

Abstract
Mitochondrial dynamics are increasingly recognized to play an important role in regulating mitochondrial function in response to diverse stimuli. Given the overlap in the physiological processes influenced by mitochondria and the physiological processes disrupted in tumor cells, we speculate that tumor cells alter mitochondrial shape to promote the tumorigenic phenotype. Here, we briefly review the evidence linking changes in mitochondrial fusion and fission to a number of key tumorigenic processes, including metabolic rewiring, inhibition of cell death, cell migration, cell proliferation and self-renewal capacity. The role of mitochondrial dynamics in tumor growth is an important emerging area of research, a better understanding of which may lead to promising new therapeutic options for the treatment of cancer.
Introduction
The development of a tumor is driven by a series of physiological changes both within the tumor cells and the cells of the surrounding tissue that disarm the multiple mechanisms in place to maintain normal tissue homeostasis. These physiological changes, famously summarized by Hanahan and Weinberg [1], are driven initially by discrete genetic alterations that activate or disable key control mechanisms that drive a proliferative phenotype and inhibit key cell death and growth arrest fail-safes. As the tumor develops, evolutionary pressures select for additional changes, both genetic and non-genetic, that allow it to avoid immune detection, to outgrow its fuel supply and to escape the tissue of origin and colonize additional organs. The challenges of combating cancer lie in the complexity of mechanisms through which these physiological changes arise and the difficulty inherent in selectively and safely targeting these changes while minimizing damage to non-tumor cells. While great progress has been made over the past several decades in both understanding and combatting tumor growth, there is still a great deal of work to accomplish in order to successfully and consistently combat this disease.
An emerging area of research that has the potential to significantly alter our understanding of tumor biology and our ability to successfully treat patients is mitochondrial dynamics. While mitochondria have long been postulated to play a role in tumor growth [2], recent years have seen an explosion in research demonstrating links between key oncogenic signaling pathways and mitochondria [3,4]. Furthermore, it is clear that mitochondrial changes can allow cells to adapt to the unique and rapidly changing microenvironment.
The more we understand about mitochondrial function, the clearer it becomes that their function lies at the heart of many of the physiological changes driving tumor initiation and all stages of tumor progression [4]. To better understand how the mitochondria can play so many different roles, it is important that we understand the myriad ways that mitochondrial functions are regulated. Changes in the expression and import efficiency of mitochondrial proteins, changes in the post-translational modification of key mitochondrial enzymes and alterations in the lipid content of both the inner and outer mitochondrial membranes can all influence mitochondrial behavior, and are all potential mechanisms by which tumors will coopt mitochondrial function for their own benefit [4]. In addition, a wealth of recent evidence is revealing how changes in mitochondrial shape can profoundly influence mitochondrial function. This review will highlight several of the key physiological changes associated with tumorigenesis and how changes in mitochondrial shape, through the regulation of fusion and fission, may promote the tumorigenic process (Figure 1).
Figure 1. Mitochondrial dynamics can contribute to multiple tumorigenic processes.
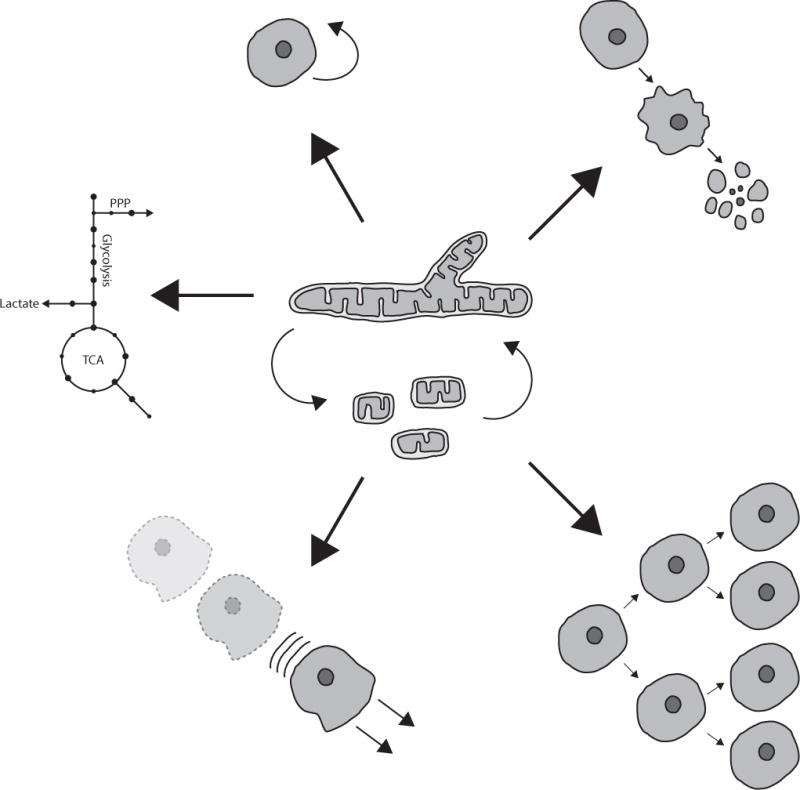
Changes in mitochondrial shape contribute to the regulation of a number of processes dysreulated in human tumors, including self-renewal, apoptosis, proliferation, cell migration and metabolic reprogramming.
