

Abstract
Osteosarcoma (OS) is an aggressive bone tumor primarily affecting children and adolescents. The etiology of OS is not fully understood. Thus, there is a great need to obtain a better understanding of OS development and progression. Alterations in miRNA expression contribute to the required molecular alterations for neoplastic initiation and progression. This study is the first to investigate miRNA expression in OS in a large discovery and validation cohort comprising a total of 101 OS samples. We established the signature of altered miRNA expression in OS by profiling the expression level of 752 miRNAs in 23 OS samples using sensitive LNA‐enhanced qPCR assays. The identified miRNA expression changes were correlated with gene expression in the same samples. Furthermore, miRNA expression changes were validated in a second independent cohort consisting of 78 OS samples. Analysis of 752 miRNAs in the discovery cohort led to the identification of 33 deregulated miRNAs in OS. Twenty‐nine miRNAs were validated with statistical significance in the second cohort comprising 78 OS samples. miRNA/mRNA targets were determined, and 361 genes with an inverse expression of the target miRNA were identified. Both the miRNAs and the identified target genes were associated with multiple pathways related to cancer as well as bone cell biology, thereby correlating the deregulated miRNAs with OS tumorigenesis. An analysis of the prognostic value of the 29 miRNAs identified miR‐221/miR‐222 to be significantly associated with time to metastasis in both cohorts. This study contributes to a more profound understanding of OS tumorigenesis, by substantiating the importance of miRNA deregulation. We have identified and validated 29 deregulated miRNAs in the – to our knowledge – largest discovery and validation cohorts used so far for miRNA analyses in OS. Two of the miRNAs showed a promising potential as prognostic biomarkers for the aggressiveness of OS.
Keywords: epigenetics, metastasis, miRNA, osteosarcoma
Abbreviations
- FF
fresh frozen
- FFPE
formalin‐fixed paraffin‐embedded
- HE
hematoxylin/eosin
- IPA
Ingenuity Pathway Analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- miRNA
microRNA
- MSC
mesenchymal stem cell
- OB
osteoblast
- OS
osteosarcoma
- PCA
principal component analysis
- QC
quality control
- UTR
untranslated region
1. Introduction
Osteosarcoma (OS) is an aggressive tumor of the bone most frequently affecting children and adolescents. OS is often diagnosed at a very late stage, as the first indication of the malignancy is pain, which is often mistaken for more common conditions such as ‘growing pains’ (McCarville, 2009). The late identification of OS leads to 15–25% of the patients exhibiting distant metastases at the time of diagnosis (Bielack et al., 2002). The five‐year survival rate of OS patients with no detectable metastases at the time of diagnosis is 65–70% (Bielack et al., 2002), whereas patients with relapsing disease or detectable metastases at the time of diagnosis have only a five‐year survival rate of 10–40% (Kager et al., 2003; Meyers et al., 1993).
OS is characterized by diverse cytogenetic alterations and exhibits changes in multiple different pathways with substantial cell‐to‐cell variation (Bridge et al., 1997). The etiology of OS is not well understood and the only prognostic markers are the absence of metastases at the time of diagnosis and the response to neoadjuvant chemotherapy measured as the extent of tumor necrosis (Wang, 2005). However, the characteristic onset of high‐grade neoplasms in young people indicates a consistent, but yet undetected, alteration or group of alterations defining OS.
Cancer initiation and progression is controlled by both genetic and epigenetic events (Hanahan and Weinberg, 2011). Epigenetic changes are particularly important for childhood cancers, which are often characterized by a low mutational load. Molecular subtypes, clinical heterogeneity, and in some cases the prediction of the disease course have been associated with epigenetic changes. This has been demonstrated for ependymomas (Mack et al., 2014) and more recently Ewing sarcoma (Sheffield et al., 2017), which are characterized by no or the same recurrent genetic aberration, respectively. The genetic components of OS have been intensively studied (Bishop et al., 2016; Kansara et al., 2014); however, the elucidation of the epigenetic changes is still in its early stage. Gene expression is regulated by numerous mechanisms and molecules, including microRNAs (miRNAs). These are small noncoding RNA molecules (22–25 nucleotides), regulating gene expression post‐transcriptionally by blocking mRNA translation and/or altering the stability of the mRNA by binding perfectly or with mismatches to the 3′‐untranslated region (UTR) of the mRNA (Stark et al., 2003). Each miRNA can potentially directly or indirectly regulate more than 100 target genes and thereby affect the expression of both tumor suppressors and oncogenes (Brennecke et al., 2005). Furthermore, miRNAs function as classical tumor suppressors and oncogenes/oncomiRs as specific miRNA alterations are associated with carcinogenesis. Deregulation of miRNA expression has been identified in numerous cancers emphasizing the importance of miRNAs in carcinogenesis and their potential as diagnostic, prognostic, and therapeutic means (Calin and Croce, 2006; Negrini et al., 2009).
