

Abstract
Interactions between lung epithelium and interstitial fibroblasts are increasingly recognized as playing a major role in the progression of several lung pathologies, including cancer. Three-dimensional in vitro co-culture systems offer tissue-relevant platforms to study the signaling interplay between diseased and healthy cell types. Such systems provide a controlled environment in which to probe the mechanisms involved in epithelial-mesenchymal crosstalk. To recapitulate the native alveolar tissue architecture, we employed a cyst templating technique to culture alveolar epithelial cells on photodegradable microspheres and subsequently encapsulated the cell-laden spheres within poly(ethylene glycol) (PEG) hydrogels containing dispersed pulmonary fibroblasts. A fibroblast cell line (CCL-210) was co-cultured with either healthy mouse alveolar epithelial primary cells or a cancerous alveolar epithelial cell line (A549) to probe the influence of tumor-stromal interactions on proliferation, migration, and matrix remodeling. In 3D co-culture, cancerous epithelial cells and fibroblasts had higher proliferation rates. When examining fibroblast motility, the fibroblasts migrated faster when co-cultured with cancerous A549 cells. Finally, a fluorescent peptide reporter for matrix metalloproteinase (MMP) activity revealed increased MMP activity when A549s and fibroblasts were co-cultured. When MMP activity was inhibited or when cells were cultured in gels with a non-degradable crosslinker, fibroblast migration was dramatically suppressed, and the increase in cancer cell proliferation in co-culture was abrogated. Together, this evidence supports the idea that there is an exchange between the alveolar epithelium and surrounding fibroblasts during cancer progression that depends on MMP activity and points to potential signaling routes that merit further investigation to determine targets for cancer treatment.
Keywords: Alveolar epithelium, Fibroblast, Co-culture, Hydrogel, Cancer, Matrix metalloproteinases
1. Introduction
Lung cancer kills more people in the U.S. than any other type of cancer underscoring the need for better treatments. To better understand and effectively treat lung cancer, the complexity of the tumor microenvironment, needs to be considered. In particular, the influence of the physical cues together with biochemical cues between cells in the lungs merits further investigation.
Given the three-dimensional architecture of alveolar tissue and tumor masses, more physiologically relevant models must employ ECM mimics that support the growth and culture of 3D multicellular tissue structures. While many techniques exist to form dense tumor spheroids (e.g., the hanging drop method) [1,2], the cyst-like alveolus structure is notoriously difficult to achieve and manipulate in vitro with primary alveolar epithelial cells, especially in synthetic ECM mimics. Recently, we demonstrated the use of photolabile microspheres as templates for patterning hollow, spherical model alveoli within peptide-modified poly(ethylene glycol) (PEG) hydrogels [3]. These hydrogels capture several key features of the native ECM (e.g., high water content; lung tissue appropriate elasticity; enzymatically degradable crosslinkers that enable local remodeling by cell-secreted proteases; introduction of integrin binding sites, such as the fibronectin-derived RGDS sequence), with the added advantage of precise user control over matrix properties (e.g., elastic modulus, scaffold geometry, tethered biochemical cues) [4]. To complement this approach, our lab has also developed a PEG crosslinker that cleaves upon exposure to selected light wavelengths (365-420 nm) under cytocompatible conditions. These materials have been used to synthesize microspheres of discrete size ranges that are completely degradable upon exposure to light, and have found applications for templating of multicellular cyst-like structures (50-200 μm) [3,5,6]. In the work presented here, our cyst templating technique was used to create model epithelial alveoli that were subsequently encapsulated in a PEG hydrogel laden with pulmonary fibroblasts.
This approach allowed the culture of two distinct lung cell types in a platform that captures physiological aspects, such as soft matrix modulus and 3D architecture, of the lung tissue to study the effects of paracrine signaling. In cancer, paracrine signaling has been shown to be a key regulator in tumor formation and invasion. As in many carcinomas, there appears to be a reciprocal exchange of signals between pulmonary fibroblasts and epithelial-derived lung cancer cells. For example, alveolar epithelium-derived adenocarcinoma cells increase α-SMA and matrix metalloproteinase production in fibroblasts [7,8]. Fibroblasts also signal to back to epithelial cells. Cancer-associated fibroblasts (CAFs) increase epithelial tumor proliferation, migration, and drug resistance [9,10]. Although in vitro co-culture models have proven to be useful tools for studying such crosstalk between cell types, the intricacies of epithelial-mesenchymal crosstalk during disease progression warrants further investigation in 3D platforms that better mimic the in vivo physical cues surrounding these cells [7,11–19].
Here, the results report on two types of epithelial cells: primary mouse alveolar epithelial cells to represent a healthy epithelium and an adenocarcinoma cell line (A549) to represent lung tumor cellular structures. These epithelial cells were co-cultured with a pulmonary fibroblast cell line (CCL-210) using the 3D cyst templating technique. The co-cultured cells were analyzed for signs of disease progression by measuring for proliferation, migration, and MMP activity. These measurements are especially relevant in a 3D context in which cells need to interact with and degrade their matrix to migrate and proliferate. Our goal was to test whether a diseased epithelium would influence the surrounding fibroblasts by increasing their proliferation and migration, and whether these changes relate to overall MMP activity. Interestingly, our results suggest a more complex feedback loop between diseased and healthy cells, in which cancer cell proliferation is increased in the presence of healthy fibroblasts.
2. Materials and methods
2.1 Microsphere synthesis
Photodegradable microspheres [6] were formed by inverse suspension polymerization via base-catalyzed Michael addition of a photodegradable diacrylate (PEGdiPDA; Mn~4070 Da) with a poly(ethylene glycol) tetrathiol (PEG4SH; Mn~5000 Da). The PEGdiPDA was synthesized as previously described [20], and PEG4SH was purchased from JenKem Technology. An aqueous phase consisting of 6.9 wt% PEGdiPDA, 4.2 wt% PEG4SH, CRGDS peptide (1.5 mM final concentration), 300mM triethanolamine (Sigma-Aldrich) in pH 8.0 phosphate buffered saline (PBS, Sigma-Aldrich) was pipetted and triturated twice into an organic phase comprised of 3:1 sorbitan monooleate (Span 80, Sigma-Aldrich) and PEG-sorbitan monooleate (Tween80, Sigma-Aldrich) dissolved at 30 mg surfactant per mL in hexanes (EMD Millipore). The aqueous droplet suspension was immediately stirred at a rate of ~200 rpm with a 1cm magnetic stir bar in a 20 mL glass scintillation vial overnight, protected from light. Polymerized particles were then retrieved via centrifugation and washed consecutively with hexanes, isopropanol, and sterile phosphate-buffered saline (PBS).
Non-degradable, fluorescently labeled microspheres were used as a control and reference point for cell tracking calculations in fibroblast migration experiments. Briefly, AlexaFluor-488 C5 maleimide (Life Technologies; <1 mM) was pre-reacted with PEG4SH for ~30 minutes in the dark. Poly(ethylene glycol) diacrylate (PEGDA; Mn~4000) was synthesized as previously described [21]. Fluorescently tagged microspheres were synthesized as above with an aqueous phase consisting of 6.9 wt% PEGDA, 4.2 wt% AlexaFluor-488-labeled PEG4SH, 300 mM triethanolamine in pH 8.0 PBS.
2.2 Cell culture
A549 human adenocarcinoma cells (CCL-185, ATCC) were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies); and human normal lung fibroblasts (CCL-210, ATCC) were cultured in low glucose DMEM. All cell line growth medium was supplemented with 10% fetal bovine serum (FBS, Life Technologies), 1% penicillin/streptomycin (Life Technologies), and 0.2% fungizone (Life Technologies). Cells were cultured at 37 °C with 5% CO2 and passaged until use in experiments.
2.3 Primary ATII cell isolation
All procedures and protocols were reviewed and approved by the Animal Care and Use Committee at the University of Colorado, Boulder. Primary mouse alveolar epithelial type II (ATII) cells were isolated as previously described [22]. Briefly, FVB/NJ mice (6 weeks old) were obtained from The Jackson Laboratories, immediately euthanized by CO2 asphyxiation and the chest cavity was opened to expose the heart-lung block. Sterile heparin was injected into the left ventricle and the inferior vena cava was cut. The pulmonary vasculature was then flushed with 1% heparin in sterile PBS. The heart-lung block was excised, and lung tissue was carefully dissected from the heart, trachea, and connective tissue.
