

Abstract
CRISPR-Cas systems depend on the Cas1-Cas2 integrase to capture and integrate short foreign DNA fragments into the CRISPR locus, enabling adaptation to new viruses. We present crystal structures of Cas1-Cas2 bound to both donor and target DNA in intermediate and product integration complexes, as well as a cryo-electron microscopy structure of the full CRISPR locus integration complex including the accessory protein Integration Host Factor (IHF). The structures show unexpectedly that indirect sequence recognition dictates integration site selection by favoring deformation of the repeat and the flanking sequences. IHF binding bends the DNA sharply, bringing an upstream recognition motif into contact with Cas1 to increase both the specificity and efficiency of integration. These results explain how the Cas1-Cas2 CRISPR integrase recognizes a sequence-dependent DNA structure to ensure site-selective CRISPR array expansion during the initial step of bacterial adaptive immunity.
Main Text
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated) bacterial adaptive immune systems store fragments of viral DNA in the CRISPR array, a genomic locus comprising direct sequence repeats separated by virally-derived spacer sequences, both of approximately 20–50 base pairs in length (1–4). In most systems, a transcriptional promoter located in an AT-rich leader sequence preceding the first CRISPR repeat gives rise to precursor CRISPR transcripts that are processed and used to recognize viral nucleic acids by base pairing with complementary sequences. Bacteria acquire immunity to new viruses when the CRISPR integrase, a heterohexameric complex of four Cas1 and two Cas2 proteins, inserts new viral DNA at the first CRISPR repeat following the leader sequence (5–7). Integration involves nucleophilic attack by the 3′ ends of the viral DNA fragment, called a protospacer, at each end of the repeat (Fig. 1a) (7). Half-site intermediates form when one of the two protospacer DNA ends attacks the CRISPR locus integration site, and can either progress to full-site integration products or be disintegrated, leaving the target sequence intact (7, 8).
Fig. 1. Half-site binding by Cas1-Cas2.

(A) Cartoon of steps of integration by Cas1-Cas2. Crystallography substrates are shown next to the corresponding reaction intermediate, with nucleotide lengths indicated. Red stars represent integration events. (B) Cartoon and surface representations of half-site substrate bound by Cas1-Cas2. DNA is colored as in (A). A substrate schematic is shown above, with disordered regions shown as dashed lines. (C) Close-up of backbone interactions between Cas1-Cas2 and half-site repeat DNA. Polar contacts are shown as dotted lines. (D) Hydroxyl radical footprinting of radiolabeled half-site DNA. Input is untreated DNA. The substrates are shown above the gel, with the radiolabel indicated with a red circle. Regions of the gel corresponding to the leader, repeat, spacer, and protospacer (pspacer) are indicated alongside the gel. The inverted repeat regions of the repeat are shown as boxes.
To ensure effective acquisition of new immunity and avoid deleterious insertions into the genome, integration by Cas1-Cas2 must be highly specific for the CRISPR locus. In the type I CRISPR system from E. coli, acquisition requires sequences spanning the leader-repeat junction as well as an inverted repeat motif in the repeat (8–11). IHF (Integration Host Factor), a histone-like protein, binds in the leader and assists in recruiting Cas1-Cas2 to the leader-proximal repeat, possibly involving a secondary upstream binding site (10, 12, 13). The mechanism by which Cas1-Cas2 recognizes these sequences is not yet known.
Here we present structures of the Cas1-Cas2 CRISPR integrase bound to both substrate and target DNA in intermediate and product integration states. We also present a structure of the entire natural integration complex including Cas1-Cas2, the DNA substrate and a 130-base pair DNA target sequence in complex with IHF. These structures show how specificity for the CRISPR repeat relies on target DNA deformation to allow access to both Cas1 integrase active sites. In addition to recruiting a secondary recognition site, IHF sharply bends the target DNA adjacent to the integration site, favoring integrase binding to this locus and thereby suppressing off-target integration. These results suggest an unexpected mechanism of target recognition with implications for the engineering of the CRISPR integrase as a genome-tagging tool.