The regulation of mitochondrial dynamics
The appearance of the mitochondrial network varies significantly in different cell types, both for cells grown in culture and for intact tissues [5]. There is much more mitochondrial morphology data available from tissue culture cells, most of which exhibit a reticular mitochondrial morphology consisting of a mix of elongated tubules and shorter fragments that extend throughout the cytoplasm. This phenotype can vary depending on cell type, and manipulations of culture conditions can also elicit changes, resulting in more netlike or highly fragmented morphologies [5,6]. Data on intact tissues are more limited, but the evidence is consistent with this range of morphologies observed in cultured cells that is highly dependent on cell type and environment [7–9]. These various morphologies arise through the delicate balance of the opposing activities of a set of dynamin related GTPases that fuse or divide mitochondrial tubules. Fission of mitochondria occurs when Dynamin related protein 1 (Drp1) is recruited to the outer mitochondrial membrane by a set of integral membrane adapter proteins, including MFF and Mid49/51 [10–13]. Drp1 oligomerizes to form a ring or spiral around the outer membrane [14]. Assembly of this oligomeric structure, along with constriction of the ring induced by GTP hydrolysis, induces constriction of the mitochondrial membrane [15–17]. Following this membrane constriction, the classical dynamin, dynamin 2, is recruited to the membrane through an unknown mechanism to complete the scission process [18]. The fission process is regulated at a number of different steps. Sites of fission, at least under certain conditions, occur at a subset of Endoplasmic reticulum (ER) mitochondria contact sites, where the ER wraps around the mitochondrial tubule to provide the initial constriction that allows Drp1 oligomers to form [19]. The process is also linked to replication of mitochondrial DNA, as newly formed nucleoids preferentially localize to the tips of newly divided mitochondria [20].
The recruitment and activity of Drp1 are also highly regulated by posttranslational modifications. Phosphorylation of Drp1 on S616 promotes Drp1-dependent fission. This site integrates a variety of upstream signals and can be targeted by multiple kinases, including Cdk1 [21], Cdk5 [22,23], Erk1 [24], Erk2 [9], PKCδ [25], CaMKII [26]. Conversely, S637 is phosphorylated by the cAMP-dependent protein kinase (PKA) to inhibit Drp1 activity [27]. This site can be dephosphorylated by the phosphatase calcineurin to promote mitochondrial fragmentation [28]. In addition to phosphorylation, Drp1 can be activated by sumoylation [29–31], ubiquitination [32–34] and s-nitrosylation [35].
This fission activity is balanced by mitochondrial fusion, which is likewise regulated by numerous upstream signals. Outer mitochondrial membrane fusion occurs following homo- or heterodimerization of two large GTPases (Mfn1 or Mfn2), which span the outer mitochondrial membrane and tether the two opposing membranes in close proximity [36]. Once tethered, the exact mechanism of fusion remains poorly understood, but is proposed to require GTPase activity [37]. Inner membrane fusion is thought to involve a similar mechanism, with the tethering provided by the large GTPase Opa1 [38]. Opa1 also plays an important role in maintaining cristae morphology [39]. The regulation of fusion activity remains poorly understood and is an area of active research. Mitofusin activity can be altered by a number of posttranslational modifications, including phosphorylation, acetylation and ubiquitination and these signals converge on this machinery in response to a variety of environmental signals, including bioenergetics stress and loss of mitochondrial membrane potential [37,40–42]. Opa1 is primarily regulated by proteolytic cleavage by two inner membrane peptidases YME1L and OPA1 [43]. Current models suggest that various stress stimuli, including mitochondrial dysfunction or respiratory deficiency, promote cleavage of Opa1 from its fusion competent long form, to a short form that not only lacks fusion activity, but actively promotes fission [44].
While still not completely understood, it is speculated that mitochondrial fusion is important to maintain mitochondrial health, perhaps through content mixing or dilution of toxic molecules [37]. Further, it has been proposed that fusion is able to protect mitochondria from autophagic clearance and promote more efficient ATP generation through oxidative phosphorylation [45,46]. A better understanding both of the regulation of fusion and the physiological benefits of a fused mitochondrial network will be critical as we seek to understand the consequences of disrupted mitochondrial dynamics in tumors.
