

SUMMARY
Myosin-based motility utilizes catalysis of ATP to drive the relative sliding of F-actin and myosin. The earliest detailed model based on cryo-electron microscopy (cryoEM) and X-ray crystallography postulated that higher actin affinity and lever arm movement were coupled to closure of a feature of the myosin head dubbed the actin-binding cleft. Several studies since then using crystallography of myosin-V and cryoEM structures of F-actin bound myosin-I, – II and – V have provided details of this model. The smooth muscle myosin II interaction with F-actin may differ from those for striated and non-muscle myosin II due in part to different lengths of important surface loops. Here we report a ~6 Å resolution reconstruction of F-actin decorated with the nucleotide-free recombinant smooth muscle myosin-II motor domain (MD) from images recorded using a direct electron detector. Resolution is highest for F-actin and the actin-myosin interface (3.5–4 Å) and lowest (~6–7 Å) for those parts of the MD at the highest radius. Atomic models built into the F-actin density are quite comparable to those previously reported for rabbit muscle actin and show density from the bound ADP. The atomic model of the MD, is quite similar to a recently published structure of vertebrate non-muscle myosin II bound to F-actin and a crystal structure of nucleotide free myosin-V. Larger differences are observed when compared to the cryoEM structure of F-actin decorated with rabbit skeletal muscle myosin subfragment 1. The differences suggest less closure of the 50 kDa domain in the actin bound skeletal muscle myosin structure.
Keywords: electron microscopy, molecular motor, ATPase, single particle
INTRODUCTION
Myosins form a family of motor proteins currently comprising over 30 identified classes (Odronitz and Kollmar, 2007) that function within cells to move different types of cargo along F-actin while converting the energy of ATP into work. The myosin-II class, the only filament forming class, plays a central role in muscle contraction where binding to F-actin, accelerates its ATP hydrolysis rate to produce filament sliding and sarcomere shortening (Sweeney and Houdusse, 2010).
Muscle myosins are primarily of class II and consist of a pair of heavy chains (HC) each of which has a pair of bound light chains, the regulatory light chain (RLC) and the essential light chain (ELC). In smooth muscle myosin-II (smM-II), the first ~850 HC residues constitute the head which is folded into a globular motor domain (MD) containing the catalytic and actin binding properties, followed in turn by a small folded domain called the converter and a long α-helix to which the light chains bind (Holmes and Geeves, 2000). This light chain-binding domain or LCD constitutes the lever arm, the motion of which causes movement of the cargo, in this case the thick filament, relative to the actin filament to produce sarcomere shortening. Following the head is a long α-helix in the form of an α-helical coiled-coil, the first ~1/3 of which, the S2 domain, causes the HC to form a dimer and the rest forms the thick filament backbone. A recent 6 Å cryo-electron microscopy (cryoEM) 3-D reconstruction of the thick filaments from the flight muscles of Lethocerus indicus has revealed the details of myosin II rods within the backbone in unprecedented detail (Hu et al., 2016).
Historically, the myosin head has been described as comprising three major proteolytically derived domains that are named after their molecular weights, the N-terminal 25-, the 50-, and the C-terminal 20-kDa segments of the heavy chain (Mornet et al., 1981). The crystal structure of myosin-II from vertebrate skeletal muscle showed that the 25-kDa domain contains an SH3 motif and that the 50-kDa domain is separated into lower and upper domains by a distinct cleft (Rayment et al., 1993). The 20-kDa domain begins with a helix associated with the lower 50-kDa domain and includes the converter and the HC component of the lever arm. Situated at the center of these four domains is a seven-stranded β-sheet that connects them via a couple of loops and helices.
The actin-based motility of myosin consists of repetitive kinetic cycles in which myosin produces a high rate of ATP hydrolysis when interacting with actin. As a product-inhibited ATPase (Holmes and Geeves, 2000), the actin-induced conformational changes enable myosin to release the hydrolysis products, rebind ATP and continue the cycle. This mechanism was initially elucidated using muscle myosin and evolved into the Lymn-Taylor kinetic model (Lymn and Taylor, 1971). Several classes of non-muscle myosins work in a similar way, but with modifications of the different rate constants producing different functional adaptations (Houdusse and Sweeney, 2016). Thus, this cyclic myosin-actin interaction has become a general ATP hydrolysis mechanism of myosin. During this cycle, the myosin head bridges the separation between thick and thin filaments, initially weakly, followed by conformational changes, some linked to product release, that alter both the position of the lever arm and the affinity for actin. The process results in force generation, the so-called power-stroke, and is now referred to as the swinging lever-arm hypothesis (Geeves and Holmes, 2005).
In the myosin catalytic cycle, not only are large conformational changes observed in the major subdomains, but also subtle changes occur in the connectors, which might have close associations with the accelerated rate of ATP hydrolysis and the swinging of lever arm. It has been reported initially in myosin V (Coureux et al., 2003) and later in myosin II (Yang et al., 2007) that the seven-stranded β-sheet is more twisted in the nucleotide-free state than in the actin-detached transition states. Both of these structures differ from other nucleotide free crystal structures in having a closed actin-binding cleft that is associated with the rigor complex of actomyosin. In the transition state, a structure called the relay helix is kinked but it is straight in the rigor state (Geeves and Holmes, 2005). The actin binding cleft is open in the weak binding states (Dominguez et al., 1998), or half-closed in the prepower-stroke state (Smith and Rayment, 1996), or entirely closed in the rigor state when attached to actin (Behrmann et al., 2012; Holmes et al., 2003). The positions of key structures at the catalytic site, switch 1, switch II, and P-loop, highly depend on the biochemical state of myosin (Coureux et al., 2004).
