

Abstract
Malignant pleural mesothelioma (MPM) is an aggressive malignancy with very limited therapeutic options. Fibroblast growth factor (FGF) signals play important roles in mesothelioma cell growth. Several FGFs and FGF receptors (FGFRs) are predicted targets of the miR‐15/16 family, which is downregulated in MPM. The aim of this study was to explore the link between the miR‐15/16 family and the FGF axis in MPM. Expression analyses via RT‐qPCR showed downregulation of the FGF axis after transfection with miR‐15/16 mimics. Direct interaction was confirmed by luciferase reporter assays. Restoration of miR‐15/16 led to dose‐dependent growth inhibition in MPM cell lines, which significantly correlated with their sensitivity to FGFR inhibition. Treatment with recombinant FGF2 prevented growth inhibition and further reduced the levels of FGF/R‐targeting microRNAs, indicating a vicious cycle between miR‐15/16 down‐ and FGF/FGFR signaling upregulation. Combined inhibition of two independent miR‐15/16 targets, the FGF axis and Bcl‐2, resulted in additive or synergistic activity. Our data indicate that post‐transcriptional repression of FGF‐mediated signals contributes to the tumor suppressor function of the microRNA‐15/16 family. Inhibiting hyperactivated FGF signals and Bcl‐2 might serve as a novel therapeutic combination strategy in MPM.
Keywords: fibroblast growth factor, fibroblast growth factor receptor; malignant pleural mesothelioma; microRNA‐15; microRNA‐16
Abbreviations
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- MPM
malignant pleural mesothelioma
1. Introduction
Malignant pleural mesothelioma (MPM) is a very aggressive malignancy of the pleural linings with dismal outcome (median survival of 9–17 months) and limited, mostly palliative therapeutic options (van Zandwijk et al., 2013). MPM is highly related to asbestos exposure, and due to the long latency period and the widespread use of asbestos, the worldwide incidence is expected to further increase within the next decades (Linton et al., 2012; Robinson, 2012).
As classical malignant drivers such as mutated EGFR or Raf are uncommon in MPM, treatments specifically targeting these mutations are ineffective in this disease (Dubey et al., 2010; Garland et al., 2007; Govindan et al., 2005). Instead, genomic analysis has revealed genetic deletions in MPM largely in genes considered to be tumor suppressive, such as P16/CDKN2A and NF2 (Jaurand and Fleury‐Feith, 2005), which are not easily targetable. We and others have previously identified fibroblast growth factors (FGFs) and their receptors (FGFRs) as signaling molecules being overexpressed and driving malignant growth in MPM (Marek et al., 2014; Schelch et al., 2014). Hyperactivation of FGF receptors and downstream pathways has been associated with tumor progression, therapy resistance, and poor prognosis (Korc and Friesel, 2009). However, unlike, for instance, FGFR1 in non‐small‐cell lung cancer (NSCLC) and squamous cell lung cancer (Weiss et al., 2010) or FGF19 in hepatocellular carcinoma (HCC) (Sawey et al., 2011), recurrent gene amplification of FGFs or FGFRs was not detected in MPM and thus cannot account for the frequent overexpression of several FGF/R family members found in MPM (Marek et al., 2014).
MicroRNAs (miRNAs) are small noncoding RNAs post‐transcriptionally regulating gene expression via interaction with sites in the 3′UTRs of target mRNAs (He and Hannon, 2004). They are known to be important regulators of cell signaling proteins during cancer development and progression (Garzon et al., 2009). In particular, the miR‐15/16 microRNA family that targets numerous cancer‐relevant factors including the anti‐apoptotic protein Bcl‐2 has been shown to function as a tumor suppressor in various tumors (Bandi et al., 2009; Bhattacharya et al., 2009; Bonci et al., 2008), and we have shown that this is also the case in MPM (Reid et al., 2013).
Recently, miR‐16 was shown to directly interact with FGF2 in nasopharyngeal carcinoma (He et al., 2016), and several other members of the FGF/R family are predicted targets of miR‐15/16. Here, we experimentally validated the targeting of FGF1, FGF2, and FGF18 as well as FGFR1 and FGFR4 by the miR‐15/16 family in MPM, suggesting a potential mechanism contributing to the overexpression of the FGF axis in MPM. We demonstrate that miR‐15/16 replacement downregulates FGF/FGFRs and provide evidence that inhibition of FGF/FGFR‐dependent signals contributes to the growth suppressive effects of miR‐15/16 mimics. Furthermore, we identified a vicious cycle between FGF signaling and miR‐15/16 downregulation, driving malignant growth. Combined inhibition of two independent miR‐15/16 targets, FGFR1 and the Bcl‐2, resulted in synergistic growth inhibition, suggesting microRNA replacement as a novel therapy for FGFR‐dependent MPM and other FGFR‐dependent tumors.
2. Material and methods
2.1. Cell culture and cell lines
All cell lines were maintained in RPMI‐1640 or DMEM medium (Thermo Fisher, Waltham, MA, USA) containing 10% heat‐inactivated FBS in a humidified atmosphere of 5% CO2 at 37 °C. Cell lines were regularly checked for mycoplasma contamination (MycoFluor, Thermo Fisher) and STR profiling was used to confirm identity (GenePrint 10, Promega, Madison, WI, USA) once per year. All used cell lines, the histological MPM subtype they were derived from, their standard growth medium as well as their source are listed in Table S1.
2.2. MicroRNA mimics and siRNAs
MicroRNA mimics as well as validated negative control mimics (nc) were dissolved in ultrapure H2O and transfected as described below. For siRNAs, a nontargeting control siRNA (c‐81) was used. SiRNA specific for RRM1 served as control for transfection efficacy. All used microRNA mimics and siRNAs, their sequences/IDs and sources are listed in Table S2.
2.3. Cytokines and drugs
Recombinant FGF2 or FGFR/Bcl‐2 inhibitors were directly added to cells at the indicated concentrations. As control, equal amounts of solvent (PBS for FGF2, DMSO for all inhibitors) were added. Treatments, their targets and sources are summarized in Table S3.
2.4. Transfection with microRNA mimics and siRNAs
Reverse transfection with microRNA mimics or siRNAs was carried out as per the pipetting scheme in Table S4. First, mimics or siRNAs were diluted in serum‐free RPMI medium to the required concentrations. Lipofectamine (RNAiMAX, Thermo Fisher) was diluted 1 : 100 in serum‐free medium. After at least 5 minutes, appropriate volumes of lipofectamine and diluted mimic/siRNA were mixed and incubated for at least 20 minutes and up to 2 h at room temperature. Meanwhile, cells were harvested, counted, and diluted as required. Finally, the lipofectamine/RNA mix was added into each well and cells were added, gently mixed, and transferred to the incubator.
2.5. RNA isolation and RT‐qPCR
Cells were transfected in 6‐well plates (1.5 × 104 cells per well) with microRNA mimics (5 nm) or siRNAs (10 nm) according to the scheme in Table S4. After the indicated time, RNA was isolated using TRIzol (Thermo Fisher) according to the manufacturer's instructions and dissolved in ultrapure H2O. Concentration and purity of the RNA were measured on a nanophotometer (Implen, Munich, Germany). For the analysis of target genes, 500 ng RNA was reverse‐transcribed with Superscript III reverse transcriptase (Thermo Fisher). To measure the expression of microRNAs, 50 ng RNA was reverse‐transcribed using MultiScribe reverse transcriptase (Thermo Fisher) and stem‐loop RT primers specific for the microRNA of interest. SYBR green‐ or TaqMan‐based real‐time quantitative PCR was performed according to the manufacturer's instructions on an ABI‐7500 (Applied Biosystems, Foster City, CA, USA) or ViiA7 (Thermo Fisher) thermocycler. 18S was used as a reference for mRNA, and RNU6B and miR‐191 as references for microRNA expression, respectively. All used TaqMan microRNA assays are listed in Table S5. Primers and sequences for SYBR green‐based PCR are in Table S6. Relative levels of mRNAs or microRNAs were calculated as previously described (Reid et al., 2013) using the 2−ΔΔCq method (Livak and Schmittgen, 2001).