Lung tissue was minced and digested in 0.1% Type-1 collagenase (Worthington Biochemical) and 1 mg/mL DNase (Sigma) for 20 minutes at 37°C. Trypsin (Fisher, 0.01% final concentration) was then added and the solution was incubated for another 20 minutes at 37°C. A solution containing 1mg/mL trypsin inhibitor (Life Technologies) and 1 mg/mL DNase was added, and this final mixture was filtered through 100 μm cell strainers and through a Nitex filter with 10 μm pore diameter. The filtered solution was centrifuged for 5 minutes at 2000 rpm, and the resultant pellet was resuspended in DMEM/F-12 medium (Sigma). This cell suspension was added to tissue culture plates coated with IgG (11 mg/cm2) and incubated for 1 hour at 37°C to separate immune cells from epithelial cells. Non-adherent cells were recovered, centrifuged for 5 minutes at 2000 rpm, resuspended in 1:1 Dulbecco’s Modified Eagle Medium:Nutrient Mixture F-12 (DEMEM/F12, Corning) supplemented with 10% FBS and 1% antibiotic/antimitotic (Life Technologies), and counted. Cells were then centrifuged once more at 2000 rpm for 5 minutes and resuspended at 500,000 cells/mL in DMEM/F-12 medium supplemented with 10% FBS, 1% antibiotic/antimitotic, hepatocyte growth factor (HGF; R&D Systems; 50 ng/mL), and Fibroblast Growth Factor 7 (FGF-7; Sigma; 10 ng/mL). Primary ATII cells were immediately used in cyst experiments.
2.4 Microsphere seeding
A549 cells at 150,000 cells/mL or primary ATII cells at 500,000 cells/mL in appropriate growth medium were combined with 40 μL of photodegradable microspheres (approximately 50,000 microspheres/mL) in an ultra-low adhesion 24-well plate (Corning). The seeding ratio was 76 cells/microsphere for the A549s and 253 cells/microsphere for the ATII cells. Plates containing pre-cysts were incubated at 37°C with 5% CO2 on an orbital shaker at 45 rpm. Prior to encapsulation in hydrogel formulations, A549 pre-cysts were incubated for 18-24 hours and primary pre-cysts were incubated for 3 days to allow for attachment and optimal microsphere coverage [3].
2.5 Cell labeling
For Click-it Plus EdU assays and co-culture migration experiments, fibroblasts were labeled with Cell Tracker Green CMFDA (Life Technologies) per manufacturer’s instructions prior to encapsulation in hydrogels. Fibroblasts were suspended in appropriate serum-free growth medium containing Cell Tracker Green (10 μM final concentration) and incubated at 37°C for 30 minutes, after which they were centrifuged and resuspended in PBS at 5 million cells/mL for encapsulation in hydrogels. For fibroblast-only migration experiments, fibroblasts were labeled with Cell Tracker Red CMPTX (Life Technologies, 20 μM final concentration) as above.
For co-culture migration experiments epithelial pre-cysts were stained with Cell Tracker Red CMPTX (Life Technologies). Pre-cysts were carefully removed from 24 well plates after the appropriate incubation period and allowed to settle by gravity. The supernatant was removed with a pipette. Appropriate serum-free growth medium containing Cell Tracker Red (20 μM final concentration) was added, and cells were incubated for 30 minutes at 37°C. Cell Tracker Red medium was removed, and cysts were resuspended in PBS for encapsulation in hydrogels.
2.6 Gel formation/cell encapsulation
2.6.1 Materials
8-arm poly(ethylene glycol) norbornene (PEG-Nb Mw ~40,000) and photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) were both synthesized as previously published [23,24]. Briefly, 8-arm PEG-OH was reacted with 5-norbornene-2-carboxylic acid (16 eq. to PEG-OH), N,N′-diisopropylcarbodiimide (16 eq. to PEG-OH), and 4-dimethylaminopyridine (1 eq. to PEG-OH) in dichloromethane overnight on ice. The product was precipitated in cold diethyl ether, dialyzed in deionized water, and lyophilized. An enzymatically-cleavable di-cysteine peptide (KCGPQG↓IWGQCK) and an integrin-binding peptide (CRGDS) were purchased commercially from American Peptide Company, Inc. A non-degradable di-cysteine peptide (KCGPQGIdWGQCK) containing the unnatural D isoform of isoleucine was synthesized using solid phase peptide synthesis on a Tribute Protein Synthesizer (Protein Technologies) with a Rink Amide MBHA resin (Novabiochem), as previously reported [25]. A quenched MMP-cleavable fluorogenic peptide substrate (Dabcyl-GGPQG↓IWGQK-Fluorescein-AEEAcC) was synthesized using solid phase peptide synthesis as described previously [26].
2.6.2 Proliferation Gels
For Click-iT Plus EdU assays, 8-arm 40 kDa PEG-Nb (5 wt%) was combined with dicysteine peptide crosslinker (degradable or non-degradable) (0.85:1 thiol:ene), CRGDS (1mM), and LAP (0.05 wt%) in sterile PBS and mixed by vortex to form a gel precursor solution. The pH was adjusted to 6.8-7.2 with sterile 0.1 M sodium hydroxide. A fibroblast cell suspension (final concentration 1.6 million cells/mL) and/or epithelial cysts were then added to the gel precursor solution and gently mixed with a pipette. 30 μL drops of this precursor solution were placed on Sigmacote-treated glass slides and exposed to 365 nm light at ~2 mW/cm2 for 3 minutes to initiate the radical-mediated thiol-ene polymerization reaction. Each polymerized gel was then transferred to an untreated 24-well plate (Corning) with 1 mL of DMEM/F-12 growth medium supplemented with 10% FBS and 1% antibiotic/antimitotic and incubated at 37 °C with 5% CO2. For MMP inhibition studies, 10 μM GM6001 in DMSO (Santa Cruz Biotechnology) or 0.05% DMSO was added to the media.
2.6.3 Migration Gels
For migration experiments, each well in a 24-well glass-bottomed plate (Greiner Bio-One) was washed in 95% ethanol prior to surface functionalization with thiol groups in 0.5% (v/v) (3-mercaptopropyl)trimethoxysilane in 95% ethanol (pH ~5.5) for 5 minutes. Each well was then washed in 95% ethanol and allowed to air dry. To make the gel precursor solution, 8-arm 40kDa PEG-Nb (3 wt%), di-cysteine MMP-degradable crosslinker (0.75:1), CRGDS (1 mM), and LAP (0.05 wt%) in sterile PBS were combined and mixed by vortex. Sodium hydroxide (0.1 M) was used to adjust the pH of the pre-cursor solution to 6.8-7.2. A fibroblast cell suspension (final concentration 1 million cells/mL) and either epithelial cysts or fluorescently-labeled non-degradable microspheres were added to the gel precursor solution and gently pipetted to mix. A 6 mm diameter circle was cut from square, 1 mm tall rubber gaskets with a biopsy punch. The gaskets were sealed to the bottom of the wells in the thiolated 24-well glass-bottom plate and subsequently filled with 30 μL drops of the precursor solution. To initiate the thiol-ene polymerization, the plate was exposed to 365 nm light at ~2 mW/cm2 for 4 minutes. After polymerization, the rubber gaskets were carefully removed. To each of the wells, DMEM/F-12 growth medium supplemented with 10% FBS and 1% antibiotic/antimitotic was added and incubated at 37 °C with 5% CO2. For MMP inhibition studies, 10 μM GM6001 (Santa Cruz Biotechnology) was added to the media.
2.6.4 MMP Sensor Peptide Gels
To evaluate MMP activity, single cell fibroblasts, epithelial cysts, or fibroblast/cyst combinations were encapsulated in MMP degradable hydrogels containing a quenched MMP-cleavable fluorogenic peptide substrate with the same sequence used to crosslink the hydrogels (Dabcyl-GGPQG↓fWGQK-Fluorescein-AEEAcC), as described previously [26,27]. MMP sensor gels consisted of 8-arm 40k PEG-Nb (5 wt%), di-cysteine MMP-degradable peptide crosslinker (0.85:1 thiol:ene), fluorogenic MMP sensor peptide (0.25 mM), CRGDS (1 mM), and LAP photoinitiator (0.05 wt%) in sterile PBS. The pH was adjusted to 6.8-7.2 with sterile, 0.1 M sodium hydroxide. A fibroblast cell suspension (final concentration 1.6 million cells/mL) and/or epithelial cysts were then added to the gel precursor solution and gently mixed with a pipette. Hydrogels were polymerized on top of thiol-functionalized glass coverslips (12 mm, Fisher Scientific) to create a covalent linkage between the hydrogel and the coverslip. Glass coverslips were flame cleaned and functionalized as previously described via emersion in silane solution (0.5% (3-mercaptopropyl) trimethoxysilane (Sigma Aldrich)) in 95% ethanol/water, pH~5.5), rinsed in 95% ethanol in water, and dried. 50 μL of precursor solution was pipetted into a rubber gasket with a 6 mm inner diameter centered on a coverslip. Gels were then polymerized under 365 nm light at an intensity of ~2 mW/cm2 for 3 minutes. Rubber gaskets were removed and gels covalently attached to coverslips were placed into wells of a 24-well plate with 1 mL DMEM/F-12 growth medium. Media was changed one hour after encapsulation to remove any excess fluorogenic peptide, and samples were incubated for 22 to 46 hours after encapsulation. For MMP inhibition studies, 10 or 100 μM GM6001 (Santa Cruz Biotechnology) or 0.5% DMSO was added to the media.