Knowledge about the structure of myosin in different catalytic steps has accumulated gradually from the crystal structures of myosin subfragments dissociated from F-actin combined with spectral analysis and electron microscopy of actin filaments decorated with myosin subfragments (Criddle et al., 1985; Gourinath et al., 2003; Himmel et al., 2002; Milligan and Flicker, 1987). To date, structures of three actin-detached catalytic intermediates of myosin-II have been solved; the prepower-stroke, also called the transition state (Dominguez et al., 1998; Gourinath et al., 2003), post-rigor (Menetrey et al., 2008), and a so-far unique ADP-bound state with an unusual lever arm position (Houdusse et al., 2000). Previously structures of F-actin-myosin-II complexes were determined from cryoEM only to medium resolution (Holmes et al., 2003; Rayment et al., 1993; Volkmann et al., 2003; Volkmann et al., 2005). Recently, 3-D images of the actin-myosin complex from a non-muscle class I and class II myosin have been reported at near atomic resolution (Behrmann et al., 2012; von der Ecken et al., 2016). The least well-characterized step is the transition between the weakly attached, prepower-stroke and the strongly bound power-stroke despite it being the most critical step of the Lymn-Taylor cycle.
Here we report a sub-nanometer actin-bound smooth muscle myosin-II motor domain (smMD) complex in the nucleotide-free state at an average resolution ~6 Å obtained using iterative helical real space reconstruction (IHRSR). The reconstruction is very similar to the structure of the non-muscle myosin II class bound to actin in most essentials, particularly in the actin subunit structure, the actin-myosin interface as well as the transducer β-sheet. The nucleotide free myosin-V crystal structure was also an excellent fit to the density map even though that structure was in an actin-free state (Wulf et al., 2016). Conversely, the myosin-V transition state crystal structure from the same work was, as might have been predicted, a poor fit. When our density map and atomic model are compared with the recent 5.2 Å structure of F-actin decorated with nucleotide free rabbit skeletal muscle myosin II subfragment 1 (Fujii and Namba, 2017), large differences are seen in the N-terminal 25 kDa domain and upper 50 kDa domains.
EXPERIMENTAL PROCEDURES
Specimen Preparation
The smooth motor domain consisted of residues 1-L790, followed by a FLAG-tag to facilitate affinity purification. Sf9 cells were infected with recombinant baculovirus encoding for the heavy chain, harvested 72 hours later, and purified by FLAG-affinity chromatography (Sigma-Aldrich) essentially as described in (Trybus, 2000).
Actin was prepared from chicken muscle acetone powder (Pardee and Spudich, 1982) with the modification that the chromatography step was done on a Superdex 200 column. Actin was stored as G-actin in a −80°C freezer, thawed as needed. It was then polymerized to 1.5 mg/ml F-actin (with 10mM Imidazole, 10 mM KCl, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, pH 7.4) for 1 hour and diluted to 0.1 mg/ml just before use (with 10 mM Imidazole, 10 mM NaCl, 0.5 mM MgCl2, 0.5 mM DTT, pH 7.4)
Specimens were made for cryo-EM by applying 4 µl of actin to the grid bar side of a 2/1 Quantifoil grid (2 µm holes separated by 1 µm of carbon matrix) for 1 minute, rinsing with MD dilution buffer consisting of 10 mM imidazole, 10 mM KCl, 1.0 mM MgCl2, 1.0 mM EGTA, 0.5 mM DTT, pH 7.0 and applying 3 µl of MD solution for ~5 minutes. Some grids were prepared in a 3 °C cold room by manually blotting for 3–4 seconds followed by plunging into liquid ethane. Other grids were frozen at the University of Vermont in a Gatan CP-3 freezing device operated at 100% relative humidity at room temperature.
Data Collection and Preliminary Analysis
Approximately 4,000 low dose images were collected automatically using the Leginon software package (Suloway et al., 2005) on a Titan Krios electron microscope (FEI, Hillsboro, OR) equipped with a field emission gun and operated at 300 keV. Images were recorded with a DE-20 direct electron detector. The defocus mean and standard deviation was 3.6 ± 0.7 µm underfocus; the pixel size was 0.9861, scaled to the known rise/actin subunit of 27.6 Å. Each micrograph consisted of a 43-frame movie, with total dose of 60 e−/Å2.
We used the Appion software package (Lander et al., 2009) to manage the data, perform damage compensated motion correction, CTF determination, and particle picking. The damage compensated motion correction process (Wang et al., 2014) was used to correct for beam induced specimen motion and accumulated electron dose. Defocus was first searched using ACE (Mallick et al., 2005) and then refined by CTFFIND3 (Mindell and Grigorieff, 2003). The filaments were manually selected, divided into 384 × 384 pixel boxes, then extracted and normalized using the DoG picker utility within Appion (Lander et al., 2009). Each “particle” consisted of a filament segment masked to a length of 210 Å, or slightly more than 7 actin subunits of 27.6 Å separation. Adjacent filament segments overlapped by ~6 subunit repeats (~84% overlap). A total of 346,395 filament segments were selected from 1,417 of the best micrographs. Appion software was used to create a metadata (.star file) having all the positional, orientation and defocus information of the segments and was supplied to RELION (Scheres, 2012) for further processing.
Three-Dimensional Reconstruction
The specific version used was RELION 1.2 implemented by Z. Hong Zhou’s group (Clemens et al., 2015), which included the Iterative Helical Real Space Reconstruction (IHRSR) package (Egelman, 2007). A small set of particles was subjected to 2D classification in order to eliminate bad particles but unfortunately, none of the class averages showed obvious bad particle assemblies. Hence, hierarchical 3D classification was carried out in order to identify “shiny particles” for further analysis.
To reduce the computational burden, the particle stack was divided into two halves and 3D classification was performed on each of them separately. The 8 Å cryo-EM map of the rigor (nucleotide-free) actin-tropomyosin-myosin complex (EMD-1987) (Behrmann et al., 2012) was low-pass filtered to 100Å and used as initial model for 3D refinement of both the particle sets. To increase the speed of the process of selecting good particles by interactive 3D classification, each stack was first binned by a factor of 4. The first particle stack, consisting of 154,559 particles, was subjected to 3D classification from which 82,661 good particles were selected. From the second stack, consisting of 191,836 initial particles, only 104,221 good particles were identified. Then the 186,882 selected good particles were combined, binned by a factor of 2 and subjected to 25 more classification iterations. Particles were randomly divided into four classes and four reconstructions calculated. A set of projections were generated for each reconstruction, and used to reassign each of the 186,882 particles to one of the four groups according to which projection it most closely resembled. After 25 cycles of 3D classification, the final good-looking classes were selected and combined leaving 101,976 segments for “shiny particle” data analysis.
