

Abstract
Mutations and splice variants in the estrogen receptor (ER) gene, ESR1, may yield endocrine resistance in metastatic breast cancer (MBC) patients. These putative endocrine resistance markers are likely to emerge during treatment, and therefore, its detection in liquid biopsies, such as circulating tumor cells (CTCs) and cell‐free DNA (cfDNA), is of great interest. This research aimed to determine whether ESR1 mutations and splice variants occur more frequently in CTCs of MBC patients progressing on endocrine treatment. In addition, the presence of ESR1 mutations was evaluated in matched cfDNA and compared to CTCs. CellSearch‐enriched CTC fractions (≥5/7.5 mL) of two MBC cohorts were evaluated, namely (a) patients starting first‐line endocrine therapy (n = 43, baseline cohort) and (b) patients progressing on any line of endocrine therapy (n = 40, progressing cohort). ESR1 hotspot mutations (D538G and Y537S/N/C) were evaluated in CTC‐enriched DNA using digital PCR and compared with matched cfDNA (n = 18 baseline cohort; n = 26 progressing cohort). Expression of ESR1 full‐length and 4 of its splice variants (∆5, ∆7, 36 kDa, and 46 kDa) was evaluated in CTC‐enriched mRNA. It was observed that in the CTCs, the ESR1 mutations were not enriched in the progressing cohort (8%), when compared with the baseline cohort (5%) (P = 0.66). In the cfDNA, however, ESR1 mutations were more prevalent in the progressing cohort (42%) than in the baseline cohort (11%) (P = 0.04). Three of the same mutations were observed in both CTCs and cfDNA, 1 mutation in CTCs only, and 11 in cfDNA only. Only the ∆5 ESR1 splice variant was CTC‐specific expressed, but was not enriched in the progressing cohort. In conclusion, sensitivity for detecting ESR1 mutations in CTC‐enriched fractions was lower than for cfDNA. ESR1 mutations detected in cfDNA, rarely present at the start of first‐line endocrine therapy, were enriched at progression, strongly suggesting a role in conferring endocrine resistance in MBC.
Keywords: cell‐free DNA, circulating tumor cells, endocrine resistance, ESR1 mutations
Abbreviations
- AI
aromatase inhibitor
- cfDNA
cell‐free DNA
- CTC
circulating tumor cell
- dPCR
digital PCR
- HBD
healthy blood donor
- MBC
metastatic breast cancer
- PD
progressive disease
- SD
standard deviation
- VAF
variant allele frequency
1. Introduction
Endocrine therapy is the mainstay of treatment for estrogen receptor (ER)‐positive metastatic breast cancer (MBC) patients. However, 40% of these patients obtain no clinical benefit from first‐line endocrine therapy, and virtually all of the patients in whom the tumor initially responds will eventually develop resistance (Pritchard, 2013). Several mechanisms have been linked to endocrine resistance (De Marchi et al., 2016), but none of these have been implemented in daily clinical practice because their clinical value could not be confirmed, or was not strong enough. One recently revealed mechanism for acquired resistance is the emergence of mutations in the gene coding for ER, ESR1, yielding a constitutively activated ER. Functional studies have suggested that tumor cells with these mutations are less responsive to estrogen deprivation as induced by aromatase inhibitors (AIs) (Robinson et al., 2013; Toy et al., 2013), but may still experience growth inhibition by ER‐blocking agents such as tamoxifen and fulvestrant (Jeselsohn et al., 2014; Robinson et al., 2013; Toy et al., 2013). This was recently supported in a retrospective clinical analysis, in which a modest progression‐free survival benefit was observed for MBC patients with an ESR1 mutation who were treated with fulvestrant, when compared to the AI exemestane (Fribbens et al., 2016). These results have further emphasized the potential for the determination of ESR1 mutations to guide treatment decision making in ER‐positive MBC (Angus et al., 2017).
Another mechanism that potentially contributes to acquired endocrine therapy resistance is the occurrence of ESR1 mRNA splice variants. ESR1 splice variants have been described as having various effects on the transcriptional activity of the ER (Taylor et al., 2010), and are heterogeneously expressed in primary breast cancers (Poola and Speirs, 2001). The ERα∆5 splice variant is of particular interest, as preclinical experiments have reported that this variant exerts constitutional transcriptional activity (Bollig and Miksicek, 2000; Fuqua et al., 1991). However, to date, the putative role of ESR1 splice variants with regard to endocrine resistance in MBC has not been assessed.
ESR1 mutations and mRNA splice variants are likely to emerge during treatment and can therefore only be observed in tumor cells obtained during or after treatment. Thus, these investigations require metastatic tumor tissue obtained through biopsies, which can be technically challenging, or even impossible.
Circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) are alternative and minimally invasive means of assessing the characteristics of metastatic cancer cells. Theoretically, each acts as a different substrate for DNA, with DNA from CTCs coming from intact cancer cells, and ctDNA [which is part of the total cell‐free DNA (cfDNA)] is thought to originate mainly from apoptotic tumor cells (Haber and Velculescu, 2014). The introduction of very sensitive digital polymerase chain reaction (dPCR) assays has opened new avenues to determine the presence of mutations in ctDNA and in CTC‐derived DNA of patients with cancer. Although promising results have been achieved with the detection of ESR1 mutations in cfDNA using dPCR (Chu et al., 2015; Fribbens et al., 2016; Guttery et al., 2015; Schiavon et al., 2015; Takeshita et al., 2015, 2016; Wang et al., 2016), the important advantage of using CTCs over cfDNA is that multiple parameters in multiple dimensions (DNA, RNA, and protein) can be measured in the same sample and can be associated with, for example, endocrine resistance. This implies that besides assessing mutations in CTC‐derived DNA, the characterization of RNA from CTCs permits the assessment of splice variants.
