

Abstract

The past decade has seen the discovery of four different classes of radical S-adenosylmethionine (rSAM) methyltransferases that methylate unactivated carbon centers. Whereas the mechanism of class A is well understood, the molecular details of methylation by classes B–D are not. In this study, we present detailed mechanistic investigations of the class C rSAM methyltransferase TbtI involved in the biosynthesis of the potent thiopeptide antibiotic thiomuracin. TbtI C-methylates a Cys-derived thiazole during posttranslational maturation. Product analysis demonstrates that two SAM molecules are required for methylation and that one SAM (SAM1) is converted to 5′-deoxyadenosine and the second SAM (SAM2) is converted to S-adenosyl-l-homocysteine (SAH). Isotope labeling studies show that a hydrogen is transferred from the methyl group of SAM2 to the 5′-deoxyadenosine of SAM1 and the other two hydrogens of the methyl group of SAM2 appear in the methylated product. In addition, a hydrogen appears to be transferred from the β-position of the thiazole to the methyl group in the product. We also show that the methyl protons in the product can exchange with solvent. A mechanism consistent with these observations is presented that differs from other characterized radical SAM methyltransferases.
Introduction
For many decades, methyl transfer in nature utilizing S-adenosyl-l-methionine (SAM) as the methyl donor was thought to occur exclusively by SN2-like processes.1 However, early metabolic labeling studies suggested that methyl groups derived from methionine, are also transferred to non-nucleophilic carbons.2−5 Subsequent characterization of the biosynthetic gene clusters6−9 and bioinformatic and biochemical analyses of the proteins involved in these reactions10,11 showed that they are part of the radical SAM (rSAM) superfamily of enzymes,12 which comprise the largest enzyme superfamily known.13,14 In the years since this groundbreaking discovery, four distinct classes of rSAM methyltransferases (MTs) have been identified that differ in their cofactor/cosubstrate requirements and their proposed reaction mechanisms.15−18 The mechanism of RNA base methylases (class A) has been well-studied (Figure S1),19−23 but the mechanisms of the other three classes are considerably less understood. Class B consists of cobalamin-dependent methyltransferases, including TsrM,24,25 Fom3,10,26,27 GenK,28 CysS,29 ThnK,11 Sven0516,30 and PoyC.31 Class C are cobalamin-independent and are involved in the biosynthesis of several natural products,32−36 whereas class D enzymes use methylenetetrahydrofolate as the methyl group donor.37 In this study, we undertook a detailed mechanistic investigation of a class C rSAM MT (TbtI) involved in the biosynthesis of the thiopeptide thiomuracin (Figure 1a), and showed that it uses a fundamentally different mechanism from other characterized classes of rSAM MTs.
Figure 1.

Product analysis for the methyl transfer reaction catalyzed by TbtI. (a) Structure of thiomuracin A1. The methyl group that is introduced by TbtI is shown in red. (b) Regioselective methylation of the thiazole at position 4 of the TbtA core peptide by TbtI. The Val-Gly-Ala sequence at the N-terminus of peptide 1 originates from the leader peptide.36 (c) Time-dependent formation of the methylated TbtA peptide, 5′-dA, and SAH. Reaction mixtures included the following components: TbtA hexazole (50 μM), [4Fe–4S] cluster reconstituted TbtI (10 μM), SAM (1 mM), flavodoxin (10 μM), flavodoxin reductase (10 μM), NAPDH (2 mM) in reaction buffer (50 mM Tris–HCl, pH 7.5).
Results
TbtI Contains One Iron–Sulfur Cluster and Does Not Require Additional Cysteines
We recently reported the in vitro reconstitution of TbtI, which regioselectively C-methylates one of six thiazoles during the biosynthesis of thiomuracin (Figure 1b).36 Although sequence analysis does not place TbtI among class A rSAM MTs, we nevertheless first assessed whether TbtI might employ a similar mechanism. Class A rSAM MTs utilize an unusual mechanism in which a methyl group from SAM is first transferred to a Cys on the protein.20 Then, reductive cleavage of a second SAM molecule occurs, mediated by a reduced iron–sulfur [4Fe–4S] cluster, which generates a 5′-deoxyadenosyl radical (5′-dA•). The 5′-dA• species then abstracts a hydrogen atom from the methylcysteine to initiate the methyl transfer process (Figure S1). We first analyzed the sequence of TbtI and its orthologs from various thiopeptide gene clusters36 and identified five conserved Cys residues (Cys14, Cys18, Cys21, Cys254, and Cys343; Figure S2). Three of these appear in a canonical motif (CxxxCxxC), which typically supply the ligands for a characteristic rSAM [4Fe–4S] cluster.12 TbtI variants in which these Cys residues were replaced by Ala (C14A, C18A, and C21A) were inactive (Figure S3), but substitution of the remaining Cys residues (C89A, C254A, and C343A) did not adversely affect the activity of TbtI (Figure S3), suggesting that they are functionally dispensable.
Thus, these data show that unlike class A rSAM MTs, which require at least two additional Cys other than the three used to bind the [4Fe–4S] cluster,22 TbtI does not require additional Cys residues for catalysis. This conclusion is further supported by the fact that a TbtI homologue involved in nocathiacin biosynthesis (NocN) contains only one additional Cys other than the three involved in [4Fe–4S] cluster coordination.15 We considered that a methyl group from SAM could be transferred to an amino acid residue other than Cys. To test this possibility, the enzyme was treated with 14CH3-SAM and the peptide substrate without addition of reducing agents. The enzyme was isolated and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by Coomassie staining or autoradiographic imaging. We did not observe a radioactive band for the protein, suggesting that the methyl group is not initially transferred to the enzyme. This finding could also be explained by a methyl group already being present on the enzyme upon isolation from Escherichia coli; however, single turnover experiments presented below argue against this possibility. We cannot completely rule out that transfer of the methyl group from SAM to an amino acid residue only occurs in the presence of all reaction components including reductants. TbtI lacks the cobalamin-binding motif characteristic of class B rSAM MTs,11,12,24,29,31,38−41 as well as the tetrahydrofolate-binding domain observed in a recently discovered class D rSAM MT.37 All of these factors point toward a distinct mechanism for TbtI.