“Shiny particle” selection utilized 25 additional hierarchical 3D classifications of the 101,976 good particles. This time, one of the best looking class averages from the final iteration of the previous classifications was chosen as the reference image and filtered to 20 Å. The 101,976 particles were divided into four classes containing 26,683, 17,698, 40,847 and 16,748 filament segments. All of the four class averages appeared good and 3D auto refinement was carried out on each of the classes separately. This process revealed an acto-MD density with an estimated resolution of ~7 Å for all four classes. Each of the four reconstructions were compared with one another in Chimera and the three most homogeneous classes (1–3) were combined to produce the final reconstruction. A combined total of ~85,000 particles were subjected to 3D auto-refinement. One of the good reconstructions from the previous individual auto-refinement scheme was low-pass–filtered to 60 Å and used as the starting model for the final reconstruction. The 3D auto-refinement converged in 24 cycles.
The resolution based on the gold standard FSC (0.143 criterion) (Scheres and Chen, 2012) showed an average resolution of ~6 Å for the final acto-MD electron density map (Supplemental Figure S1). The temperature factors are calculated using EM-BFACTOR (Fernandez et al., 2008) and the F-actin-myosin map was sharpened using a temperature factor of −390.86 Å2. Local resolution of the full reconstructed volume computed using Resmap (Kucukelbir et al., 2014) revealed a resolution gradient of ~4.0 Å in the actin core region, ~5 Å in the central part of the map and ~6.5 Å at the outer myosin domains.
Atomic Model Fitting
The starting models of myosin used for homology modeling came from scallop, Argopecten irradians, myosin (PDB 1DFK) and slime mold, Dictyostelium discoideum, myosin (PDB 1FMV). First, a homology model of the chicken smooth muscle myosin sequence was built by MODELLER (Fiser and Sali, 2003) using both PDBs as input. Because several large loops, such as loop 2, are not determined in all the myosin head S1 atomic models, those loops were deleted after homology modeling, in order to avoid clashes in the real space flexible fitting. The real space flexible fitting was performed using Relax in Rosseta (DiMaio et al., 2015) at a resolution of 5.5 Å. Because the converter domain has a large conformational change, the SH3 domain, which is located near the converter domain, was not fit well into the density by the real space flexible fitting. So after the converter domain was fit, the SH3 domain was manually fit into the density and another real space flexible fitting of myosin was performed.
The starting model of actin was taken from the actin-tropomyosin filament structure (PDB 3J8A). The actin species in 3J8A is from rabbit, Oryctolagus cuniculus, skeletal muscle α-actin (NCBI Reference Sequence: NP_001026234.1). The α-actin atomic model was four residues short of the actual C-terminus. When we found that the γ-actin atomic model (von der Ecken et al., 2016) fit our actin density quite well, we built in the remaining four residues basing their placement on the γ-actin structure. As of this writing, the subsequent flexible fitting has not been completed. The real space flexible fitting was performed using Relax in Rosseta at resolution 4.0 Å. In the above fitting, only one myosin MD and one actin subunit are considered. Next, the combination of myosin-actin (MD-myosin-actin) was refined using rosetta_scripts in Rosseta (DiMaio et al., 2015) with asymm_refine.xml. Next, the actin-smMD contact was refined using rosetta_scripts with symm_refine.xml. Finally, ADP was fit into density map with the actin main chain fixed using Relax. The reconstruction and atomic model fitting statistics are summarized in Supplemental Table 1.
RESULTS and DISCUSSION
Motion corrected images recorded on the DE-20 showed F-actin with a high degree of saturation of actin subunits with smMD, which are individually resolved (Fig. 1). A significant fraction of the filaments appeared bundled and thus only a small fraction of filaments were suitable for further analysis. In addition to the heavily decorated F-actin, a significant fraction of filaments were completely undecorated, a phenomenon typical of F-actin decorated with myosin heads (Behrmann et al., 2012).
Figure 1.

Electron micrograph of F-actin decorated with the smooth muscle myosin motor domain. Segments were taken only from the filament region marked by the line. Arrowheads point to bundled filaments. Arrows point to actin filaments completely undecorated with myosin heads.
The reconstruction procedures to find the best-preserved segments of decorated actin eliminated 75% of the segments. The remaining 25% of segments produced a density map with variable resolution that depends roughly on the distance from the helical axis (Fig. 2A). The local resolution computed with RESMAP shows regions with ~3.5 Å but these occur mostly near the actin filament. On the smMD, the local resolution is mostly in the 5–7 Å range (Fig. 2B), being best near the actin and worst near the converter and SH3 domains. Some regions within the smMD show right-handed α-helices and their bulky side chains rather than cylindrical shapes in those places where α-helices are expected consistent with a resolution of 4.5–5 Å (Fig. 2C). Except around the actin subunits, other places in the reconstruction do not show density corresponding to amino acid side chains with clarity.
Figure 2.
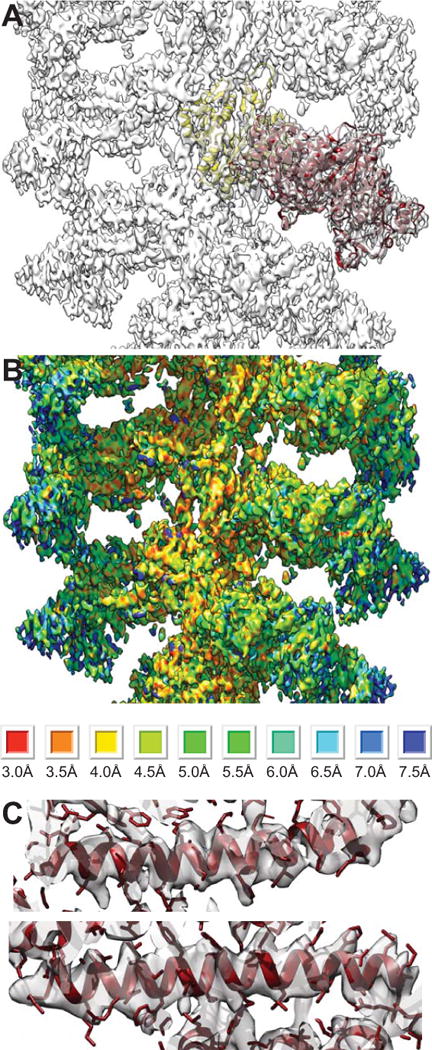
Overview and resolution of the reconstruction of F-actin decorated with smMD. (A) Overview showing the atomic models of the actin subunit (yellow) and the smMD (dark red). (B) RESMAP image of the reconstruction. Resolution is clearly highest close to the filament axis and lowest at the high radius where the converter and SH3 domain are positioned. RESMAP color ranges are shown at the bottom. (C) Images of a pair of long α-helices from the acto-smMD reconstruction (purple). Density corresponding to large side chains is clearly visible. Top panel helix comprises residues 477–506; bottom panel helix comprises residues 420–450.