The current study set out to evaluate ESR1 mutations and splice variants in CellSearch‐enriched CTCs of MBC patients before the start of first‐line endocrine therapy, and during progression under any line of endocrine therapy. The main objective was to determine whether these putative mechanisms for endocrine resistance are enriched in patients progressing on endocrine therapy. To this end, a cohort of MBC patients before the beginning of first‐line endocrine therapy for MBC was defined, as well as a cohort of MBC patients progressing under any line of endocrine therapy. Additionally, in a subgroup of these patients, the ESR1 mutation status in CTCs was compared with patient‐matched cfDNA.
2. Materials and methods
2.1. Patients and treatment
The patients evaluated in this study were selected from two CTC studies comprising patients receiving endocrine therapy (study 06‐248 (Mostert et al., 2015; Onstenk et al., 2015b; Sieuwerts et al., 2011) and study 09‐405 (Reijm et al., 2016)). Six centers in the Netherlands and Belgium participated in these studies from February 2008 through March 2015. The patients were included in these studies if they had MBC, and a new line of endocrine therapy was begun. Blood was sampled before the start of endocrine therapy and/or at the time of progression to palliative endocrine treatment. At both of these time points, 10 mL of blood was drawn for CTC enumeration, and another 10 mL of blood was drawn for CTC characterization. In each participating center, the institutional board approved the study protocols (Erasmus MC ID MEC‐06‐248 and MEC‐09‐405). All patients provided written informed consent.
Two cohorts of patients were defined for the current study: a cohort starting first‐line endocrine therapy for MBC and a separate cohort progressing under any line of palliative endocrine therapy. Further eligibility criteria required that the patient had ≥5 CTCs/7.5 mL of blood at the time of the blood draw, to allow for the characterization of CTCs.
2.2. Enumeration and isolation of DNA and RNA from CTCs and cfDNA and ESR1 mutation determination
Details regarding the CTC enumeration and isolation of DNA/RNA from CTCs have been reported previously (Mostert et al., 2015; Onstenk et al., 2015b; Reijm et al., 2016; Sieuwerts et al., 2011). Briefly, in each patient, 10 mL of blood was drawn in CellSave tubes (Janssen Diagnostics, Raritan, NJ, USA) for CTC enumeration, which was performed on 7.5 mL of blood within 96 h of the blood draw using the CellSearch system (Janssen Diagnostics). Another 10 mL of blood was drawn into EDTA tubes for CTC characterization, and CTCs were isolated from 7.5 mL of blood within 24 h using the CellSearch system with the CellSearch profile kit (Janssen Diagnostics) (Fig. S1). Subsequently, DNA and RNA were isolated from enriched CTCs using the AllPrep DNA/RNA Micro Kit (Qiagen, Germantown, MD, USA) (Sieuwerts et al., 2011). For cfDNA analyses, the remainder of the EDTA blood (maximum of 2.5 mL) was centrifuged to isolate plasma within 24 h after the blood draw. Cell‐free DNA (cfDNA) was isolated from a total of 200 μL of plasma using the QIAamp circulating nucleic acid kit (Qiagen).
DNA from the CellSearch‐enriched CTC fractions and cfDNA from plasma were quantified using the Quant‐iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The DNA (0.1–1 ng·μL−1) was subjected to an ESR1 target‐specific amplification of 15 cycles with TaqMan PreAmp Master Mix (Thermo Fisher Scientific), as recommended by the manufacturer, using the ESR1 PreAmp primer combination (Table S1) at a final concentration of 400 nm each. The resulting pre‐amplified 136 base pair product covering the positions of all four ESR1 hotspot mutation sites (D538G and Y537S/C/N) was diluted 10‐fold, and quantified via regular quantitative PCR (qPCR) for wild‐type (WT) ESR1 using the same primers. The resulting Cq value was used to control the number of WT copies to be loaded onto the chips for dPCR analyses. The variant allele frequencies (VAF) of the studied mutations for ESR1 were evaluated with mutation‐specific TaqMan assays (the primer and probe sequences are given in Table S1, and the reproducibility of these assessments in Fig. S2) via chip‐based dPCR (QuantStudio 3D; Thermo Fisher Scientific) according to the manufacturer's instructions. Positive and negative control DNA was always included in each dPCR run, and all of the analyzed DNA samples (CTC and cfDNA) were evaluated in duplicate.
Digital PCR was performed for four ESR1 hotspot mutation sites (D538G and Y537S/C/N). Ten healthy blood donors were used to specify the cutoffs for the presence of ESR1 mutations in CellSearch‐enriched samples. Seven of them had sufficient plasma available, and these samples were used to specify the cutoffs for the presence of ESR1 mutations in cfDNA. The cutoff for the positivity for each individual assay was set at the highest VAF in the healthy blood donors plus 2.58 standard deviations (SD) (99% confidence interval) (Figs S3 and S4). The cutoffs were as follows: D538G = 0.6% (CTCs) and 1.0% (cfDNA), Y537S = 0.3% (for both CTCs and cfDNA), Y537N = 0.3% (CTCs) and 1.65% (cfDNA), Y537C = 0.5% (CTCs) and 0.65% (cfDNA). Both of the duplicate ESR1 mutation measurements had to be above the cutoffs for a sample to be considered positive for a specific ESR1 mutation.