miRNAs are also important regulators of osteogenic signaling pathways and are implicated in normal osteoblast (OB) growth and differentiation (Inose et al., 2009; Lian et al., 2012). Hence, changes in this balance will affect proliferation of bone cells and may contribute to OS development. The impact of alterations in miRNA expression patterns in OS development has been addressed in several studies (Duan et al., 2011; Jones et al., 2012; Lulla et al., 2011; Maire et al., 2011; Namlos et al., 2012; Thayanithy et al., 2012; Won et al., 2013; Zhang et al., 2015). However, none of the results are consistent across the studies, which may reflect different methodologies and normalization strategies, as well as low sample numbers. Most studies have evaluated a single miRNA, but a few studies have established a miRNA expression profile of OS by profiling several hundred miRNAs (Duan et al., 2011; Jones et al., 2012; Lulla et al., 2011; Maire et al., 2011; Namlos et al., 2012; Won et al., 2013; Zhang et al., 2015). However, all studies have analyzed a limited number of OS samples ranging from 2 to 18.
In this study, we established the signature of miRNA expression alterations in OS by profiling the expression of 752 miRNAs in 23 OS samples using a highly sensitive qPCR technology. Significantly deregulated miRNAs were validated in an independent cohort comprising 78 OS samples. Both the miRNAs and identified target mRNAs were evaluated for their relation to cancer development and progression, as well as bone cell biology.
2. Materials and methods
2.1. OS samples
Fresh‐frozen diagnostic biopsies of primary tumors from 23 patients diagnosed with OS were collected from the archives of the Institute of Pathology, Aarhus University Hospital (Table 1). The biopsies were immediately snap‐frozen and stored at −80 °C. 25–50 tissue sections of 10 μm were used for RNA extraction for each sample. Tissue sections were hematoxylin/eosin (HE)‐stained for every fifth 10‐μm tissue section cut to confirm the presence of tumor cells.
Table 1.
Clinical and pathological data for 23 fresh‐frozen and 78 FFPE primary OS samples analyzed
| Patient characteristics | Number of patients (%) | |
|---|---|---|
| Discovery cohort | Validation cohort | |
| Total | 23 (100) | 78 (100) |
| Gender | ||
| Male | 11 (48) | 43 (55) |
| Female | 12 (52) | 35 (45) |
| Age (years) | ||
| Median age | 17 | 25.0 |
| Mean age | 27.9 | 32.0 |
| Range | 9–76 | 5–82 |
| Primary site | ||
| Femur | 8 (35) | 27 (35) |
| Tibia | 10 (44) | 22 (28) |
| Fibula | 1 (4) | 2 (2) |
| Others | 4 (17) | 27 (35) |
| Metastasis at diagnosis | ||
| Yes | 8 (35) | 33 (42) |
| No | 15 (65) | 45 (58) |
| Histology | ||
| Osteoblastic | 9 (39) | 30 (39) |
| Chondroblastic | 9 (39) | 22 (28) |
| Telangiectasic | 3 (13) | 4 (5) |
| Othera | 2 (9) | 22 (28) |
| Chemotherapy treatment | ||
| < 40 years: Cisp., doxo., meth. | 15 (65) | 44 (56) |
| > 40 years: Cisp., doxo. | 2 (9) | 7 (9) |
| Unknown | 6 (26) | 27 (35) |
| Tumor necrosis after chemotherapy | ||
| Good responders (> 90% necrosis)b | 9 (39) | 17 (22) |
| Poor responders (< 90% necrosis)c | 8 (35) | 34 (43) |
| Unknown | 6 (26) | 27 (35) |
| Survival (months) | ||
| Mean survival | 75.5 | 81.7 |
| Range | 1–175 | 1–282 |
| Long‐term survival | ||
| < 5 years | 7 (30) | 30 (39) |
| > 5 years | 12 (52) | 42 (54) |
| Not evaluatedd | 4 (18) | 6 (7) |
| Type of sample analyzed | ||
| Biopsy | 23 (100) | 66 (85) |
| Larger tumor specimen | 0 (0) | 12 (15) |
Cisp., cisplatin; doxo., doxorubicin; meth., methotrexate.
All OS samples were high‐grade tumors except for two of the FFPE samples, which were parosteal OS.
Of the good responders, one patient in the discovery cohort and one in the validation cohort were > 40 years.
Of the poor responders, one patient in the discovery cohort and six in the validation cohort were > 40 years.
Patients with < 5 years since diagnosis or who died of other causes than OS.