2.7 Microparticle template erosion
Gels containing fibroblasts and/or cysts were cultured for one day to allow cell attachment to the encapsulating hydrogel. Following this 24-hour incubation period, all samples were exposed to 365 nm light at ~10 mW/cm2 for 15 minutes to cleave the photo-labile moiety in the microspheres and fully erode the cyst templates. Gels were then incubated for ~1 hour at 37°C with 5% CO2 before changing growth medium. Growth medium was then exchanged daily until completion of each experiment (between 1 and 5 days).
2.8 Click-iT EdU assay and quantifying proliferation
Three biological replicates of each cell combination were studied, and images of the proliferating nuclei were quantified using MATLAB. Cell proliferation was detected using a Click-iT Plus EdU AlexaFluor-594 imaging kit (Life Technologies). A copper-catalyzed covalent “click reaction” between a picolyl azide on an AlexaFluor dye and an alkyne on the thymidine nucleoside analog, 5-ethynyl-2′-deoxyuridine (EdU), allowed for fluorescent staining of proliferating nuclei. EdU was added to growth media at a final concentration of 10 μM on day 1 (24 hours post encapsulation) or day 4 (96 hours post encapsulation). Samples were then incubated for 17 hours at 37°C with 5% CO2 to allow for EdU incorporation into DNA during active DNA synthesis, per manufacturer instructions. Samples were then fixed with 4% paraformaldehyde in PBS on day 2 and day 5 for 15 minutes at room temperature and washed in 3% bovine serum albumin (BSA) in PBS. Cells were permeabilized in 1% TritonX-100 for one hour and washed with 3% BSA in PBS. Click-it Plus reaction cocktail containing the AlexaFluor 594 dye was prepared as per manufacturer instructions, added to each sample, and incubated for 3 hours at room temperature, rocking. Samples were then washed with 3% BSA in PBS. Finally, nuclei were stained with DAPI (1:2000, Life Technologies) for one hour and washed in PBS. All samples were imaged on a confocal microscope (Zeiss LSM 710) with a 20* water-dipping objective (Plan Appochromat; NA = 1.0). Image stacks (10 per condition) were taken from the top to bottom of individual cysts, or in the center of the gel for fibroblast-only conditions (z step = 10 μm, average stack size ~500 μm).
For quantification, four categories of cell nuclei were counted using MATLAB: fibroblast nuclei, epithelial nuclei, proliferating fibroblast nuclei, and proliferating epithelial nuclei. TIFF images of each z-slice in each of three channels: DAPI, AF-594, and Cell Tracker Green, were created in Image-J and processed in MATLAB. Separate masks were made of objects (nuclei) in the DAPI channel and AlexaFluor 594 channels, and objects were dilated by two pixels with a Disk Structuring Element. The mean intensity within each dilated object in the Cell Tracker Green channel was evaluated. Objects with a mean intensity above a given threshold were counted as fibroblasts; if the mean intensity fell below this threshold, the object was counted as an epithelial cell. Cell nuclei centroids were tracked and checked for their appearance in successive slices to account for double counted nuclei. Double counts were subtracted to give final cell counts and the percent of proliferating cells. Object counts were pooled from all ten image stacks, and the mean percent positive for EdU was calculated from three biological replicates. Statistical analysis was performed using two-way ANOVA followed by Bonferroni posttests. All error bars represent standard error of the mean (SEM).
2.9 Tracking cell migration
The 3D migration of fibroblasts in co-culture hydrogels was observed with an Operetta High Content Imaging System (Perkin Elmer) using Harmony High Content Imaging and Analysis software (Perkin Elmer) for automated image collection in real-time. As mentioned previously, co-culture hydrogels were formed and allowed to swell for 24 hours before cyst templates were eroded and fresh growth medium was introduced. During each experiment, the 10x long WD objective was used with the optical model set to confocal. Two channels, AlexaFluor 594 and AlexaFluor 488, were selected and exposure times were adjusted for each experiment. The layout selection for the images taken in each well was defined as nine fields of view in a 3×3 square in the center of the well. 400 μm z-stacks beginning at a height of 25 μm from the glass surface were collected for each image. Images were taken at 30-minute intervals for 24 hours. A live cell chamber was used to maintain the temperature at 37 °C and 5% CO2.
Fibroblast migration in 3D was analyzed with Volocity 3D Image Analysis Software (Perkin Elmer). The protocol tracked the fibroblasts in the appropriate channel (AlexaFluor 488 for cyst co-cultures, AlexaFluor 594 for fibroblast-only gels) within a spherical region of interest (ROI) around a cyst or microsphere with a radius of 125 μm greater than the object. This radius was chosen to limit ROI overlap between neighboring cysts/microspheres, which were spaced about 250 μm apart, on average. Either the epithelial cyst in the AlexaFluor 594 channel or the microsphere in the AlexaFluor 488 channel was tracked as a single object to be used as a reference point. The centroids of the tracked reference point and fibroblasts were exported for subsequent analysis in MATLAB to determine cell speeds, displacements, directionality, and the percent of migrating cells. To compensate for drift in each ROI analyzed, the fibroblast centroid data was normalized to the corresponding tracked reference point. Fibroblast speeds were calculated as the average of the individual speeds between subsequent time points on the cell path. Displacement (distance between start and end points) was used to determine if a fibroblast was migrating towards or away from a cyst, where a positive displacement was traveling towards a cyst and negative displacement was traveling away from a cyst relative to the centroid of the cyst. The directionality (displacement/total distance traveled) was a measure of the straightness of a cell path analogous to persistence time, with values close to 1 indicating a perfectly linear path [28]. The fraction of migrating cells was defined as the number of fibroblasts migrating divided by the total number of fibroblasts tracked, where a cell was deemed migrating if the maximum distance away from the starting position was greater than one cell body length (15 μm) at any point during the 24 hour period, a criterion used in previous migration studies [28]. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn’s Multiple Comparison test. All error bars represent standard error of the mean (SEM).
2.10 Quantification of MMP activity and metabolic activity
After 22 or 46 hours post-encapsulation, 10% PrestoBlue (Invitrogen) was added to each well to detect metabolic activity. Two hours later, fluorescence intensity readings were conducted using a Synergy H1 microplate reader (BioTek) at 560 nm excitation/590 nm emission for PrestoBlue, and 494 nm excitation/521 nm emission for the fluorogenic peptide. After fluorescence readings, cells were immediately fixed with 4% PFA for 15 minutes and washed thrice with PBS. Cells were then stained with Hoechst (1:1000, ThermoScientific) for 1 hour and subsequently washed with PBS.
Gels were imaged using an Operetta High Content Imaging System (Perkin Elmer) using Harmony High Content Imaging software, with the 10X long WD objective in confocal mode. 300 μm z-stack images were taken (31 images 20μm apart) in 9 fields of view for each gel. Harmony Analysis software was then used to count cell nuclei. Fluorogenic peptide signal (MMP activity) and PrestoBlue signal (metabolic activity) were each normalized to cell number. Statistical analysis was preformed using two-way ANOVA with Bonferroni posttests. Error bars represent standard error of the mean (SEM).
3. Results
Using a previously developed cyst-forming technique [3], a 3D co-culture system for lung cells was created with alveolar epithelial cysts embedded in a synthetic polymer hydrogel and surrounded by low-density pulmonary fibroblasts (Fig. 1). This in vitro culture system enabled us to probe cellular behavior in response to co-culture with a diseased cell type in the appropriate structural context in an attempt to elucidate the role of epithelial-mesenchymal crosstalk in disease progression. To accomplish this goal, three cell types were used: healthy alveolar epithelial cells (primary mouse cells), lung tumor epithelial cells (A549 cell line), and pulmonary fibroblasts (CCL-210 cell line). While the primary epithelial cells attached and spread to form single-cell layers on the microsphere templates, the cancerous A549 cells tended to form multilayered structures either on the microspheres or as detached aggregates, both of which were embedded in the encapsulating hydrogel. Therefore, the A549 results presented here include both the cyst-like structures, as well as the higher density aggregates, with no significant differences found between the two structure types in the assays studied here. We believe that this phenomenon is relevant to the tumor structures found in cancer progression, and many of the 3D model systems presented in the literature make use of tumor spheroids [1,7,15,29]. The advantage with the presented co-culture system is that we can study both normal alveolar epithelial cells in their native tissue structure, as well as tumor aggregates, and how they respond in the presence of fibroblasts distributed around the epithelial surface.