Tumor cell metabolism and mitochondrial dynamics
Tumor cells have much different metabolic demands than the differentiated, non-dividing cells from which they arise. Rapid proliferation requires increased production of molecular building blocks required to build new cells. While several different strategies have been described to support this increase in anabolic metabolism, the most well understood mechanism is an increase in the uptake of glucose and the utilization of glucose derived carbon for a variety of biosynthetic pathways, rather than its complete oxidation to generate ATP [47,48]. While on the surface this metabolic shift in glucose utilization suggests a diminished role for mitochondrial metabolism, more recently it has become clear that metabolic function of mitochondria still plays a critical role in these cells. Not only do many TCA cycle intermediates feed into important biosynthetic pathways, but the relative decrease of glucose-derived carbon used for mitochondrial energy production can be compensated by relative increases in the oxidation of alternative carbon sources such as glutamine and lipids {DeBerardinis:2008gk}. As a result, mitochondrial ATP production in many cancer cells has been shown to be comparable to that of non-transformed cells {Vyas:2016kg}.
How might alteration of mitochondrial shape contribute to this metabolic shift? Studies from a number of different systems indicate how changes in mitochondrial shape can affect cellular metabolism. For example, under starvation conditions, it was shown that elongated mitochondria exhibited increased dimer formation of ATP synthase, leading to higher efficiency [45]. Furthermore, if elongation of mitochondria inhibits mitophagy, as has been proposed [46], it will lead to increased mitochondrial mass in the absence of a compensatory decrease in mitochondrial biogenesis, further increasing oxidative capacity. Given this, it is tempting to speculate that mitochondrial fragmentation will lead to diminished oxidative capacity and potentially contribute to a more glycolytic phenotype (Figure 2). Consistent with this, extensive mitochondrial fragmentation is observed in a number of different tumor types, including lung adenocarcinoma, pancreatic ductal adenocarcinoma, melanoma, colon carcinoma, and glioma [9,24,49–51] and Drp1-dependent mitochondrial fission was shown to promote increased glycolysis in high grade neuroblastoma [52]. Furthermore, oncogenic Ras signaling and activation of MAPK signaling, both of which are known inducers of the glycolytic switch 100,101, promote Drp1-dependent mitochondrial fragmentation {Kashatus:2015eq, Serasinghe:2015kc}.
Figure 2. Highly fragmented mitochondria can contribute to a glycolytic metabolism in multiple ways.
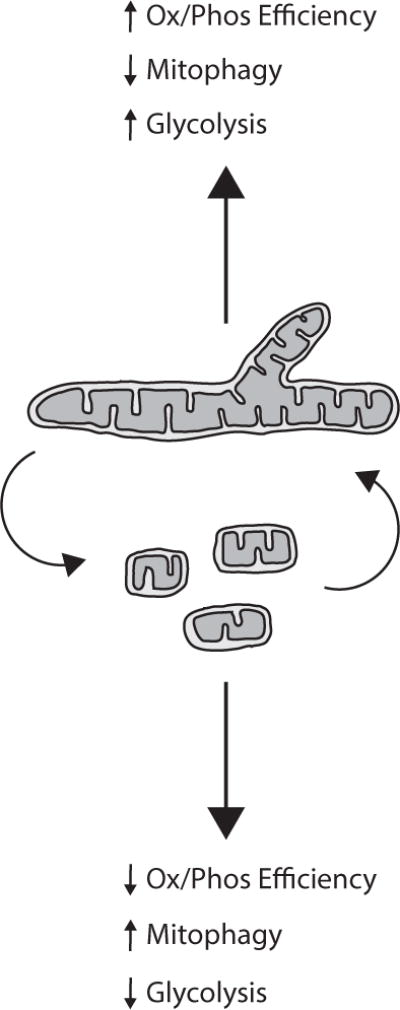
Studies from a variety of systems have shown that mitochondrial fragmentation is associated with increased mitophagy and decrease efficiency of ATP synthesis, suggesting that mitochondrial fragmentation may be a mechanism through which tumor cells reprogram their metabolism.
A great deal of work over the past several years has highlighted the many disparate mechanisms through which cancer cells alter their metabolism, including through alteration of key metabolic enzymes such as PKM {Yang:2012fh} and the mitochondrial pyruvate carrier (MPC) {Schell:2014dg, Vacanti:2014ei}, and through changes in the transporters that bring nutrients such as glucose and glutamine into the cell {Altman:2016hj}. Because of this, the full extent to which mitochondrial shape per se contributes to a glycolytic shift will require more careful analysis. Further, it remains to be determined how the fusion and fission machinery impacts glucose metabolism independently of mitochondrial shape, and how fusion-fission dynamics impact other key metabolic adaptations of tumor cells, such as altered amino acid metabolism and lipid metabolism. However, despite our incomplete knowledge at this point, the growing links between mitochondrial dynamics and nutrient utilization [53,54] suggest that mitochondrial shape will likely a key mechanism through which tumor cells promote the metabolic shifts required for their unabated growth.