2.6. Luciferase reporter assay
Fragments of the 3′UTRs of FGF1, FGF2, FGF5, FGF18, FGFR1, and FGFR4 containing binding sites for miR‐15a, miR‐15b, and miR‐16 were cloned from SPC212 cDNA. Total RNA was reverse‐transcribed (MMLV RT kit, Promega) and fragments were amplified using AmpliTaq Gold 360 (Promega) with specific forward and reverse primers (Table S7). PCR products were first cloned into the TOPO TA vector (Thermo Fisher) and then subcloned into the pSiCheck2 plasmid (Promega). The mutated FGFR1 constructs were generated by site‐directed mutagenesis. Briefly, specific, completely overlapping primers harboring a mutation in the microRNA binding site were designed (Table S7) and a PCR was carried out using PfuUltra II Fusion HS DNA Polymerase (Agilent, Santa Clara, CA, USA) followed by DpnI (NEB, Ipswich, MA, USA) digest. Sequences were confirmed by Sanger sequencing carried out at the Ramaciotti Centre (UNSW, Sydney). The resulting reporter constructs (1 μg), together with microRNA mimics or controls (5 nm), were used to transfect 2 × 105 cells in 6‐well plates. A dual luciferase assay (Promega) was carried out as per the manufacturer's protocol 48 h after transfection.
2.7. Protein isolation and western blot
For protein analysis, 4 × 105 cells in T25 flasks were transfected with 2.5 nm microRNA mimics. After 96 h, protein was isolated in lysis buffer (150 mm NaCl, 50 mm HEPES, 10% glycerol, 1 mm EDTA, 0.5 mm Na3VO4, 10 mm NaF, 1% Triton X‐100, and 1.5 mm MgCl2) and concentration was measured using a Bradford protein assay (Bio‐Rad, Hercules, CA, USA). 20 μg of protein per lane was separated by SDS/PAGE (precast, NuSep) and blotted onto PVDF membranes. Immunodetection was performed using antibodies against FGFR1 (D8E4 XP, #9740, Cell Signaling; 1 : 1000) and FGFR4 (H‐121, sc‐9006, Santa Cruz, Dallas, TX, USA; 1 : 1000). Beta‐actin (#A5441, Sigma, St. Louis, MO, USA; 1 : 10 000) served as loading control.
2.8. Growth inhibition assay
Triplicate of 2.5 × 103 MPM cells were transfected with microRNA mimics, siRNAs, or various combinations as indicated and seeded in a 96‐well plate with a total volume of 120 μL per well. Usually, mimics were used at final concentrations of 0.2 nm, 1 nm, and 5 nm and siRNAs at 10 nm. In case of additional treatment with cytokines or drugs, 80 μL of medium containing the compound at the required concentrations or vehicle was added to a total volume of 200 μL per well. To test the effects of drugs alone, 2 × 103 cells per 96‐well in a volume of 100 μL medium were seeded in triplicate. On the next day, cells were treated with 100 μL of medium containing drugs single or in combination as indicated. At the indicated time points, the medium was discarded and plates were frozen at −80 °C. For analysis, plates were thawed and 200 μL lysis buffer (10 mm Tris/HCl pH = 8, 2.5 mm EDTA, 0.1% Triton X‐100) containing SYBR green (10 000×, Thermo Fisher, 1 : 8000) was added. Plates were incubated overnight at 4 °C in the dark. Fluorescence was measured at 485/520 nm on a FLUOstar OPTIMA microplate reader (BMG Labtech, Offenburg, Germany).
2.9. MTT assay
Triplicate of 2 × 103 cells in 100 μL medium were seeded into a 96‐well plate. On the next day, 100 μL of medium containing treatments was added. DMSO was used as solvent control. After 72 h, cell viability was measured by MTT assay according to the manufacturers' protocol (EZ4U, Biomedica, Vienna, Austria).
2.10. Colony formation assay
Cells (2.5 × 103) were transfected with mimics in 96‐well plates as described above. On the next day, cells from each well were harvested and half transferred into each of two wells of a 6‐well plate, resulting in approximately 1000–1500 cells per well, and returned to the incubator. After 7–14 days, cells were fixed with methanol/acetic acid (3 : 1) and clones were stained with crystal violet and air‐dried and pictures were taken. For quantification, clones were destained in 2% SDS, and absorbance was read at 562 nm.
2.11. Statistical analysis
If not stated otherwise, all data are presented as means ± SEM of at least three independent experiments performed in triplicate. Statistical significance between control and treatment groups was calculated using Prism7 (GraphPad, San Diego, CA, USA) using one‐way ANOVA with Dunnett's multiple comparison test for comparison of multiple groups. Pearson's correlation was used to investigate correlations between the effect of microRNAs re‐expression on cell growth and sensitivity to pharmacological FGFR inhibition. A P‐value <0.05 was considered statistically significant. AUCs were calculated with Prism7. Combination indices (CI values) were calculated using the compusyn software (CompuSyn Inc., Paramus, NJ, USA). Predicted values (PV) for additive effects represent the arithmetic products of the % viability of each single treatment.
3. Results
3.1. Downregulation of the microRNA‐15/16 family corresponds with an upregulation of the FGF axis in MPM cell lines
Recently, our work has shown hyperactivation of FGF signals due to overexpression of FGFR1, FGF2, and FGF18 in MPM (Schelch et al., 2014). As there are no gene amplifications or mutations reported, we reasoned that microRNAs might play a role in the aberrant FGF/R expression. We used target prediction (TargetScanHuman 7.1, Agarwal et al., 2015) and found that the tumor‐suppressive miR‐15/16 microRNA family, which is frequently downregulated in tumors including MPM (Reid et al., 2013), is predicted to target several members of the FGF axis.
To investigate the relationship between the miR‐15/16 family and the FGF axis, we first measured the expression of miR‐15a, miR‐15b, and miR‐16 as well as several members of the FGF/FGFR family in a panel of seven MPM cell lines (plus five drug‐resistant derivatives) via RT‐qPCR. MeT‐5A, a nonmalignant transformed mesothelial cell line, was used as a reference. We found a prominent overexpression of FGFR1, FGF2, and FGF18 (Fig. 1A) as well as reduced expression of the microRNAs miR‐15a, 15b, and 16 in the MPM cell lines compared with MeT‐5A (Fig. 1B).
Figure 1.
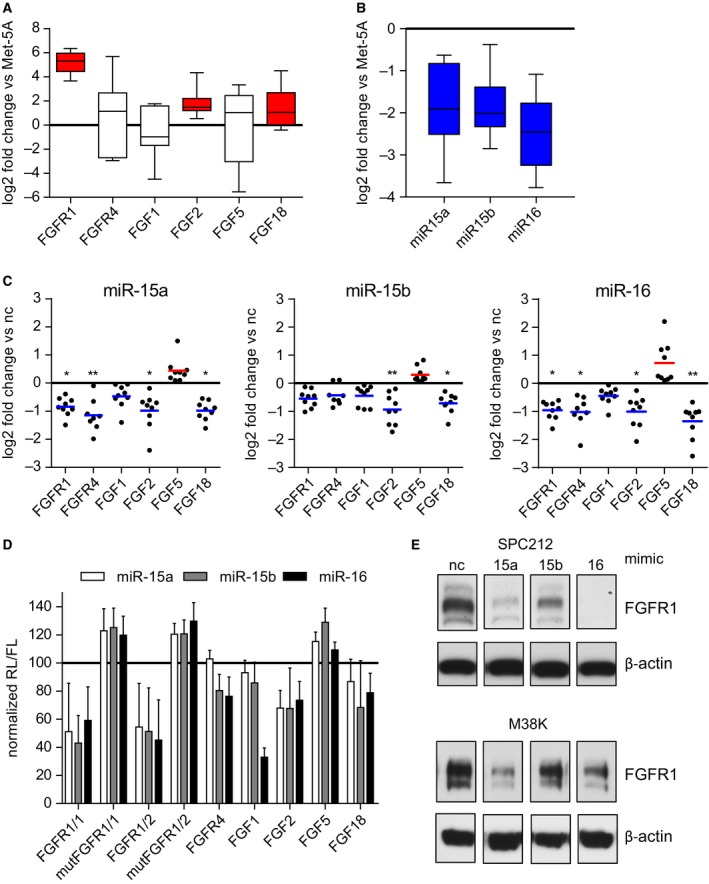
The microRNAs 15a, 15b, and 16 are downregulated in mesothelioma cells and target several members of the FGF family. Expression of (A) several members of the FGF axis and (B) the microRNA‐15/16 family compared to MeT‐5A, analyzed by RT‐qPCR. (C) Cells were transfected with microRNA mimics (5 nm) and target gene expression was checked after 24 h via RT‐qPCR and compared with noncoding (nc) control mimics. Each dot represents one cell line and is depicted as the mean of three independent experiments. (D) Normalized percentage of luciferase activity (renilla/firefly ratio, RL/FL) 48 h after transfection with microRNA or control mimics (5 nm) in SPC212 cells. (E) Immunoblot analysis of FGFR1 expression 96 h after transfection with microRNA or control mimics (2.5 nm). *P < 0.05, **P < 0.01, ***P < 0.001; otherwise, P > 0.05.