Target binding in the half-site intermediate
To determine the mechanism by which Cas1-Cas2 recognizes its target sequence, we crystallized the integrase bound to DNA substrates representing a half-site integration intermediate as well as the full-site integration product (Fig. 1a). The full-site product mimic, which we term the pseudo-full-site substrate, was designed with a break in the middle of the protospacer to allow Cas1-Cas2 to access the repeat (Fig. 1a). Both substrates bound to Cas1-Cas2 with high affinity (Fig. S1). The half-site-bound structure, refined at 3.9 Å resolution, revealed an overall complex architecture similar to that of the previously-solved protospacer-bound structures (Fig. 1b, Fig. S2, Table S1) (14, 15). A Cas2 dimer sits at the center of two Cas1 dimers, with the protospacer DNA stretching across the flat back of the complex. The first 18 base pairs of the repeat sequence bind across a central channel formed by Cas2 and the non-catalytic Cas1 monomers, with the leader-repeat junction positioned across a Cas1 active site (Fig. 1b; Fig. S3a,b). Seven nucleotides of the spacer-proximal repeat are unresolved, while the repeat-spacer junction binds at the distal Cas1 active site. Basic residues on both Cas2 (K38, R40) and the non-catalytic Cas1 monomers (K12, K259) are positioned to contact the phosphate backbone of the mid-repeat DNA (Fig. 1b, c) (15). Charge-swap mutations of these residues reduce or eliminate acquisition of new spacers in vivo, confirming their importance for the CRISPR integration reaction (Fig. S4a).
Although earlier work suggested that inverted sequence motifs in the repeat might form a cruciform structure during target recognition, our structure shows that the center of the repeat remains a canonical duplex at this intermediate stage of integration (7, 16, 17). Although the inverted repeat sequences are critical for spacer acquisition, we found no evidence of sequence-specific contacts in these motifs (Fig. 1c) (9, 11, 18). Contacts between the mid-repeat DNA and the integrase proteins are limited to nonspecific backbone interactions, with no regions of Cas1 or Cas2 positioned to interrogate either the major or minor groove. To test for contacts in solution, we performed hydroxyl radical footprinting of the half-site substrate bound by the complex (Fig. 1d). Protection of the backbone is clearly seen in the protospacer, including in the single-stranded end where the DNA binds in a channel of Cas1. Only weak protection occurs near the ends of the repeat on the non-integrated target strand and largely does not overlap with the inverted repeats. Several hypersensitive nucleotides are apparent at the beginning of the second inverted repeat even in the absence of protein, suggesting that these nucleotides exhibit increased flexibility or a distorted conformation in solution. Although direct sequence readout could involve a distinct but transient binding mode prior to half-site integration, our data suggest that integrase recognition of the repeat sequence likely relies on a mechanism other than base-specific hydrogen-bonding.
Leader sequence recognition in the pseudo-full-site structure
The pseudo-full-site-bound structure was solved at 2.9 Å and reveals more details of the interaction between Cas1 and the target DNA (Table S1). The nucleotides at both the leader-adjacent and spacer-adjacent integration sites are clearly resolved, while the middle of the repeat was disordered, suggesting that the repeat disengages from Cas2 following full integration (Fig 2a, Fig. S3c, d). Previous crystal structures suggested that the Cas1 α-helix 7 might interact with target DNA, and we indeed observe insertion of this helix into the minor groove of both the leader and spacer regions of the target DNA (Fig. 2b) (14). The terminal residues of the leader sequence contribute to integration efficiency, and our structure reveals that several residues make hydrogen bonds with the minor-groove face of leader bases (8, 18–20). Cas1 R146 hydrogen bonds with A-3 and T-4 and is essential for integration in vivo, suggesting that it may also stabilize binding through interactions with the phosphate backbone (Fig. 2b, c, Fig. S4b). Cas1 S143 interacts with T-3 of the non-integrated target strand, though it is dispensable for in vivo activity (Fig. 2b, c, Fig. S4b).
Fig. 2. Pseudo-full-site binding by Cas1-Cas2.

(A) Overview of pseudo-full-site substrate binding by Cas1-Cas2. In the second view, the expected path of the disordered DNA is shown as dashed lines. A schematic of the substrate is shown, with the disordered region as dashed lines. (B) A view of minor groove insertion by α-helix 7. Dotted lines in close-up show polar contacts. The sequence of the leader-repeat junction and residue numbering are shown. (C) Agarose gel of a representative in vivo acquisition assay with indicated Cas1 mutants and wild-type Cas2. Acquisition results in expansion of the CRISPR array, which is visible as larger bands above the parental locus. The H208A active-site mutant is used as a negative control.
Integration requires DNA distortion
Notably, both the half-site and the pseudo-full-site structures reveal significant distortion of the target DNA. The DNA exhibits a sharp kink at both integration sites, with the bases on either side of the leader-repeat and repeat-spacer junction forming a nearly 30° angle (Fig. 3a). The repeat-spacer junction of the half-site substrate exhibits a similar kink, which indicates that the distortion occurs not as a result of integration but instead upon Cas1-Cas2 binding to the target. Binding across the Cas2 dimer surface also forces a bend in the repeat, mostly localized to the region directly over Cas2 (Fig. 3b).