Fig. 1.
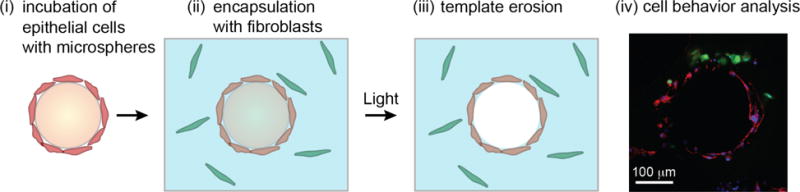
Cross-sectional schematic of the co-culture set-up. (i) Epithelial cells (red) were incubated on an orbital shaker with photodegradable microspheres (orange) containing RGDS peptides to allow for cellular attachment to the surface of the microsphere. (ii) Pre-cysts were co-encapsulated with a single cell suspension of fibroblasts (green) in a poly(ethylene glycol) (PEG) hydrogel (blue) containing pendant RGDS for cell adhesion and an enzymatically-degradable peptide crosslinker to allow for local matrix remodeling. (iii) One day after encapsulation, cytocompatible 365 nm light at ~10 mW/cm2 for 15 minutes was applied to completely erode the microsphere templates, leaving a shell of epithelial cells surrounding a liquid-filled lumen. (iv) Cells were cultured for 1-5 days before being analyzed for proliferation, migration, or protease activity. In this confocal image slice, normal fibroblasts labeled with Cell Tracker Green were co-cultured with primary epithelial cells, which were subsequently stained for an alveolar epithelial type 1 (ATI) phenotype marker. Green = Cell Tracker (fibroblasts), Blue = DAPI (nuclei), Red = T1α (ATI).
3.1 Proliferation
Since cancer is characterized by unchecked cell division, we aimed to determine changes in cell proliferation using our co-culture model. Here, we used a commercially available fluorescent EdU assay to measure the proliferation of each cell type after a 17-hour incubation period initiated on days 1 and 4 post-encapsulation. Days 1 and 4 were both measured to account for changes in proliferation over time. Automated analysis of confocal image slices (Fig. 2A) was performed to quantify the percent of nuclei positive for EdU.
Fig. 2.
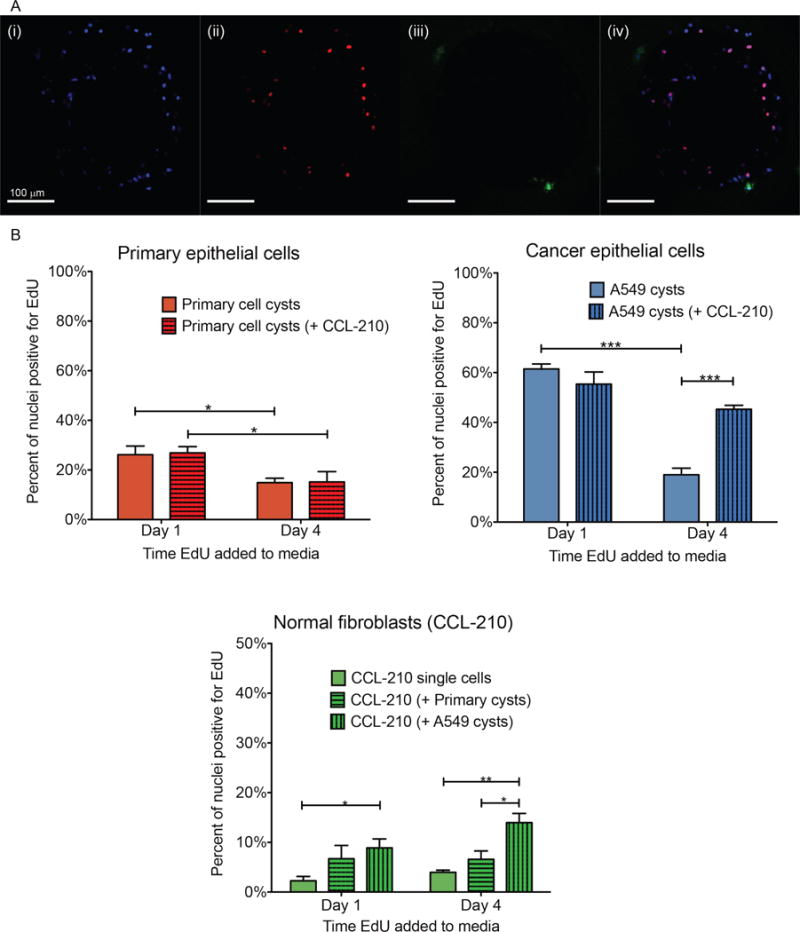
Click-iT Plus EdU proliferation assay. (A) Example confocal image slices used for automated nuclei counts. Each object found in the blue and red channels was measured for mean intensity in the green channel, and objects above a certain threshold were counted as fibroblasts. (i) Blue = DAPI (all nuclei). (ii) Red = EdU (proliferating nuclei). (iii) Green = Cell Tracker (fibroblasts). (iv) Merged image of previous three channels. (B) Plots depict percent of nuclei positive for EdU at the two time points in monoculture and both co-cultures, separated by cell type. Results are presented as means ± SEM of three biological replicates of each condition. *p < 0.05, **p < 0.01, ***p < 0.001
The results for both healthy and cancerous epithelial cell types demonstrated a significant decrease in proliferation between day 1 and day 4, whether alone or in co-culture with fibroblasts (Fig. 2B). This could be a result of increased cell number causing increased cell-cell contact thereby preventing further proliferation. The primary epithelial cells had no difference in proliferation between co-culture and monoculture on either day indicating that paracrine signaling between between the healthy epithelial cells and fibroblasts does not affect healthy epithelial cell proliferation. In cancer cells, however, co-culture caused changes in proliferation. On day 1, the A549 cancer cells also showed no difference between monoculture and co-culture with fibroblasts but, on day 4 when A549 cells were co-cultured with the fibroblasts the epithelial cells showed increased proliferation compared with monoculture. Signaling between the cancerous epithelial cells and fibroblasts appeared to take longer than 1 day to affect proliferation. When comparing primary healthy epithelial cell proliferation to A549 proliferation, as expected, A549 cells had significantly higher percentages of EdU-positive nuclei on day 1 for monoculture and both co-culture conditions (Supplementary Fig. 1). On day 4, however, A549 proliferation was significantly higher only when co-cultured with normal fibroblasts, contrary to the hypothesis that signaling from healthy fibroblasts prevents cancer cell growth.
The fibroblasts had a much lower proliferation rate than either epithelial cell type, and this rate did not appear to change over time (Fig. 2B). Interestingly, the fibroblasts had a significantly higher percentage of EdU-positive nuclei when co-cultured with A549 cells on both days compared to monoculture, and on day 4 when compared to co-culture with primary cysts. This indicates that epithelial cells signal back to fibroblasts to increase fibroblast proliferation.
3.2 Fibroblast migration
Increased motility of both fibroblasts and epithelial cells has been used as an indicator of disease progression since motility enables cancer metastasis [30–33]. In cancer metastasis, it has been proposed that tumor cells detach from the main tumor and migrate through the surrounding matrix to reach the blood stream [34]. In addition, tumors are thought to recruit and transform local fibroblasts into activated fibroblasts that promote tumor growth [35,36]. These cancer-associated fibroblasts (CAFs) have been shown to facilitate tumor cell invasion by creating tracks through the ECM, along which cancer cells can migrate [35,37,38]. Key parameters of migration including speed and directionality have been shown to be better indicators of malignant potential than endpoint migration assays [39]. We thus measured the effect of co-culturing on a variety of motility parameters such as speed, directionality, and fraction of cells migrating to determine whether crosstalk between epithelial and mesenchymal cells could influence cancer progression through changes in migration.
In this study, we fluorescently labeled each cell type and used live cell microscopy to record confocal image stacks every 30 minutes for 24 hours, starting one-day post encapsulation (Fig. 3A). Using automated software and analysis codes, we tracked cell position in three dimensions over time and calculated key migration measurements, namely the fraction of cells migrating per analyzed cyst, the fraction of cells migrating toward the reference cyst, the speed, and the directionality. While no significant movement was detected in the epithelial cells, we did observe fibroblast migration in these gels. Cells that traveled at least one cell body length away from their starting position at any point during the 24-hour experiment were considered to be migrating. All other measurements were calculated for the migrating cells only. Cells considered to be moving toward their reference cyst had final positions closer to the cyst than their starting positions.