Mitochondrial dynamics and stemness
Most tissues in the human body are comprised primarily of differentiated cells that lack the capacity for self renewal but a small number of stem cells resident within those tissues allows for the replacement of old or damaged cells and maintenance of tissue homeostasis [55]. Whether this hierarchical structure exists for tumors is still a matter of some debate, but it is clear that many tumor types consist of a mix of cells with differing self renewal capacity and that the existence of tumor stem cells or stem-like cells can pose challenges for effective therapeutic intervention [56]. For example, in glioma, as few as 100 glioma cells expressing the cell surface antigen CD133 are able to form tumors when injected into immunocompromised mice, while 105 CD133-negative cells isolated from the same population of tumor cells lack tumor forming capabilities [57]. The existence of these tumor cell populations with self-renewal capacity was first demonstrated for liquid tumors such as leukemia [58], but has since been demonstrated for a number of solid tumors, including breast [59], colorectal cancer [60,61], pancreatic cancer [62,63] and melanoma [64,65], among others [56]. The emerging importance of this phenomenon in a wide variety of tumor types underscores the need to understand the pathways that promote and maintain this self renewal capacity.
A number of recent studies have suggested a role for mitochondrial morphology in the self renewal and stemness. Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) was shown to require an Erk and Drp1-dependent mitochondrial fragmentation event that occurs following forced expression of Oct4, Sox2, Klf4 and c-Myc [66]. Consistent with this, differentiation from embryonic stem cells into cardiomyocytes requires the mitochondrial fusion activity of Mfn2 and Opa1 [67]. In contrast, Mfn2 is required for the maintenance of haematopoietic stem cells, and deletion of Opa1 or Mfn1 and Mfn2 impaired self-renewal capacity of neural stem cells, suggesting cell type specificity in the role of mitochondrial shape in maintaining stemness [68,69]. Studies of cancer stem cells are more limited, but mitochondrial fragmentation was observed in brain tumor initiating cells (BTICs), and inhibition of Drp1-dependent fission led to their apoptosis and inhibited tumor growth [23]. The changes in mitochondrial morphology observed during stem cell differentiation likely represent the change in metabolic requirements [70], a more complete understanding of which is critical given the clinical importance of directly targeting the stem cell compartment for so many tumor types.
Regulation of cell death by mitochondrial dynamics
An important part of maintaining normal tissue homeostasis is the ability of damaged or otherwise abnormal cells to undergo programmed cell death. In the absence of additional mutations, aberrant activation of oncogenic signaling pathways, excessive genetic damage and disruption of normal tissue architecture will activate signaling pathways that can lead to the orderly destruction and clearance of the effected cells. For a tumor to develop requires disruption of this process and tumor cells accomplish this in a number of different ways. Most of these cell death pathways converge on the mitochondria, where, in the case of apoptotic cell death, pores are formed on the OMM to allow the release of a number of factors that initiate a cascade of proteolytic cleavage events resulting in the orderly destruction of the cell [71]. Not surprisingly, alterations in the levels or activities of the pore forming proteins, or their endogenous antagonists, are associated with tumor progression [71].
Intriguingly, many of these key regulators of cell death physically associate with the mitochondrial fusion and fission machinery and the initiation of cell death is invariably associated with dramatic changes in mitochondrial morphology [72–74]. Thus, it stands to reason that changes in mitochondrial shape, or the activities of mitochondrial shape-changing proteins, can impact the ability of tumor cells to survive. The most consistent observation related to the relationship between mitochondrial shape and apoptosis is that mitochondrial outer membrane permeabilization (MOMP) is accompanied by extensive mitochondrial fragmentation, but the exact relationship between these events remains enigmatic and seems to be context dependent [75]. Under certain conditions, inhibition of fission is sufficient to delay or inhibit cytochrome C release from mitochondria [10,11,76–79], potentially through effects on cristae remodeling [80–82] or membrane curvature [83–85]. However, it is also clear that in certain cell types and in response to certain stimuli, loss of Drp1 or inhibition of fission has little or no effect on MOMP and the initiation of apoptosis [78,86–89]. Furthermore, Mfn1 deficient MEFs, which exhibit highly fragmented mitochondria, are resistant to certain apoptotic stimuli, and knockdown of Drp1 was sufficient to re-sensitize the cells [85].