Fig. 3. Integration involves DNA distortion.

(A) View of kink introduced at leader-repeat junction. The kink in the pseudo-full-site structure is highlighted with a dashed line showing the central axis of the DNA. The inset shows the bases before and after the integration site. Part of the backbone is omitted for clarity, and the angle formed by adjacent bases is shown with dashed lines. (B) Representation of the half-site repeat bending over the Cas2 dimer. The DNA trajectory is fit with a dashed line to show the localized bending. (C) Modeled B-form DNA fails to connect resolved regions of the half-site repeat. Modeled bases are shown with bases as sticks rather than rings. The (+) strand and (−) strand are shown in dark and light blue, respectively, to show that the modeled DNA does not properly join with the spacer-proximal DNA. (D) Urea-PAGE gel of integration assay with radiolabeled protospacer. The substrate and expected products are shown as cartoons with the radiolabel represented with a red circle. Their expected positions are indicated. The repeat sequences are shown above, with the mutated regions highlighted in red. Timepoints were taken at 0, 1, 5, 15, and 30 minutes. (E) Urea-PAGE gel of second-site integration assay using mutant repeat sequences. The substrate and expected product are schematized with the radiolabel indicated with a red circle, and their expected positions are indicated on the gel. The mutant repeats are the same as in (D). Timepoints were taken at 0, .5, 1, 2, 10, 30, and 60 mintues. (F) Integration assay with radiolabeled protospacer and mismatched repeats. Mismatches were introduced in the region of the repeat highlighted in red in the wild-type sequence above the gel. The positions of the mismatches are schematized above each time course, with the red circles representing the highlighted mid-repeat nucleotides. Timepoints were taken at 0, 1, 5, 15, and 30 minutes.
Both structures show that the repeat must also undergo twist deformation to be properly positioned in both active sites. Modeling B-form DNA into the disordered regions of the repeat results in the incorrect backbone being positioned in the spacer-side active site (Fig. 3c). Connecting the resolved regions of DNA requires that the missing region be under-wound by approximately one third of a turn relative to canonical B-form DNA. It is unclear how this distortion is distributed across the disordered region, and the lack of order might indicate that the DNA adopts a range of conformations to accommodate the strain. The required bending and under-winding of the repeat, together with the lack of sequence-specific contacts in the repeat, suggests that Cas1-Cas2 recognize the target through indirect readout based on the repeat’s sequence-dependent deformability. The poly-G stretches in the inverted repeat motifs in particular may facilitate the adoption of strained conformations to allow binding across both active sites (21, 22).
To investigate whether these motifs are required for the DNA to be coordinated at opposing active sites, we performed in vitro integration assays using repeats with mutations known to prevent acquisition in vivo (Fig. 3d) (9). The mutations did not significantly affect leader-side integration, but they prevented integration at the repeat-spacer junction. Half-site substrates bearing the same mutations were unable to be converted to full-site products, despite supporting binding and disintegration, while wild-type half-sites were readily converted to full-site products (Fig. 3e, Fig. S5). These results confirm that the repeat sequence is important not for binding and recruitment of Cas1-Cas2 but instead for determining the ability of the target to reach the spacer-side active site.
To further investigate the importance of DNA deformation for spacer-side integration, we performed integration assays using targets with single- or double-base mismatches between the inverted repeats (Fig. 3f). The introduction of a mismatch is expected to disrupt the DNA duplex and generate a flexible hinge in the middle of the repeat. Mismatches immediately before the second inverted repeat increased the rate of spacer-side integration, indicating that increasing the deformability of the repeat at specific sites enhances full-site integration. These data support the model that sequence-dependent distortion is necessary for recognition and integration at the repeat. Notably, both G→C and G→A transitions in the inverted repeats prevented full-site integration, suggesting that the necessary deformation of the repeat depends on factors other than or in addition to GC content, such as specific purine-pyrimidine steps in the region where mismatches favor integration.