We fit an atomic model to the density using Rosseta but starting with a homology model based on the prepower stroke transition state of scallop adductor muscle myosin II. Despite the rather large difference in conformation between the starting model and the final fitted model, the agreement is quite striking with the atomic model from the higher resolution reconstruction of F-actin decorated with vertebrate non-muscle myosin II motor domain (nmMD)(von der Ecken et al., 2016). The structures of the actin subunit are nearly identical (Fig. 3A). At the C-terminus, our reconstruction indicated a helical arrangement of the last four residues that were missing in the α-actin that we started the fitting with (Fig. 3B). The non-muscle γ-actin atomic model (von der Ecken et al., 2016) is a very good fit to this feature so we manually built the last four residues into the density using the γ-actin model as a guide. At the N-terminus of our actin structure, density extends only as far as T5. Two other minor departures occur at G168 (G167 for γ-actin) where the γ-actin chain falls out of the density envelope slightly and T324 (T323 for γ-actin) where both models appear to fit the density equally well. We conclude that at our resolution, the α-actin used in the present study and the γ-actin used for non-muscle myosin II are nearly indistinguishable.
Figure 3.
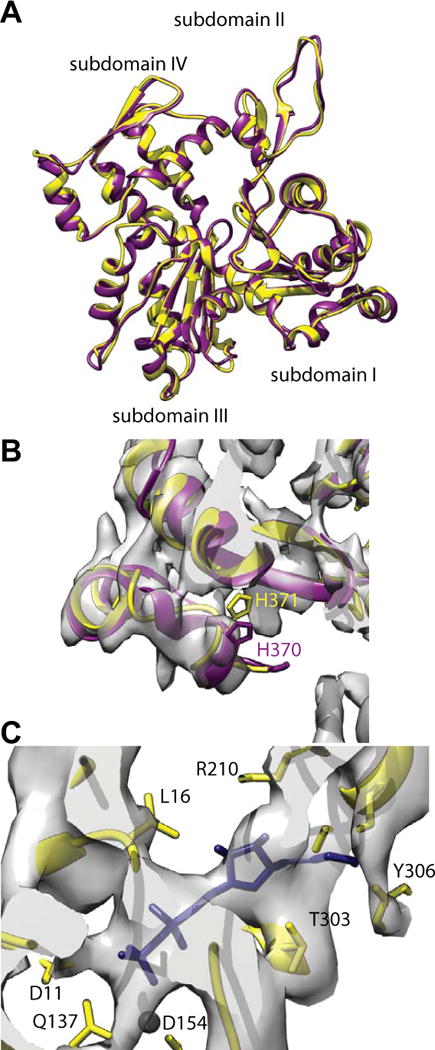
Comparison of vertebrate non-muscle γ-actin and skeletal muscle α-actin subunits. (A) Overlay of the fitted actin subunits. Skeletal muscle α-actin is colored yellow and the non-muscle γ-actin subunit colored dark magenta. (B) Region near the C-terminus. The rabbit α-actin subunit used for initiating the fitting did not include four residues at the C-terminus. These were added in later but have not been energy minimized. The C-termini of the non-muscle γ-actin subunit fits the density very well indicating that after refinement, the muscle α-actin C-terminus will likely be very similar. (C) Region near the ADP binding site. Substantial density is present where the nucleotide binds. The black sphere is a magnesium ion for which clear density is not visible. Its presence provides a useful landmark.
Our density map where F-actin is located also shows strong density in the ADP binding pocket (Fig. 3C). We therefore fit ADP into the density. The conformation obtained is similar but not identical to that obtained for γ-actin (von der Ecken et al., 2016). The differences are probably not significant at our resolution.
For the smMD, the atomic model begins at residue D2 but at the optimal contour threshold (0.016), the model does not enter the density until N22. The topology of the SH3 domain is the same as the nmMD, but the chains are displaced by about half the spacing between β-strands (Fig. 4A,B). The converter domains, the other feature of interest at high radius, have a similar but not identical topology and do not overlap. This could be influenced by the fact that the nmMD has 7 turns of the lever arm α-helix, which are missing in the smMD construct (Fig. 4B). The most noticeable difference in the converter occurs between residues E735 and G749. This segment of polypeptide chain is extended and without secondary structure and differs significantly in the path that the two chains follow.
Figure 4.