2.3. Short tandem repeat analysis on patient‐matched CTC‐DNA and cfDNA
In a subset of samples with ≥ 10 CTCs and a high enough DNA content (≥ 30 ng) for which not all CellSearch‐enriched DNA was used for ESR1 mutation analysis, a short tandem repeat (STR) analysis was performed to confirm that the CellSearch‐enriched DNA and cfDNA were indeed from the same donor. The PowerPlex 16 System (Promega, Madison, WI, USA), in combination with an ABI PRISM 3130xl Genetic Analyzer (Thermo Fisher Scientific) and genemarker v1.91 software (Softgenetics LLC, State College, PA, USA), was used to genotype the DNA, as recommended by the manufacturer's instructions.
2.4. ESR1 splice variants and expression in RNA from enriched CTCs
The measured ‘splice variant gene panel’ consisted of full‐length (FL) ESR1 and ESR1 splice variants ∆5, ∆7, 36 kDa, and 46 kDa. In addition, reference genes and epithelial genes were evaluated. Two microlitre of complementary DNA was pre‐amplified in 15 cycles with TaqMan assays and TaqMan PreAmp Master Mix (Thermo Fisher Scientific), as recommended by the manufacturer, using the gene panel combination given in Table S1. After pre‐amplification, each gene was individually measured via qPCR with the same TaqMan assay used in the pre‐amplification. Positive and negative controls were included in each individual experiment to monitor the reproducibility of the measurements (for reproducibility, see also Fig. S5).
The splice variants were assessed in CellSearch‐enriched fractions of 10 healthy blood donors to evaluate the possible leukocyte expression of FL ESR1 and splice variants. The splice variant gene panel was always evaluated in duplicate, and the averages of the duplicate measurements were used for further calculation. Only those samples with sufficient mRNA signal (reference genes average ∆Cq<26.5) and epithelial signal (KRT19/EPCAM average ∆Cq<26.5), as described previously (Onstenk et al., 2015a; Sieuwerts et al., 2009, 2011), were used for further evaluation of splice variants. The ∆Cq values for the splice variants were calculated relative to the FL ESR1. In those cases where no expression could be measured for both the splice variant and the FL ESR1, the sample was excluded from the analysis.
2.5. Statistical considerations
The primary objective of this research was to investigate whether ESR1 mutations were more frequently observed in CTCs of MBC patients progressing on endocrine therapy, than in those patients starting first‐line endocrine therapy. Based on data from the literature (Robinson et al., 2013; Toy et al., 2013), it was hypothesized that ESR1 mutations in CTCs would be detectable in 30% of MBC patients experiencing progressive disease (PD) during palliative endocrine therapy and that ESR1 mutations in CTCs would be present in 5% of those patients beginning palliative first‐line endocrine therapy. In order to detect this difference (α = 0.05 and β = 0.2), 44 MBC patients progressing on palliative endocrine therapy and 44 MBC control patients initiating first‐line endocrine therapy were needed.
Secondary objectives included (a) an assessment of ESR1 mutations in cfDNA samples, and a comparison between the detection of ESR1 mutations in cfDNA versus CTC; (b) an exploration of whether ESR1 mutations measured in cfDNA are enriched under endocrine therapy; (c) an exploration of whether ESR1 splice variants are more prevalent in those patients experiencing PD than in patients beginning first‐line endocrine therapy for MBC; and (d) an exploration of whether certain clinical factors are associated with the presence of ESR1 mutations and/or splice variants.
Differences in the prevalence of ESR1 mutation and splice variants between the baseline cohort and the progressing cohort were calculated using Fisher's exact test (two‐sided), while those patients with matched samples in the baseline and the progressing cohort were excluded from this analysis. Correlations were tested using Kendall's tau correlation coefficient, and the differences in splice variant ∆Cq values between groups were tested using the Kruskal–Wallis test. All of the analyses were performed using Stata/SE version 12 (StataCorp LP, College Station, TX, USA), and all of the data obtained from this study are available in Doc. S1.
3. Results
3.1. Patient characteristics
For the baseline cohort, a total of 43 patient samples were included, while the progressing cohort contained a total of 40 patient samples (Table 1). Most of the patients in the baseline cohort were not treated with any adjuvant chemotherapy (79%); however, 17 patients (40%) had been treated with adjuvant endocrine therapy. Samples in the progressing cohort originated mainly from patients progressing on first‐line (55%) or second‐line (30%) palliative endocrine therapy. Prior to the PD sample, 37 patients (93%) had received at least one line of AI treatment. Most patients (81%) in the baseline cohort experienced PD on endocrine therapy during the time of follow‐up. For six of these patients, matched samples from the baseline cohort and progressing cohort were available; however, for the other 29 patients, no PD sample was available, mainly because it was not collected. The median CTC count was higher in the baseline cohort (81 CTCs/7.5 mL) than in the progressing cohort (21 CTCs/7.5 mL).
Table 1.