3.2. The miR‐15/16 family targets the FGF axis in MPM
In order to directly assess the impact of the miR‐15/16 family members on FGF/FGFR expression in MPM cells and to validate the prediction of FGF/FGFRs as miR‐15/16 targets in MPM, we reintroduced each of the three miRNAs into the MPM cell lines via transfection with mimics and measured target gene expression via RT‐qPCR. All three miRNA mimics led to downregulation of the expression of FGFR1 and FGFR4, which are both predicted targets of the respective miRNAs. Among the ligands, FGF1, FGF2, and FGF18 are predicted targets and all three were also reduced upon mimic expression (Fig. 1C). FGF5 is not a target of the miR‐15/16 family and, as expected, was not downregulated. Luciferase reporter assays confirmed the direct interaction between miR‐15a, 15b, and 16 and their predicted target sites in the 3′UTR of FGFR1 (FGFR1/1, FGFR1/2), FGFR4, FGF1, FGF2, and FGF18, but no interaction with FGF5 or mutated target sites of FGFR1/1 or FGFR1/2 was found. (Fig. 1D). We also observed a downregulation of FGFR1 on the protein level after transfection with miRNA mimics (Fig. 1E). This was more pronounced with miR‐15a and miR‐16, reflecting the changes in the mRNA level. These findings establish several FGF/FGFR members as experimentally validated targets of the miRNA 15/16 family in mesothelioma.
3.3. Growth repression by miRNA mimics correlates with sensitivity to FGFR1 inhibition in MPM cells
In line with our previous findings, transfection with miRNA mimics led to a dose‐dependent growth repression of MPM cells (Fig. 2A). In agreement with the effects on target gene expression, miR‐15b also had the weakest impact on MPM cell growth. The strongest effect was seen in MSTO, SPC212, and P31 cells, while VMC20, M38K, SPC111 as well as the control mesothelial cell lines MeT‐5A and LP9 were only moderately affected. Restoration of miR‐15/16 also dose dependently reduced the ability to form colonies in MPM cells when plated at low density (Figs 2B and C, S1).
Figure 2.
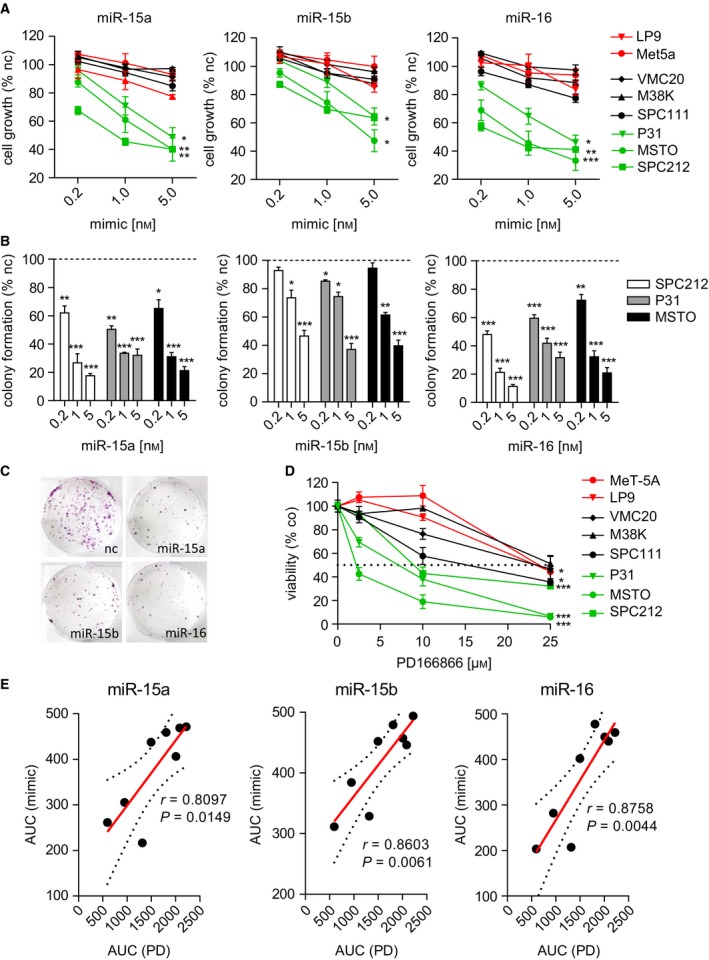
Growth repression by microRNA mimics correlates with sensitivity to FGFR1 inhibition. (A) Growth inhibition determined by SYBR green staining 72 h after transfection with mimics compared with noncoding control (nc). (B) Quantification of colony formation assays of the three sensitive cell lines after transfection with mimics as indicated. (C) Representative colony formation pictures of MSTO cells transfected with 5 nm microRNA or control mimics as indicated. (D) Dose–response curves of MPM cell lines 72 h after treatment with PD166866 or DMSO (co), determined by MTT assay. (E) Growth inhibition shown as area under curve (AUC) of mimics (5 nm) in correlation (Pearson) effects of PD166866, calculated from MTT dose–response curves in E. Each dot represents one cell line. *P < 0.05, **P < 0.01, ***P < 0.001; otherwise, P > 0.05.
We previously reported that FGFR1 inhibition reduces MPM tumor growth in vitro and in vivo (Schelch et al., 2014). In our panel of MPM cell lines, MTT assays using the small‐molecule‐specific FGFR1 inhibitor PD166866 showed a dose‐dependent decrease in cell viability reflecting the effects seen by miRNA mimics (Fig. 2D). There was a significant correlation (Pearson) between the effectiveness of growth repression by mimic expression and sensitivity to PD166866 (Fig. 2E). The fact that those cell lines which are sensitive to miR‐15/16 are also more responsive to FGFR1 inhibition suggests that blockade of FGF/FGFR‐dependent growth/survival signals is an important player in growth regulation by this microRNA family.
3.4. Stimulation with FGF2 can rescue mimic‐induced growth repression of MPM cells at early but not late time points
We reasoned that in MPM cells dependent on FGF signals for cell growth and survival, growth suppression upon mimic‐induced downregulation of FGFs would be prevented by stimulation with exogenous FGF2 as long as FGFRs are sufficiently expressed. Therefore, to further characterize the interaction between miR‐15/16 and the FGF axis, we stimulated the cells at two different time points: 24 and 96 h after transfection. Indeed, FGF2 treatment 24 h after transfection could reduce/prevent the growth inhibition effects caused by mimics (Figs 3A and S2). In contrast, when added 96 h after transfection, FGF2 had no effect on cell growth (Figs 3B and S2). This is most likely explained by reduced FGFR1 protein at this late time point as shown in Fig. 1E.
Figure 3.