Product Analysis
To gain insight into the mechanism of methyl transfer, we first determined the products of the reaction that are derived from SAM. Out of a concern that the strong reductant dithionite might react with the initial products of the enzymatic reaction, we replaced dithionite in our original assay36 with flavodoxin/flavodoxin reductase and NADPH to deliver the reducing equivalents to TbtI (see the Experimental Section). As anticipated on the basis of other rSAM proteins,42 5′-deoxyadenosine (5′-dA) was detected in slightly more than one equivalent compared to the methylated TbtA product (Figure 1c). Additionally, S-adenosyl-l-homocysteine (SAH) was detected in near stoichiometric quantities, and 5′-dA and SAH were formed at very similar rates (Figure 1c). When the reaction was conducted with CD3-SAM (96% CD3 by ESI-MS; see the Supporting Information, Figure S4), ∼90% of the 5′-dA contained a single deuterium (Figure S4b), suggesting that the 5′-dA• generated from SAM1 may abstract a hydrogen atom from another SAM molecule (SAM2) during catalysis. It also suggests that the small amount of unlabeled 5′-dA produced was the result of uncoupled SAM cleavage, as has been observed in other rSAM enzymes.42 Control experiments showed that production of SAH and 5′-dA was dependent on the presence of both TbtI and the reducing system (Figure S5). It was recently reported that another class C rSAM MT involved in nosiheptide biosynthesis (NosN) produces thioadenosine in its reaction.34 We therefore investigated if TbtI generated thioadenosine; however, we were unable to observe its production by LC-MS, whereas an authentic standard was readily detected at the concentrations expected to be produced (Figure S5). Another recent study on NosN also concluded that thioadenosine is not a product of the reaction.35 On the basis of the products we observed, we formulated a series of working hypotheses for the enzymatic mechanism of TbtI (Figure 2).
Figure 2.

Proposed potential reaction mechanisms for TbtI. 5′-dA• is generated from SAM1 in a process mediated by the reduced [4Fe–4S] cluster. The 5′-dA• then abstracts a hydrogen atom from the methyl group of SAM2, and the resulting radical adds to the thiazole. An active site base deprotonates radical 3, leading to the elimination of SAH, yielding radical 4. Three different pathways (a–c) can provide the product and reset the enzyme. In pathway a, the radical is reduced to the anion 5 and protonated. In pathway b, radical 4 abstracts a hydrogen atom from an active site amino acid (X–H). X–H could be the protonated base B–H. In pathway c, radical 4 abstracts a hydrogen atom from the methyl group of 5′-dA. Ade = adenine.
Isotope Labeling Studies Involving the Methyl Group of SAM
To evaluate the proposed mechanisms, we first investigated whether thiazole methylation involves transfer of the intact methyl group of SAM or a methylene group. Therefore, reactions with TbtI were carried out using CD3-SAM. Owing to the large molecular weight of the TbtA hexazole core substrate even after removal of most of the leader peptide (three residues remain, Val-Gly-Ala) with endoproteinase GluC (C71H85N19O19S6, [M + H]+ = 1700 Da), the numerous isotopologues visible by mass spectrometry rendered isotope labeling studies difficult to interpret. Therefore, we prepared the TbtA hexazole substrate from E. coli grown in minimal media using 13C-depleted glucose and 15N-depleted (NH4)2SO4 as the sole carbon and nitrogen sources, and again removed the majority of the leader peptide by treatment with endoproteinase GluC. The resulting TbtA hexazole core peptide 1 (Figure 1b) was analyzed by electrospray ionization mass spectrometry (ESI-MS) and exhibited a much-simplified spectrum (Figure S6). Use of CD3-SAM in a 16 h methylation assay with TbtI and peptide 1 resulted in an unexpectedly complicated mass spectrum for the product in which ions were observed consistent with the addition of CH3, CH2D, and CHD2 groups (Figure 3a, black spectrum). These findings indicated that there was either an intermediate that underwent considerable solvent exchange or the product exchanged with solvent after initial formation (or both). To test the latter possibility, we repeated the experiment with CD3-SAM and quenched the reaction after only 1 h. The product was analyzed by ESI-MS, and an ion was observed that was consistent with the transfer of a CD2H group (Figure 3a, blue spectrum). This product was purified by high performance liquid chromatography (HPLC), exposed to the assay conditions for the extended reaction time (16 h), reisolated, and then again analyzed by ESI-MS. In agreement with the original findings, a complicated mixture of product peptide was observed that corresponded to apparent transfer of CH3, CH2D, and CHD2 (Figure S7). This exchange was dependent on the presence of TbtI and SAM; replacing SAM with SAH did not result in exchange. Thus, these experiments show that the initial product of the reaction of TbtI with CD3-SAM and 1 mainly contains a CD2H group but that the enzyme can exchange the deuterium labels with protium from solvent. Unfortunately, the large amounts of 5′-dA produced from the abortive cleavage of SAM during the 16 h reactions precluded our ability to determine if deuterium from the methyl group is transferred to the 5′-dA during the exchange process.
Figure 3.
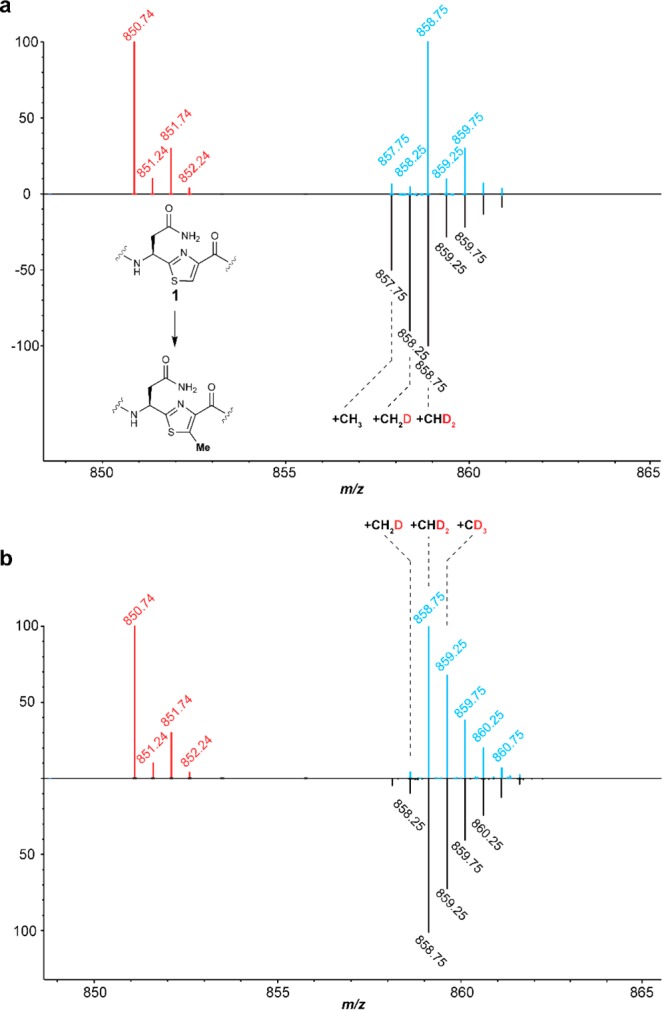
ESI mass spectra to investigate the origin of the methyl protons. 13C- and 15N-depleted substrate peptides were obtained as described (see the Experimental Section). (a) Spectra showing the doubly charged ion for the hexazole-containing core peptide substrate 1 (red) and the methylated product obtained with CD3-SAM in H2O after 1 h (blue) or 16 h (black) reactions. (b) Spectra showing substrate (red) and product obtained with CD3-SAM in D2O (blue) and CD3-5′,5′,4′,3′-D4-SAM in D2O (black) after 1 h. See Table S2 for calculated and observed masses.