Baseline characteristics
| Parameter | Description | Baseline cohort (n = 43) | PD cohort (n = 40) |
|---|---|---|---|
| Age at sample draw | Median age (range) | 72 (37–83) | 63 (35–88) |
| Adjuvant endocrine therapy (%) | No | 26 (60) | 26 (65) |
| Yes, tamoxifen only | 10 (23) | 9 (23) | |
| Yes, tamoxifen + AI | 5 (12) | 4 (10) | |
| Yes, AI only | 2 (5) | 1 (2) | |
| Adjuvant chemotherapy (%) | No | 34 (79) | 28 (70) |
| Yes | 9 (21) | 12 (30) | |
| Neoadjuvant therapies (%) | No | 43 (100) | 40 (100) |
| Number of previous lines endocrine therapy lines for MBC (%) | 0 | 43 (100) | |
| 1 | 22 (55) | ||
| 2 | 12 (30) | ||
| ≥3 | 6 (15) | ||
| Endocrine therapy after start (BL cohort) or before PD (PD cohort) (%) | AI | 30 (70) | 25 (63) |
| Tamoxifen | 13 (30) | 7 (17) | |
| Fulvestrant | 8 (20) | ||
| Previous endocrine therapy lines for MBC (in case of inclusion at PD on ≥second‐line endocrine therapy) (%) | Yes, AI only | 9 (23) | |
| Yes, AI + tamoxifen | 6 (15) | ||
| Yes, tamoxifen only | 3 (7) | ||
| Progression on the current line (%) | Yes | 35 (81) | 40 (100) |
| CTC count | Median count (range) | 81 (6–32492) | 21 (5–2837) |
3.2. ESR1 mutations in CTCs and matched cfDNA
In the six matched samples from the baseline and progressing cohorts, no ESR1 mutations were detected. ESR1 mutations were observed in the CTCs of two (5%) baseline cohort samples (2× Y537N) and three (8%) progressing cohort samples (2× D538G, 1× Y537S) (P = 0.66) (Table 2). One of the patients in the baseline cohort with an ESR1 mutation had received prior adjuvant treatment with tamoxifen, while the other patient had not received any prior adjuvant therapy. Two of the ESR1 mutations in CTCs from patients in the progressing cohort, occurring after palliative first‐line therapy, were observed in one patient who had been treated with an AI and in one patient who had been treated with tamoxifen. The third ESR1 mutation was observed in a patient progressing on fulvestrant as second‐line palliative endocrine therapy, who had received an AI as her first‐line treatment.
Table 2.
Observed ESR1 mutations in CTC and cfDNA samples. All patients in whom a mutation was called in either CTCs or cfDNA, along with clinical information. Shown percentages are variant allele frequencies. Called mutations are depicted in boldface
| CTC code | Baseline CTCs | Baseline cfDNA | Adjuvant therapy | PD CTCs | PD cfDNA | Progression on therapy | Prior therapies for MBC |
|---|---|---|---|---|---|---|---|
| CTC798a | D538G (0.14%) | D538G (1.93%) | None | Not available | Not available | ||
| CTC1581 | Y537S (0.39%)b | Y537S (0.47%) | None | Not available | Not available | ||
| Y537N (0.42%) | Y537N (0.05%) | ||||||
| CTC1571 | Y537N (3.77%) | Not available | Tamoxifen | Not available | Not available | ||
| CTC1007a | Not available | Not available | None | Y537S (0.01%) | Y537S (9.26%) | Fulvestrant | AI |
| CTC1364a | Not available | Not available | None | D538G (0.25%) | D538G (40.05%) | Tamoxifen | AI |
| CTC1565a | Not available | Not available | Tamoxifen + AI | D538G (0.14%) | D538G (5.14%) | Fulvestrant | AI |
| CTC1569 | Not available | Not available | None | Y537N (0.25%) | Y537N (1.96%) | AI | |
| CTC1352 | Not available | Not available | None | D538G (0.47%) | D538G (20.93%) | AI | Tamoxifen |
| CTC1567 | Not available | Not available | None | Y537S (1.98%) | Y537S (1.21%) | Tamoxifen | |
| CTC1360 | Not available | Not available | None | D538G (0.52%) | D538G (2.86%) | AI | |
| CTC1587 | Not available | Not available | Tamoxifen | D538G (0.84%) | D538G (15.98%) | Fulvestrant | AI |
| CTC1406 | Not available | Not available | Tamoxifen | D538G (1.13%) | D538G (10.18%) | AI | |
| CTC1393 | Not available | Not available | None | D538G (0.18%) Y537C (0.23%) | D538G (27.1%)Y537C (12.96%) | AI | |
| CTC1410 | Not available | Not available | Tamoxifen | D538G (0.37%) | D538G (23.84%) | AI |
a STR analysis confirmed that the CTC‐DNA and cfDNA samples were from the same patient. For other samples, not enough DNA available for STR analysis. bAverage VAF positive, but negative in duplicate analysis.
Matched cfDNA and CTCs from the same time point were available from a subset of the patients in the baseline cohort (n = 18) and the progressing cohort (n = 26) (Table S2). Two ESR1 mutations (1× D538G and 1× Y537S) (11%) were observed in cfDNA of the baseline cohort, and 12 ESR1 mutations were observed in 11 patients (42%) in cfDNA of the progressing cohort (8× D538G, 2× Y537S, 1× Y537N, 1× Y537C) (P = 0.04) (Table 2). In the four matched cfDNA samples from the baseline and progressing cohorts, no ESR1 mutations were detected. Neither of the mutations found in cfDNA from the baseline cohort were observed in the CTCs (Table 2). In one of these patients, however, an Y537N mutation was observed in CTCs, but not in cfDNA. Neither of the patients with ESR1 mutations in cfDNA from the baseline cohort had received any adjuvant therapy.