Active site geometry
To better understand Cas1 active site geometry, we grew pseudo-full-site-bound crystals in the presence of Ni2+, which does not support catalysis but should allow for Mg2+-like coordination geometry, and solved the structure to 3.3 Å resolution (Fig. S6, Table S1). We observed density and peaks in the anomalous difference map for a single Ni2+ located at each of the four Cas1 active sites, though the metals are at lower occupancy in the substrate-engaged active sites, potentially due to lower solvent accessibility at these sites (Fig. 4a, Fig. S7a–d). At the non-catalytic active sites, the metal is coordinated by H208 and D221, as previously described (14, 23). In the post-integration active sites, the phosphate of the newly-formed phosphodiester bond bridging the protospacer and the repeat coordinates the metal, and the free 3′ OH of the cleaved leader or spacer is in close proximity. E141, which has been annotated as a metal-coordinating residue, had poor side-chain density in all monomers and appeared to be outside the range of a favorable interaction with the metal (Fig. S7e, f). The absolute requirement of E141 for activity suggests that it may play another role in catalysis, perhaps acting as a proton donor for the leaving 3′ hydroxyl (6, 23).
Fig. 4. Full-site integration requires a basic clamp around the active site.
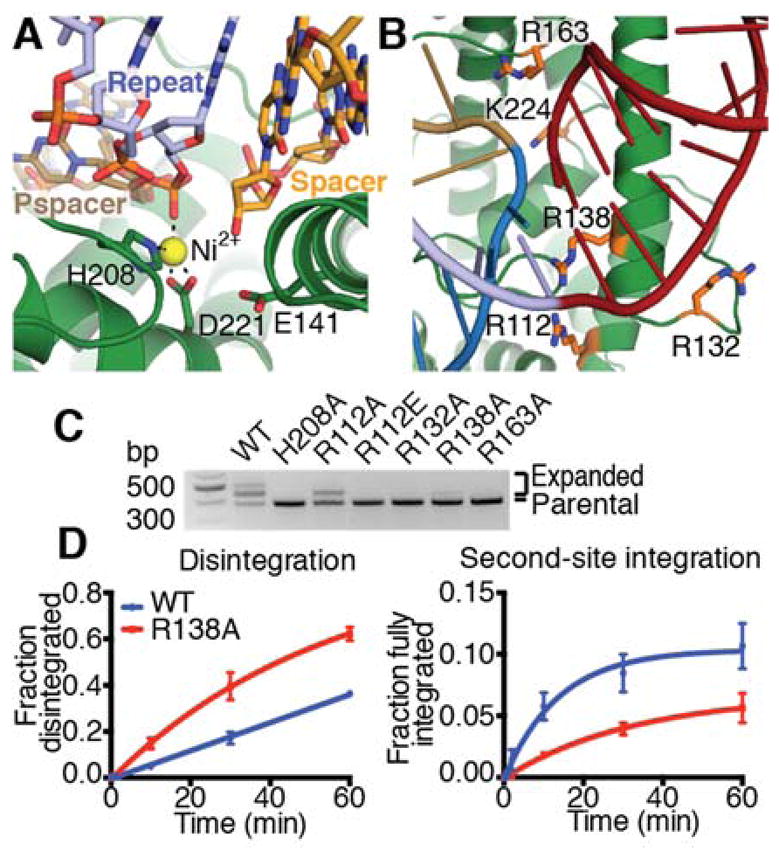
(A) Metal coordination in the spacer-side active site. Active site residues, repeat, spacer, and protospacer (pspacer) are labeled, and coordination is shown as dotted lines. (B) View of basic residues surrounding leader-repeat junction. Basic residues in close proximity to the target DNA backbone on either side of the integration site are shown as sticks and colored orange. (C) Agarose gel of in vivo acquisition assay with indicated Cas1 mutants. H208A Cas1 is used as a negative control. (D) Quantification of disintegration and second-site integration time-course assays by wild-type and R138A Cas1. Mean and standard deviation of three independent experiments are plotted. Representative gels are shown in Figure S8.
In vivo CRISPR integration assays to test the role of basic residues in the integrase that might contact either side of the DNA integration site showed that alanine mutants of Cas1 R132, R138, and R163 eliminate or nearly eliminate acquisition (Fig. 4b, c). The R112A Cas1 mutant maintained some activity, but the R112E mutation prevented acquisition. The importance of all of these residues may reflect the need for a strong network of favorable contacts to capture the DNA in a strained conformation. To test this hypothesis, we performed disintegration and second-site integration assays with an R138A Cas1 mutant. This mutation reduced the rate of second-site integration by 50%, but R138A Cas1 exhibited wild-type-like binding and enhanced disintegration activity, likely due to faster product release or the reduced rate of the competing forward reaction (Fig. 4d, Fig. S8). These data confirm that R138 is dispensable for catalysis but is important for trapping the DNA at the distal active site.