Because of the solvent exchange observed at extended reaction times, all subsequent assays were performed for 1 h. We first investigated whether the generation of the product containing a CHD2 group when using CD3-SAM in H2O involves the incorporation of a proton from solvent. Thus, the reaction was performed using CD3-SAM in D2O. Analysis of the product by ESI-MS (Figure 3b) showed that the major product still contained CHD2, although the relative amount of CD3 transfer increased. This observation suggests that a hydrogen in an initially nonexchangeable position migrates to the methyl group, possibly with the intermediacy of a partially solvent exchangeable intermediate. The mechanisms in Figure 2 provide three possible sources for this hydrogen. First, the thiazole hydrogen on the β-carbon of 1 that must be removed during the methylation process could migrate via an active site base to the methyl group of the product (pathway a). Alternatively, a hydrogen that originates from the protein that does not exchange could be transferred to the methyl group (pathway b). On the basis of literature precedent, an example of the latter could be the α-hydrogens of Gly.43,44 Lastly, the hydrogen could originate from the methyl group of 5′-dA (pathway c).
Isotope Labeling Studies Involving the 5′-Position of SAM and the Substrate Thiazole
We first considered 5′-dA as the source for the additional hydrogen in the methyl group of the product. Abstraction of a hydrogen atom from 5′-dA by radical 4 (pathway c, Figure 2) would explain transfer of CHD2 when employing CD3-SAM. We therefore prepared SAM from Met-CD3 and 5′,5′,4′,3′-D4-ATP (∼92% CD3-5′,5′,4′,3′-D4-SAM by ESI-MS, Figure S8). The resulting CD3-5′,5′,4′,3′-D4-SAM was then reacted with TbtI and the TbtA hexazole 1 in D2O. The reaction products were analyzed by ESI-TOF MS. The amount of product that contained CHD2 was similar to that observed when the 5′ position of SAM was unlabeled (Figure 3b). Hence, the hydrogen in this product does not originate from the 5′ position of the adenosine, ruling out pathway c under these conditions.
We next considered pathway b to account for the product hydrogen, in which a nonexchangeable hydrogen on the protein is transferred to the product methyl group in the first turnover; this hydrogen would be replaced by a solvent-derived hydrogen in subsequent turnovers. This behavior has previously been observed, such as the transfer of Cα hydrogens from a Gly residue.43,44 In principle, hydrogen transfer could also originate from other amino acids or very tightly hydrogen bonded positions that would only exchange upon reaction. Because TbtI contains several conserved Gly residues (Figure S2), we reacted a 20-fold excess of substrate 1 and CD3-SAM with TbtI in buffered D2O and analyzed the product by ESI-MS (Figure S9). The major product again contained a CHD2 group, arguing against a protein-derived hydrogen that is transferred to the methyl group.
Having provided evidence against pathways b and c, we next replaced the Cβ protium of the thiazole with deuterium to interrogate pathway a (Figure 2). We prepared TbtA hexazole using an E. coli strain that is auxotrophic for Cys and provided β-D2-Cys to the 13C/15N-depleted minimal media. Unfortunately, the expression level of the peptide under these conditions was very low. During optimization of the expression conditions, we noted that improved production could be achieved upon addition of a limited number of unlabeled amino acids to the culture media (see the Experimental Section for details). Analysis by tandem ESI-MS confirmed the desired deuterium labeling of the six Cys residues in the TbtA precursor (Figure S10). The resulting peptide (∼95% D2 at Cys4 by ESI-MS-MS) was then converted to the corresponding hexazole-containing core peptide 2in vitro as previously reported.45 Owing to the need to add additional amino acids (which were not 13C- and 15N-depleted), the monoisotopic purity of the hexazole 2 was lower than that of the substrate 1. Nevertheless, an ion consistent with the hexa-deuterated TbtA hexazole core peptide was the major product observed by ESI-MS.
With peptide 2 in hand, we conducted a series of single turnover experiments. If the hydrogen in the transferred CHD2 was protein-derived in Figure 3b, then a single turnover experiment in D2O with substrate 2 and CD3-SAM should still result in CHD2 transfer. However, when the experiment was performed with a 3-fold excess of TbtI over substrate 2, CD3 transfer was observed as the major product (Figure 4a). This result provided additional data against pathway b: a nonexchangeable hydrogen from the enzyme was not the source of hydrogen in the methyl group of the product. We tentatively propose that the small amount of CHD2 transfer observed may originate in part because of incomplete deuteration of the reagents. We then repeated the single turnover experiment with CD3-SAM in D2O but employed substrate 1. A considerable amount of CD3 transfer was observed, indicating that solvent exchange occurred in the single turnover experiments with excess enzyme (Figure 4b), but importantly, the relative amount of CD3 transfer declined considerably compared to experiments conducted with substrate 2. Collectively, these data provide strong support for pathway a being operative during TbtI catalysis. These experiments also argue against TbtI already containing a covalently bound methyl group introduced during expression in E. coli; this methyl group would have been unlabeled, and therefore, we would have observed a considerable amount of product containing CH3 and CH2D.
Figure 4.

A solvent exchangeable proton migrates from the β-position of thiazole 4 to the methyl group in the product. (a) Spectra showing the doubly charged core peptide of the TbtA hexazole substrate 2 that is deuterated at the β-carbon of each thiazole (red) and the corresponding product obtained with CD3-SAM in D2O (blue) in a single turnover reaction. (b) Spectra showing the doubly charged core peptide of the TbtA hexazole substrate 1 (red) and the corresponding product obtained with CD3-SAM in D2O (blue) in a TbtI-catalyzed single turnover reaction. See Table S2 for calculated and observed masses.