Fig. 3.
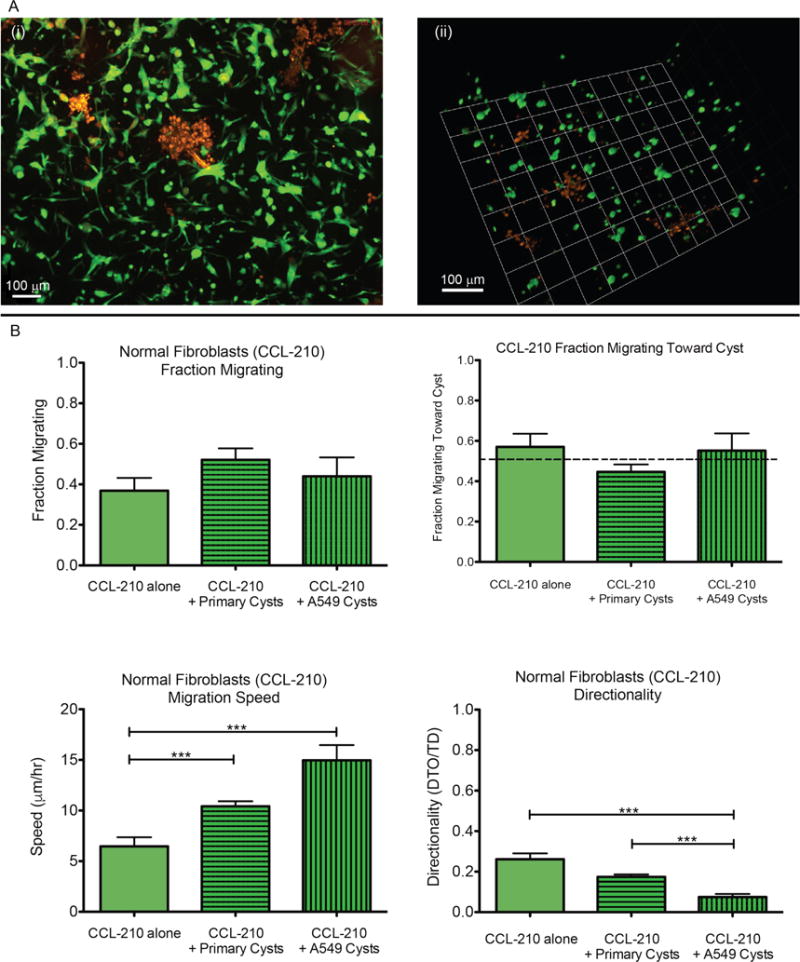
Normal fibroblast migration analysis. (A) Example images of fibroblasts (green) in co-culture with epithelial cysts (red) used in cell tracking. (i) Max z-projection of 400 μm confocal stack. (ii) 3D rendering of 200 μm confocal stack using Volocity (Perkin Elmer). (B) Plots show fraction migrating, fraction migrating toward cyst, migration speed, and directionality (Distance-To-Origin/Total Distance) of migrating normal fibroblasts (CCL-210) in monoculture and co-culture with both epithelial cell types. Data in fraction migrating plot represent means ± SEM of cells migrating per cyst. Data in the remaining plots represent means ± SEM of all migrating cells. ***p < 0.001
The normal fibroblasts (CCL-210, Fig. 3B) had no statistically significant difference in cells migrating per cyst when co-cultured with either A549 cancer cells (44%) or primary cysts (52%) compared to monoculture (37%). Likewise, in all conditions the normal fibroblasts showed no directional preference toward or away from the reference cyst. Interestingly, the average speed of the migrating CCL-210 cells in monoculture was ~6 μm/h, while their speed in co-culture with either epithelial cell type was significantly faster (10 μm/h with primary cysts and 15 μm/h with A549 cells). With regards to directionality (distance to origin/total distance), where a value of 1 represents a completely straight path, the normal fibroblasts in all conditions exhibited relatively random migration. However, co-culture with A549 cells resulted in significantly less directed motion (0.07) than in monoculture (0.26) or in co-culture with primary cysts (0.17).
3.3 Matrix metalloproteinase (MMP) activity
Increased protease activity is associated with cancer progression, enabling tumor outgrowth and angiogenesis [40]. To measure global MMP activity in our gels, a recently developed fluorogenic MMP-sensitive peptide (Dabcyl-GGPQG↓fWGQK-Fluorescein-AEEAcC) was covalently tethered to the hydrogel network enabling fast and in situ measurement of MMP activity by taking a fluorescence area scan of the gel using a standard plate reader [26,27]. Overall metabolic activity was measured concurrently by adding a resazurin-based assay to each well. The fluorescence signals from the MMP sensor peptide and from the metabolic activity assay were both normalized to cell count as estimated by imaging cell nuclei across the bulk of each gel.
In both co-cultures, primary epithelial cells with the normal fibroblasts and cancer epithelial cells with the normal fibroblasts, on both days overall metabolic activity appeared to be synergistically increased over the calculated weighted average of the two cell types cultured alone (Fig. 4A). The weighted average is the level expected in the co-culture if there were no crosstalk between the cell types influencing metabolism or MMP expression. The primary cell co-cultures had an average normalized metabolic activity on both days of 0.20 compared to a weighted average of 0.13 on both days. The cancer cell co-cultures had an average normalized metabolic activity of 0.25 and 0.27 on days 1 and 2, respectively, compared with weighted averages of 0.17 and 0.20.
Fig. 4.
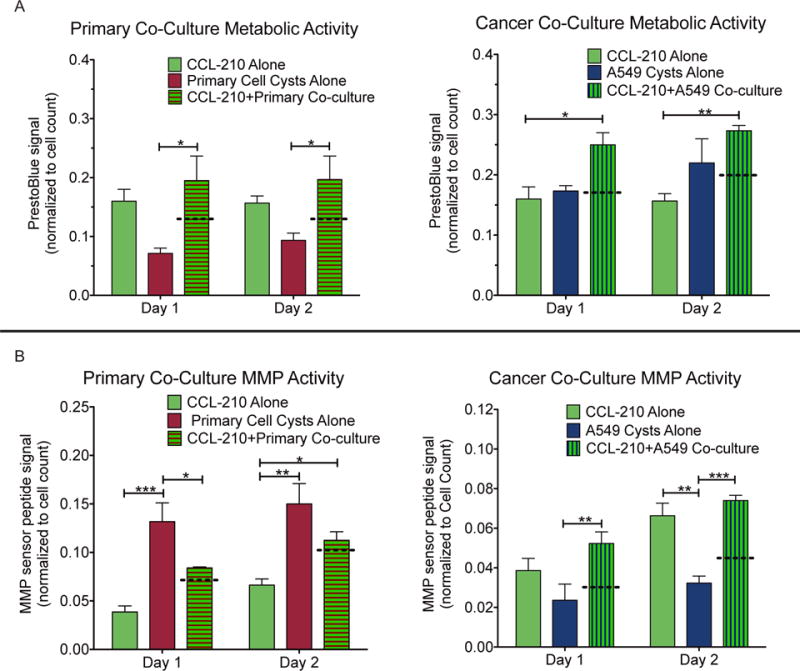
(A) Metabolic activity on days 1 and 2 after encapsulation as measured by resorufin fluorescence normalized to cell count for each cell type cultured alone and in co-culture. (B) MMP activity on days 1 and 2 after encapsulation as measured by an MMP-sensitive fluorescent sensor peptide normalized to cell count for each cell type cultured alone and in co-culture. Dotted lines represent the weighted average of the two cell types cultured alone, which would be the expected value for the co-culture barring any crosstalk. Data represents means ± SEM for 3 biological replicates of each condition. *p < 0.05, **p < 0.01, ***p < 0.001
In contrast, only the cancer cell co-cultures demonstrated a synergistic increase in MMP activity on both days (Fig. 4B). The primary cell co-cultures had an average normalized MMP activity of 0.08 and 0.11 on days 1 and 2, respectively, compared with weighted averages of 0.07 and 0.10. However, the cancer cell co-cultures had day 1 and day 2 MMP activities of 0.05 and 0.07, respectively, versus weighted averages of 0.03 and 0.04.