Formalin‐fixed, paraffin‐embedded (FFPE) samples from 78 primary tumors from patients diagnosed with OS were collected from the archives of the Institute of Pathology, Aarhus University Hospital (Table 1). A complete outline of the different procedures and analyses performed with each cohort is shown in Fig. 1.
Figure 1.
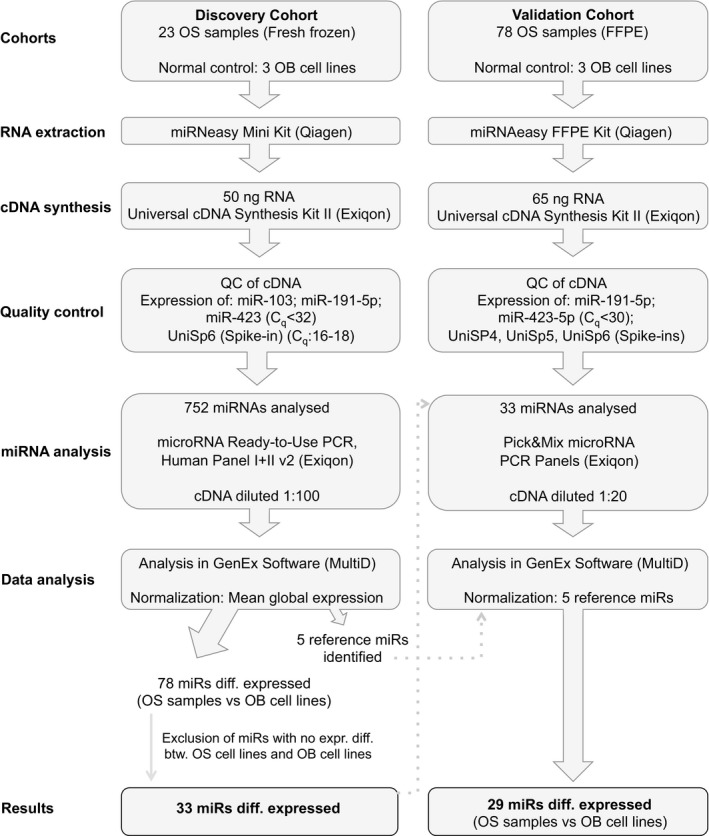
A detailed outline of the different processes and analyses performed for the OS discovery and validation cohort. QC, quality control; diff., differential; btw., between.
The Local Ethical Committee, The Central Denmark Region, approved this study (journal number 1‐10‐72‐521‐12).
2.2. Cell lines
Five OS and three OB cell lines were used in this study. The OS cell lines CRL‐1543, CRL‐1427, CRL‐2098, HTB‐85, and HTB‐96 and the OB cell line CRL‐11372 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The OB cell line C‐12720 was purchased from PromoCell (Heidelberg, Germany), and the OB cell line HO‐f‐4610 was purchased from SanBio (Uden, The Netherlands). HTB‐96 and HTB‐85 were cultured in McCoy's 5a medium modified. CRL‐1427 and CRL‐1543 were cultured in Eagle's minimum essential medium. CRL‐2098 was cultured in RPMI‐1640. All OS cell lines were supplemented with 40 mg·mL−1 streptomycin, 240 mg·mL−1 penicillin (400 000 U·mL−1), and 10% fetal bovine serum, except for HTB‐85, which was supplemented with 15% fetal bovine serum. CRL‐11372 was cultured in 1 : 1 mixture of Ham's F12 medium and Dulbecco's modified Eagle's medium without phenol red, supplemented with 0.3 mg·mL−1 G‐418 and 10% fetal bovine serum. C‐12720 was cultured in OB growth medium + supplement mix (PromoCell). HO‐f was cultured in OB medium (ScienCell Research Laboratories, Carlsbad, CA, USA), and the flasks were coated with poly‐L‐lysine solution bioreagent 0.01% (Sigma Aldrich, St Louis, MO, USA).
All eight cell lines were cultured in a humidified atmosphere of 5% CO2 at 37 °C, except for CRL‐11372, which was cultured at 34 °C.
2.3. Total RNA extraction from fresh‐frozen OS samples and cell lines
Total RNA including miRNAs was extracted from the 23 fresh‐frozen OS samples and the eight cell lines. The tissue was homogenized using a TissueLyser bead mill and one tungsten carbide bead (3 mm) per sample. RNA extraction was performed using the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions until each sample was separated into three phases. The upper aqueous phase was transferred to a new tube and total RNA was purified using a QIAcube automated device (Qiagen) according to the manufacturer's instructions including an on‐column DNase digest. RNA was quantified using a NanoDrop ND‐2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and the quality was assessed using the RNA 6000 Nano Kit (Agilent Technologies Inc., Santa Clara, CA, USA) on an Agilent 2100 BioAnalyzer.