When the mutations in cfDNA from the progressing cohort samples were compared with the mutation status of the CTCs, three of three mutations observed in CTCs were confirmed in cfDNA. With one exception, variant allele frequencies (VAFs) of the mutations were much higher in cfDNA than in CTCs (Table 2). In addition, nine mutations in eight patients were observed in the cfDNA, but not in the CTCs. The mutations found in cfDNA of the progressing cohort occurred after first‐line endocrine therapies (n = 6), namely AIs (n = 5) and tamoxifen (n = 1), and after second‐line endocrine therapies (n = 5), namely fulvestrant (n = 3) and tamoxifen (n = 2). All of these latter patients had received an AI as first‐line palliative endocrine treatment.
From four patients with matched CTC‐cfDNA samples and discordant CTC versus cfDNA ESR1 mutation results, unamplified DNA was available to perform STR analyses (Table 2). These analyses showed that both of the DNA fractions originated from the same patient, and thus excluded sample swapping.
3.3. ESR1 splice variants in CTCs
In order to assess the presence of ESR1 splice variants in CTCs, RNA was extracted from CellSearch‐enriched CTCs and analyzed for the expression of four ESR1 splice variants relative to full‐length ESR1. In the baseline cohort, 10 (23%) of the 43 samples were excluded from further analysis, because of insufficient quality of mRNA (n = 4) or lack of an epithelial signal (n = 6). In the progressing cohort, 17 (43%) of 40 samples had to be excluded because of insufficient quality of the mRNA (n = 2), lack of an epithelial signal (n = 6), or unavailable RNA (n = 9).
ESR1 splice variant ∆Cq values relative to full‐length ESR1 were not correlated with CTC counts (Fig. S6). ∆Cq values of the ∆5 splice variant relative to full‐length ESR1 were significantly higher in patients than in healthy blood donors (HBDs) (Fig. 1A), but the ∆5 splice variant was not enriched in the progressing cohort, when compared to the baseline cohort (P = 0.39). When four matched samples, taken from the baseline and progressing cohorts, were analyzed from patients receiving first‐line AI treatment, the ∆5 splice variant was enriched at PD in two of the patients (Fig. S7). The ∆7 and 36 kDa splice variants were similarly expressed in patient samples and HBDs (Fig. 1B,C). Nevertheless, for the four matched samples from the baseline and progressing cohorts, the ∆7 and 36‐kDa splice variants were enriched at PD in one and three patients, respectively (Fig. S7). The 46‐kDa splice variant was only observed in patient samples and not in HBDs; however, this did not reach statistical significance (Fig. 1D).
Figure 1.
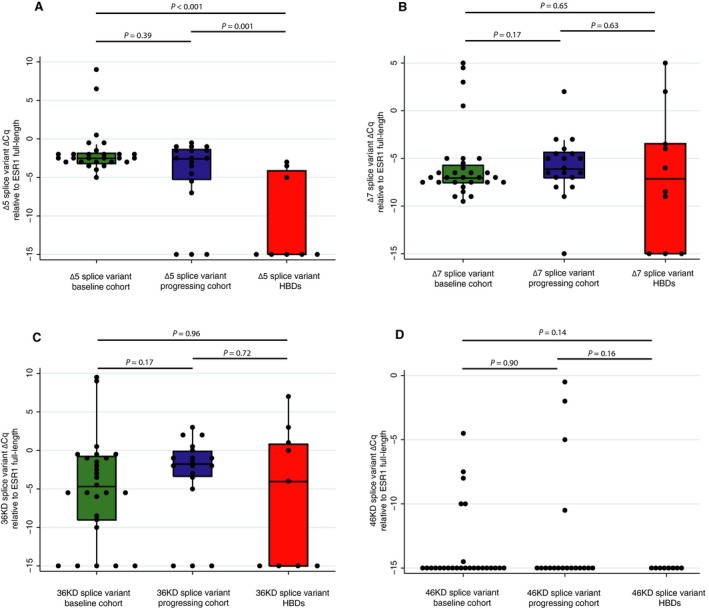
Occurrence of splice variants in the baseline cohort, the progressing cohort, and healthy blood donors (HBDs). Boxes demonstrate median and IQR; lines represent adjacent values (1.5*IQR). Observations were binned at ∆Cq of 0.5.
4. Discussion
The current study evaluated whether ESR1 mutations and splice variants were enriched in CTCs from MBC patients progressing under endocrine therapy. No enrichment of any of these putative resistance mechanisms in CTCs was observed after endocrine therapy. However, cfDNA analyses did reveal an enrichment of ESR1 mutations at the time of progression on endocrine therapy, when compared with before the initiation of first‐line endocrine treatment.