How does this relationship between mitochondrial fragmentation and apoptosis affect tumorigenesis? One possibility is that the fragmented mitochondrial morphology observed in most tumors represents an adaptation that promotes tumor cell survival [3]. It is also possible, however, the tumor cell is trading an increased sensitivity to apoptosis for the other physiological benefits of a constitutively fragmented mitochondrial network. A couple of important questions arise from this latter conclusion. First, can this increased sensitivity be exploited for therapeutic purposes? Second, can manipulation of mitochondrial shape increase the selectivity and efficacy of chemotherapeutic drugs designed to kill cancer cells? A better understanding of how MOMP is influenced both by mitochondrial membrane curvature and the specific interactions between Bcl2 family members and the mitochondrial dynamics machinery will be required to devise the appropriate strategies for exploiting the tumor associated changes in mitochondrial shape to kill cancer cells.
Mitochondrial dynamics and cell migration
Cancer mortality is rarely caused by the primary tumor, but more often a result of metastasis to distant organs [90]. The ability to invade and increased migratory behavior are essential for a tumor cell to exit the primary tumor, escape into the blood stream, and colonize the site of metastasis. Migration and invasion are energetically demanding processes that require exquisite spatial and temporal coordination of a number of key cellular processes. Mitochondria are increasingly recognized to play an important role in this regulation and changes in mitochondrial shape and localization are key to that role [91].
One consistent observation in the analysis of the role of mitochondrial dynamics in migration and invasion is that mitochondria accumulate at the leading edge, where there is extensive actin remodeling and high energy demand (Figure 3). In most model systems analyzed to date, this localization is accompanied by mitochondrial fragmentation. For example, metastatic breast cancer cells exhibit increased fission activity compared with non-metastatic cells, and inhibition of Drp1 or forced expression of Mfn1 is sufficient to inhibit lamellipodia formation and metastatic potential [92–94]. Importantly, mitochondrial function is required for this increased metastatic ability, as inhibition of mitochondrial function also led to decreased lamellipodia formation and migratory behavior [92]. Similar finding have been reported for glioma [95,96], where inhibition of Drp1-dependent mitochondrial fission inhibited pseudopodia and microvilli formation as well as cell invasion. Interestingly, the increased Drp1 activity in these cells is potentially downstream of the kinase NIK, representing a novel NF-κB independent role for this kinase [96]. Furthermore, Drp1 may impact actin dynamics directly in glioma cells, in addition to facilitating proper mitochondrial localization, through direct interactions with the GTPase RhoA [95]. The requirement for mitochondrial fission has also been observed in oncocytic thyroid tumors, where inhibition of Drp1 blocked migration and invasion ability [97]. The model arising from these studies is that mitochondrial function is required to fuel the energetically demanding actin remodeling at the leading edge of migrating and invading cancer cells and that mitochondrial fission facilitates the rapid relocalization of mitochondria along microtubule tracks. However, in at least some instances, fission may not be required for this process. Indeed, in certain lung cancer and glioblastoma cell lines, mitochondrial tubules extend to the cell periphery in a manner dependent on mitofusin activity, and inhibition of Mfn1 or Mfn2 was sufficient to inhibit both mitochondrial trafficking and cell invasion [98]. Importantly, mitochondrial function at the leading edge, ie - localized ATP production, was still important for the migratory behavior, suggesting that mitochondrial function, but not dynamics per se, is what is important for invasion and migration, and that cellular context will be critical in determining how changes in mitochondrial shape affect this important tumorigenic process.
Figure 3. Mitochondrial localization to the leading edge promotes migration and invasion.
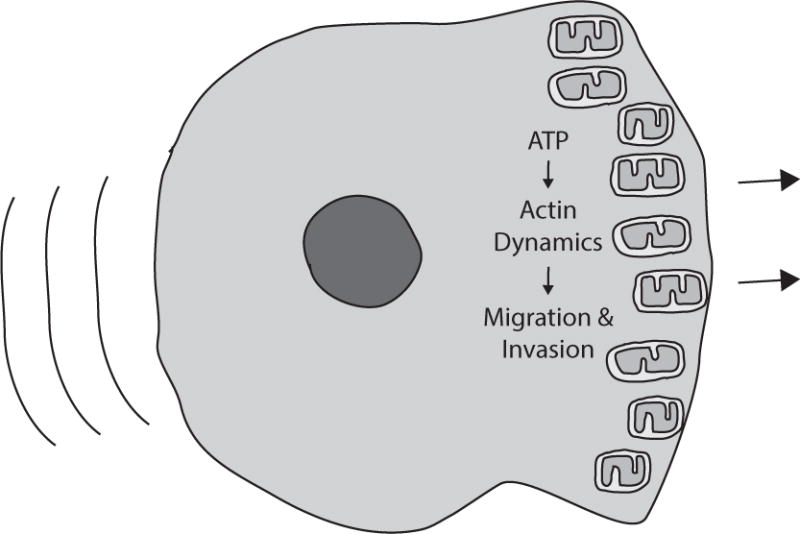
Studies in a variety of tumor types demonstrate that mitochondrial function is required at the leading edge of migrating cells to facilitate multiple energetically demanding processes.