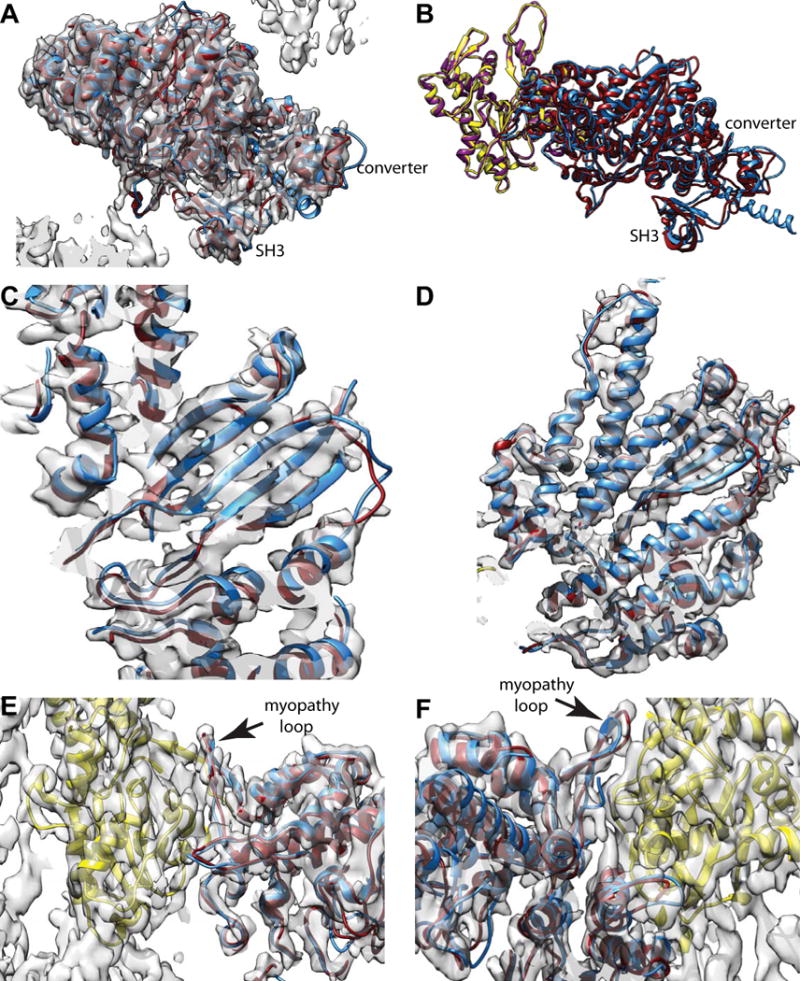
Comparison of vertebrate non-muscle and smooth muscle myosin MDs when bound to F-actin. Vertebrate smooth muscle MD is colored dark red; the vertebrate non-muscle MD is colored dodger blue, muscle α-actin is yellow, non-muscle γ-actin is colored dark magenta. (A) View showing the relative difference between converter and SH3 domains plus the reconstruction envelope. (B) Slightly different view from (A) showing the actin subunits and the converter in better profile without the map. (C) View showing five strands of the transducer β-sheet with both the smooth muscle MD (dark red) and non-muscle MD (dodger blue). (D) Similar view as (C) but showing four major α-helices, which align well to the smooth muscle MD density and atomic model. (E) Actin-myosin interface from the “front”. (F) Actin-myosin interface from the back. In all these views, the vertebrate smooth muscle acto-MD and the vertebrate non-muscle acto-MD atomic models are nearly superimposable at this resolution.
In the region of the transducer β-sheet, the two structures are virtually superimposable as are most of the α-helices in that neighborhood (Fig. 4B,C). The helix from R507-W546 is tilted out of alignment at its beginning but otherwise the differences in this area seem insignificant and the alignment seems quite good.
At the actin-myosin interface, the similarity is more striking than the differences (Fig. 4E,F). The most noticeable difference occurs in the loop from R530-G536. The corresponding loop in the nmMD is R543-G549. In fact the fit of our smMD in this region is not good, but is closer than is the nmMD. Density corresponding to loop 2 is not visible in our reconstruction, nor is it visible in the nmMD structure. Other than this, the topology in the actin-myosin interface is very similar.
We also compared our reconstruction with crystal structures of the myosin-V MD with ADP strongly bound (PDB 4ZG4) (Wulf et al., 2016) and nucleotide free (PDB 1OE9) (Coureux et al., 2003). Here differences were observed when ADP is strongly bound. Because there is little sequence homology between the smMD and the myosin-V MD, we fit both myosin-V MD atomic coordinates to our map as rigid bodies using the fitinmap utility of Chimera (Pettersen et al., 2004). The initial fit done this way for nucleotide-free myosin-V MD was entirely satisfactory and could not be visually improve by manual adjustment. The nucleotide-free myosin-V coordinates overlapped the smMD atomic model quite well and fell almost entirely within the density map envelope (Fig. 5A–E).
Figure 5.
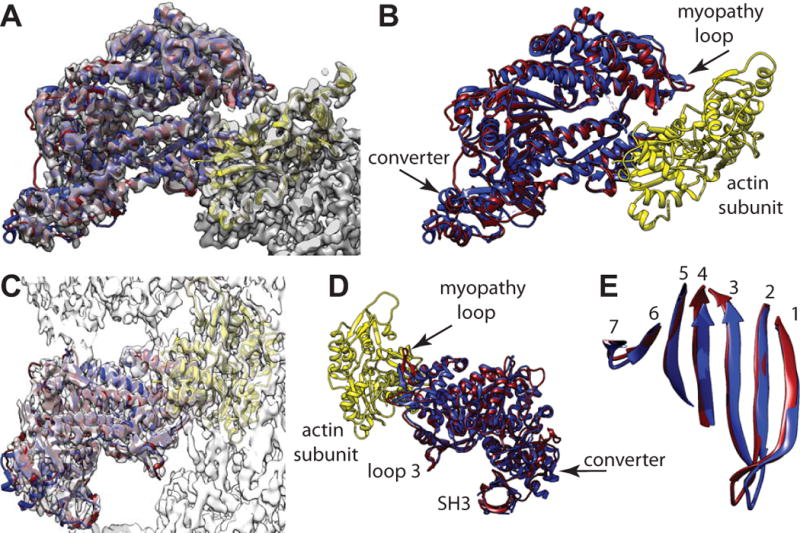
Comparison of the actin bound, smMD and the nucleotide-free myosin-V MD crystal structure (PDB 1OE9), which has been aligned to the reconstruction using Chimera’s fitinmap utility. Coloring scheme has the actin subunit yellow, the smMD dark magenta, and the myosin-V MD blue. (A) View down the actin binding cleft showing the excellent fit of the myosin-V crystal structure even though not bound to actin. The myosin-V converter domain has a very similar position and orientation as the smMD converter. (B) Same view direction as panel A but with the map removed to shown the excellent alignment of the myosin-V helices with the corresponding smMD helices and loops. (C) View perpendicular to the helix axis showing the fit of the myosin-V coordinates within the acto-smMD reconstruction. (D) View from the opposite side without the map showing alignment of the myopathy loop, loop 3 and the SH3 domains. (E) View showing the excellent alignment of the 7-stranded transducer β-sheets.