IHF sharply bends the integration locus and recruits an upstream binding site
To investigate the mechanism by which IHF recruits Cas1-Cas2 to the leader-proximal repeat, we purified the Cas1-Cas2 and IHF bound to a half-site substrate with an extended leader sequence (Fig. S9). Negatively stained samples were used to generate an initial low-resolution reconstruction that showed additional density attached to Cas1-Cas2 module that we could assigned to IHF (Fig. S10). We then used cryo-EM to solve the structure at a final resolution of 3.6 Å (Fig. S11–S13). We generated a complete model of the Cas1-Cas2-IHF-DNA holo-complex by first fitting the crystal structure of half-site-bound Cas1-Cas2 solved in this work and the published atomic model of the IHF module (PDB:1IHF) into the cryo-EM map, followed by manually rebuilding the models to fit the density. The DNA substrates were manually built ab initio and the resulting complete model was improved by real-space refinement (Fig. S14).
Compared to the holo-complex, Cas1-Cas2 and the repeat are overall in the same conformation as in the half-site crystal structure, and disorder of the spacer end of the complex again prevented building the DNA across to the distal active site. The structure shows how IHF binds the leader immediately upstream of Cas1-Cas2 and induces a 180° turn in the DNA, directing it back toward the Cas1-Cas2 complex (Fig. 5a) (24). The upstream binding motif interacts with one of the non-catalytic Cas1 protomers, with the loop between α6 and α7 inserting into the minor groove. R117 and Q136 interact with the phosphate backbone, and R131 and R132 are positioned to hydrogen bond with the minor groove face of bases in the conserved recognition region (Fig. 5b). R132 is essential for integration in vivo, but it is difficult to assess the importance of its role in upstream readout given that R132 on the catalytic Cas1 protomer is implicated in the basic clamp described above (Fig. 4b, 5c). R131 and Q136 also contribute significantly to DNA binding, as alanine mutations of either reduce acquisition. Mutation of the conserved upstream sequence as a block eliminated acquisition, as previously noted, and single nucleotide mutations revealed G-53, which is recognized by R131, as particularly important for recognition (Fig. 5d) (12).
Fig. 5. Upstream sequence recognition by Cas1.

(A) Cryo-EM structure of Cas1-Cas2 with IHF and extended leader. The atomic model is shown as a cartoon, and the electron density is shown as a transparent surface. Density is shown using an 8 σ threshold. (B) View of upstream sequence readout by Cas1. Electron density is shown as a transparent surface using an 8 σ threshold. Relevant Cas1 residues are labeled. Bases in the conserved recognition sequence are labeled, with numbering such that the final residue of the leader is −1. (C) Acquisition assay with wild-type Cas2 and the indicated Cas1 mutants. H208A Cas1 is used as a negative control. (D) Acquisition assay with wild-type proteins and the noted mutations in the leader sequence. Single-nucleotide mutations in the conserved recognition region are highlighted in red. “IHF flip” denotes the leader sequence with the IHF binding sequence reversed in place. H208A Cas1 is used as a negative control. (E) Integration assay with radiolabeled protospacer and targets with variable leaders. “Upstream mutant” substrate has the “GGTAG→CCATC” mutation in the conserved recognition motif, while the “Truncated” substrate begins at residue −46, after the recognition motif. Time points were taken at 0, 1, 5, 15, and 30 minutes. (F) Quantification of integration assays with limiting protospacer and limiting target. Mean and standard deviation of three independent experiments are shown. A representative gel of the limiting target experiment is shown in Figure S14.
To determine how much the IHF-dependent recruitment of Cas1-Cas2 depends on upstream sequence recognition as opposed to nonspecific stabilizing interactions, we performed in vitro integration assays with targets containing leaders with mutations in the upstream binding region or leaders truncated prior to the upstream interaction region (Fig. 5e). Mutations in the binding site reduced the rate of leader-side integration three-fold when target is limiting (Fig. 5f, Fig. S15). The rate effect is masked when the target is in excess over protospacer-bound complex, but a higher level of off-target integration is observed (Fig. S15). The increased importance of the upstream sequence for in vivo acquisition suggests that it may be important for initial identification of the target in context of genomic DNA, while it is dispensable when the correct target is saturating and no competitor is present. Truncation of the leader had a much more significant effect, with the rate of leader-side integration reduced ~100-fold when target was limiting (Fig. 5f). Spacer-side integration was also affected by the truncation, as indicated by the appearance of a second band consistent with misplaced integration within the repeat (Fig. 5e). These results show that nonspecific interactions with the leader DNA are critical for robust Cas1-Cas2 activity and specificity, while the sequence-specific interactions aid in efficient recognition.