Mitochondrial dynamics and cell proliferation
A defining feature of a tumor cell is the ability to rapidly proliferate, even in the absence of external growth signals [1]. The decision to proliferate is influenced by multiple factors, including many of the physiological processes described above. Progression through the cell cycle ultimately requires activation of a series of well defined signaling cascades that in normal cells are highly responsive to incoming signals from outside of the cell as well as each of the cell’s various compartments. This communication ensures that cell division only occurs if conditions are appropriate for it to do so. Division puts a number of demands on a cell: energetic, biosynthetic and structural. To help meet all of these demands, the regulation of mitochondrial dynamics is tightly coupled with the cell cycle machinery. During the energetically demanding S-phase, mitochondria elongate to increase efficiency of ATP production [99]. However, during the more structurally demanding mitotic phase, the mitochondria fragment to ensure equal distribution to daughter cells [21]. These dramatic shifts in mitochondrial morphology are coordinated with cell cycle progression through reciprocal interactions between the fusion and fission machinery and the cyclin dependent kinases [100]. How do changes in mitochondrial morphology impact the reciprocal regulation between the mitochondrial dynamics machinery and the cell cycle machinery? Do oncogene-induced changes in mitochondrial shape promote cell cycle progression, or is the loss of cell cycle control of mitochondrial structure another checkpoint tumor cells have to overcome?
A number of studies from tissue culture cells suggest that alterations in mitochondrial structure throughout the cell cycle are due to direct and indirect manipulation of fission activity by cyclin dependent kinases and other cell cycle regulated activities. During mitosis, Drp1 is activated by Cyclin B/Cdk1 phosphorylation on S616 in a process that requires several other factors, including the small GTPase RalA and the kinase Aurora A [21,101]. Drp1 activity is further enhanced during mitosis through de-sumoylation by the Sumo protease SenP5 [102]. Interestingly, while inhibition of mitotic mitochondrial fission does not completely block progression through mitosis, it can delay mitotic entry and slow proliferation underscoring the importance of this fragmentation for proper cell cycle regulation [101,102]. After mitosis, fragmentation is reversed through degradation of Drp1, mediated by the APC/C-Cdh1 ubiquitin ligase [103]. The loss of Drp1 allows reformation of the mitochondrial network and presumably contributes to the hyperfusion of mitochondria observed as the cell builds to the G1/S transition [99]. In addition to increasing ATP production, this hyperfusion promotes buildup of Cyclin E, which in turn promotes S-phase entry [99,104]. How does this regulation play out in tumor cells, which often lack proper control of the cell cycle machinery? In normal tissues, both hyperfusion and excessive fragmentation can result in aneuploidy, DNA damage, disrupted proliferation and loss of normal tissue homeostasis [104–106]. Based on this, it seems likely that oncogene induced mitochondrial fragmentation (or hyperfusion, in the case of some Myc-driven tumors [107]) can potentially promote further tumor progression through the induction of genomic instability, but also represents a potential roadblock for a tumor cell to rapidly progress through the cell cycle. In this regard, it is important to consider the additional benefits, highlighted above, that a tumor cell gains from oncogene induced changes in mitochondrial morphology, as well as the fact that the loss of tumor suppressors such as p53 and Rb, major regulators of cell cycle progression, can potentially override the regulation of cell cycle progression by the mitochondrial fusion and fission machinery.
Concluding thoughts
The progression from tumor initiation to metastasis requires the dysregulation of a number of normal physiological processes. Each tumor represents a unique set of genetic lesions that drive these changes, making it difficult to develop therapeutic approaches that can broadly impact the growing population of cancer patients. The identification of common mechanisms that underlie many of these physiological processes would thus have the potential to have a very positive impact. As mitochondrial are so central to so much of cellular physiology, they represent an emerging area of research in the cancer community. This review highlights just a few of the many physiological processes dysregulated in tumors that may be impacted by tumor-associated changes in mitochondrial shape. In each of the examples, mitochondrial shape will represent just one of the many factors influencing the process and will also be tightly integrated with cellular signaling, redox status, transcriptional regulation, and a number of other potential influences. In addition, mitochondrial shape changes will be impacting multiple processes simultaneously, each of which will to some extent be integrated with the others. The challenge of future research lies in untangling these inherent complexities. This will require the collaboration of scientists across a wide range of disciplines, from cell biologists to geneticists to systems biologists in order to fully comprehend how changes in mitochondrial shape contribute to the complex physiological changes that occur during tumor growth. This is an exciting time to be studying mitochondrial dynamics, as it has become increasingly clear that dynamic changes to mitochondrial shape, through fusion, fission and movement, have significant impact on their function. A clearer understanding of mitochondrial dynamics has the potential to greatly improve our ability to both understand and treat human cancer.