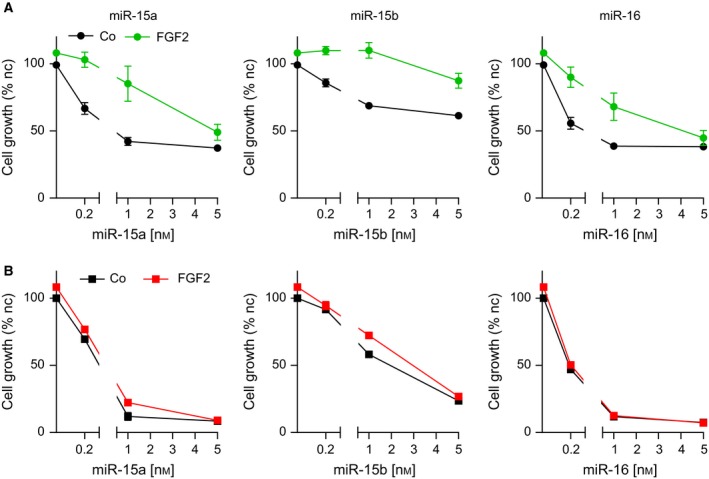
Stimulation with FGF2 reduces mimic‐induced growth inhibition of MPM cells at early but not late time points. Growth inhibition assay of SPC212 cells after transfection with mimics at indicated concentrations in combination with FGF2 treatment (10 ng·mL−1) (A) 24 h after mimic transfection or (B) 96 h after transfection. For the latter, each well was split 1 : 3 on the day before treatment to avoid complete confluence. Assays were quantified 72 h after FGF2 treatment. *P < 0.05, **P < 0.01, ***P < 0.001; otherwise, P > 0.05.
3.5. MicroRNAs targeting the FGF axis are regulated by FGFR‐mediated signals in a vicious cycle
Our data suggest that one of the functions of the miR‐15/16 family is to keep FGF signals in check. As regulation of growth signals often involves feedback loops to maintain homeostasis, we next investigated whether activated FGF signals would in turn enhance microRNA expression. We determined the expression of miR‐15a, miR‐15b, miR‐16, and their respective precursor pri‐miRs via Taqman‐based RT‐qPCR after 24‐h stimulation with recombinant FGF2 and found further downregulation of miR‐15/16 in the majority of cell lines (Fig. 4A, Table S8). However, there was no significant correlation between expression changes and sensitivity to mimics/FGFR inhibition (data not shown). In addition to miR‐15a, ‐15b, and ‐16, miR‐195, and miR‐424—additional members of this family—as well other microRNAs predicted to target various members of the FGF axis—miR‐23a, miR‐24, and miR‐223—were also downregulated upon FGF pathway activation. In contrast, miR‐93, miR‐103, miR‐137, and miR‐193a‐3p, which are not involved in the regulation of the FGF axis, remained largely unchanged (Fig. 4B, Table S8). These data suggest that a feed‐forward cycle is operational in the mutual regulation between the miR‐15/16 family and the FGF axis in MPM cells. Once triggered, this feed‐forward loop could result in a vicious cycle contributing to enhanced cell growth in MPM. This model would suggest that, conversely, inhibition of FGF signals could partially rescue miR‐15/16 expression. Therefore, we treated the three sensitive cell lines with the FGFR1 inhibitor PD166866 as well as the multikinase inhibitor ponatinib, which inhibits FGFR1‐4, Abl, PDGFRA, and Src, and assessed the expression of miR‐15/16. Indeed, after 48 h, we observed with both treatments an upregulation of miR‐15a, ‐15b, and ‐16 (Fig. 4C).
Figure 4.
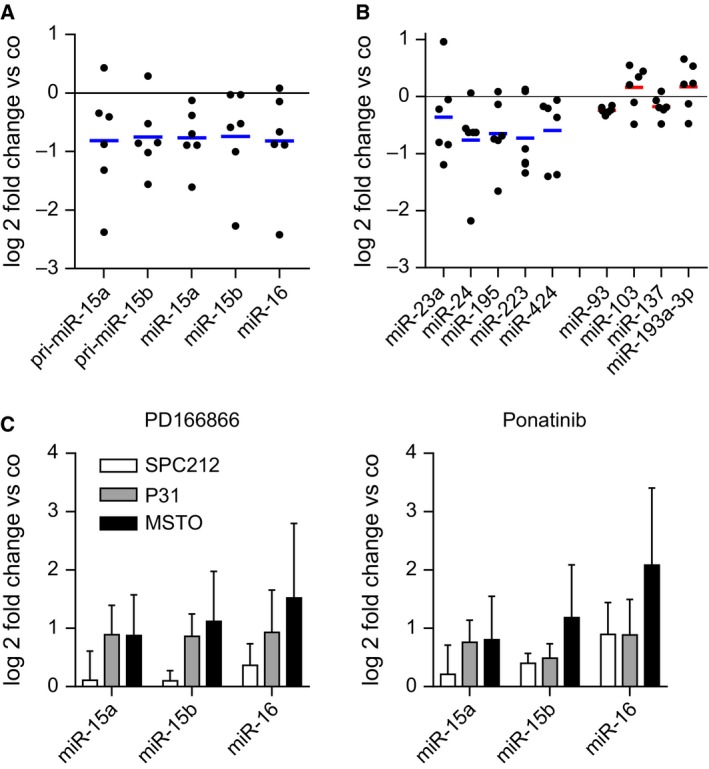
Inhibition or stimulation of FGF signals regulates the microRNA‐15/16 family. (A) Expression of the miR‐15a, miR‐15b, miR‐16, and their respective pri‐miRs or (B) miR‐23a, miR‐24, miR‐195, miR‐223, and miR‐424 as well as miR‐93, miR‐103, miR‐137, and miR‐193a‐3p after treatment with FGF2 (10 ng·mL−1) for 24 h compared with control cells was determined by qPCR. Each dot represents a cell line and is depicted as the mean of three experiments performed in duplicate. (C) MicroRNA expression determined by qPCR after 24 h treatment with PD166866 (10 μm) or ponatinib (500 nm) and respective vehicle controls (Co). *P < 0.05, **P < 0.01, ***P < 0.001; otherwise, P > 0.05.
3.6. Competition between microRNA restoration and FGFR inhibition confirms interaction between miR‐15/16 and the FGF axis
As our data suggest that mutual regulation between miR‐15/16 and FGF signals drives enhanced growth of MPM cells, interference with either one or both mechanisms could prove therapeutically beneficial. Indeed, we have previously demonstrated that restoration of miR‐16 is feasible in MPM models and results in growth suppression in vitro and in vivo (Reid et al., 2013) and several groups including our own have established FGFR inhibition as viable anti‐MPM strategy (Blackwell et al., 2016; Marek et al., 2014; Schelch et al., 2014). Thus, we tested the effect of combining microRNA replacement with pharmacological FGFR inhibition. The combination of mimic expression with the FGFR1 inhibitor PD166866 reached a higher total growth inhibition compared with each treatment alone. However, with higher doses of mimics, the combinations showed reduced effects compared with the single treatments, indicating target competition (Fig. 5A). Comparable results, showing effects weaker than the predicted value (PV, indicated as white ticks within the black bars) of additive interactions, were obtained when we combined miRNA mimics with siRNA against FGFR1, FGFR4, or a combination of both (Figs 5B, S3 and S4).
Figure 5.