Suppression of off-target integration by IHF
We also investigated whether IHF contributes to Cas1-Cas2 recruitment by mechanisms other than juxtaposition of the upstream binding site. Our structure reveals that Cas1 and the alpha protomer of IHF (IHF-α) are in close proximity, with a solvent-inaccessible surface of 200 Å2 between the two proteins (Fig. 6a). However, there is no significant continuous electron density between the proteins. Mutations of IHF-α residues near the interface with Cas1 identified E10 and D14 as important for acquisition (Fig. 6b). These residues might interact favorably with Cas1 R131 or R132 to aid in Cas1 recruitment. However, reversing the orientation of the IHF binding site in the leader, which should position IHF-β rather than IHF-α to interact with Cas1, did not severely impact acquisition, suggesting that any interaction that occurs is not highly specific (Fig. 5d).
Fig. 6. Interactions between Cas1 and IHF.

(A) Surface and cartoon representations of the interface between Cas1 and IHF-α. In the inset, residues at the interaction surface are shown as sticks, and residues of interest are labeled. Electron density is shown as a surface with an 8 σ threshold. (B) Acquisition assay with wild-type Cas1 and Cas2 and the indicated IHF-α mutants. H208A Cas1 is used as a negative control. (C) Integration assays with radiolabeled protospacer and targets with truncated leaders. IHF is included unless otherwise noted. Mutant substrates have 1, 2, or 5 base pairs inserted between the IHF recognition sequence and the Cas1 recognition sequence of the leader. Time points were taken at 0, 1, 5, 15, and 30 minutes. (D) Quantification of leader-side integration with radiolabeled protospacer and truncated targets. Mean and standard deviation of three independent replicates are shown.
To further investigate the role of IHF, we performed integration assays with and without IHF, using a truncated leader to prevent contribution from upstream interactions (Fig. 6c). In the absence of IHF, off-target integration occurs in the leader, demonstrating a role for IHF in limiting spurious integration events. Shifting the IHF binding site one to five nucleotides farther away from the leader-repeat junction led to a modest decrease in the efficiency of leader-side integration, though the site of integration was unaltered (Fig. 6c, d). This supports the model that contacts between IHF and Cas1 contribute to specific and efficient CRISPR locus expansion, though recruitment of the upstream binding site appears to be the more important contribution.
Conclusions
These data show that the type I Cas1-Cas2 from E. coli relies heavily on active site positioning and structural features of the DNA, rather than direct sequence recognition, to localize DNA integration to the CRISPR locus (Fig. S16). The ability of the DNA substrate duplex to access both Cas1 active sites regulates recognition of the CRISPR repeat, with the GC-rich inverted repeats allowing for twist deformation while the mid-repeat sequence acts as a hinge, and IHF aids in recruitment at the leader by providing a secondary binding surface for the complex. The lack of direct sequence recognition might reflect the evolutionary origins of Cas1 as a more promiscuous transposase (25–27). DNA target site bending is a common feature in transposases and integrases, where it disfavors the disintegration reaction by ejecting DNA from the integrase active sites once integration is achieved (28, 29). While Cas1-Cas2 may use a similar mechanism, as suggested by the displacement of the mid-repeat upon full-site integration, CRISPR systems appear to have exploited the requirement for DNA bending to provide sequence specificity for the integration reaction. The role played by IHF also represents a surprising variation on a feature sometimes seen in transposases. In both λ and μ phage mobilization pathways, IHF, or the related protein HU, are involved in bringing recognition sequences on the viral DNA into contact with the integrase (29, 30). Notably, in the phage pathways, IHF aids in the recognition of donor DNA, while in CRISPR acquisition it is important for recognition of the target DNA, highlighting the shift in substrate selectivity from donor to target that was essential for the “domestication” of Cas1 for use in immunity (25, 26).
The unique substrate preferences of the CRISPR integrase could make it useful as a molecular recording device for barcoding genomes or generating locus-specific sequence insertions (31). Bacterial transposases including Tn5 and MuA provide robust tools for DNA tagging, insertion and deletion, but they are promiscuous in their target selection and require sequence-specific interactions with the donor DNA that limit their use in some systems (32–34). While the CRISPR integrase shares the reaction chemistry of other transposases, its unique substrate sequence independence coupled with its selectivity for target DNA sequences may enable a complementary set of applications. The architecture of the CRISPR integration complexes presented here suggests that subtle adjustment of the distance between Cas1 active sites could reprogram the CRISPR integrase to recognize different integration target sites. Changes in integrase architecture could thereby be exploited for genome tagging applications and may also explain natural divergence of CRISPR arrays in bacteria.