2.4. RNA extraction from FFPE samples
Four tissue sections of 10 μm from each FFPE sample were used for RNA extraction. RNA was extracted using the miRNeasy FFPE Kit (Qiagen) according to the manufacturer's instructions (Fig. 1). One microlitre Spike‐in (UniSp2, UniSp4, and UniSp5), from the RNA Spike‐in kit, UniRT (Exiqon, Vedbæk, Denmark), was added to the PKD buffer during the purification.
2.5. cDNA synthesis and miRNA expression analysis of fresh‐frozen OS samples
For the initial miRNA profiling analysis, 50 ng of total RNA from 23 fresh‐frozen OS samples, two OS, and three OB cell lines was reverse‐transcribed using the miRCURY LNA™Universal RT microRNA PCR kit (Exiqon) in a Mastercycler® pro S (Eppendorf, Hamburg, Germany) in a 50 μL reaction volume, comprising the Universal cDNA synthesis kit II, RNA Spike‐in kit (Exiqon) (Fig. 1). Quality control (QC) was performed according to the manufacturer's instructions using the ExiLENT SYBR® Green master mix kit (Exiqon), using 1 : 80 diluted cDNA. Quality of the cDNA synthesis and PCR amplification were evaluated through the detection of miR‐103, miR‐191‐5p, miR‐423, and UniSp6 at stable C q levels (C q < 32) (Fig. 1).
The miRNA profile was established using 45 ng cDNA from each sample by qPCR using the microRNA Ready‐to‐Use PCR, Human panel I and panel II v2 analyzing 752 miRNAs (Exiqon) on a Lightcycler® 480 V2 Real‐Time PCR System (Roche Applied Science, Penzberg, Germany) (Fig. 1). The plates were set up using a Bravo Automated Liquid Handling Platform (Agilent Technologies Inc.).
2.6. Validation of miRNA expression in OS FFPE samples
A total of 65 ng RNA (5 ng·μL−1) from the OS FFPE samples and the cell lines was reverse‐transcribed in 20 μL reactions using the Universal cDNA Synthesis kit II (Exiqon) and a 2720 Thermal Cycler (Thermo Fisher Scientific) (Fig. 1). Reverse transcription was performed in duplicate for each sample. All the subsequent amplifications were performed using a Lightcycler® 480 Real‐Time PCR System (Roche Applied Science). QC of the cDNA synthesis and PCR amplification were performed according to the manufacturer's instructions (Exiqon), with slight modifications; the cDNA was diluted 1 : 20 to ensure the detection of lowly expressed miRNAs. Spike‐ins were included in the RNA purification and RT step using the RNA Spike‐in kit, UniRT (Exiqon). hsa‐miR‐191‐5p, hsa‐miR‐423‐5p, UniSp4, UniSp5, UniSp6, and Cel‐miR‐39‐3p were used for QC of the RNA purification and cDNA synthesis (Fig. 1). UniSp4 and UniSp5 RNA spike‐in controls were used for QC of RNA purifications. All primary OS FFPE samples, except one, had C q values < 37 for UniSp5. UniSp6 and Cel‐miR‐39‐3p spike‐in control primers were used for QC of the cDNA synthesis and to rule out PCR inhibition, as a higher concentration of cDNA was used in the analysis, than described in the manufacturer's instructions (65 ng RNA instead of 20 ng for the cDNA synthesis and 1 : 20 dilution of the cDNA instead of 1 : 100). These conditions were optimized for these amounts to ensure adequate amplification from the FFPE samples. All primary OS FFPE samples had C q values between 16.5 and 18 for UniSp6. Negative controls included in the cDNA synthesis were a no RNA template and a no RT enzyme.
miRNA expression analysis was performed for 78 FFPE primary OS samples, five OS, and the three OB cell lines using 5 μL 1 : 20 diluted cDNA from each sample using a custom Pick&Mix microRNA PCR Panel, 384 well Ready‐to‐Use (Exiqon) (Fig. 1). Five reference miRNAs, hsa‐miR‐148b‐3p, hsa‐miR‐185‐5p, hsa‐miR‐191‐5p, hsa‐miR‐423‐5p, and hsa‐miR‐425‐3p, were selected based on the results from the discovery cohort, using both geNorm (Vandesompele et al., 2002) and NormFinder (Andersen et al., 2004). All 78 OS FFPE samples had C q values < 30 for the reference miRNAs.