The observation that ESR1 mutations were more frequently observed in cfDNA than in CTCs suggests that cfDNA is a more sensitive substrate for the analysis of ESR1 mutations than CTCs enriched by the FDA‐approved CellSearch system. This is also reflected by the VAFs in the CTCs, which were generally low (range: up to 3.8%), as opposed to the VAFs in the cfDNA, which were generally much higher (range: up to 40%). One explanation for this difference could be the presence of contaminating leukocytes following the CellSearch enrichment of CTCs, which we had previously reported to be around 1000 leukocytes (Sieuwerts et al., 2009), thereby decreasing the sensitivity for the detection of ESR1 mutations in CTCs. However, our experiments suggesting those amounts of leukocytes after CellSearch profile were conducted in healthy donors in perfect circumstances with quick processing. For the current study, materials from patients were used which were sometimes shipped from distant sites and processed within 24 h, which may have resulted in a higher number of contaminating leukocytes. Therefore, the numbers of leukocytes that are present after CellSearch enrichment may be even higher than 1000 leukocytes in some samples, which is likely to decrease sensitivity for detecting ESR1 mutations even more in those samples. Although cfDNA analysis is also challenged by the contamination of wild‐type DNA, our results suggest that this is less of an issue in cfDNA than in CTCs.
The stringency of the cutoffs for ESR1 mutations, now arbitrarily set at the highest VAF observed in HBDs plus 2.58xSD (representing the 99% confidence interval), could have played a role in the limited sensitivity of ESR1 mutation detection in CTCs. When less stringent cutoffs based on the highest VAF in HBDs were explored (data not shown), slightly more ESR1 mutations were observed in CTCs; however, the majority of these mutations were not observed in cfDNA, suggesting that relaxing the cutoffs for ESR1 mutation positivity may lead to false‐positive findings. This stresses the need to include HBDs, and to be stringent with setting the cutoff value for ESR1 mutation positivity. Interestingly, the current study observed one ESR1 mutation exclusively present in CTCs, but not in cfDNA. This finding suggests that some ESR1 mutations may be missed by cfDNA analysis only, albeit this observation may be merely anecdotal.
The current study is among the first to assess ESR1 mutations in a cohort of patients beginning first‐line endocrine treatment for MBC. While it has already been recognized that primary breast cancers rarely harbor ESR1 mutations (Jeselsohn et al., 2014; Toy et al., 2013), most studies thus far have evaluated patients who had been pretreated with palliative endocrine therapy, suggesting that these mutations become enriched during treatment with AIs (Schiavon et al., 2015). Here, it has been confirmed that ESR1 mutations are not frequently present in MBC patients before first‐line endocrine therapy, and are enriched in MBC patients progressing under endocrine therapy.
Most of the patients in this study having an ESR1 mutation progressed on AI treatment or had previously been treated with an AI. In three of the patients, ESR1 mutations were observed after progression on fulvestrant, suggesting that although it has been reported that fulvestrant is more effective than AIs in ESR1‐mutant patients (Fribbens et al., 2016; Spoerke et al., 2016), mutant subclones can still be observed at PD on fulvestrant therapy. Of further note is the fact that in the current study the observed mutations in the baseline cohort occurred in those patients who were not pretreated with AIs, or who received no pretreatment with endocrine therapy at all. In addition, an ESR1 mutation was observed in CTCs and cfDNA of one patient progressing on first‐line palliative tamoxifen therapy, but who had not received any AI treatment, also not in the adjuvant setting. These findings are in line with the observations of multiple groups (Guttery et al., 2015; Jeselsohn et al., 2014; Takeshita et al., 2015), who reported ESR1 mutations in metastatic biopsies or cfDNA of patients who had only received tamoxifen, or no pretreatment at all. This could also fit with the observations by Wang et al. (2016), who reported that ESR1 mutations were sometimes present in primary breast cancers of patients at extremely low VAFs.
In the current study, the ESR1 splice variant ∆5 was expressed at higher levels in the CellSearch‐enriched samples from MBC patients than in HBD samples; however, we found no enrichment of this splice variant during endocrine therapy for MBC. The ∆7, 36‐kDa, and 46‐kDa splice variants were not significantly more highly expressed in patients versus HBDs. The fact that full‐length ESR1 and splice variants were also measured in a subset of HBDs suggests that leukocytes, which are known to express ESR1 (Scariano et al., 2008), may also express these splice variants. This clearly complicates the analysis of ESR1 splice variants measured in CellSearch‐enriched CTC fractions, where one thousand‐fold of leukocytes is still present. In metastatic prostate cancer, the presence of the androgen receptor (AR) splice variant V7 in CTCs was previously demonstrated to be strongly associated with resistance to endocrine agents (Antonarakis et al., 2014), but not to chemotherapy (Antonarakis et al., 2015; Onstenk et al., 2015a; Scher et al., 2016). It should, however, be noted that splice variants of ESR1 in breast cancer differ importantly from splice variants of the AR, as ESR1 splice variants are also expressed in healthy breast tissue (Poola and Speirs, 2001), and full‐length AR and splice variants are typically absent in CellSearch‐enriched fractions of HBDs (Onstenk et al., 2015a). It should also be kept in mind that, in the current study, only a limited number of samples could be evaluated for the presence of splice variants. However, given that the ESR1 splice variant ∆5 has been linked to endocrine resistance (Bollig and Miksicek, 2000; Fuqua et al., 1991), is CTC‐specific expressed, and that we found anecdotal evidence of enrichment of this splice variant in paired samples, further research of this splice variant in CTCs is warranted.