To identify residues that may mediate the transfer process, we substituted conserved residues Tyr8 and Glu105 of TbtI with Ala (Figure S2). The corresponding residues are near the SAM-binding site in HemN,46,47 a rSAM decarboxylase from the heme biosynthetic pathway that is homologous to TbtI. Replacement of either residue resulted in TbtI variants with severely decreased activity (Figure S11). Two additional conserved residues (Trp23 and Tyr200) were targeted for Ala substitution given that they could participate in electron or hydrogen atom transfer; however, the activity of the resulting TbtI variants was not decreased, suggesting that they are not involved in catalysis (Figure S3C).
Discussion
A combination of product analysis and isotope labeling studies has allowed us to formulate a mechanism for TbtI that is consistent with all of our available data. The observation of 5′-dA as a product suggests that the TbtI-catalyzed reaction is initiated in a manner similar to canonical rSAM proteins: electron transfer from the reduced [4Fe–4S] cluster results in the reductive cleavage of SAM1 to form methionine and 5′-dA• (Figure 2).42 We propose that the 5′-dA• then abstracts a hydrogen atom from the methyl group of SAM2. This hypothesis is supported by the observation that use of CD3-SAM resulted in the incorporation of one deuterium into 5′-dA. This reaction appears irreversible, since we never observed multiple deuteriums in 5′-dA (Figure S4). Use of two molecules of SAM is also consistent with (i) the observed stoichiometry of 5′-dA and SAH to the methylated product, (ii) the previously reported crystal structure of HemN with two bound SAM molecules,46,47 and (iii) recent results reported for the class C rSAM MT NosN.35 Although HemN and TbtI catalyze different reactions (decarboxylation and methylation, respectively), the use of two molecules of SAM might be a general property of class C rSAM enzymes.
Upon hydrogen atom abstraction from the methyl group of SAM2, we propose that the resulting methylene radical adds to the electrophilic C=C bond of the thiazole, generating resonance-stabilized radical 3 (Figure 2). This may trigger deprotonation from the β-position by an active site base B to eliminate SAH and form resonance-stabilized radical 4 that partially restores thiazole aromaticity (Figure 2). This proton is adjacent to an enoxyradical, and previous studies on 2-hydroxyglutaryl-CoA dehydratase48 have suggested that the pKa values of such protons are on the order of ∼14. One candidate residue for the active site base is Glu105, which upon substitution with Ala severely decreases TbtI activity (Figure S11). The equivalent Glu in HemN (Glu145) is near the SAM-binding site;46,47 thus, an alternative role for Glu105 could be to bind SAM. Reduction of radical 4, possibly by the [4Fe–4S] cluster, would produce resonance-stabilized anion 5, which can then be protonated by the conjugate acid of the active site base, possibly Glu105. This pathway would explain the observed deuterium transfer from the β position of the thiazole to the methyl group in the product (Figure 4).
A variation of this mechanism features abstraction by radical 4 of a hydrogen atom from a redox active amino acid in the active site (e.g., Tyr/Trp/Gly; Cys residues can be ruled out, since their replacement with Ala had no effect on activity, Figure S3). The amino acid radical would then be reduced and protonated to reset the enzyme for another turnover (Figure 2, pathway b). The single turnover experiments argue against the possibility that this hydrogen is in a nonexchangeable position in the first turnover but becomes a solvent-derived hydrogen in subsequent turnovers. Hence, the involvement of Gly radicals is unlikely, but other redox active amino acids cannot yet be ruled out. For instance, a tyrosinate could serve as the base that deprotonates radical 3 and the resulting phenol moiety could then provide a hydrogen atom to radical 4. Tyr8 is conserved in the class C rSAM proteins (Figure S2). In HemN, the equivalent Tyr56 π-stacks with the adenine of SAM2. Substitution of TbtI Tyr8 with Ala severely decreased activity (Figure S11), and hence, Tyr8 might be a hydrogen atom donor that generates the final product. We ruled out the possibility, at least in vitro, that the final hydrogen atom donor is 5′-dA by conducting experiments with SAM that was deuterated at both the methyl group and the 5′ position (Figure 2, pathway c). Use of this deuterated SAM analogue did not result in an increase in the deuterium content of the final product. However, from an efficiency perspective, hydrogen atom abstraction from 5′-dA would be attractive, as it would directly regenerate the 5′-dA• and would not require the additional electron required for pathways a and b. Furthermore, as shown by the SAM-dependent solvent exchange of the methyl group in the product, 5′-dA• may abstract a hydrogen atom from the methyl group, which would be the microscopic reverse of the last step of pathway c. Hence, while pathway c clearly is not consistent with our in vitro findings, perhaps under the right conditions pathway c might be operational in vivo.
The proposed mechanism accounts for the observed solvent exchange upon prolonged reaction times by realization that the methylated peptide product, SAM1, and SAH can likely still bind to the active site, since these three molecules can occupy nearly identical positions as the substrate peptide, SAM1, and SAM2 at the start of catalysis (the only difference is that the methyl group of SAM2 is transferred to the peptide). Reductive cleavage of SAM1 would generate 5′-dA•, and the methyl group of the product peptide would be in a similar position as the methyl group of SAM2 during productive turnover. Thus, hydrogen atom abstraction could again generate radical 4 and either pathway a or b would result in hydrogen transfer by an active site residue, thus affecting the observed exchange (Figure S12a). Alternatively, the exchange could occur by a heterolytic deprotonation/reprotonation sequence that requires bound SAM (Figure S12b).
In summary, our study suggests that methyl transfer catalyzed by TbtI involves two SAM molecules. Our data are consistent with a mechanism in which one SAM is reductively cleaved to 5′-dA• as in other rSAM enzymes. The 5′-dA• then abstracts a hydrogen atom from a second SAM, and the resulting carbon-based methylene radical attacks the thiazole at position 4 of the peptide substrate. This SAM-derived radical is unprecedented in rSAM enzymology. We note that, like the class A rSAM MTs and a recent report on the class C MT NosN,35 TbtI formally transfers a methylene group from SAM to the substrate and hence might not be considered a true methyl transferase. Regardless, the manner by which this is accomplished adds a fundamentally different use of SAM in this class of enzymes, and highlights their versatility and catalytic diversity in natural product biosynthesis. Additional biochemical and structural studies on TbtI are underway to gain further insights into the mechanism of this remarkable transformation.