2.7. Gene expression analysis
Genomic RNA expression analysis was performed using the SurePrint G3 Human 8X60K One color Microarrays (Agilent), which analyzes 27958 Entrez Gene RNAs and 7419 lincRNAs. Hundred nanogram RNA from each sample of the 23 fresh‐frozen OS primary tumors, five OS cell lines, and three OB cell lines was analyzed as per the manufacturer's instructions. The Agilent G3 Microarrays were scanned using the NimbleGen MS200 scanner with Multi‐TIFF as File Settings Control, a single scan area was selected, and the entire feature area was captured. Two‐micrometre resolution was selected. The red channel was omitted from the analysis. Grid alignment was determined manually. Data were extracted using the Agilent Feature Extraction 11.5.1.1. The raw data were preprocessed using ArrayStar v12.1.0 build 134 (DNASTAR, Madison, WI, USA) and normalized using the quantile normalization method and log2‐transformed. Genes were considered differentially expressed when the difference between OS samples and OB cell lines was > 2‐fold and with a Bonferroni‐corrected P‐value < 0.05. Only genes with an average normalized probe intensity of log2 > 6 in OB cell lines were considered expressed.
2.8. Data analysis
miRNA expression analysis was performed using the genex software version 5.4.2.128 (MultiD, Göteborg, Sweden). Raw data (C q values) for the 752 miRNAs analyzed using the discovery cohort were imported and merged (Table S1). Each sample has been run with two 384‐well plates containing different miRNAs. Hence, the raw data contain numerous NA values for the miRNAs not present in the respective plates. The mean global expression was used for normalization of the Human Panel I+II v2. The limit for a detectable miRNA was set to C q < 37. Log2‐normalized data are available in Table S2. Pick&Mix microRNA PCR Panels were normalized using five selected stably expressed reference genes (hsa‐miR‐148b‐3p, hsa‐miR‐185‐5p, hsa‐miR‐191‐5p, hsa‐miR‐423‐5p, and hsa‐miR‐425‐3p) (Fig. 1). Raw data (C q values) for the 33 miRNAs analyzed in the validation cohort are available in Table S3. The raw data have been imported and merged using the genex software version 5.4.2.128 (MultiD). Log2‐normalized data for the validation cohort are available in Table S4.
The statistical software r v.3.0.0 (R Core Team 2013) was used for unsupervised hierarchical clustering using the ‘Ward’ method as distance measure with an in‐house‐developed script. Pathway and network analyses were performed using Ingenuity® Pathway Analysis (IPA®; Qiagen), the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000; Kanehisa et al., 2014), and DIANA miRPath.v3 (Vlachos et al., 2015). For the IPA core analysis, the KEGG pathway analysis, and the DIANA pathway analysis, human was selected as the species analyzed.
Validated miRNA gene targets were established using miRWalk2.0 (Dweep and Gretz, 2015) with the identification of miRNA binding sites in the 3′UTR. The generated gene lists of validated miRNA gene targets were compared with a list of genes comprising > 2‐fold change (P‐value < 0.05) in expression between 23 OS samples and three OB cell lines using Venn analysis (G. B. Andersen, T. E. Kjeldsen, F. Busato, C. Dong, J. Q. D. Tran, A. Daunay, D. B. Hussmann, F. Jehan, V. Geoffroy, J. F. Deleuze, H. Hager, E. Willerslev, L. L. Hansen & J. Tost, in preparation). Boxplots, Mann–Whitney U‐tests, and Kaplan–Meier analysis were made with the graphpad prism 7 Software (GraphPad Software Inc., La Jolla, CA, USA). For Kaplan–Meier analysis, the log‐rank (Mantel–Cox) test was used for the determination of P‐values. For the prognostic analyses (survival and time to metastasis), median expression was used to subdivide the samples into high and low expression groups.
2.9. Data accessibility
All miRNA data generated and analyzed during this study are included in this published article (Tables S1–S4 including both raw and log2‐normalized data). The normalized log2 expression data for the 361 genes analyzed in this study are provided in Table S5. The raw gene expression dataset used in the current study is part of another ongoing study and will be published and deposited in a public repository separately. Meanwhile, these data are available from the corresponding author upon request.
3. Results
In the present study (Fig. 1), we investigated changes in miRNA expression of 752 miRNAs in primary OS and OS cell lines compared to the miRNA expression levels of OB cell lines. OB cell lines were used as a reference as OBs/osteoprogenitor cells are considered to represent the cell of origin for OS (Abarrategi et al., 2016). The 33 miRNAs identified as differentially expressed in the OS discovery cohort were validated in an independent validation cohort comprising 78 primary OS samples.
3.1. Overall miRNA expression
The relative expression levels of 752 miRNAs were evaluated in 23 primary OS samples, two OS, and three OB cell lines. Following normalization, using the global mean, a total of 339 and 202 miRNAs were reliably detected in more than 40% and 90% of the samples, respectively. The frequency of missing data was calculated based on all samples (both OS samples and OB cell lines). Further analyses were performed using the dataset with the expression of 339 miRNAs to ensure analysis of all miRNAs important for OS etiology. If using a 90% threshold, miRNAs not expressed in OB cell lines or miRNAs downregulated/depleted in OS samples could be removed, thereby excluding biologically relevant miRNAs.