The fit using the ADP bound myosin-V MD was poor (data not shown). When fit as a rigid body into the density, many features were displaced out of the density and poorly aligned with the smMD coordinates. Although the topology of both MD atomic models is similar, most features are displaced or otherwise modified. Almost nothing overlapped exactly.
We also compared our reconstruction with the recent structure of rabbit striated muscle α-actin decorated with subfragment 1 of rabbit skeletal muscle myosin II (Fujii and Namba, 2017), hereafter referred to as the skMD since only the motor domain can be compared with the smMD. Here we found significant differences. We tried three alignment methods. The first used the actin subunit coordinates to drive the alignment using the Matchmaker utility of Chimera. Done this way, the actin subunit structures are superimposable and matched well and with them a large part of the lower 50 kDa domain, i.e. residues F469-E487, F517-N565, F581-P604 and R657-T668 using the smMD sequence. However, nothing else matched well except for those features, like the myopathy loop, T404-T419 and residues N364-T382 (smMD sequence), that contact actin. We also fit the actin-skMD coordinates as a single rigid body into the acto-smMD density map. This fit was slightly different giving some improvement to the parts that fit poorly using the actin subunit as the alignment driver, i.e. the upper 50 kDa domain, but at the expense of those features that previously fit well such as the lower 50 kDa domain. The third, a rigid body fit using only the skMD coordinates, was also slightly different giving again a small improvement to the upper 50 kDa domain at the expense of the lower 50 kDa domain. We preferred the fit using the actin subunit coordinates as the alignment driver because the differences are more easily visualized. The other alignments did not eliminate these differences; it only reduced them. The following discussion is based on the fit using the actin coordinates as the alignment driver.
When the smMD and the skMD are compared within the density map, two things stand out. First, the so-called loop 3 feature of the skMD is located completely outside of density (Fig. 6A). Its position is similar to that of the homology model of the transition state from which our smMD fitting started. Note that the nucleotide-free myosin-V MD structure was a good fit at loop 3 (Fig. 5C,D). Second, many of the upper 50 kDa domain helices are positioned outside of density (Fig. 6A) in a way that places them further from the axis of the F-actin. When the map is left out and the models viewed from the other side to show the lower 50 kDa domain, it matches well but the upper 50 kDa domain does not (Fig. 6B). We also looked at the position of the transducer β-sheet, which revealed significant differences. Generally, the length of the peptides that conformed to the β-sheet secondary structure were longer in the smMD atomic model than the skMD model with the first three strands displaced to the right (Fig. 6C). The transducer β-sheet is also twisted differently (Fig. 6D). When the map is superimposed at the same time, the first four strands are not positioned within the density envelope (Fig. 6E). When the two atomic models are viewed looking through the actin binding cleft, the general impression is that with the exception of the lower 50 kDa helices, which fit very well, the upper 50 kDa and N-terminal 25 kDa domains are shifted outwards of the smMD atomic model (Fig. 6F). This gives the impression that the actin-binding cleft is more open in the skMD structure than in the smMD structure.
Figure 6.
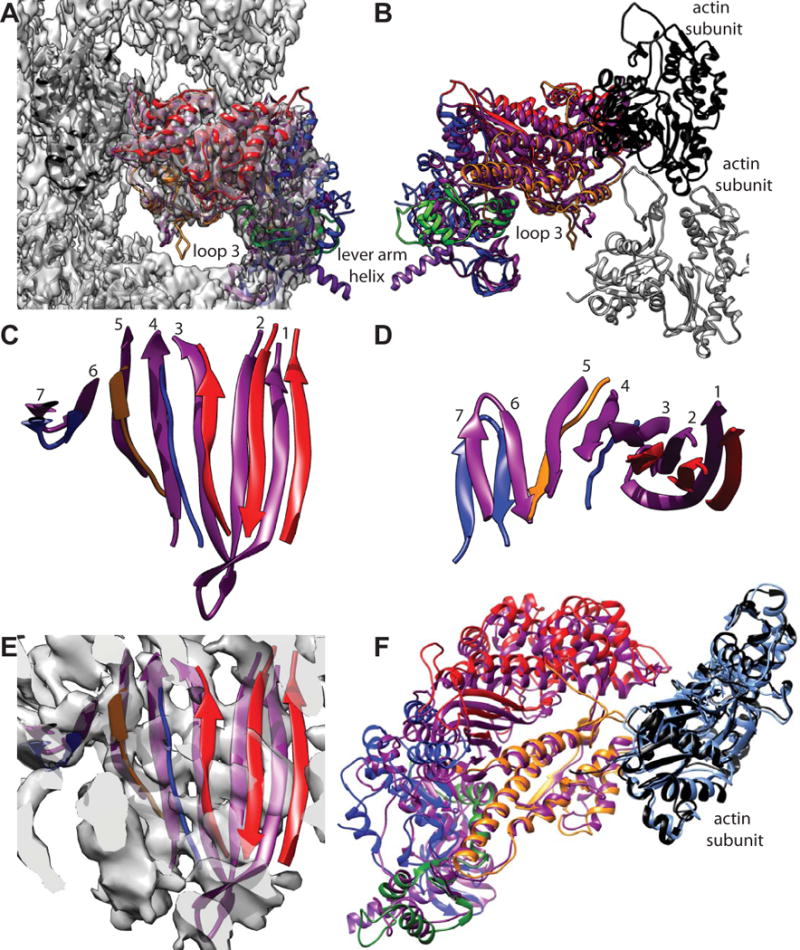
Comparison of acto-smMD with acto-skMD. (A) The two reconstructions shown with the acto-smMD reconstruction. The acto-skMD was aligned to the acto-smMD using the actin subunit coordinates from the acto-skMD atomic model (PDB 5H53), which is colored black, to drive the alignment. There is little difference between the actin subunit atomic models from the two reconstructions. The smMD atomic model is colored purple. The acto-skMD atomic model is colored according to the MD subdomains, which are N-terminal 25 kDa domain (blue), upper 50 kDa domain (red), lower 50 kDa domain (orange), converter domain (green) and the lever arm (magenta). Note that the smMD does not have the lever arm helix. (A) Both atomic models shown within the reconstruction envelope. Many features of the skMD atomic model fall outside of the density envelope of the smMD. The most obvious difference is the position of loop 3 (skeletal residues K567–F579), which falls clearly outside the reconstruction envelope. Loop 3 is part of the lower 50 kDa domain. (B) The atomic models of the skMD and the smMD shown with a pair of actin subunits, one black, the other gray. This view from the opposite direction from that of panel A. (C) Comparison of the transducer β-sheet with the smooth muscle structure shown in purple and the skeletal muscle sheet colored according to subdomain origin. Since the sheet itself is curved, the displacements for strands 1 and 2 are the most obvious. This view direction is from outside the MD looking in towards the actin-binding cleft. The relative displacement has the skeletal β-sheet to the side and on the outside of the smooth β-sheet (roughly looking from the top of panel F towards the bottom). (D) View looking down from the top of panel C. (E) Same view direction as panel C but with the reconstruction envelope showing. Note that the skMD β-sheet mostly falls outside of the corresponding density envelope. (F) View looking down the actin binding cleft showing the actin subunit atomic models from the two reconstructions as well as their MD atomic models. The actin atomic model from the acto-smMD reconstruction is shown in sky blue. Note that the helices of the lower 50 kDa domains overlap well, whereas features of the upper 50 kDa domains overlap poorly. The 25 kDa domains also overlap poorly.