5. Conclusion
ESR1 mutations and splice variants in CellSearch‐enriched CTCs were not enriched in MBC patients progressing on palliative endocrine therapy, but ESR1 mutations were enriched in those patients when they were assessed in cfDNA. Therefore, cfDNA appears to be a more sensitive and robust source for detecting ESR1 mutations than DNA from CellSearch‐enriched CTCs. However, the use of other CTC enrichment methods might yield better results (Denis et al., 2016). To improve the sensitivity and specificity of detecting mutations and splice variants, and to really exploit the potential power of CTCs, characterization of pure CTCs with single cell isolation systems is probably required (Swennenhuis and Terstappen, 2015). Until that has been proven feasible and superior to analysis of cfDNA, the detection of ESR1 mutations in cfDNA rather than CTCs is recommended. The increased incidence of ESR1 mutations in cfDNA at the time of progression on endocrine therapy further adds to the evidence that emergence of ESR1 mutations is involved in resistance to endocrine therapy in MBC.
Data Accessibility
Dataset S1. Overview of all data from this study.
Author contributions
NB, AMS, MPJ, JAF, JWM, and SS designed the study; NB, AS, NMV, WO, SRV, and MD performed the laboratory experiments. JK, MPJ, and JWM supervised the experiments; LYD, PH, FEJ, AJ, and CMS included patients in the clinical study for this analysis; NB, WO, and AB collected the clinical data; NB analyzed the data and compiled statistics; NB and AMS wrote the manuscript, which was reviewed, edited, and approved by all authors.
Supporting information
Fig. S1. Flowchart of study procedures.
Fig. S2. Reproducibility of ESR1 mutation measurements in CTCs and cfDNA.
Fig. S3. Cut‐offs for ESR1 mutations in CTCs.
Fig. S4. Cut‐offs for ESR1 mutations in cfDNA.
Fig. S5. Reproducibility of splice variant measurements in T47D cell line.
Fig. S6. Correlation between ESR1 splice variant delta Cq values and CTC counts.
Fig. S7. Dynamics of splice variants in 4 matched samples at baseline and PD.
Table S1. Primer and probe sequences.
Table S2. Spike‐in experiments with and without pre‐amplification.
Table S3. Characteristics of patients in cfDNA subgroup analysis.
Acknowledgements
This work was supported by a grant from Pink Ribbon (Project WO 61) and by a grant from the Cancer Genomics Netherlands (CGC.nl)/Netherlands Organization for Scientific Research (NWO).
References
- Angus L, Beije N, Jager A, Martens JW and Sleijfer S (2017) ESR1 mutations: moving towards guiding treatment decision‐making in metastatic breast cancer patients. Cancer Treat Rev 52, 33–40. [DOI] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA et al (2015) Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration‐resistant prostate cancer. JAMA Oncol 1, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL et al (2014) AR‐V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollig A and Miksicek RJ (2000) An estrogen receptor‐alpha splicing variant mediates both positive and negative effects on gene transcription. Mol Endocrinol 14, 634–649. [DOI] [PubMed] [Google Scholar]
- Chu D, Paoletti C, Gersch C, VanDenBerg D, Zabransky D, Cochran R, Wong HY, Valda Toro P, Cidado J, Croessmann S et al (2015) ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin Cancer Res 22, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi T, Foekens JA, Umar A and Martens JW (2016) Endocrine therapy resistance in estrogen receptor (ER)‐positive breast cancer. Drug Discov Today 21, 1181–1188. [DOI] [PubMed] [Google Scholar]
- Denis JA, Patroni A, Guillerm E, Pepin D, Benali‐Furet N, Wechsler J, Manceau G, Bernard M, Coulet F, Larsen AK et al (2016) Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery. Mol Oncol 10, 1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribbens C, O'Leary B, Kilburn L, Hrebien S, Garcia‐Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S, Loibl S et al (2016) Plasma ESR1 mutations and the treatment of estrogen receptor‐positive advanced breast cancer. J Clin Oncol 34, 2961–2968. [DOI] [PubMed] [Google Scholar]
- Fuqua SA, Fitzgerald SD, Chamness GC, Tandon AK, McDonnell DP, Nawaz Z, O'Malley BW and McGuire WL (1991) Variant human breast tumor estrogen receptor with constitutive transcriptional activity. Cancer Res 51, 105–109. [PubMed] [Google Scholar]
- Guttery DS, Page K, Hills A, Woodley L, Marchese SD, Rghebi B, Hastings RK, Luo J, Pringle JH, Stebbing J et al (2015) Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor‐positive metastatic breast cancer. Clin Chem 61, 974–982. [DOI] [PubMed] [Google Scholar]
- Haber DA and Velculescu VE (2014) Blood‐based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov 4, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric‐Bernstam F, Gonzalez‐Angulo AM, Ferrer‐Lozano J, Perez‐Fidalgo JA, Cristofanilli M, Gomez H et al (2014) Emergence of constitutively active estrogen receptor‐alpha mutations in pretreated advanced estrogen receptor‐positive breast cancer. Clin Cancer Res 20, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostert B, Sieuwerts AM, Kraan J, Bolt‐de Vries J, van der Spoel P, van Galen A, Peeters DJ, Dirix LY, Seynaeve CM, Jager A et al (2015) Gene expression profiles in circulating tumor cells to predict prognosis in metastatic breast cancer patients. Ann Oncol 26, 510–516. [DOI] [PubMed] [Google Scholar]