An unsupervised hierarchical clustering was performed using the log2‐transformed expression values for the 339 miRNAs (Fig. 2). The samples separated into two main clusters, of which one contained 17 OS samples and the two OS cell lines. Even though the OS cell lines were in a cluster with OS samples, the cell lines kept the closest mutual resemblance, indicating a cell line similarity stronger than the cell origin. The other cluster contained six OS samples and the three OB cell lines. There was no difference between tumor content or tumor/patient characteristics in these six OS samples compared with the rest of the OS cohort.
Figure 2.
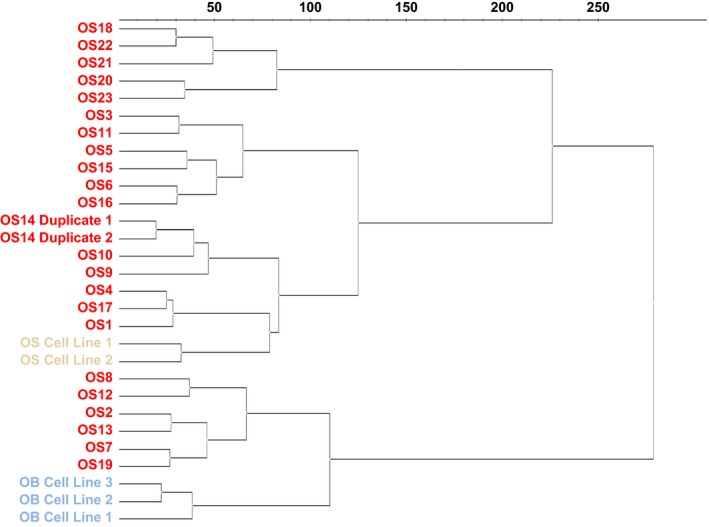
Unsupervised hierarchical cluster analysis. The expression value (log2) for each detectable miRNA (C q < 37) was used to determine the clustering of the samples. Red: primary OS samples; yellow: OS cell lines; blue: OB cell lines.
The two observed clusters could not be explained with the available clinical and histopathological data as no separation in terms of the primary tumor site, histology, age, gender, or metastatic status was observed, even though a smaller group containing five OS samples separated from the other OS samples (Fig. S1). These samples had a slightly higher miRNA detection level, which might explain the separation. Of note, these samples were different from the six OS samples grouping apart in the cluster.
3.2. Identification of differentially expressed miRNAs in OS
When comparing expression profiles between OS samples and OB cell lines, 76 miRNAs were identified as differentially expressed (P‐value < 0.05) (Table S6). As OB cell lines were used as the normal control, miRNAs with no expression change between OS and OB cell lines were excluded to avoid expression changes caused by cell culture and not as a result of cell origin. This provided a list of 33 miRNAs with deregulated expression levels, of which 26 were significantly downregulated and seven were upregulated in the OS samples compared to OB cell lines (Table 2, Fig. 3 and Fig. S2A,B). Four of these miRNAs (miR‐181a‐5p, miR‐181b‐5p, miR‐30e‐3p, and miR‐92b‐3p) did not exhibit an expression difference between OS and OB cell lines, but were, nonetheless, selected for further validation as miR‐181a‐5p, miR‐181b‐5p, and miR‐92b‐3p have previously been associated with OS development and miR‐30e‐3p has been shown to be deregulated in several other cancers (Duan et al., 2011; Jones et al., 2012; Ning et al., 2016; Zhu et al., 2014). Of the 33 differentially expressed miRNAs, 12 miRNAs have not previously been described as differentially expressed in OS (Table S7). Of the previously described miRNAs, 16 were identified with the same direction of the expression change as in this study, while five miRNAs showed an opposite expression change (Table S7). On the other hand, previous studies using microarrays or sequencing to investigate miRNA expression changes in OS identified 14 miRNAs, which were not part of the 33 validation miRNAs found in this study (each miRNA was identified in three or more studies) (Table S8). However, 10 of the miRNAs were identified as both up‐ and downregulated in different profiling studies. A further investigation of these 14 miRNAs in the OS discovery cohort analyzed in this study showed that six of the miRNAs did exhibit a differential expression in OS samples when compared to OB cell lines, but no difference was observed between OS and OB cell lines; four of the miRNAs did not exhibit a significant difference between OS samples and OB cell lines, and four miRNAs were not present in our normalized data set.
Table 2.