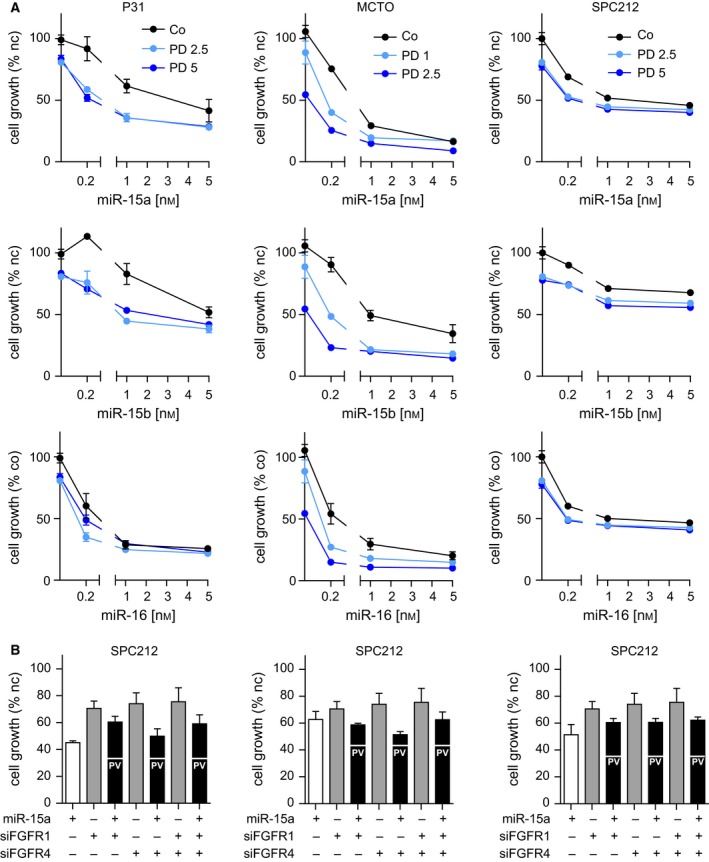
Combination of microRNA restoration and FGFR inhibition or knockdown shows effects of target competition. (A) Growth inhibition assay of MPM cells after transfection with microRNA or noncoding control mimics in combination with PD166866. Treatment was for 72 h with the indicated concentrations (1 μm, 2.5 μm, or 5 μm). DMSO was used as solvent control. (B) Growth inhibition assay 72 h after cells were transfected with mimics (5 nm) and/or siRNAs (10 nm) and/or respective controls (nc). Predicted values of additive interaction (PV) are indicated as white lines within the black combination bars. *P < 0.05, **P < 0.01, ***P < 0.001; otherwise, P > 0.05.
3.7. Combined inhibition of the miR‐15/16 targets FGFR1 and Bcl‐2 has synergistic effects in MPM cells
We reasoned that inhibition of two independent targets of miR‐15/16 might avoid the target competition seen in Fig. 5. A well‐established target of the miR‐15/16 family is the anti‐apoptotic protein Bcl‐2, for which both the clinically relevant specific inhibitor venetoclax and the broadly active Bcl‐2 family inhibitor obatoclax are available. First, we confirmed the downregulation of Bcl‐2 upon transfection with microRNA mimics in our panel of cell lines via RT‐qPCR. As expected, Bcl‐2 levels were reduced in all cell lines (Fig. 6A). Therefore, we next tested the FGFR1 inhibitor PD166866 as well as the multikinase inhibitor ponatinib in combination with venetoclax and obatoclax. Used as single agents, obatoclax was highly effective in the nanomolar range; venetoclax, however, required much higher concentrations to inhibit growth in MPM cells (Fig. 6B). Importantly, in combination with low‐dose FGFR inhibitors, we found enhanced effects of both obatoclax and venetoclax. The combination of obatoclax and ponatinib showed synergistic activity in all three cell lines tested, with combination indices (CI) below 1, especially at the lower concentrations (Fig. 6C). In the other combination settings, where calculation of CI values was not possible, effects stronger than the predicted value (PV, indicated as red bars) of additive interactions were observed at concentrations of 1 μm PD166866 or 100 nm ponatinib in combination with 50 nm obatoclax or 10 μm venetoclax (Figs 6C and S5).
Figure 6.
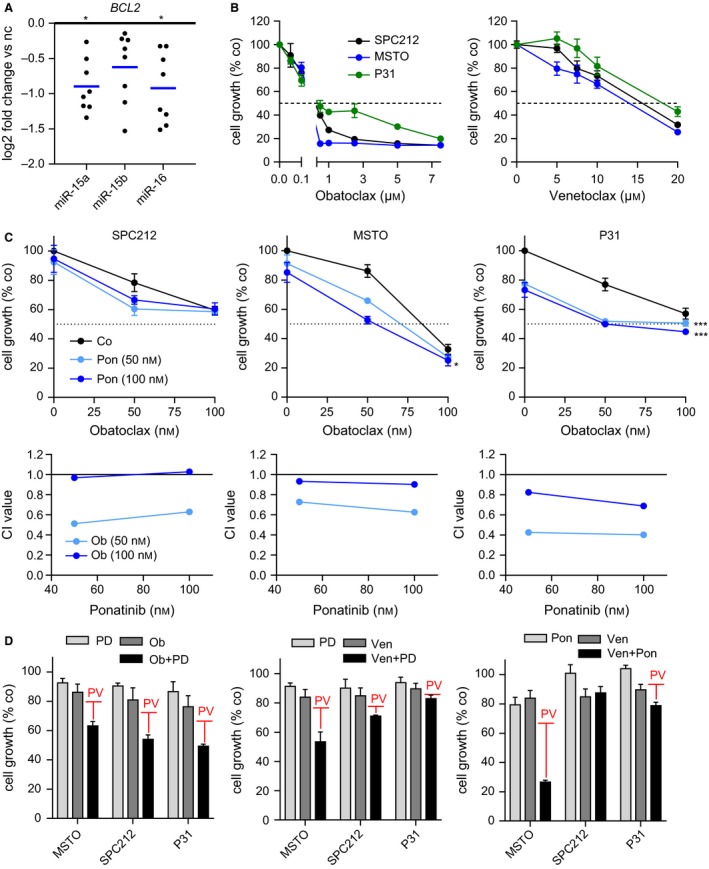
Pharmacological inhibition of the FGF axis in combination with Bcl‐2 has synergistic effects in MPM. (A) Expression analysis of BCL2 via qPCR 24 h after transfection with microRNA mimics (5 μm) compared with noncoding (nc) control mimics. (B) Growth inhibition assays using obatoclax and venetoclax as single agents at indicated concentrations for 72 h. DMSO was used as solvent control. (C) Growth inhibition of MPM cells after treatment with ponatinib (Pon, 50 nm, 100 nm) in combination with obatoclax (Ob, 50 nm, 100 nm) for 72 h. DMSO was used as solvent control. Combination indices (CI values) were calculated using CompuSyn. (D) Growth inhibition of PD166866 (PD, 1 μm) or ponatinib (Pon, 100 nm) in combination with obatoclax (Ob, 50 nm) or venetoclax (Ven, 10 μm) for 72 h. The red bars represent the predicted value of additive interaction (PV) of the two respective single treatments. *P < 0.05, **P < 0.01, ***P < 0.001; otherwise, P > 0.05.
4. Discussion
The translation of specific molecularly targeted approaches into clinical practice, often in combination with conventional chemo‐ or radiotherapy, has become a promising strategy, which is currently improving the treatment for various other cancers (Iams et al., 2017; Lemjabbar‐Alaoui et al., 2015). However, MPM remains an exception. Apart from a modest increase in median survival by the addition of bevacizumab to the standard pemetrexed/platinum combination, this asbestos‐related cancer (Zalcman et al., 2015), with increasing worldwide incidence, continues to be renowned for its treatment resistance. Therefore, novel insight into the molecular basis of treatment resistance and more effective treatment strategies are urgently needed. Our previous work characterized the FGF axis as a potent malignant driver in MPM, and we also described the tumor‐suppressive role of the miR‐15/16 family, which is frequently downregulated in MPM (Reid et al., 2013; Schelch et al., 2014).
So far, the tumor suppressor function of miR‐15/16, which targets important drivers of malignant growth such as FGF2 and the anti‐apoptotic protein Bcl‐2 (He et al., 2016; Willimott and Wagner, 2010), has been well established in various tumor types including prostate, lung, ovarian, and MPM (Bandi et al., 2009; Bhattacharya et al., 2009; Bonci et al., 2008; Reid et al., 2013). We demonstrate here that several members of the FGF axis (FGFR1, FGFR4, FGF1, FGF2, and FGF18) are prominent miR‐15/16 targets, downregulated in MPM cells upon microRNA restoration via mimics.