Experimental Section
General Methods
All polymerase chain reactions (PCRs) were carried out on a C1000 thermal cycler (Bio-Rad). DNA sequencing was performed by ACGT, Inc. MALDI-TOF MS was carried out on a Bruker Daltonics UltrafleXtreme MALDI TOF/TOF instrument (Bruker). ESI-MS/MS analyses were performed using a SYNAPT ESI quadrupole TOF Mass Spectrometry System (Waters) equipped with an ACQUITY Ultra Performance Liquid Chromatography (UPLC) system (Waters). HiTrap columns for Ni-NTA affinity chromatography were purchased from GE Healthcare.
Materials
Reagents used for molecular biology experiments were purchased from New England BioLabs (Ipswich, MA), Thermo Fisher Scientific (Waltham, MA), or Gold Biotechnology Inc. (St. Louis, MO). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Plasmid inserts were sequenced at ACGT Inc. (Wheeling, IL). Oligonucleotides were purchased from Integrated DNA Technologies Inc. (Coralville, IA). Deuterium-labeled reagents were purchased from Cambridge Isotope Laboratories.
Strains and Plasmids
E. coli DH5α and E. coli BL21 (DE3) cells were used as a host for cloning and plasmid propagation and as a host for protein expression, respectively. Coexpression vector pRSFDuet-1 was obtained from Novagen. E. coli JW3582-2 that is auxotrophic for cysteine was purchased from the Coli Genetic Stock Center at Yale University, https://cgsc2.biology.yale.edu/Strain.php?ID=108920.
Construction of TbtI Variants
Site directed mutagenesis (SDM) was performed using the QuikChange method (Agilent) as per the manufacturer’s instructions using PfuTurbo DNA polymerase. The primers for each mutant are listed in Table S1. The mutations were verified by sequencing using a custom MBP forward primer or the T7 reverse primer (Table S1).
Expression, Purification, and Assays of TbtI
Expression, purification, and in vitro reconstitution of the [4Fe–4S] cluster of TbtI were carried out as previously described.36 Briefly, the in vitro methylation reaction mixtures generally included the following components: TbtA hexazole substrate (50–100 μM), purified TbtI from E. coli heterologous expression after in vitro reconstitution of the [4Fe–4S] cluster (10 μM), S-adenosyl-l-methionine (SAM, 1 mM), flavodoxin (10 μM), flavodoxin reductase (10 μM), reduced nicotinamide adenine dinucleotide phosphate (NADPH, 2 mM) in reaction buffer (50 mM Tris–HCl, pH 7.5). The reaction was allowed to proceed for 1 h at 25 °C in an anaerobic chamber (Coy Laboratory).
For reactions performed in D2O, all reagents that participate in the reaction were first dissolved in D2O, lyophilized, and redissolved in D2O followed by adjusting of the pD. The buffer containing the proteins was exchanged with D2O storage buffer (50 mM Tris, 300 mM NaCl, pD 7.5) by repeated concentration by ultrafiltration and dilution (four times) before addition into the reaction. The reaction was allowed to proceed in D2O reaction buffer (50 mM Tris, pD 7.5) for 1 h at 25 °C in the anaerobic chamber.
For single turnover reactions, a reaction mixture generally included the following components: TbtA hexazole substrate (20 μM), purified TbtI after in vitro reconstitution of the [4Fe–4S] cluster (60 μM), S-adenosyl-l-methionine (SAM, 1 mM), flavodoxin (30 μM), flavodoxin reductase (30 μM), reduced nicotinamide adenine dinucleotide phosphate (NADPH, 2 mM) in reaction buffer (50 mM Tris–HCl, pH 7.5). The reaction was allowed to proceed for 10 min at 25 °C in the anaerobic chamber.
To determine the specific activity of the TbtI used, the enzyme (10 μM) was incubated with the hexazole substrate peptide (100 μM), S-adenosyl-l-methionine (SAM, 1 mM), flavodoxin (20 μM), flavodoxin reductase (20 μM), reduced nicotinamide adenine dinucleotide phosphate (NADPH, 2 mM) in reaction buffer (50 mM Tris–HCl, pH 7.5). Aliquots were removed at set time points (30, 60, 90, and 120 s), and the reaction was allowed to proceed at 25 °C in an anaerobic chamber. At 120 s, the conversion was 15%. Product formation over time was linear with a velocity of 15.6 × 10–3 μmol/min/mg of TbtI (i.e., 0.72 min–1). To ensure that under these conditions the reaction catalyzed by TbtI was rate limiting, the amounts of flavodoxin and flavodoxin reductase were doubled, and no change in velocity was observed. Similarly, to ensure that all substrates were saturating, the concentrations of SAM, NADPH, and peptide were doubled without observing a change in velocity.
Generation of 13C- and 15N-Depleted TbtA Hexazole
Isotopically depleted TbtA hexazole was produced in E. coli cultured in modified M9 minimal media.49E. coli BL21(DE3) cells containing a pTrc99 plasmid encoding maltose binding protein (MBP)-tagged TbtF, MBP-tagged TbtG, and TbtE and a pRSFDuet-1 plasmid encoding the His6-TbtA were grown overnight at 37 °C in lysogeny broth (LB) containing 25 μg/mL kanamycin and 50 μg/mL ampicillin. After harvesting the cells, the supernatant was discarded and the cells were washed with 5 mL of wash buffer (22 mM KH2PO4, 42 mM Na2HPO4, 8.5 mM NaCl, pH 7.4). Following washing, the cells were resuspended in wash buffer and used to inoculate (1:100) the modified M9 minimal media, with the following composition per 100 mL: 10 mL of a 10× stock of minimal media (220 mM KH2PO4, 420 mM Na2HPO4, 85 mM NaCl, pH 7.4), 0.3 mL of 40% aqueous (14NH4)2SO4 (Sigma-Aldrich), 2 mL of 20% aqueous 13C-depleted glucose (Cambridge Isotope Laboratories), 0.1 mg of FeSO4, 10 μg of thiamine, 200 μL of 1 M MgSO4, 10 μL of 1 M CaCl2, and 75 μL of a trace element solution (5 mM CaCl2, 1.25 mM ZnCl2, 260 μM CuCl2·H2O, 252 μM CoCl2·6H2O, 250 μM Na2MoO4·2H2O, pH 7.4). The media also contained 50 μg/mL ampicillin and 25 μg/mL kanamycin. The cells were grown at 37 °C and induced at OD600 = 0.6–0.8 by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and grown for an additional 16–18 h at 18 °C before harvesting. Purification of TbtA hexazole was carried out as previously described.50
Generation of TbtA Hexazole Deuterium Labeled at Each Thiazole
MBP-tagged TbtA containing β-D2-Cys was generated using E. coli strain JW3582-2. A lysogenization step was performed to JW3582-2 before production of MBP-tagged TbtA so that the host strain could be used to express target genes cloned in T7 expression vectors. Lysogenization was performed using the λDE3 Lysogenization Kit (Novagen) as per the manufacturer’s instructions. The lysogenized JW3582-2 was transformed with a pET28 plasmid containing the ampicillin resistance gene and encoding MBP-tagged TbtA.