Supplementary Material
Highlights.
Mitochondrial fusion and fission can influence mitochondrial function.
Mitochondrial function underlies a number of physiological changes associated with cancer.
Mitochondrial dynamics represent a potential mechanism cancer cells use to promote tumorigenic growth.
Acknowledgments
DK is supported by grant #1R01CA200755-01 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.On respiratory impairment in cancer cells. 1956;124:269–270. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=13351639&retmode=ref&cmd=prlinks. [PubMed] [Google Scholar]
- 3.Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. 2017:1–19. doi: 10.1007/s00018-016-2451-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuznetsov A, Hermann M, Saks V, Hengster P, Margreiter R. The cell-type specificity of mitochondrial dynamics. The International Journal of Biochemistry & Cell Biology. 2009;41:1928–1939. doi: 10.1016/j.biocel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial Morphology Transitions and Functions: Implications for Retrograde Signaling? Am J Physiol Regul Integr Comp Physiol. 2013;304:R393–R406. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarron JG, Wilson C, Sandison ME, Olson ML, Girkin JM, Saunter C, et al. From structure to function: mitochondrial morphology, motion and shaping in vascular smooth muscle. J Vasc Res. 2013;50:357–371. doi: 10.1159/000353883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 2014;289:12005–12015. doi: 10.1074/jbc.M113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol Cell. 2015;57:537–551. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011 doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Liu T, Jin S, Wang X, Qu M, P.U.E. n et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. Embo J. 2011:1–17. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fröhlich C, Grabiger S, Schwefel D, Faelber K, Rosenbaum E, Mears J, et al. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. Embo J. 2013 doi: 10.1038/emboj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, et al. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA. 2013;110:E1342–51. doi: 10.1073/pnas.1300855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 2016 doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. 2016;353:aaf5549–aaf5549. doi: 10.1126/science.aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 22.Cho B, Cho HM, Kim HJ, Jeong J, Park SK, Hwang EM, et al. CDK5-dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp Mol Med. 2014;46:e105. doi: 10.1038/emm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015:1–12. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial Division Is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol Cell. 2015;57:521–536. doi: 10.1016/j.molcel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol Biol Cell. 2011;22:256–265. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu S, Wang P, Zhang H, Gong G, Cortes NGutierrez, Zhu W, et al. CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. Nat Commun. 2016;7:13189. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 28.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harder Z, Zunino R, Mcbride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Current Biology. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–1022. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. Embo J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karbowski M, Neutzner A. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007 doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho D, Nakamura T, Fang J, Cieplak P, Godzik A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009 doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003;134:333–344. doi: 10.1093/jb/mvg150. [DOI] [PubMed] [Google Scholar]
- 37.Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to Metabolism. Mol Cell. 2016;61:683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 38.Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–176. doi: 10.1016/s0014-5793(02)02985-x. [DOI] [PubMed] [Google Scholar]
- 39.Pernas L, Scorrano L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu Rev Physiol. 2015;78 doi: 10.1146/annurev-physiol-021115-105011. annurev–physiol–021115–105011. [DOI] [PubMed] [Google Scholar]
- 40.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Dorn GW. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gegg ME, Cooper JM, Schapira AHV, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE. 2009;4:e4756. doi: 10.1371/journal.pone.0004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacVicar T, Langer T. OPA1 processing in cell death and disease – the long and short of it. J Cell Sci. 2016:jcs.159186. doi: 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 44.Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diaz-Ruiz R, Uribe-Carvajal S, Devin A, Rigoulet M. Tumor cell energy metabolism and its common features with yeast metabolism. Biochim Biophys Acta. 2009;1796:252–265. doi: 10.1016/j.bbcan.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 49.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. Faseb J. 2012;26:2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoue-Yamauchi A, Oda H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem Bioph Res Co. 2012;421:81–85. doi: 10.1016/j.bbrc.2012.03.118. [DOI] [PubMed] [Google Scholar]
- 51.Arismendi-Morillo G. Electron microscopy morphology of the mitochondrial network in human cancer. Int J Biochem Cell Biol. 2009;41:2062–2068. doi: 10.1016/j.biocel.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013;32:4748–4757. doi: 10.1038/onc.2012.500. [DOI] [PubMed] [Google Scholar]
- 53.Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol. 2015;33C:111–118. doi: 10.1016/j.ceb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Westermann B. Bioenergetic role of mitochondrial fusion and fission, Biochim. Biophys Acta. 2012;1817:1833–1838. doi: 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 55.Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281–1242281. doi: 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nassar D, Blanpain C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu Rev Pathol. 2016;11:47–76. doi: 10.1146/annurev-pathol-012615-044438. [DOI] [PubMed] [Google Scholar]