The loss of post‐transcriptional control of the FGF axis due to downregulation of microRNAs is a potential mechanism underlying the high expression of the FGF axis in tumors such as MPM, which do not show gene amplifications or mutations (Marek et al., 2014). However, a lack of a significant correlation between basal FGF/R levels and miR‐15/16 expression suggests that the levels of this microRNA family contribute to, but do not completely control, regulation of FGF/R expression. Besides miR‐15/16, other microRNAs have also been shown to impact on FGF signals in malignant and nonmalignant diseases, including miR‐99a, miR‐214, or miR‐216 (Jiang et al., 2014; Wang et al., 2008, 2013). Interestingly, miR‐99a was found at lower levels in MPM samples compared with normal pleura (Andersen et al., 2014) and differentially expressed between epithelioid and sarcomatoid tumors (Pass et al., 2010). Similarly, miR‐214 was shown to be downregulated in MPM samples compared with controls in two independent studies (Amatya et al., 2016; Balatti et al., 2011). In addition, we found that FGF signaling suppressed the expression of miR‐23a and miR‐223, both of which are altered in MPM and predicted to target components of the FGF axis (Birnie et al., 2015; Cheng et al., 2013).
Previously, high levels of both amplified and nonamplified FGFRs have been linked to response to FGFR inhibition in various cancers (Goke et al., 2015; Nakanishi et al., 2014; Wynes et al., 2014; Zhang et al., 2014). In accordance with this, we have recently shown that nonmalignant mesothelial cells express much lower levels of FGF/R and are more resistant to FGFR inhibition than MPM cells (Schelch et al., 2014). However, among the MPM cell lines tested, neither FGF/R nor miR‐15/16 basal expression correlated with sensitivity to FGFR inhibition or mimics. Also, comparable to MeT‐5A, some MPM cell lines did not respond to growth inhibition. This suggests that the actual signals which trigger cell proliferation and survival and therefore the extent of dependency on the FGF axis are controlled by a combination of multiple factors including transcriptional and translational regulation and other effectors acting downstream of the FGF receptors. MicroRNAs targeting specific components of the FGF family represent one part of this regulatory system, which is supported by effects of target competition when we combined microRNA mimics with FGFR inhibitors. Also, it is well known that the FGF axis is part of a complex signaling network, which interacts with numerous other pathways and is influenced by various feedback and feed‐forward loops (Ornitz and Itoh, 2001). We have uncovered a vicious cycle in MPM where FGF signaling is capable of downregulating the miR‐15/16 family and other FGF/R‐targeting microRNAs, thereby further driving their growth‐promoting effects by inhibiting their own microRNA‐mediated post‐transcriptional regulation.
The involvement of the same tumor suppressor microRNAs in the regulation of multiple cancer‐related signaling pathways highlights the potential of microRNA mimics as novel cancer treatment approach. In addition, it is unlikely that microRNA‐based therapy is negatively affected by mutation‐induced resistance usually occurring with tyrosine kinase inhibitors. MicroRNA replacement has been evaluated in preclinical and clinical studies (Bader, 2012; Liang et al., 2016; Reid et al., 2013; Takeshita et al., 2010). In MPM, a miR‐16‐based mimic has shown early signs of clinical activity (Kao et al., 2015). As the underexpression/loss of members of the miR‐15/16 family is not a unique characteristic of MPM and considering the important role of the FGF axis in tumor development and progression, it is more than likely that the deregulation of FGF signals also contributes to the tumor‐promoting effects of miR‐15/16 loss in other tumor types. Therefore, the miR‐16 suppletion approach needs further investigation for tumors characterized with mutated or dysregulated FGF signaling.
We demonstrate here that selecting microRNA target proteins which (a) have a well‐documented role in driving malignant growth and (b) can be targeted by pharmacological inhibition represents a rational approach for combination treatment in MPM. This approach enabled us to identify that FGFR1 and Bcl‐2 represent novel synergistic treatment targets in MPM. Multiple members of the miR‐15/16 family may be targeted with microRNA mimics, whereas FGFRs and Bcl‐2 have the advantage of being druggable. FGFR inhibition has shown promising effects in various preclinical studies (Dey et al., 2010; Maruyama‐Takahashi et al., 2008; Metzner et al., 2011; Pattarozzi et al., 2017; Qing et al., 2009; Schelch et al., 2014) and is currently being tested in patients with solid tumors, especially in those with known FGFR overexpression or genomic alterations (Katoh and Nakagama, 2014). In mesothelioma, a phase lll study using the multi‐RTK inhibitor nintedanib (NCT01907100) and a phase lb trial using the FGFR ligand trap GSK3052230 in combination with first‐line chemotherapy (NCT01868022) are ongoing. Similarly, inhibition of Bcl‐2 also showed promising effects in various cancers including MPM (Hoda et al., 2016; Li et al., 2008; Schwartz et al., 2016; Xiong et al., 2016). Other combined treatment approaches have been tested in MPM (Kanteti et al., 2014; Ou et al., 2016), and a trial of FAK and PD1 inhibition (NCT02758587) is ongoing. With the lack of targetable mutations in MPM, novel treatments focusing on gene expression and genomic alterations are gaining increasing attention. For example, a trial of the EZH2 inhibitor in the context of BAP1 loss‐of‐function mutation (NCT02860286) is currently recruiting. Similarly, our characterization of the impact of microRNA expression changes on MPM biology led us to identify the synergistic effect of combining inhibitors of FGFR1 and Bcl‐2, two independent targets of the miR‐15/16 pathway. With the limited progress in MPM treatment over the last decade, continuing use of alternative approaches for the identification of novel MPM treatment targets will be needed to improve the outlook for patients.
In conclusion, our study suggests that loss of the miR‐15/16 family plays a role in the overexpression of the FGF axis in MPM. We identified a vicious cycle of malignant growth between FGF signals and miR‐15/16 and show that cells which are more sensitive to FGFR inhibition also respond better to microRNA mimics, providing evidence for microRNA replacement as an alternative therapeutic approach in MPM and other tumors harboring FGF/R mutations or amplifications. Furthermore, we demonstrate that combined inhibition of two miR‐15/16 targets, FGFR1 and Bcl‐2, has synergistic effects on tumor cell growth encouraging further consideration as novel combination strategy in MPM.
Author contributions
GR, MG, NvZ, and KS designed the study. KS, MBK, YYC, and MW conducted experiments and performed data analysis. KS, MBK, NvZ, and GR wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig. S1. Representative pictures of MSTO cells in a colony formation assay after transfection with microRNA mimics or non‐coding control.
Fig. S2. Growth inhibition assays of P31 and MSTO cells after transfection with mimics in combination with FGF2 treatment.
Fig. S3. Target gene and protein expression analysis after transfection with siRNAs via qPCR or western blot.
Fig. S4. Growth inhibition assays of P31 and MSTO cells 72 h after transfection with microRNA mimics and/or siRNAs or respective controls.
Fig. S5. Growth inhibition assays of SPC212, MSTO and P31 cells 72 h after treatment with PD166866 (PD) or ponatinib (Pon) in combination with obatoclax or venetoclax, or respective controls, at the indicated doses.
Table S1. List of used cell lines, the histological MPM subtype they were derived from, standard growth media and sources.
Table S2. List of mimics and siRNA, their sequences/IDs and sources.
Table S3. List of cytokines and drugs, their targets and sources.
Table S4. Transfection schemes for different assay formats.
Table S5. List of primers and probes used in TaqMan‐based reverse transcription and qPCR.
Table S6. Primers and their sequences used for SYBR green‐based qPCR.
Table S7. Primers and their sequences used for Luciferase reporter assays.
Table S8. Fold change, standard deviation and P‐values for microRNAs of individual cell lines after treatment with FGF2.