3.4 Matrix metalloproteinase (MMP) inhibition
To investigate whether the observed increase in MMP activity in the cancer cell co-culture might be influencing the increased cancer cell proliferation and fibroblast migration speed seen earlier, we tested the influence of a broad spectrum small molecule MMP inhibitor (GM6001) on proliferation and migration in our co-culture model. GM6001 binds with the zinc active site on MMPs and has been shown to inhibit the bioactivity of MMPs 1, 2, 3, 8, and 9, among others [40,41]. Furthermore, this inhibitor has previously been used with A549 cells to effectively block invasion into a 3D matrix and has been shown to reduce the fluorescence signal from the MMP sensor peptide used in this work [26,27,42]. To confirm the effectiveness of GM6001 with our co-culture model, we repeated the MMP activity assay with 10 μM GM6001 in the media and observed a reduction by half in the normalized signal on both days compared with the DMSO control group (Supplementary Fig. 2).
We repeated the proliferation assay with cancer cells alone and in co-culture with normal fibroblasts and added 10 μM GM6001 to the media, comparing it to a DMSO control group as well as our original data (Fig. 5). In addition we performed the same assay in non-degradable gels, which contained a crosslinker with the same di-cysteine peptide sequence as before except the isoleucine at the cleavage site was switched to the unnatural D isoform, rendering it insensitive to MMP cleavage [25]. In all conditions, the cancer cells alone demonstrated statistically similar levels of proliferation on day 4, around 22%. For the co-cultures, both the original degradable gel and the DMSO control had statistically higher proliferation levels on day 4 than the cancer cells alone, around 43%. Both the MMP-inhibited and non-degradable gel co-cultures exhibited a reduction in proliferation on day 4 that was statistically lower than the co-culture controls and statistically similar to the cancer cells alone, 24% and 29% respectively.
Fig. 5.
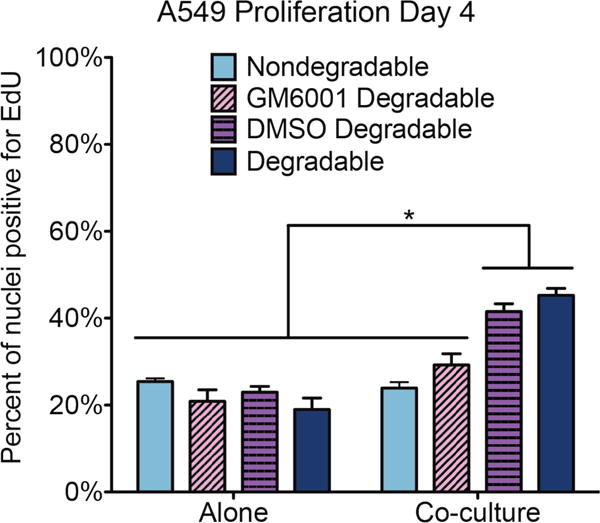
Click-iT Plus EdU proliferation assay. Plot depicts percent of A549 cell nuclei positive for EdU on day 4 in monoculture and the A549/CCL-210 co-cultures, separated by gel and media type. The nondegradable gels contained a peptide crosslinker insensitive to MMP cleavage. GM6001 was added at 10 μM, and the DMSO control media contained 0.05% DMSO. The degradable bars refer to the original experiment in MMP-degradable gels with regular growth media. Results are presented as means ± SEM of three biological replicates of each condition. *p < 0.05
The corresponding fibroblast proliferation results (Supplementary Fig. 3) on day 4 in the MMP-inhibited and DMSO control co-cultures showed no change from the original (14%) and were significantly higher than the fibroblasts alone (4%). The fibroblasts in the non-degradable gel co-cultures were not statistically different from either group (10%).
We also repeated the migration assay with MMP inhibitor in the media for both the fibroblasts alone and the fibroblasts in co-culture with the cancer epithelial cells. In both cases, the inhibitor completely blocked migration, so we were unable to calculate migration speed or directionality.
4. Discussion
Epithelial-mesenchymal crosstalk is a key regulator during lung development and normal wound healing processes, and growing evidence suggests that altered paracrine signaling between the alveolar epithelium and interstitial fibroblasts may lead to disease progression in multiple pathologies [8,9,34,43–45]. To study these interactions, in vitro co-culture systems, and particularly biomaterial matrices, have evolved to serve as valuable tools for controlling the cell types present, and their physical cues from the surrounding matrix, with the ability to mix healthy with diseased cells [7,12–15]. In the 3D model system presented here, alveolar epithelial cysts were surrounded by pulmonary fibroblasts in an encapsulating hydrogel matrix that recapitulates aspects of the basic tissue architecture of the distal lung. Compliant 3D culture networks have been shown to more closely represent cell behavior in vivo than traditional flat and typically stiff surfaces (e.g., tissue culture plastic ware is ~6 orders of magnitude stiffer than lung tissue), and are especially influential in the areas of contact-inhibited growth [46,47] and migration mechanisms [48,49] relevant to this work. Advantageously, the spatial arrangement of cells in this platform better reflects many aspects of the in vivo lung structure, but this also limits the types of analysis that can be performed on individual cell populations. Other platforms, like transwell inserts, are better for maintaining cell separation to use downstream applications such as RT-qPCR and western blots, but with a transwell system, spatial and temporal information is lost. Therefore, imaging-based measurements were used to visualize specific cell populations and to quantify cell behavior in 3D over time.
In the healthy adult lung, alveolar epithelial cell turnover is slow compared to many other tissues; the replacement time is reported to be approximately one month [50–52]. During wound healing, proliferation increases dramatically to repopulate the epithelium in a few days, followed by cell cycle arrest and apoptosis of undifferentiated cells [50,52,53]. Tumor cells, on the other hand, exhibit unchecked proliferation, and the ATCC reports that the adenocarcinoma cell line A549 has a doubling time of just 22 hours. Therefore, it is not surprising that in our culture system the percent of nuclei positive for EdU one day after encapsulation was significantly higher for A549 cysts than primary cysts. Both epithelial cell types demonstrated a dramatic decrease in proliferation over time, which we attribute to contact inhibition [47,54] and physical constraint by the encapsulating hydrogel. While the gel network is degradable by various matrix-metalloproteinases (MMPs; including 1, 2, 3, 8, and 9) [55,56], their expression by these cells appears to be low, especially for the cancer cells, and limited outgrowth into the surrounding hydrogel is observed. The fibroblasts exhibit very low levels of proliferation, which does not change over time or in the presence of healthy epithelial cells, matching the normally quiescent phenotype seen in vivo [45,50,57].
The exception to these trends is seen in the A549/CCL-210 co-culture condition, which shows significantly higher proliferation of both cell types four days after encapsulation, as well as higher proliferation of the normal fibroblasts on day 1. Profibrotic factors secreted from the A549 epithelial cells (osteopontin, PDGF, TGFβ, etc.) [43,58,59] may be one cause of this immediate increase in fibroblast proliferation. Enhanced proliferation is one marker used to distinguish CAFs from normal fibroblasts in the tumor stroma [35]. Therefore, these results may indicate the onset of fibroblast transformation by the cancer cells in our co-culture model. While the cancer cells alone show a decrease in proliferation over time in our gels, co-culture with normal fibroblasts enables the cancer cells to maintain a high level of proliferation on day 4. One possible explanation is that the increased MMP activity observed in the cancer cell-fibroblast co-culture leads to increased local matrix degradation, which reduces contact inhibition and allows more space for the cancer cells to continue proliferating. This hypothesis is supported by the fact that switching to an MMP-insensitive peptide crosslinker, as well as blocking global MMP activity with a broad-spectrum small molecule MMP inhibitor, abrogates the enhanced proliferation seen on day 4 in co-culture.
Normally, the alveolar epithelium produces paracrine signals, such as prostaglandin E-2 (PGE2), that keep the interstitial fibroblasts in a quiescent state, suppressing migration, proliferation, and ECM-production [44]. Conversely, in cancer the malignant epithelium produces a multitude of profibrotic and pro-inflammatory signals, such as PDGF, TGFβ, and IL-1, and these signals are thought to contribute to increased fibroblast migration, proliferation, and secretion of tumor-promoting signals seen in lung cancer patients [9,35,36]. In the migration studies presented here, the observed speeds are much slower than previously reported values for human dermal fibroblasts in similar hydrogels (~40 μm/h) [28], but this may be due to the higher crosslinking density of our hydrogels. Regardless, the normal fibroblasts migrated faster in culture with A549 cells than they migrated in monoculture, although this faster motion was significantly less directed than in the other conditions. As with the increased proliferation seen in this same co-culture condition, increased protease activity may contribute to faster migration of fibroblasts, due to local degradation of the matrix. Alternatively, the cancerous A549 cells may already be producing one or many of the profibrotic signals listed above or secreting less of the quiescent signal PGE2. Reduction of MMP activity by a broad-spectrum inhibitor was enough to prevent any migration of fibroblasts through these gels, likely due to the need for local network degradation to create space for the cells to migrate through. However, it is still unclear what influence MMPs have on fibroblast migration speed and directionality in this co-culture model. Alternative peptide crosslinkers in the gel network susceptible to cleavage by specific MMPs may enable more investigation into this mechanism Further, we did not observe any invasion of the A549 cells into the surrounding matrix despite the increase in local matrix degradation. A recent study showed that healthy lung fibroblasts may decrease the metastatic potential of lung cancer cells through an undefined secreted factor [60]. Therefore, we may be observing a similar protective effect.