Supplementary Material
Acknowledgments
We thank G. Meigs and the Advanced Light Source 8.3.1 beamline staff and D. Tzanko, A. Gonzalez and the Stanford Synchrotron Radiation Light Source 9-2 beamline staff for assistance with data collection, and A. East-Seletsky for input on the manuscript. Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599 and Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The EM data was collected in the HHMI EM facility located in UC Berkeley. We thank D. B. Toso and P. Grob for expert electron microscopy assistance, A. Chintangal for computational support, and members of Nogales lab for helpful discussions in EM data processing. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). This project was funded by US National Science Foundation grant no. 1244557 (J.A.D.) and National Institute of General Medicine Sciences grant no. 1P50GM102706-01 (J.H. Cate). A.V.W. and K.W.D. are supported by a US National Science Foundation Graduate Research Fellowship, and G.J.K. is funded by Howard Hughes Medical Institute. J.A.D. and E.N. are investigators of the Howard Hughes Medical Institute and a member of the Center for RNA Systems Biology. Atomic coordinates and structure factors for the reported crystal structures have been deposited at the Protein Data Bank under accession codes 5VVJ (half-site-bound) 5VVK (pseudo-full-site-bound) and 5VVL (pseudo-full-site-bound with Ni2+). The cryo-EM structure and map have been deposited at the Protein Data Bank under accession code 5WFE and the Electron Microscopy Data Bank under accession code EMD-8827. A patent was filed by the University of California for the use of Cas1-Cas2 for integrating DNA into genomes. J.A.D. is a cofounder and Scientific Advisory Board member of Caribou Biosciences and Intellia Therapeutics and a cofounder of Editas Medicine, all of which develop CRISPR-based technologies. Correspondence and requests for materials should be addressed to J.A.D. (doudna@berkeley.edu).
Footnotes
References
- 1.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 2.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology (Reading, Engl) 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 3.Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology (Reading, Engl) 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 4.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nuñez JK, et al. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat Struct Mol Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nuñez JK, Lee ASY, Engelman A, Doudna JA. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015:1–17. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rollie C, Schneider S, Brinkmann AS, Bolt EL, White MF. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. eLife. 2015;4:e08716. doi: 10.7554/eLife.08716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goren MG, et al. Repeat Size Determination by Two Molecular Rulers in the Type I-E CRISPR Array. CellReports. 2016;16:2811–2818. doi: 10.1016/j.celrep.2016.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuñez JK, Bai L, Harrington LB, Hinder TL, Doudna JA. CRISPR Immunological Memory Requires a Host Factor for Specificity. Molecular Cell. 2016;62:824–833. doi: 10.1016/j.molcel.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 11.Moch C, Fromant M, Blanquet S, Plateau P. DNA binding specificities of Escherichia coli Cas1–Cas2 integrase drive its recruitment at the CRISPR locus. Nucleic Acids Res. 2016:gkw1309–10. doi: 10.1093/nar/gkw1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoganand KNR, Sivathanu R, Nimkar S, Anand B. Asymmetric positioning of Cas1–2 complex and Integration Host Factor induced DNA bending guide the unidirectional homing of protospacer in CRISPR-Cas type I-E system. Nucleic Acids Res. 2017;45:367–381. doi: 10.1093/nar/gkw1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fagerlund RD, et al. Spacer capture and integration by a type I-F Cas1–Cas2–3 CRISPR adaptation complex. Proc Natl Acad Sci. 2017;23:201618421–17. doi: 10.1073/pnas.1618421114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nuñez JK, Harrington LB, Kranzusch PJ, Engelman AN, Doudna JA. Foreign DNA capture during CRISPR–Cas adaptive immunity. Nature. 2015:1–13. doi: 10.1038/nature15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, et al. Structural and Mechanistic Basis of PAM-Dependent Spacer Acquisition in CRISPR-Cas Systems. Cell. 2015:1–27. doi: 10.1016/j.cell.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Babu M, et al. A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Molecular Microbiology. 2010;79:484–502. doi: 10.1111/j.1365-2958.2010.07465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arslan Z, Hermanns V, Wurm R, Wagner R, Pul U. Detection and characterization of spacer integration intermediates in type I-E CRISPR-Cas system. Nucleic Acids Res. 2014;42:7884–7893. doi: 10.1093/nar/gku510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang R, Li M, Gong L, Hu S, Xiang H. DNA motifs determining the accuracy of repeat duplication during CRISPR adaptation in Haloarcula hispanica. Nucleic Acids Res. 2016:gkw260–12. doi: 10.1093/nar/gkw260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGinn J, Marraffini LA. CRISPR-Cas Systems Optimize Their Immune Response by Specifying the Site of Spacer Integration. Molecular Cell. 2016:1–20. doi: 10.1016/j.molcel.2016.08.038. [DOI] [PMC free article] [PubMed]