Acknowledgements
This work was supported by the Marietta Blau Scholarship, OeAD, Federal Ministry of Science and Research, Austria (KS), grants of the Herzfelder Foundation and the Initiative‐Krebsforschung of the Medical University of Vienna, Austria (MG), and a Translational Program Grant (11/TPG/3‐06) from the Cancer Institute NSW, Australia to NvZ and GR. We thank M. Eisenbauer for help with establishing VMC20, R. Stahel for SPC111 and SPC212 cells, K. Grankvist for P31, V.L. Kinnula for M38K, and the University of Queensland Thoracic Research Centre (The Prince Charles Hospital, Brisbane, Australia) for kindly providing MM05 cells.
References
- Agarwal V, Bell GW, Nam J‐W and Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatya VJ, Mawas AS, Kushitani K, Mohi El‐Din MM and Takeshima Y (2016) Differential microRNA expression profiling of mesothelioma and expression analysis of miR‐1 and miR‐214 in mesothelioma. Int J Oncol 48, 1599–1607. [DOI] [PubMed] [Google Scholar]
- Andersen M, Grauslund M, Ravn J, Sorensen JB, Andersen CB, and Santoni‐Rugiu E (2014) Diagnostic potential of miR‐126, miR‐143, miR‐145, and miR‐652 in malignant pleural mesothelioma. J Mol Diagn 16, 418–430. [DOI] [PubMed] [Google Scholar]
- Bader AG (2012) miR‐34 – a microRNA replacement therapy is headed to the clinic. Front Genet 3, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatti V, Maniero S, Ferracin M, Veronese A, Negrini M, Ferrocci G, Martini F and Tognon MG (2011) MicroRNAs dysregulation in human malignant pleural mesothelioma. J Thorac Oncol 6, 844–851. [DOI] [PubMed] [Google Scholar]
- Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, Hasan L, Kappeler A, Brunner T and Vassella E (2009) miR‐15a and miR‐16 are implicated in cell cycle regulation in a Rb‐dependent manner and are frequently deleted or down‐regulated in non‐small cell lung cancer. Cancer Res 69, 5553–5559. [DOI] [PubMed] [Google Scholar]
- Bhattacharya R, Nicoloso M, Arvizo R, Wang E, Cortez A, Rossi S, Calin GA and Mukherjee P (2009) MiR‐15a and MiR‐16 control Bmi‐1 expression in ovarian cancer. Cancer Res 69, 9090–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnie KA, Yip YY, Ng DC, Kirschner MB, Reid G, Prele CM, Musk AW, Lee YC, Thompson PJ, Mutsaers SE et al (2015) Loss of miR‐223 and JNK signaling contribute to elevated stathmin in malignant pleural mesothelioma. Mol Cancer Res 13, 1106–1118. [DOI] [PubMed] [Google Scholar]
- Blackwell C, Sherk C, Fricko M, Ganji G, Barnette M, Hoang B, Tunstead J, Skedzielewski T, Alsaid H, Jucker BM et al (2016) Inhibition of FGF/FGFR autocrine signaling in mesothelioma with the FGF ligand trap, FP‐1039/GSK3052230. Oncotarget, 7, 39861–39871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, D'Urso L, Pagliuca A, Biffoni M, Labbaye C et al (2008) The miR‐15a‐miR‐16‐1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med 14, 1271–1277. [DOI] [PubMed] [Google Scholar]
- Cheng YY, Kirschner MB, Cheng NC, Gattani S, Klebe S, Edelman JJ, Vallely MP, McCaughan BC, Jin HC, van Zandwijk N et al (2013) ZIC1 is silenced and has tumor suppressor function in malignant pleural mesothelioma. J Thorac Oncol 8, 1317–1328. [DOI] [PubMed] [Google Scholar]
- Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ and Hynes NE (2010) Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res 70, 4151–4162. [DOI] [PubMed] [Google Scholar]
- Dubey S, Janne PA, Krug L, Pang H, Wang X, Heinze R, Watt C, Crawford J, Kratzke R, Vokes E et al (2010) A phase II study of sorafenib in malignant mesothelioma: results of Cancer and Leukemia Group B 30307. J Thorac Oncol 5, 1655–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland LL, Rankin C, Gandara DR, Rivkin SE, Scott KM, Nagle RB, Klein‐Szanto AJ, Testa JR, Altomare DA and Borden EC (2007) Phase II study of erlotinib in patients with malignant pleural mesothelioma: a Southwest Oncology Group Study. J Clin Oncol 25, 2406–2413. [DOI] [PubMed] [Google Scholar]
- Garzon R, Calin GA and Croce CM (2009) MicroRNAs in cancer. Annu Rev Med 60, 167–179. [DOI] [PubMed] [Google Scholar]
- Goke F, Franzen A, Hinz TK, Marek LA, Yoon P, Sharma R, Bode M, von Maessenhausen A, Lankat‐Buttgereit B, Goke A et al (2015) FGFR1 expression levels predict BGJ398 sensitivity of FGFR1‐dependent head and neck squamous cell cancers. Clin Cancer Res 21, 4356–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan R, Kratzke RA, Herndon JE 2nd, Niehans GA, Vollmer R, Watson D, Green MR and Kindler HL (2005) Gefitinib in patients with malignant mesothelioma: a phase II study by the Cancer and Leukemia Group B. Clin Cancer Res 11, 2300–2304. [DOI] [PubMed] [Google Scholar]
- He L and Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5, 522–531. [DOI] [PubMed] [Google Scholar]
- He Q, Ren X, Chen J, Li Y, Tang X, Wen X, Yang X, Zhang J, Wang Y, Ma J et al (2016) miR‐16 targets fibroblast growth factor 2 to inhibit NPC cell proliferation and invasion via PI3K/AKT and MAPK signaling pathways. Oncotarget 7, 3047–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoda MA, Pirker C, Dong Y, Schelch K, Heffeter P, Kryeziu K, van Schoonhoven S, Klikovits T, Laszlo V, Rozsas A et al (2016) Trabectedin is active against malignant pleural mesothelioma cell and xenograft models and synergizes with chemotherapy and Bcl‐2 inhibition in vitro. Mol Cancer Ther 15, 2357–2369. [DOI] [PubMed] [Google Scholar]
- Iams WT, Sosman JA and Chandra S (2017) Novel targeted therapies for metastatic melanoma. Cancer J 23, 54–58. [DOI] [PubMed] [Google Scholar]
- Jaurand MC and Fleury‐Feith J (2005) Pathogenesis of malignant pleural mesothelioma. Respirology 10, 2–8. [DOI] [PubMed] [Google Scholar]
- Jiang H, Qu L, Wang Y, Cong J, Wang W and Yang X (2014) miR‐99a promotes proliferation targeting FGFR3 in human epithelial ovarian cancer cells. Biomed Pharmacother 68, 163–169. [DOI] [PubMed] [Google Scholar]
- Kanteti R, Dhanasingh I, Kawada I, Lennon FE, Arif Q, Bueno R, Hasina R, Husain AN, Vigneswaran W, Seiwert T et al (2014) MET and PI3K/mTOR as a potential combinatorial therapeutic target in malignant pleural mesothelioma. PLoS One 9, e105919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SC, Fulham M, Wong K, Cooper W, Brahmbhatt H, MacDiarmid J, Pattison S, Sagong JO, Huynh Y, Leslie F et al (2015) A significant metabolic and radiological response after a novel targeted MicroRNA‐based treatment approach in malignant pleural mesothelioma. Am J Respir Crit Care Med 191, 1467–1469. [DOI] [PubMed] [Google Scholar]
- Katoh M and Nakagama H (2014) FGF receptors: cancer biology and therapeutics. Med Res Rev 34, 280–300. [DOI] [PubMed] [Google Scholar]
- Korc M and Friesel RE (2009) The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets 9, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar‐Alaoui H, Hassan OU, Yang YW and Buchanan P (2015) Lung cancer: biology and treatment options. Biochim Biophys Acta 1856, 189–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Viallet J and Haura EB (2008) A small molecule pan‐Bcl‐2 family inhibitor, GX15‐070, induces apoptosis and enhances cisplatin‐induced apoptosis in non‐small cell lung cancer cells. Cancer Chemother Pharmacol 61, 525–534. [DOI] [PubMed] [Google Scholar]
- Liang G, Zhu Y, Jing A, Wang J, Hu F, Feng W, Xiao Z and Chen B (2016) Cationic microRNA‐delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther 23, 829–838. [DOI] [PubMed] [Google Scholar]