Protease activity is important for normal ECM turnover and matrix remodeling after wound closure [40,58]. However, an imbalance in MMP and TIMP levels favoring matrix destruction has been associated with tumor invasiveness, angiogenesis, and metastasis [9,40]. Fromigué, et al. found an increase in RNA expression and protein levels of specific MMPs (2, 9, and 11) in the conditioned media from direct co-cultures of A549 cancer cells and CCL-210 pulmonary fibroblasts, which matches our observation of increased general MMP activity for co-cultures of the same cell lines [8]. Interestingly, this synergistic increase in MMP activity is not seen in the healthy primary cell co-culture, suggesting that paracrine signaling from the cancer cells may be causing this increase in MMP production through pro-inflammatory signals such as TGFβ.
While the results presented here point to an increase in MMP activity as being influential in tumor-promoting cell behavior (e.g. increased fibroblast migration and cancer cell proliferation), the complex signaling pathways involved in this epithelial-mesenchymal crosstalk remain elusive. Targeted MMP inhibitors are actively being developed as anti-cancer drugs based on tumor cell expression profiles; however, this evidence suggests the need to probe further into the source of these MMPs (e.g. tumor-adjacent stromal cells) as well as the upstream signaling cues causing the increase in MMP production. In situ hybridization might be used in this co-culture system to determine specific MMP gene expression profiles for the two cell types and discover the origin of this upregulation in MMPs in co-culture versus monoculture. Since protease activity is essential for normal tissue homeostasis and wound healing throughout the body, further investigation of the tumor-stromal signaling pathways leading to this increase in MMPs could reveal potential targets for the development of novel therapeutics, limiting the deleterious side effects seen with current broad-spectrum MMP-inhibiting drugs. Cellular outcomes pertinent to anti-cancer drugs, such as matrix remodeling and 3D migration, inherently necessitate the engineering of 3D, adaptable environments for co-culture of multiple cell types and testing the efficacy of drugs. Therefore, once therapeutics have been identified, this unique co-culture model could provide a spatially-organized, alveolar tissue-relevant platform for screening before attempting to use more expensive and complex mouse models.
5. Conclusions
The 3D in vitro co-culture system used in this study provided an innovative platform for studying the interactions between alveolar epithelial cysts and dispersed pulmonary fibroblasts and investigating cell functions related to disease progression. The results presented here support the growing body of evidence in the literature that crosstalk between the alveolar epithelium and interstitial fibroblasts influences their behavior in terms of proliferation, migration, and protease activity. Co-culture of tumor-derived epithelial cells with normal pulmonary fibroblasts led to an increase in MMP activity, cancer cell and fibroblast proliferation, and fibroblast migration speed. Future investigation into the signals contributing to the differences in cell behavior discovered here would provide needed insight into possible pathways conducive to drug development. For example, MMP activity appears to be influential in fibroblast motility and proliferation of tumor cells, furthering the importance of developing targeted MMP inhibitors for anti-cancer therapy. Moreover, this spatially-relevant co-culture model could be a useful starting point for drug screening trials by capturing key geometric aspects of cell behavior in alveolar tissue before moving to more complex in vivo models.
Supplementary Material
Acknowledgments
The authors would like to thank Sharon Ryan for teaching us the ATII mouse cell isolation procedure, Kyle Kyburz for discussions concerning live cell tracking experiments and analysis, Emi Tokuda for providing the MMP sensor peptide and many helpful discussions, and Chun Yang for providing the non-degradable peptide crosslinker. Funding for this work was provided by the Howard Hughes Medical Institute, the National Science Foundation (CTS1236662), and the NIH Biophysics training grant (T32 GM-065103).
References
- 1.Friedrich J, Ebner R, Kunz-Schughart LA. Experimental anti-tumor therapy in 3-D: spheroids–old hat or new challenge? Int J Radiat Biol. 2007;83:849–871. doi: 10.1080/09553000701727531. [DOI] [PubMed] [Google Scholar]
- 2.Weiswald LB, Bellet D, Dangles-Marie V. Spherical Cancer Models in Tumor Biology. Neoplasia. 2015;17:1–15. doi: 10.1016/j.neo.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis KJR, Tibbitt MW, Zhao Y, Branchfield K, Sun X, Balasubramaniam V, Anseth KS. In vitro model alveoli from photodegradable microsphere templates. Biomater Sci. 2015;3:821–832. doi: 10.1039/C5BM00034C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis KJR, Anseth KS. Hydrogel scaffolds to study cell biology in four dimensions. MRS Bull. 2013;38:260–268. doi: 10.1557/mrs.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tibbitt MW, Han BW, Kloxin AM, Anseth KS. SFB Student Award Winner in the Ph.D. Category: Synthesis and application of photodegradable microspheres for spatiotemporal control of protein delivery. J Biomed Mater Res Part A 100A. 2012:1647–1654. doi: 10.1002/jbm.a.34107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amann A, Zwierzina M, Gamerith G, Bitsche M, Huber JM, Vogel GF, Blumer M, Koeck S, Pechriggl EJ, Kelm JM, Hilbe W, Zwierzina H. Development of an innovative 3D cell culture system to study tumour–stroma interactions in non-small cell lung cancer cells. PLoS One. 2014;9:e92511. doi: 10.1371/journal.pone.0092511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fromigué O, Louis K, Dayem M, Milanini J, Pages G, Tartare-Deckert S, Ponzio G, Hofman P, Barbry P, Auberger P, Mari B. Gene expression profiling of normal human pulmonary fibroblasts following coculture with non-small-cell lung cancer cells reveals alterations related to matrix degradation, angiogenesis, cell growth and survival. Oncogene. 2003;22:8487–8497. doi: 10.1038/sj.onc.1206918. [DOI] [PubMed] [Google Scholar]
- 9.Bhowmick N, Neilson E, Moses H. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. http://www.nature.com/nature/journal/v432/n7015/abs/nature03096.html (accessed December 1, 2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S, Yano S. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–8. doi: 10.1158/1078-0432.CCR-09-1001. [DOI] [PubMed] [Google Scholar]
- 11.Prasad S, Hogaboam CM, Jarai G. Deficient repair response of IPF fibroblasts in a co-culture model of epithelial injury and repair. Fibrogenesis Tissue Repair. 2014;7:7. doi: 10.1186/1755-1536-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Kaplan DL. Hormone-responsive 3D multicellular culture model of human breast tissue. Biomaterials. 2012;33:3411–3420. doi: 10.1016/j.biomaterials.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horie M, Saito A, Mikami Y, Ohshima M, Morishita Y, Nakajima J, Kohyama T, Nagase T. Characterization of human lung cancer-associated fibroblasts in three-dimensional in vitro co-culture model. Biochem Biophys Res Commun. 2012;423:158–63. doi: 10.1016/j.bbrc.2012.05.104. [DOI] [PubMed] [Google Scholar]
- 14.Fang X, Sittadjody S, Gyabaah K, Opara EC, Balaji KC. Novel 3D Co-Culture Model for Epithelial-Stromal Cells Interaction in Prostate Cancer. PLoS One. 2013;8:1–11. doi: 10.1371/journal.pone.0075187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SA, Lee EK, Kuh HJ. Co-culture of 3D tumor spheroids with fibroblasts as a model for epithelial–mesenchymal transition in vitro. Exp Cell Res. 2015;335:187–196. doi: 10.1016/j.yexcr.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Majety M, Pradel LP, Gies M, Ries CH. Fibroblasts Influence Survival and Therapeutic Response in a 3D Co-Culture Model, PLoS One. 2015;10:e0127948. doi: 10.1371/journal.pone.0127948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puperi DS, Balaoing LR, O’Connell RW, West JL, Grande-Allen KJ. 3-Dimensional spatially organized PEG-based hydrogels for an aortic valve co-culture model. Biomaterials. 2015;67:354–364. doi: 10.1016/j.biomaterials.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci. 2016;113:E7–E15. doi: 10.1073/pnas1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting Organ-Level Lung Functions on a Chip. Science (80-) 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kloxin AM, Tibbitt MW, Anseth KS. Synthesis of photodegradable hydrogels as dynamically tunable cell culture platforms. Nat Protoc. 2010;5:1867–87. doi: 10.1038/nprot.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cruise GM, Scharp DS, Hubbell JA. Characterization of permeability and network structure of interfacially photopolymerized poly(ethylene glycol) diacrylate hydrogels. Biomaterials. 1998;19:1287–1294. doi: 10.1016/S0142-9612(98)00025-8. [DOI] [PubMed] [Google Scholar]