Expression of MBP-tagged TbtA containing β-D2-cys was performed in modified M9 minimal media as described above, except that amino acids were added into the culture (final concentrations: 0.5 mM Arg, 1 mM His, 1 mM Leu, 1 mM Lys, 0.5 mM Met, 2 mM Pro, 3 mM Thr, 0.4 mM Trp, and 1 mM of β-D2-Cys). Purification and in vitro cyclodehydration of the β-D2-cysteine-labeled MBP-tagged TbtA were carried out as previously described.45
Leader Peptide Removal
Proteolytic removal of the leader peptide to generate TbtA-hexazole core peptides for TbtI reactions was carried out as previously described.50
Preparation of SAM
E. coli strain DM22-(pK8), which overexpresses E. coli SAM synthetase, was constructed by G. D. Markham et al. (Fox Chase Cancer Center, Philadelphia, PA).51 DM22-(pK8) was grown in LB containing 30 μg/mL tetracycline at 37 °C while being shaken and harvested by centrifugation 12 h after inoculation. The cells (∼12 g) were resuspended in 36 mL of 0.1 M Tris–HCl (pH 8.0) containing 1 mM ethylenediaminetetraacetic acid (EDTA) and 50 μg/mL lysozyme and gently stirred at room temperature for 30 min. Phenylmethylsulfonyl fluoride (PMSF) was added to a final concentration of 1 mM, and cells were lysed with a MultiFlex C3 Homogenizer (Avestin). The cell lysate was centrifuged for 30 min at 26,500 × g at 4 °C. The cleared lysate was dialyzed against 4 L of 100 mM Tris–HCl (pH 8.0), 1 mM EDTA, in 2 × 30 mL 20,000 molecular weight cutoff (MWCO) Slide-A-Lyzer dialysis cassettes (Pierce) at 4 °C overnight, and was divided into three portions, frozen in liquid nitrogen, and stored at −80 °C.
SAM was generated enzymatically by incubating an aliquot of the lysate in a buffered solution (0.1 L) containing 0.1 M Tris–HCl (pH 8.0), 50 mM KCl, 1 mM EDTA, 20% acetonitrile, 26 mM MgCl2, 13 mM adenosine triphosphate (ATP), and 10 mM methionine. After the mixture had been gently stirred for 5 h at room temperature, the reaction was quenched by adjusting the pH to 5.0 with HCl. The mixture was placed on ice for 15 min and centrifuged at 4000 × g for 30 min at 4 °C to remove precipitated material. The supernatant was diluted to 1 L with 1 mM sodium acetate (pH 5.0) and loaded on a CM-52 cation exchange resin (Whatman) that had been charged with 0.2 M sodium acetate (pH 5.0) and equilibrated in 1 mM sodium acetate (pH 5.0). The column was rinsed with 1 L of 1 mM sodium acetate (pH 5.0), and SAM was eluted with 0.5 L of 40 mM H2SO4. The presence of SAM was monitored by UV–vis absorption at 260 nm, and fractions containing SAM were combined. The solution was concentrated by rotovap to a volume of 4 mL, and was divided into several fractions and stored at −80 °C. Deuterium-labeled SAM was prepared using the same procedure but with labeled methionine or ATP.
Radioactive Assay with 14CH3-SAM
The assay to determine whether the SAM methyl group is transferred onto TbtI was performed with 14CH3-SAM (0.02 mCi/mL, 300 μM). The in vitro reconstitution of the [4Fe–4S] cluster of MBP-TbtI was performed as previously described,36 followed by addition of TbtA hexazole inside an anaerobic chamber. After incubation at room temperature for 1 h, the reaction mixture was taken out of the anaerobic chamber and moved into an anaerobic bag filled with nitrogen gas, and 1:1 14CH3-SAM/unlabeled CH3-SAM mixture was then added into the reaction inside the anaerobic bag. The reaction mixture included the following components: MBP-TbtI (10 μM), TbtA hexazole (100 μM), 1:1 14CH3-SAM/unlabeled CH3-SAM mixture (300 μM) in a final volume of 100 μL. The reaction was incubated at room temperature for 1 h, and 30 μL of the reaction mixture was loaded on a 4–20% polyacrylamide gel. Electrophoresis was performed for 45 min at 150 V (constant voltage), and then the gel was stained with Coomassie blue staining solution (40% methanol, 10% acetic acid, 0.1% Coomassie blue R250 in H2O) for 30 min and destained in destain buffer (20% methanol, 10% acetic acid in H2O) for 3 h. After drying the gel overnight with a gel-drying kit (Promega) in accordance with the manufacturer’s procedure, the dried gel was exposed on a phosphorimager cassette for 4 days. The film was finally visualized with a STORM840 Phosphorimager Scanner (Amersham Biosciences). Whereas the Coomassie stained gel clearly showed the presence of the protein, no radioactivity was observed in the gel. A parallel experiment with unlabeled SAM and including the reducing system demonstrated full enzyme activity.
Chitin Binding Domain (CBD)-Fused Flavodoxin (Flv)/Flavodoxin Reductase (Flx) Overexpression and Purification
E. coli BL21(DE3) cells were transformed with an intein based expression vector pTYB1 plasmid encoding the CBD fused Flv/Flx.52 Cells were grown for 24 h on LB agar plates containing 100 μg/mL ampicillin at 37 °C. Single colonies were picked to inoculate 10 mL of LB containing the same concentration of antibiotic and grown at 37 °C for 16–18 h. This culture was used to inoculate 1 L of LB supplemented with the same concentration of antibiotic and grown to an OD600 of 0.6 before being placed on ice for 15 min. Protein expression was then induced with the addition of 0.5 mM IPTG and supplemented with flavin adenine mononucleotide (FMN) for overexpression of Flv and flavin adenine dinucleotide (FAD) for overexpression of Flx to a final concentration of 10 μM, respectively. Expression was allowed to proceed for 12 h at 18 °C. Cells were harvested by centrifugation at 4000 × g for 20 min, washed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4), and harvested by centrifugation. The cells were flash-frozen with liquid nitrogen and stored at −80 °C for a maximum of 1 week before use.