- Linton A, Vardy J, Clarke S and van Zandwijk N (2012) The ticking time‐bomb of asbestos: its insidious role in the development of malignant mesothelioma. Crit Rev Oncol Hematol 84, 200–212. [DOI] [PubMed] [Google Scholar]
- Livak KJ and Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Marek LA, Hinz TK, von Massenhausen A, Olszewski KA, Kleczko EK, Boehm D, Weiser‐Evans MC, Nemenoff RA, Hoffmann H and Warth A et al (2014) Nonamplified FGFR1 is a growth driver in malignant pleural mesothelioma. Mol Cancer Res 12, 1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama‐Takahashi K, Shimada N, Imada T, Maekawa‐Tokuda Y, Ishii T, Ouchi J, Kusaka H, Miyaji H, Akinaga S, Tanaka A et al (2008) A neutralizing anti‐fibroblast growth factor (FGF) 8 monoclonal antibody shows anti‐tumor activity against FGF8b‐expressing LNCaP xenografts in androgen‐dependent and ‐independent conditions. Prostate 68, 640–650. [DOI] [PubMed] [Google Scholar]
- Metzner T, Bedeir A, Held G, Peter‐Vorosmarty B, Ghassemi S, Heinzle C, Spiegl‐Kreinecker S, Marian B, Holzmann K, Grasl‐Kraupp B et al (2011) Fibroblast growth factor receptors as therapeutic targets in human melanoma: synergism with BRAF inhibition. J Invest Dermatol 131, 2087–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Sakata K, Sase H, Isobe T, Morikami K, Shindoh H, Mio T et al (2014) The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther 13, 2547–2558. [DOI] [PubMed] [Google Scholar]
- Ornitz DM and Itoh N (2001) Fibroblast growth factors. Genome Biol 2, REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou WB, Lu M, Eilers G, Li H, Ding J, Meng X, Wu Y, He Q, Sheng Q, Zhou HM et al (2016) Co‐targeting of FAK and MDM2 triggers additive anti‐proliferative effects in mesothelioma via a coordinated reactivation of p53. Br J Cancer 115, 1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pass HI, Goparaju C, Ivanov S, Donington J, Carbone M, Hoshen M, Cohen D, Chajut A, Rosenwald S, Dan H et al (2010) hsa‐miR‐29c* is linked to the prognosis of malignant pleural mesothelioma. Cancer Res 70, 1916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattarozzi A, Carra E, Favoni RE, Wurth R, Marubbi D, Filiberti RA, Mutti L, Florio T, Barbieri F and Daga A (2017) The inhibition of FGF receptor 1 activity mediates sorafenib antiproliferative effects in human malignant pleural mesothelioma tumor‐initiating cells. Stem Cell Res Ther 8, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing J, Du X, Chen Y, Chan P, Li H, Wu P, Marsters S, Stawicki S, Tien J, Totpal K et al (2009) Antibody‐based targeting of FGFR3 in bladder carcinoma and t(4;14)‐positive multiple myeloma in mice. J Clin Invest 119, 1216–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, Williams M, Wright C, Edelman JJ, Vallely MP et al (2013) Restoring expression of miR‐16: a novel approach to therapy for malignant pleural mesothelioma. Ann Oncol 24, 3128–3135. [DOI] [PubMed] [Google Scholar]
- Robinson BM (2012) Malignant pleural mesothelioma: an epidemiological perspective. Ann Cardiothorac Surg 1, 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, Zhao A, Busuttil RW, Yee H, Stein L et al (2011) Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 19, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelch K, Hoda MA, Klikovits T, Munzker J, Ghanim B, Wagner C, Garay T, Laszlo V, Setinek U, Dome B et al (2014) Fibroblast growth factor receptor inhibition is active against mesothelioma and synergizes with radio‐ and chemotherapy. Am J Respir Crit Care Med 190, 763–772. [DOI] [PubMed] [Google Scholar]
- Schwartz J, Niu X, Walton E, Hurley L, Lin H, Edwards H, Taub JW, Wang Z and Ge Y (2016) Synergistic anti‐leukemic interactions between ABT‐199 and panobinostat in acute myeloid leukemia ex vivo. Am J Transl Res 8, 3893–3902. [PMC free article] [PubMed] [Google Scholar]
- Takeshita F, Patrawala L, Osaki M, Takahashi RU, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG, Brown D et al (2010) Systemic delivery of synthetic microRNA‐16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell‐cycle genes. Mol Ther 18, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel‐Duby R and Olson EN (2008) The endothelial‐specific microRNA miR‐126 governs vascular integrity and angiogenesis. Dev Cell 15, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li J, Wang X, Zheng C and Ma W (2013) Downregulation of microRNA‐214 and overexpression of FGFR‐1 contribute to hepatocellular carcinoma metastasis. Biochem Biophys Res Commun 439, 47–53. [DOI] [PubMed] [Google Scholar]
- Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A et al (2010) Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2, 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willimott S and Wagner SD (2010) Post‐transcriptional and post‐translational regulation of Bcl2. Biochem Soc Trans 38, 1571–1575. [DOI] [PubMed] [Google Scholar]
- Wynes MW, Hinz TK, Gao D, Martini M, Marek LA, Ware KE, Edwards MG, Bohm D, Perner S, Helfrich BA et al (2014) FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin Cancer Res 20, 3299–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Tang Y, Liu Z, Dai J and Wang X (2016) BCL‐2 inhibition impairs mitochondrial function and targets oral tongue squamous cell carcinoma. Springerplus 5, 1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalcman G, Mazières J, Margery J, Greillier L, Audigier‐Valette C, Moro‐Sibilot D, Molinier O, Corre R, Monnet I, Gounant V et al (2015) Bevacizumab 15 mg/kg plus cisplatin‐pemetrexed (CP) triplet versus CP doublet in malignant pleural mesothelioma (MPM): results of the IFCT‐GFPC‐0701 MAPS randomized phase 3 trial. J Clin Oncol 33 (suppl), abstr 7500. [Google Scholar]
- van Zandwijk N, Clarke C, Henderson D, Musk AW, Fong K, Nowak A, Loneragan R, McCaughan B, Boyer M, Feigen M et al (2013) Guidelines for the diagnosis and treatment of malignant pleural mesothelioma. J Thorac Dis 5, E254–E307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Hylander BL, LeVea C, Repasky EA, Straubinger RM, Adjei AA and Ma WW (2014) Enhanced FGFR signalling predisposes pancreatic cancer to the effect of a potent FGFR inhibitor in preclinical models. Br J Cancer 110, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative pictures of MSTO cells in a colony formation assay after transfection with microRNA mimics or non‐coding control.
Fig. S2. Growth inhibition assays of P31 and MSTO cells after transfection with mimics in combination with FGF2 treatment.
Fig. S3. Target gene and protein expression analysis after transfection with siRNAs via qPCR or western blot.
Fig. S4. Growth inhibition assays of P31 and MSTO cells 72 h after transfection with microRNA mimics and/or siRNAs or respective controls.
Fig. S5. Growth inhibition assays of SPC212, MSTO and P31 cells 72 h after treatment with PD166866 (PD) or ponatinib (Pon) in combination with obatoclax or venetoclax, or respective controls, at the indicated doses.
Table S1. List of used cell lines, the histological MPM subtype they were derived from, standard growth media and sources.
Table S2. List of mimics and siRNA, their sequences/IDs and sources.
Table S3. List of cytokines and drugs, their targets and sources.
Table S4. Transfection schemes for different assay formats.
Table S5. List of primers and probes used in TaqMan‐based reverse transcription and qPCR.
Table S6. Primers and their sequences used for SYBR green‐based qPCR.
Table S7. Primers and their sequences used for Luciferase reporter assays.
Table S8. Fold change, standard deviation and P‐values for microRNAs of individual cell lines after treatment with FGF2.