- 20.Wright AV, Doudna JA. Protecting genome integrity during CRISPR immune adaptation. Nat Struct Mol Biol. 2016;23:876–883. doi: 10.1038/nsmb.3289. [DOI] [PubMed] [Google Scholar]
- 21.Olson WK, Gorin AA, Lu X, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein–DNA crystal complexes. Proc Natl Acad Sci. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardiner EJ, Hunter CA, Packer MJ, Palmer DS, Willett P. Sequence-dependent DNA Structure: A Database of Octamer Structural Parameters. J Mol Biol. 2003;332:1025–1035. doi: 10.1016/j.jmb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Wiedenheft B, et al. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure. 2009;17:904–912. doi: 10.1016/j.str.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 24.Rice PA, Yang SW, Mizuuchi K, Nash HA. Crystal Structure of an IHF-DNA Complex: A Protein-Induced DNA U-Turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 25.Béguin P, Charpin N, Koonin EV, Forterre P, Krupovic M. Casposon integration shows strong target site preference and recapitulates protospacer integration by CRISPR-Cas systems. Nucleic Acids Res. 2016:gkw821–10. doi: 10.1093/nar/gkw821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krupovic M, Makarova KS, Forterre P, Prangishvili D, Koonin EV. Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol. 2014;12:36. doi: 10.1186/1741-7007-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hickman AB, Dyda F. The casposon-encoded Cas1 protein from Aciduliprofundum boonei is a DNA integrase that generates target site duplications. Nucleic Acids Res. 2015;43:10576–10587. doi: 10.1093/nar/gkv1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468:326–329. doi: 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montaño SP, Pigli YZ, Rice PA. The Mu transpososome structure sheds light on DDE recombinase evolution. Nature. 2012;491:413–417. doi: 10.1038/nature11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laxmikanthan G, et al. Structure of a Holliday junction complex reveals mechanisms governing a highly regulated DNA transaction. eLife. 2016;5:1–23. doi: 10.7554/eLife.14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shipman SL, Nivala J, Macklis JD, Church GM. Molecular recordings by directed CRISPR spacer acquisition. Science. 2016:1–16. doi: 10.1126/science.aaf1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goryshin IY, Jendrisak J, Hoffman LM, Meis R, Reznikoff WS. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat Biotechnol. 1999;18:97–100. doi: 10.1038/72017. [DOI] [PubMed] [Google Scholar]
- 33.Nadler DC, Morgan SA, Flamholz A, Kortright KE, Savage DF. Rapid construction of metabolite biosensors using domain-insertion profiling. Nat Commun. 2016;7:1–11. doi: 10.1038/ncomms12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adey A, Shendure J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012;22:1139–1143. doi: 10.1101/gr.136242.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabsch W, XDS Acta Cryst. 2010;D66:125–132. doi: 10.1107/S0907444909047337. 1–8 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Cryst. 2013;D69:1204–1214. doi: 10.1107/S0907444913000061. 1–11 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diederichs K, Karplus PA. Better models by discarding data? Acta Cryst. 2013;D69:1215–1222. doi: 10.1107/S0907444913001121. 1–8 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lander GC, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang G, et al. EMAN2: An extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Shaikh TR, et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat Protoc. 2008;3:1941–1974. doi: 10.1038/nprot.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mastronarde DN. SerialEM: A Program for Automated Tilt Series Acquisition on Tecnai Microscopes Using Prediction of Specimen Position. 2003:1–2. https://doi.org/10.1017/S1431927603445911.
- 42.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimanius D, Forsberg BO, Scheres SH, Lindahl E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife. 2016;5:19. doi: 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. 1–17 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. 1–16 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crys. 2012;D68:352–367. doi: 10.1107/S0907444912001308. 1–16 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng G, Lu XJ, Olson WK. Web 3DNA--a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Research. 2009;37:W240–W246. doi: 10.1093/nar/gkp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carey M, Smale ST. Hydroxyl-Radical Footprinting. Cold Spring Harbor Protocols. 2007 doi: 10.1101/pdb.prot4810. pdb.prot4810. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.