We find that the structural differences between the nucleotide-free smMD, nmMD and myosin-V MDs when bound to actin are small whereas the differences between the smMD and the skMD when bound to actin are large. Neither our reconstruction of acto-smMD nor that of acto-skMD has sufficient resolution to make a detailed comparison at the level of amino acid side chains. The most obvious difference at the current resolution lies in the size of the actin binding cleft, reflected in the displacement outward of the upper 50 kDa domain, and the position of the transducer β-sheet, which likely correlate with the properties of the two myosin species. The myosin species used for the acto-skMD reconstruction is rabbit leg muscle.
The kinetics of the actin-activated ATPase of four muscle myosins from chicken fast and slow skeletal, cardiac and smooth, were compared in a single study (Marston and Taylor, 1980). Measurable differences were found in the rates of ATP induced dissociation from actin, the rate of reassociation with actin after ATP cleavage and rates of release of ADP when actin bound. The rate of ATP induced dissociation from actin at 20°C was slowest for smooth and too rapid to be measurable for fast skeletal myosin; at 3°C there was a 4-fold difference. Marston and Taylor concluded that ATP must induce a conformational change in myosin which we now know involves opening of the actin binding cleft (Holmes et al., 2004). The more open actin-binding cleft in the acto-skMD reconstruction, which may be interpreted as partially along the opening pathway, may offer an explanation for the difference in this rate. Since we only observe a single, nucleotide-free, actin-bound state, the reconstructions cannot offer an explanation for the rates of reassociation with actin following ATP cleavage or the differences in ADP release. However, we do point out that in smooth muscle myosin, the ADP release rate is about 20 times slower for smooth compared to fast skeletal muscle myosin (Marston and Taylor, 1980) and causes a further, 35 Å displacement of the end of the myosin lever arm toward rigor (Whittaker et al., 1995). Restraint on the lever arm movement may thus affect the rate of ADP release.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants R01 GM30598 (to KAT), P01 HL110869 (to KMT) and R01 AR53975 (to SL). Zhongjun Hu was supported by predoctoral fellowship 15PRE25090150 from the American Heart Association. The Titan Krios was partially funded by NIH Grant S10 RR25080. The DE-20 was funded by NIH grant S10 OD018142. We thank Elena Krementsova for expression and purification of the smooth muscle myosin motor domain, and Michael Rademacher and Teresa Ruiz for the use of their FEI Technai 12 and Gatan CP-3 freezing apparatus for the preparation of some of the samples used in this study. SL and KMT wish to thank Dorit Hanein and Niels Volkmann for their early contributions to the acto-smMD structure before the recent technological advances in cryoEM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
CB and ZH1 computed the reconstruction, AW collected the data, DWT prepared the cryoEM samples, KMT and SL provided the myosin motor domain, CB, ZH1, ZH2, KMT, SL & KAT wrote the paper. ZH2 performed initial reconstruction attempts using CCD data.
ACCESSION NUMBERS
The coordinates for the α-actin-smMD atomic model have been deposited in the RCSB Protein Data Bank under accession code 6BIH. The density map has been deposited in the EM Data Bank under accession code EMD-7100.