Cells were resuspended in cold lysis buffer (20 mM Tris–HCl pH 8.5, 500 mM NaCl, 1 mM TCEP, and 0.1% Triton X-100 (v/v)) containing 4 mg/mL lysozyme, 2 μM leupeptin, 2 μM benzamidine, and 2 μM E-64. Cells were further lysed by sonication (3 × 45 s with 10 min agitation periods at 4 °C). Insoluble debris was removed by centrifugation at 20,000 × g for 40 min at 4 °C. The supernatant was then applied to a column containing lysis-buffer pre-equilibrated chitin resin (NEB; 15 mL of resin/1 L of initial cell culture). The column was washed with 10–15 column volumes of wash buffer (20 mM Tris–HCl pH 8.5, 1 M NaCl, 1 mM TCEP, and either 10 μM FMN for Flv or 10 μM FAD for Flx, respectively). The column was flushed quickly with 20–25 mL of cleavage buffer (20 mM Tris–HCl pH 8.5, 500 mM NaCl, 1 mM EDTA, 25 mM DTT), and the column was incubated at 4 °C for 20–24 h. The CBP fused proteins were eluted using elution buffer (20 mM Tris–HCl pH 8.5, 500 mM NaCl), and collected in 1 mL fractions. The yellow colored fractions were pooled and concentrated using a 30 kDa MWCO Amicon Ultra centrifugal filter (EMD Millipore). A buffer exchange step was performed with protein storage buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 300 mM NaCl, 1 mM TCEP, and 5% glycerol (v/v)) prior to final concentration, freezing in liquid nitrogen, and storage at −80 °C.
UPLC MS and ESI-MS/MS
The sample was prepared as described previously.45 LC-MS was performed using a Waters SYNAPT mass spectrometer outfitted with an ACQUITY UPLC, an ACQUITY Bridged Ethyl Hybrid C8 column (2.1 × 50 mm2, 1.7 μm particle size, 200 Å; Waters), an ESI ion source, and a quadruple TOF detector. A gradient of 2–100% aq. MeCN with 0.1% formic acid (v/v) over 20 min was used. Fragmentation of the sample was performed using a collision-induced dissociation (CID) method. A ramping of cone voltage of 15–18 kV during the scan was performed to generate peptide fragments for MS/MS analysis.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute (to W.A.V.), the National Institutes of Health (GM097142 to D.A.M.), and a Seemon Pines Fellowship from the Department of Chemistry at the University of Illinois at Urbana–Champaign (to G.A.H.). Constructs for expression of flavodoxin/flavodoxin reductase were generously provided by the laboratory of Squire J. Booker (Penn State University), and constructs for SAM synthetase were provided by Vahe Bandarian (University of Utah).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10203.
Experimental details and supporting figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Takusagawa F.; Fujioka M.; Spies A.; Schowen R. L. In Comprehensive Biological Catalysis; Sinnott M., Ed.; Academic Press: New York, 1998; p 1. [Google Scholar]
- Kamigiri K.; Hidaka T.; Imai S.; Murakami T.; Seto H. J. Antibiot. 1992, 45, 781. 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T.; Hidaka T.; Kamigiri K.; Imai S.; Seto H. J. Antibiot. 1992, 45, 1812. 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- Williamson J. M.; Inamine E.; Wilson K. E.; Douglas A. W.; Liesch J. M.; Albers-Schönberg G. J. Biol. Chem. 1985, 260, 4637. [PubMed] [Google Scholar]
- Houck D. R.; Kobayashi K.; Williamson J. M.; Floss H. G. J. Am. Chem. Soc. 1986, 108, 5365. 10.1021/ja00277a063. [DOI] [Google Scholar]
- Hidaka T.; Goda M.; Kuzuyama T.; Takei N.; Kidaka M.; Seto H. Mol. Gen. Genet. 1995, 249, 274. 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- Hidaka T.; Hidaka M.; Kuzuyama T.; Seto H. Gene 1995, 158, 149. 10.1016/0378-1119(95)00101-B. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T.; Seki T.; Dairi T.; Hidaka T.; Seto H. J. Antibiot. 1995, 48, 1191. 10.7164/antibiotics.48.1191. [DOI] [PubMed] [Google Scholar]
- Nunez L. E.; Mendez C.; Brana A. F.; Blanco G.; Salas J. A. Chem. Biol. 2003, 10, 301. 10.1016/S1074-5521(03)00069-3. [DOI] [PubMed] [Google Scholar]
- Sato S.; Kudo F.; Kim S. Y.; Kuzuyama T.; Eguchi T. Biochemistry 2017, 56, 3519. 10.1021/acs.biochem.7b00472. [DOI] [PubMed] [Google Scholar]
- Marous D. R.; Lloyd E. P.; Buller A. R.; Moshos K. A.; Grove T. L.; Blaszczyk A. J.; Booker S. J.; Townsend C. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10354. 10.1073/pnas.1508615112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Nucleic Acids Res. 2001, 29, 1097. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiva E.; Brown S.; Almonacid D. E.; Barber A. E. 2nd; Custer A. F.; Hicks M. A.; Huang C. C.; Lauck F.; Mashiyama S. T.; Meng E. C.; Mischel D.; Morris J. H.; Ojha S.; Schnoes A. M.; Stryke D.; Yunes J. M.; Ferrin T. E.; Holliday G. L.; Babbitt P. C. Nucleic Acids Res. 2014, 42, D521. 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf B. J.; McCarthy E. L.; Booker S. J. Annu. Rev. Biochem. 2016, 85, 485. 10.1146/annurev-biochem-060713-035504. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; van der Donk W. A.; Liu W. Acc. Chem. Res. 2012, 45, 555. 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori D. G. Curr. Opin. Chem. Biol. 2013, 17, 597. 10.1016/j.cbpa.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerle M. R.; Schwalm E. L.; Booker S. J. J. Biol. Chem. 2015, 290, 3995. 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y.; Yu Y.; Pan H.; Guo H.; Li Y.; Liu W. Mol. BioSyst. 2010, 6, 1180. 10.1039/c005121g. [DOI] [PubMed] [Google Scholar]