- 22.Kloxin AM, Lewis KJR, Deforest CA, Seedorf G, Tibbitt MW, Balasubramaniam V, Anseth KS. Responsive culture platform to examine the influence of microenvironmental geometry on cell function in 3D. Integr Biol. 2012;4:1540–1549. doi: 10.1039/c2ib20212c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gould ST, Darling NJ, Anseth KS. Small peptide functionalized thiol-ene hydrogels as culture substrates for understanding valvular interstitial cell activation and de novo tissue deposition. Acta Biomater. 2012;8:3201–9. doi: 10.1016/j.actbio.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009;30:6702–7. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang C, Mariner PD, Nahreini JN, Anseth KS. Cell-mediated delivery of glucocorticoids from thiol-ene hydrogels. J Control Release. 2012;162:612–618. doi: 10.1016/j.jconrel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leight JL, Alge DL, Maier AJ, Anseth KS. Direct measurement of matrix metalloproteinase activity in 3D cellular microenvironments using a fluorogenic peptide substrate. Biomaterials. 2013;34:7344–7352. doi: 10.1016/j.biomaterials.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leight JL, Tokuda EY, Jones CE, Lin AJ, Anseth KS. Multifunctional bioscaffolds for 3D culture of melanoma cells reveal increased MMP activity and migration with BRAF kinase inhibition. Proc Natl Acad Sci. 2015;112:201505662. doi: 10.1073/pnas.1505662112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz MP, Rogers RE, Singh SP, Lee JY, Loveland SG, Koepsel JT, Witze ES, Montanez-Sauri SI, Sung KE, Tokuda EY, Sharma Y, Everhart LM, Nguyen EH, Zaman MH, Beebe DJ, Ahn NG, Murphy WL, Anseth KS. A quantitative comparison of human HT-1080 fibrosarcoma cells and primary human dermal fibroblasts identifies a 3D migration mechanism with properties unique to the transformed phenotype. PLoS One. 2013;8:1–24. doi: 10.1371/journal.pone.0081689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franzdóttir SR, Axelsson IT, Arason AJ, Baldursson O, Gudjonsson T, Magnusson MK. Airway branching morphogenesis in three dimensional culture. Respir Res. 2010;11:162. doi: 10.1186/1465-9921-11-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choe C, Shin YS, Kim SH, Jeon MJ, Choi SJ, Lee J, Kim J. Tumor–stromal Interactions with Direct Cell Contacts Enhance Motility of Non-small Cell Lung Cancer Cells Through the Hedgehog Signaling Pathway. Anticancer Res. 2013;33:3715–3723. http://ar.iiarjournals.org/content/33/9/3715.abstract. [PubMed] [Google Scholar]
- 31.Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M, Allgayer H. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012;130:2044–53. doi: 10.1002/ijc.26218. [DOI] [PubMed] [Google Scholar]
- 32.Suganuma H, Sato A, Tamura R, Chida K. Enhanced migration of fibroblasts derived from lungs with fibrotic lesions. Thorax. 1995;50:984–989. doi: 10.1136/thx.50.9.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vuorinen K, Gäo F, Oury TD, Kinnula VL, Myllarniemi M. Imatinib mesylate inhibits fibrogenesis in asbestos-induced interstitial pneumonia. Exp Lung Res. 2007;33:357–373. doi: 10.1080/01902140701634827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Augsten M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front Oncol. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsh T, Pietras K, McAllister SS. Fibroblasts as architects of cancer pathogenesis. Biochim Biophys Acta - Mol Basis Dis. 2013;1832:1070–1078. doi: 10.1016/j.bbadis.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott RW, Hooper S, Crighton D, Li A, Konig I, Munro J, Trivier E, Wickman G, Morin P, Croft DR, Dawson J, Machesky L, Anderson KI, Sahai EA, Olson MF. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J Cell Biol. 2010;191:169–185. doi: 10.1083/jcb.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, Hooper S, Mitter R, Feral CC, Cook M, Larkin J, Marais R, Meneguzzi G, Sahai E, Marshall CJ. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell. 2011;20:229–245. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 39.Weiger MC, Vedham V, Stuelten CH, Shou K, Herrera M, Sato M, Losert W, Parent CA. Real-Time Motion Analysis Reveals Cell Directionality as an Indicator of Breast Cancer Progression. PLoS One. 2013;8:1–12. doi: 10.1371/journal.pone.0058859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vandenbroucke RE, Dejonckheere E, Libert C. Series “Matrix metalloproteinases in lung health and disease”: A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur Respir J. 2011;38:1200–1214. doi: 10.1183/09031936.00027411. [DOI] [PubMed] [Google Scholar]
- 41.Yadav RK, Gupta SP, Sharma PK, Patil VM. Recent Advances in Studies on Hydroxamates as Matrix Metalloproteinase Inhibitors: A Review. Curr Med Chem. 2011;18:1704–1722. doi: 10.2174/092986711795471329. [DOI] [PubMed] [Google Scholar]
- 42.Wang S, Li E, Gao Y, Wang Y, Guo Z, He J, Zhang J, Gao Z, Wang Q. Study on Invadopodia Formation for Lung Carcinoma Invasion with a Microfluidic 3D Culture Device. PLoS One. 2013;8 doi: 10.1371/journal.pone.0056448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–61. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 44.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006;3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 45.Marchand-Adam S, Marchal J, Cohen M, Soler P, Gerard B, Castier Y, Leseche G, Valeyre D, Mal H, Aubier M, Dehoux M, Crestani B. Defect of hepatocyte growth factor secretion by fibroblasts in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168:1156–61. doi: 10.1164/rccm.200212-1514OC. [DOI] [PubMed] [Google Scholar]
- 46.Puliafito A, Hufnagel L, Neveu P, Streichan S, Sigal A, Fygenson DK, Shraiman BI. Collective and single cell behavior in epithelial contact inhibition. Proc Natl Acad Sci. 2012;109:739–744. doi: 10.1073/pnas.1007809109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rejniak KA, Wang SE, Bryce NS, Chang H, Parvin B, Jourquin J, Estrada L, Gray JW, Arteaga CL, Weaver AM, Quaranta V, Anderson ARA. Linking changes in epithelial morphogenesis to cancer mutations using computational modeling. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doyle AD, Wang FW, Matsumoto K, Yamada KM. One-dimensional topography underlies three-dimensional fi brillar cell migration. J Cell Biol. 2009;184:481–490. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peyton SR, Kalcioglu ZI, Cohen JC, Runkle AP, Van Vliet KJ, Lauffenburger DA, Griffith LG. Marrow-Derived stem cell motility in 3D synthetic scaffold is governed by geometry along with adhesivity and stiffness. Biotechnol Bioeng. 2011;108:1181–1193. doi: 10.1002/bit.23027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herzog EL, Brody AR, Colby TV, Mason R, Williams MC. Knowns and unknowns of the alveolus. Proc Am Thorac Soc. 2008;5:778–82. doi: 10.1513/pats.200803-028HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guillot L, Nathan N, Tabary O, Thouvenin G, Le Rouzic P, Corvol H, Amselem S, Clement A. Alveolar epithelial cells: master regulators of lung homeostasis. Int J Biochem Cell Biol. 2013;45:2568–73. doi: 10.1016/j.biocel.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res. 2001;2:33–46. doi: 10.1186/rr36. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=59567&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11:S12–S15. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 54.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 55.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A. 2003;100:5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hogan BLM, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CCW, Niklason L, Calle E, Le A, Randell SH, Rock J, Snitow M, Krummel M, Stripp BR, Vu T, White ES, Whitsett JA, Morrisey EE. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15:123–38. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selman M, Thannickal VJ, Pardo A, Zisman DA, Martinez FJ, Lynch JP. Idiopathic Pulmonary Fibrosis. Drugs. 2004;64:405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 59.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M, Kaminski N. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2:0891–0903. doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mishra DK, Compean SD, Thrall MJ, Liu X, Massarelli E, Kurie JM, Kim MP. Human Lung Fibroblasts Inhibit Non-Small Cell Lung Cancer Metastasis in Ex Vivo 4D Model. Ann Thorac Surg. 2015;100:1167–1174. doi: 10.1016/j.athoracsur.2015.05.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.