- 57.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 58.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994 doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 59.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 61.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 62.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 63.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 64.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–137. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prieto J, León M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, et al. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. 2016;7:11124. doi: 10.1038/ncomms11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kasahara A, Cipolat S, Chen Y, Dorn GW, Scorrano L. Mitochondrial Fusion Directs Cardiomyocyte Differentiation via Calcineurin and Notch Signaling. Science. 2013;342:734–737. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- 68.Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. 2016;529:528–531. doi: 10.1038/nature16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell. 2016;19:232–247. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 70.Wanet A, Arnould T, Najimi M, Renard P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015;24:1957–1971. doi: 10.1089/scd.2015.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 72.Hoppins S, Nunnari J. Mitochondrial Dynamics and Apoptosis—the ER Connection. Science. 2012:1–4. doi: 10.1126/science.1224709. [DOI] [PubMed] [Google Scholar]
- 73.Otera H, Mihara K. Mitochondrial dynamics: functional link with apoptosis. Int J Cell Biol. 2012;2012:821676–10. doi: 10.1155/2012/821676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scorrano L. Keeping mitochondria in shape: a matter of life and death. Eur J Clin Invest. 2013;43:886–893. doi: 10.1111/eci.12135. [DOI] [PubMed] [Google Scholar]
- 75.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 77.Cereghetti GM, Costa V, Scorrano L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010;17:1785–1794. doi: 10.1038/cdd.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 79.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. 2016;212 doi: 10.1083/jcb.201508099. jcb.201508099–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Germain M, Mathai JP, Mcbride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. Embo J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Große L, Wurm CA, Brüser C, Neumann D, Jans DC, Jakobs S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. Embo J. 2016 doi: 10.15252/embj.201592789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Renault TT, Floros KV, Elkholi R, Corrigan KA, Kushnareva Y, Wieder SY, et al. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol Cell. 2015;57:69–82. doi: 10.1016/j.molcel.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 88.Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 89.Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, et al. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 91.Senft D, Ronai ZA. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. 2016;39:43–52. doi: 10.1016/j.ceb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–4824. doi: 10.1038/onc.2012.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang B, Wang J, Huang Z, Wei P, Liu Y, Hao J, et al. Aberrantly upregulated TRAP1 is required for tumorigenesis of breast cancer. Oncotarget. 2015;6:44495–44508. doi: 10.18632/oncotarget.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Han XJ, Yang ZJ, Jiang LP, Wei YF, Liao MF, Qian Y, et al. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int J Oncol. 2015;46:691–700. doi: 10.3892/ijo.2014.2781. [DOI] [PubMed] [Google Scholar]
- 95.Yin M, Lu Q, Liu X, Wang T, Liu Y, Chen L. Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ROCK1 pathway. Biochem Bioph Res Co. 2016 doi: 10.1016/j.bbrc.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 96.Jung J-U, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, et al. NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr Biol. 2016;26:3288–3302. doi: 10.1016/j.cub.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ferreira-da-Silva A, Valacca C, Rios E, Pópulo H, Soares P, Sobrinho-Simões M, et al. Mitochondrial Dynamics Protein Drp1 Is Overexpressed in Oncocytic Thyroid Tumors and Regulates Cancer Cell Migration. PLoS ONE. 2015;10:e0122308. doi: 10.1371/journal.pone.0122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA. 2015;112:8638–8643. doi: 10.1073/pnas.1500722112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mitra K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. Bioessays. 2013;35:955–964. doi: 10.1002/bies.201300011. [DOI] [PubMed] [Google Scholar]
- 101.Kashatus DF, Lim K-H, Brady DC, Pershing NLK, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108–1115. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zunino R, Braschi E, Xu L, Mcbride HM. Translocation of SenP5 from the Nucleoli to the Mitochondria Modulates DRP1-dependent Fission during Mitosis. J Biol Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell. 2011;22:1207–1216. doi: 10.1091/mbc.E10-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mitra K, Rikhy R, Lilly M, Lippincott-Schwartz J. DRP1-dependent mitochondrial fission initiates follicle cell differentiation during Drosophila oogenesis. J Cell Biol. 2012;197:487–497. doi: 10.1083/jcb.201110058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Houten BV. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci. 2012;125:5745–5757. doi: 10.1242/jcs.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 107.von Eyss B, Jaenicke LA, Kortlever RM, Royla N, Wiese KE, Letschert S, et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell. 2015;28:743–757. doi: 10.1016/j.ccell.2015.10.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.