Expression level of 33 significantly differentially expressed miRNAs in OS samples compared to OB cell lines
| miRNA | Discovery cohort | Validation cohort | ||||
|---|---|---|---|---|---|---|
| Fold changea | P‐value | No. OS sign. changed (%) | Fold changea | P‐value | No. OS sign. changed (%) | |
| Downregulated miRNAs | ||||||
| miR‐100‐5p | −34.75 | 8.0 × 10−4 | 23 (100) | −12.80 | 2.34 × 10−5 | 76 (97) |
| miR‐221‐3p | −14.21 | 8.0 × 10−4 | 23 (100) | −10.79 | 2.34 × 10−5 | 78 (100) |
| miR‐29b‐1‐5p | −16.45 | 8.0 × 10−4 | 23 (100) | −17.44 | 2.34 × 10−5 | 78 (100) |
| miR‐125b‐1‐3p | −13.50 | 8.0 × 10−4 | 23 (100) | −5.31 | 9.38 × 10−5 | 76 (97) |
| miR‐29a‐5p | −33.65 | 8.0 × 10−4 | 23 (100) | −3.60 | 9.38 × 10−5 | 72 (92) |
| miR‐370‐3p | −14.89 | 0.0031 | 20 (87) | −19.62 | 9.38 × 10−5 | 77 (99) |
| miR‐299‐5p | −19.42 | 0.0015 | 22 (96) | −24.42 | 2.0 × 10−4 | 76 (97) |
| miR‐493‐5p | −25.62 | 0.0085 | 20 (87) | −32.46 | 3.0 × 10−4 | 75 (96) |
| miR‐409‐3p | −31.36 | 0.0015 | 22 (96) | −19.60 | 4.0 × 10−4 | 71 (91) |
| miR‐30e‐3p | −2.91 | 0.0015 | 21 (91) | −2.78 | 0.0012 | 67 (86) |
| miR‐431‐5p | −4.92 | 0.0031 | 19 (83) | −16.77 | 0.0012 | 69 (88) |
| miR‐432‐5p | −16.05 | 0.0031 | 22 (96) | −22.79 | 0.0016 | 74 (95) |
| miR‐410‐3p | −16.83 | 8.0 × 10−4 | 21 (91) | −14.66 | 0.0019 | 70 (90) |
| miR‐411‐5p | −36.54 | 8.0 × 10−4 | 22 (96) | −14.47 | 0.0034 | 67 (86) |
| miR‐376c‐3p | −23.90 | 0.0031 | 22 (96) | −16.57 | 0.0056 | 61 (78) |
| miR‐125b‐5p | −8.45 | 0.0015 | 22 (96) | −4.06 | 0.0064 | 61 (78) |
| miR‐335‐5p | −45.39 | 8.0 × 10−4 | 23 (100) | −5.32 | 0.0064 | 64 (82) |
| miR‐376a‐3p | −34.17 | 0.0085 | 21 (91) | −15.79 | 0.0064 | 65 (83) |
| miR‐382‐5p | −13.97 | 0.0054 | 21 (91) | −10.53 | 0.0135 | 62 (79) |
| miR‐154‐5p | −13.62 | 0.0085 | 20 (87) | −9.04 | 0.0244 | 53 (68) |
| miR‐222‐3p | −5.11 | 0.0123 | 19 (83) | −2.49 | 0.0267 | 54 (69) |
| miR‐92b‐3p | −3.19 | 0.0085 | 21 (91) | −2.01 | 0.0267 | 58 (74) |
| miR‐433‐5p | −4.16 | 0.0054 | 18 (78) | −4.76 | 0.029 | 52 (67) |
| miR‐127‐3p | −21.94 | 0.0054 | 22 (96) | −7.72 | 0.04 | 60 (77) |
| miR‐34a‐3p | −3.44 | 0.0085 | 17 (74) | −2.99 | 0.0888 | 5 (5) |
| miR‐136‐5p | −13.63 | 0.0315 | 21 (91) | −2.72 | 0.248 | 35 (45) |
| Upregulated miRNAs | ||||||
| miR‐181a‐5p | 12.04 | 8.0 × 10−4 | 23 (100) | 8.38 | 2.34 × 10−5 | 78 (100) |
| miR‐181c‐5p | 9.29 | 0.0015 | 20 (87) | 15.24 | 2.34 × 10−5 | 60 (77) |
| miR‐223‐3p | 727.23 | 8.0 × 10−4 | 23 (100) | 859.9 | 2.34 × 10−5 | 78 (100) |
| miR‐342‐3p | 12.84 | 0.0031 | 17 (74) | 3.44 | 9.38 × 10−5 | 75 (96) |
| miR‐378a‐3p | 4.20 | 0.0177 | 19 (83) | 5.49 | 3.0 × 10−4 | 74 (95) |
| miR‐128‐3p | 3.22 | 0.0015 | 22 (96) | 1.06 | 0.9328 | 16 (21) |
| miR‐181b‐5p | 4.19 | 0.0085 | 14 (61) | 2.72 | 0.1051 | 25 (32) |
Discovery cohort = 23 fresh‐frozen OS samples; validation cohort = 78 FFPE OS samples; miRNAs in bold text = significantly differentially expressed in both cohorts. No., number.