References
- Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–338. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Ge P, Lee BY, Horwitz MA, Zhou ZH. Atomic structure of T6SS reveals interlaced array essential to function. Cell. 2015;160:940–951. doi: 10.1016/j.cell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coureux PD, Wells AL, Ménétrey J, Yengo CM, Morris CA, Sweeney HL, Houdusse A. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425:419–423. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- Coureux PD, Sweeney HL, Houdusse A. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 2004;23:4527–4537. doi: 10.1038/sj.emboj.7600458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criddle A, Geeves M, Jeffries T. The use of actin labelled with N-(1-pyrenyl) iodoacetamide to study the interaction of actin with myosin subfragments and troponin/tropomyosin. Biochem J. 1985;232:343–349. doi: 10.1042/bj2320343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F, Song Y, Li X, Brunner MJ, Xu C, Conticello V, Egelman E, Marlovits TC, Cheng Y, Baker D. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat Methods. 2015;12:361–365. doi: 10.1038/nmeth.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R, Freyzon Y, Trybus KM, Cohen C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 1998;94:559–571. doi: 10.1016/s0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- Egelman EH. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J Struct Biol. 2007;157:83–94. doi: 10.1016/j.jsb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Fernandez JJ, Luque D, Caston JR, Carrascosa JL. Sharpening high resolution information in single particle electron cryomicroscopy. J Struct Biol. 2008;164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- Fujii T, Namba K. Structure of actomyosin rigour complex at 5.2-Å resolution and insights into the ATPase cycle mechanism. Nat Commun. 2017;8:13969. doi: 10.1038/ncomms13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeves MA, Holmes KC. The molecular mechanism of muscle contraction. Adv Prot Chem. 2005;71:161–194. doi: 10.1016/S0065-3233(04)71005-0. [DOI] [PubMed] [Google Scholar]
- Gourinath S, Himmel DM, Brown JH, Reshetnikova L, Szent-Györgyi AG, Cohen C. Cohen C. Crystal structure of scallop myosin S1 in the pre-power stroke state to 2.6-Å resolution: flexibility and function in the head. Structure. 2003;11:1621–1627. doi: 10.1016/j.str.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Himmel DM, Gourinath S, Reshetnikova L, Shen Y, Szent-Györgyi AG, Cohen C. Crystallographic findings on the internally uncoupled and near-rigor states of myosin: further insights into the mechanics of the motor. Proc Natl Acad Sci U S A. 2002;99:12645–12650. doi: 10.1073/pnas.202476799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KC, Geeves MA. The structural basis of muscle contraction. Philos Trans R Soc Lond B Biol Sci. 2000;355:419–431. doi: 10.1098/rstb.2000.0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KC, Schröder RR, Sweeney HL, Houdusse A. The structure of the rigor complex and its implications for the power stroke. Philos Trans R Soc Lond B Biol Sci. 2004;359:1819–1828. doi: 10.1098/rstb.2004.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KC, Angert I, Kull FJ, Jahn W, Schröder RR. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 2003;425:423–427. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- Houdusse A, Sweeney HL. How Myosin Generates Force on Actin Filaments. Trends Biochem Sci. 2016;41:989–997. doi: 10.1016/j.tibs.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdusse A, Szent-Györgyi AG, Cohen C. Three conformational states of scallop myosin S1. Proc Natl Acad Sci U S A. 2000;97:11238–11243. doi: 10.1073/pnas.200376897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Taylor DW, Reedy MK, Edwards RJ, Taylor KA. Structure of myosin filaments from relaxed Lethocerus flight muscle by cryo-EM at 6 Å resolution. Sci Adv. 2016;2:e1600058. doi: 10.1126/sciadv.1600058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymn R, Taylor EW. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry. 1971;10:4617–4624. doi: 10.1021/bi00801a004. [DOI] [PubMed] [Google Scholar]
- Mallick SP, Carragher B, Potter CS, Kriegman DJ. ACE: automated CTF estimation. Ultramicroscopy. 2005;104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Marston SB, Taylor EW. Comparison of the myosin and actomyosin ATPase mechanisms of the four types of vertebrate muscles. J Mol Biol. 1980;139:573–600. doi: 10.1016/0022-2836(80)90050-9. [DOI] [PubMed] [Google Scholar]
- Menetrey J, Llinas P, Cicolari J, Squires G, Liu X, Li A, Sweeney HL, Houdusse A. The post-rigor structure of myosin VI and implications for the recovery stroke. EMBO J. 2008;27:244–252. doi: 10.1038/sj.emboj.7601937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan R, Flicker P. Structural relationships of actin, myosin, and tropomyosin revealed by cryo-electron microscopy. J Cell Biol. 1987;105:29–39. doi: 10.1083/jcb.105.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Mornet D, Bertrand R, Pantel P, Audemard E, Kassab R. Proteolytic approach to structure and function of actin recognition site in myosin heads. Biochemistry. 1981;20:2110–2120. doi: 10.1021/bi00511a007. [DOI] [PubMed] [Google Scholar]
- Odronitz F, Kollmar M. Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 2007;8:R196. doi: 10.1186/gb-2007-8-9-r196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Cell Biol. 1982;24:271–289. doi: 10.1016/s0091-679x(08)60661-5. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Rayment I. X-ray Structure of the Magnesium(II)·ADP·Vanadate Complex of the Dictyostelium discoideum Myosin Motor Domain to 1.9 Å Resolution. Biochemistry. 1996;35:5404–5417. doi: 10.1021/bi952633+. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Houdusse A. Structural and functional insights into the myosin motor mechanism. Annu Rev Biophys. 2010;39:539–557. doi: 10.1146/annurev.biophys.050708.133751. [DOI] [PubMed] [Google Scholar]
- Trybus KM. Biochemical studies of myosin. Methods. 2000;22:327–335. doi: 10.1006/meth.2000.1085. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Ouyang G, Trybus KM, DeRosier DJ, Lowey S, Hanein D. Myosin isoforms show unique conformations in the actin-bound state. Proc Natl Acad Sci U S A. 2003;100:3227–3232. doi: 10.1073/pnas.0536510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N, Liu H, Hazelwood L, Krementsova EB, Lowey S, Trybus KM, Hanein D. The structural basis of myosin V processive movement as revealed by electron cryomicroscopy. Mol Cell. 2005;19:595–605. doi: 10.1016/j.molcel.2005.07.015. [DOI] [PubMed] [Google Scholar]
- von der Ecken J, Heissler SM, Pathan-Chhatbar S, Manstein DJ, Raunser S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 2016;534:724–728. doi: 10.1038/nature18295. [DOI] [PubMed] [Google Scholar]
- Wang Z, Hryc CF, Bammes B, Afonine PV, Jakana J, Chen DH, Liu X, Baker ML, Kao C, Ludtke SJ, Schmid MF, Adams PD, Chiu W. An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat Commun. 2014;5:4808. doi: 10.1038/ncomms5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker M, Wilson-Kubalek EM, Smith JE, Faust L, Milligan RA, Sweeney HL. A 35-Å movement of smooth muscle myosin on ADP release. Nature. 1995;378:748–751. doi: 10.1038/378748a0. [DOI] [PubMed] [Google Scholar]
- Wulf SF, Ropars V, Fujita-Becker S, Oster M, Hofhaus G, Trabuco LG, Pylypenko O, Sweeney HL, Houdusse AM, Schroder RR. Force-producing ADP state of myosin bound to actin. Proc Natl Acad Sci U S A. 2016;113:E1844–1852. doi: 10.1073/pnas.1516598113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gourinath S, Kovács M, Nyitray L, Reutzel R, Himmel DM, O’Neall-Hennessey E, Reshetnikova L, Szent-Györgyi AG, Brown JH. Rigor-like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure. 2007;15:553–564. doi: 10.1016/j.str.2007.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.