- Onstenk W, Sieuwerts AM, Kraan J, Van M, Nieuweboer AJM, Mathijssen RHJ, Hamberg P, Meulenbeld HJ, De Laere B, Dirix LY et al (2015a) Efficacy of cabazitaxel in castration‐resistant prostate cancer is independent of the presence of AR‐V7 in circulating tumor cells. Eur Urol 68, 939–945. [DOI] [PubMed] [Google Scholar]
- Onstenk W, Sieuwerts AM, Weekhout M, Mostert B, Reijm EA, van Deurzen CH, Bolt‐de Vries JB, Peeters DJ, Hamberg P, Seynaeve C et al (2015b) Gene expression profiles of circulating tumor cells versus primary tumors in metastatic breast cancer. Cancer Lett 362, 36–44. [DOI] [PubMed] [Google Scholar]
- Poola I and Speirs V (2001) Expression of alternatively spliced estrogen receptor alpha mRNAs is increased in breast cancer tissues. J Steroid Biochem Mol Biol 78, 459–469. [DOI] [PubMed] [Google Scholar]
- Pritchard KI (2013) Endocrine therapy: is the first generation of targeted drugs the last? J Intern Med 274, 144–152. [DOI] [PubMed] [Google Scholar]
- Reijm EA, Sieuwerts AM, Smid M, Vries JB, Mostert B, Onstenk W, Peeters D, Dirix LY, Seynaeve CM, Jager A et al (2016) An 8‐gene mRNA expression profile in circulating tumor cells predicts response to aromatase inhibitors in metastatic breast cancer patients. BMC Cancer 16, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana‐Sundaram S, Wang R, Ning Y, Hodges L et al (2013) Activating ESR1 mutations in hormone‐resistant metastatic breast cancer. Nat Genet 45, 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scariano JK, Emery‐Cohen AJ, Pickett GG, Morgan M, Simons PC and Alba F (2008) Estrogen receptors alpha (ESR1) and beta (ESR2) are expressed in circulating human lymphocytes. J Recept Signal Transduct Res 28, 285–293. [DOI] [PubMed] [Google Scholar]
- Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D et al (2016) Association of AR‐V7 on circulating tumor cells as a treatment‐specific biomarker with outcomes and survival in castration‐resistant prostate cancer. JAMA Oncol 2, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavon G, Hrebien S, Garcia‐Murillas I, Cutts RJ, Pearson A, Tarazona N, Fenwick K, Kozarewa I, Lopez‐Knowles E, Ribas R et al (2015) Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 7, 313ra182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieuwerts AM, Kraan J, Bolt‐de Vries J, van der Spoel P, Mostert B, Martens JW, Gratama JW, Sleijfer S and Foekens JA (2009) Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real‐time PCR. Breast Cancer Res Treat 118, 455–468. [DOI] [PubMed] [Google Scholar]
- Sieuwerts AM, Mostert B, Bolt‐de Vries J, Peeters D, de Jongh FE, Stouthard JM, Dirix LY, van Dam PA, Van Galen A, de Weerd V et al (2011) mRNA and microRNA expression profiles in circulating tumor cells and primary tumors of metastatic breast cancer patients. Clin Cancer Res 17, 3600–3618. [DOI] [PubMed] [Google Scholar]
- Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, Aimi J, Derynck MK, Chen M, Chan IT et al (2016) Heterogeneity and clinical significance of ESR1 mutations in ER‐positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 7, 11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swennenhuis JF and Terstappen L (2015) Sample preparation methods following cell search approach compatible of single‐cell whole‐genome amplification: an overview. Methods Mol Biol 1347, 57–67. [DOI] [PubMed] [Google Scholar]
- Takeshita T, Yamamoto Y, Yamamoto‐Ibusuki M, Inao T, Sueta A, Fujiwara S, Omoto Y and Iwase H (2015) Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res 166, 540–553.e542. [DOI] [PubMed] [Google Scholar]
- Takeshita T, Yamamoto Y, Yamamoto‐Ibusuki M, Inao T, Sueta A, Fujiwara S, Omoto Y and Iwase H (2016) Clinical significance of monitoring ESR1 mutations in circulating cell‐free DNA in estrogen receptor positive breast cancer patients. Oncotarget 7, 32504–32518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SE, Martin‐Hirsch PL and Martin FL (2010) Oestrogen receptor splice variants in the pathogenesis of disease. Cancer Lett 288, 133–148. [DOI] [PubMed] [Google Scholar]
- Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA et al (2013) ESR1 ligand‐binding domain mutations in hormone‐resistant breast cancer. Nat Genet 45, 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, Jonnalagadda AR, Trejo Bittar HE, Berg A, Hamilton RL et al (2016) Sensitive detection of mono‐ and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell‐free DNA of breast cancer patients. Clin Cancer Res 22, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flowchart of study procedures.
Fig. S2. Reproducibility of ESR1 mutation measurements in CTCs and cfDNA.
Fig. S3. Cut‐offs for ESR1 mutations in CTCs.
Fig. S4. Cut‐offs for ESR1 mutations in cfDNA.
Fig. S5. Reproducibility of splice variant measurements in T47D cell line.
Fig. S6. Correlation between ESR1 splice variant delta Cq values and CTC counts.
Fig. S7. Dynamics of splice variants in 4 matched samples at baseline and PD.
Table S1. Primer and probe sequences.
Table S2. Spike‐in experiments with and without pre‐amplification.
Table S3. Characteristics of patients in cfDNA subgroup analysis.
Data Availability Statement
Dataset S1. Overview of all data from this study.