.; Jervis P. J.; Cammack N. C.; Bhatt A.; Kruse U.; Bantscheff M.; Fütterer K.; Barros D.; Ballell L.; Drewes G.; Besra G. S. THPP Target Assignment Reveals EchA6 as an Essential Fatty Acid Shuttle in Mycobacteria. Nat. Microbiol. 2016, 1, 15006. 10.1038/nmicrobiol.2015.6. [DOI] [PubMed] [Google Scholar]
- Abrahams K. A.; Chung C.; Ghidelli-Disse S.; Rullas J.; Rebollo-López M. J.; Gurcha S. S.; Cox J. A. G.; Mendoza A.; Jiménez-Navarro E.; Martínez-Martínez M. S.; Neu M.; Shillings A.; Homes P.; Argyrou A.; Casanueva R.; Loman N. J.; Moynihan P. J.; Lelièvre J.; Selenski C.; Axtman M.; Kremer L.; Bantscheff M.; Angulo-Barturen I.; Izquierdo M. C.; Cammack N. C.; Drewes G.; Ballell L.; Barros D.; Besra G. S.; Bates R. H. Identification of KasA as the Cellular Target of an Anti-Tubercular Scaffold. Nat. Commun. 2016, 7, 12581. 10.1038/ncomms12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.; Pacitto A.; Bayliss T.; Cleghorn L. A. T.; Wang Z.; Hartman T.; Arora K.; Ioerger T. R.; Sacchettini J.; Rizzi M.; Donini S.; Blundell T. L.; Ascher D. B.; Rhee K.; Breda A.; Zhou N.; Dartois V.; Jonnala S. R.; Via L. E.; Mizrahi V.; Epemolu O.; Stojanovski L.; Simeons F.; Osuna-Cabello M.; Ellis L.; MacKenzie C. J.; Smith A. R. C.; Davis S. H.; Murugesan D.; Buchanan K. I.; Turner P. A.; Huggett M.; Zuccotto F.; Rebollo-Lopez M. J.; Lafuente-Monasterio M. J.; Sanz O.; Diaz G. S.; Lelièvre J.; Ballell L.; Selenski C.; Axtman M.; Ghidelli-Disse S.; Pflaumer H.; Bosche M.; Drewes G.; Freiberg G. M.; Kurnick M. D.; Srikumaran M.; Kempf D. J.; Green S. R.; Ray P. C.; Read K.; Wyatt P.; Barry C. E. III; Boshoff H. I. Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium Tuberculosis. ACS Infect. Dis. 2017, 3, 18–33. 10.1021/acsinfecdis.6b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A.-M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dümpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. Chemoproteomics Profiling of HDAC Inhibitors Reveals Selective Targeting of HDAC Complexes. Nat. Biotechnol. 2011, 29, 255–265. 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Målen H.; Pathak S.; Søfteland T.; de Souza G. A.; Wiker H. G. Definition of Novel Cell Envelope Associated Proteins in Triton X-114 Extracts of Mycobacterium Tuberculosis H37Rv. BMC Microbiol. 2010, 10, 132. 10.1186/1471-2180-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustijns P.; Wuyts B.; Hens B.; Annaert P.; Butler J.; Brouwers J. A Review of Drug Solubility in Human Intestinal Fluids: Implications for the Prediction of Oral Absorption. Eur. J. Pharm. Sci. 2014, 57, 322–332. 10.1016/j.ejps.2013.08.027. [DOI] [PubMed] [Google Scholar]
- Kansy M.; Senner F.; Gubernator K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]
- Sarathy J. P.; Zuccotto F.; Hsinpin H.; Sandberg L.; Via L. E.; Marriner G. A.; Masquelin T.; Wyatt P. G.; Ray P. C.; Dartois V. Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect. Dis. 2016, 2, 552–563. 10.1021/acsinfecdis.6b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp W.; Breithardt G.; Camm A. J.; Janse M. J.; Rosen M. R.; Antzelevitch C.; Escande D.; Franz M.; Malik M.; Moss A.; Shah R. The Potential for QT Prolongation and Proarrhythmia by Non-Antiarrhythmic Drugs: Clinical and Regulatory Implications. Report on a Policy Conference of the European Society of Cardiology. Eur. Heart J. 2000, 21, 1216–1231. 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]
- Werner T.; Sweetman G.; Savitski M. F.; Mathieson T.; Bantscheff M.; Savitski M. M. Ion Coalescence of Neutron Encoded TMT 10-Plex Reporter Ions. Anal. Chem. 2014, 86, 3594–3601. 10.1021/ac500140s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.