- Yan F.; LaMarre J. M.; Rohrich R.; Wiesner J.; Jomaa H.; Mankin A. S.; Fujimori D. G. J. Am. Chem. Soc. 2010, 132, 3953. 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boal A. K.; Grove T. L.; McLaughlin M. I.; Yennawar N. H.; Booker S. J.; Rosenzweig A. C. Science 2011, 332, 1089. 10.1126/science.1205358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F.; Fujimori D. G. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3930. 10.1073/pnas.1017781108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove T. L.; Benner J. S.; Radle M. I.; Ahlum J. H.; Landgraf B. J.; Krebs C.; Booker S. J. Science 2011, 332, 604. 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- Schwalm E. L.; Grove T. L.; Booker S. J.; Boal A. K. Science 2016, 352, 309. 10.1126/science.aad5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre S.; Guillot A.; Benjdia A.; Sandstrom C.; Langella P.; Berteau O. Nat. Chem. Biol. 2012, 8, 957. 10.1038/nchembio.1091. [DOI] [PubMed] [Google Scholar]
- Blaszczyk A. J.; Silakov A.; Zhang B.; Maiocco S. J.; Lanz N. D.; Kelly W. L.; Elliott S. J.; Krebs C.; Booker S. J. J. Am. Chem. Soc. 2016, 138, 3416. 10.1021/jacs.5b12592. [DOI] [PubMed] [Google Scholar]
- Woodyer R. D.; Li G.; Zhao H.; van der Donk W. A. Chem. Commun. 2007, 359. 10.1039/B614678C. [DOI] [PubMed] [Google Scholar]
- Allen K. D.; Wang S. C. Arch. Biochem. Biophys. 2014, 543, 67. 10.1016/j.abb.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J.; McCarty R. M.; Ogasawara Y.; Liu Y. N.; Mansoorabadi S. O.; LeVieux J.; Liu H. W. J. Am. Chem. Soc. 2013, 135, 8093. 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Schnell B.; Baumann S.; Muller R.; Begley T. P. J. Am. Chem. Soc. 2017, 139, 1742. 10.1021/jacs.6b10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inahashi Y.; Zhou S.; Bibb M. J.; Song L.; Al-Bassam M. M.; Challis G. L. Chem. Sci. 2017, 8, 2823. 10.1039/C6SC03533G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A.; Guillot A.; Benjdia A.; Chartier G.; Leprince J.; Berteau O. J. Am. Chem. Soc. 2016, 138, 15515. 10.1021/jacs.6b06697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Xu H.; Li Y.; Zhang F.; Chen X. Y.; He Q. L.; Igarashi Y.; Tang G. L. J. Am. Chem. Soc. 2012, 134, 8831. 10.1021/ja211098r. [DOI] [PubMed] [Google Scholar]
- Hiratsuka T.; Suzuki H.; Kariya R.; Seo T.; Minami A.; Oikawa H. Angew. Chem., Int. Ed. 2014, 53, 5423. 10.1002/anie.201402623. [DOI] [PubMed] [Google Scholar]
- Ding W.; Li Y.; Zhao J.; Ji X.; Mo T.; Qianzhu H.; Tu T.; Deng Z.; Yu Y.; Chen F.; Zhang Q. Angew. Chem., Int. Ed. 2017, 56, 3857. 10.1002/anie.201609948. [DOI] [PubMed] [Google Scholar]
- LaMattina J. W.; Wang B.; Badding E. D.; Gadsby L. K.; Grove T.; Booker S. J. J. Am. Chem. Soc. 2017, 139, 17438. 10.1021/jacs.7b08492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N.; Zhang Z.; Hudson G. A.; van der Donk W. A.; Mitchell D. A. J. Am. Chem. Soc. 2017, 139, 4310. 10.1021/jacs.7b00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. D.; Xu H.; White R. H. J. Bacteriol. 2014, 196, 3315. 10.1128/JB.01903-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Donk W. A. J. Org. Chem. 2006, 71, 9561. 10.1021/jo0614240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner W. J.; Allen K. D.; Hu K.; Helms G. L.; Chen B. S.; Wang S. C. Biochemistry 2011, 50, 8986. 10.1021/bi201220r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.; Alkhalaf L. M.; de Los Santos E. L.; Challis G. L. Curr. Opin. Chem. Biol. 2016, 35, 73. 10.1016/j.cbpa.2016.08.021. [DOI] [PubMed] [Google Scholar]
- Bridwell-Rabb J.; Zhong A.; Sun H. G.; Drennan C. L.; Liu H. W. Nature 2017, 544, 322. 10.1038/nature21689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Chem. Rev. 2014, 114, 4229. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shisler K. A.; Broderick J. B. Arch. Biochem. Biophys. 2014, 546, 64. 10.1016/j.abb.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat S. S.; Raushel F. M. Perspectives in Science 2015, 4, 32. 10.1016/j.pisc.2014.12.006. [DOI] [Google Scholar]
- Hudson G. A.; Zhang Z.; Tietz J. I.; Mitchell D. A.; van der Donk W. A. J. Am. Chem. Soc. 2015, 137, 16012. 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer G.; Moser J.; Heinz D. W.; Jahn D.; Schubert W. D. EMBO J. 2003, 22, 6214. 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer G.; Grage K.; Teschner T.; Schunemann V.; Breckau D.; Masoumi A.; Jahn M.; Heathcote P.; Trautwein A. X.; Jahn D. J. Biol. Chem. 2005, 280, 29038. 10.1074/jbc.M501275200. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Buckel W.; Zipse H. Angew. Chem., Int. Ed. 2003, 42, 1867. 10.1002/anie.200250502. [DOI] [PubMed] [Google Scholar]
- Miller L. M.; Chatterjee C.; van der Donk W. A.; Kelleher N. L. J. Am. Chem. Soc. 2006, 128, 1420. 10.1021/ja057203d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Hudson G. A.; Mahanta N.; Tietz J. I.; van der Donk W. A.; Mitchell D. A. J. Am. Chem. Soc. 2016, 138, 15511. 10.1021/jacs.6b08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham G. D.; DeParasis J.; Gatmaitan J. J. Biol. Chem. 1984, 259, 14505. [PubMed] [Google Scholar]
- Lanz N. D.; Grove T. L.; Gogonea C. B.; Lee K. H.; Krebs C.; Booker S. J. Methods Enzymol. 2012, 516, 125. 10.1016/B978-0-12-394291-3.00030-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.