Expression change between primary OS samples and OB cell lines.
Figure 3.
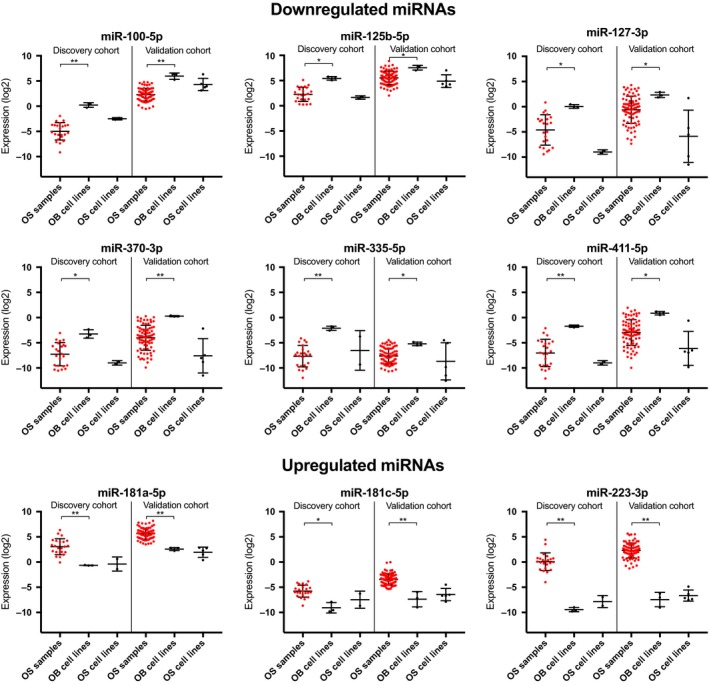
Boxplots of the expression of nine differentially expressed miRNAs. Red: OS samples. *P < 0.05; **P < 0.001.
3.3. Validation of differentially expressed miRNAs
The 33 miRNAs identified in the discovery cohort were validated in a larger, independent cohort consisting of 78 primary OS FFPE samples using a custom‐designed Pick&Mix microRNA PCR Panel.
Of the 26 miRNAs with decreased expression, 24 were confirmed in the validation cohort, and of the seven miRNAs with increased expression, five were confirmed, thereby corroborating 88% of our initial findings (Table 2, Fig. 3 and Fig. S2A,B).
3.4. Pathways associated with the deregulated miRNAs and their target genes
To determine the possible impact of the 29 differentially expressed miRNAs on OS development, enriched biological functions were explored using Ingenuity Pathway Analysis (IPA) and DIANA Tools mirPath.v3 exploring KEGG pathways. Several biological functions related to cancer development and progression were enriched for the 29 deregulated miRNAs (Table 3).
Table 3.
Biological functions associated with the 29 deregulated miRNAs in OS. (A) IPA biological functionsa and (B) KEGG pathwaysb identified by DIANA miRPath.v3
| (A) | P‐value | No. of miRNAs associated |
|---|---|---|
| Diseases and disorders | ||
| Cancerc | 4.14 × 10−13 | 18 |
| Connective tissue disorders | 4.14 × 10−13 | 12 |
| Organismal injury and abnormalities | 4.14 × 10−13 | 20 |
| Reproductive system disorders | 8.23 × 10−9 | 15 |
| Developmental disorder | 4.49 × 10−8 | 6 |
| Molecular and cellular functions | ||
| Cell death and survivalc | 2.84 × 10−4 | 5 |
| Cell‐to‐cell signaling and interactionc | 0.00342 | 1 |
| Cellular developmentc | 0.00342 | 6 |
| Cellular growth and proliferationc | 0.00570 | 6 |
| Cell cyclec | 0.0125 | 1 |
| (B)Pathway | P‐value | No. of miRNAs associated |
|---|---|---|
| Proteoglycans in cancerc | 2.93 × 10−13 | 20 |
| Prion diseases | 1.52 × 10−10 | 15 |
| ECM/receptor interactionc | 9.02 × 10−10 | 20 |
| Other types of O‐glycan biosynthesis | 6.83 × 10−8 | 13 |
| Hippo signaling pathwayc | 1.49 × 10−7 | 20 |
| Gliomac | 5.18 × 10−6 | 19 |
| Signaling pathways regulating pluripotency of stem cellsc | 5.18 × 10−6 | 21 |
| Steroid biosynthesis | 5.38 × 10−6 | 9 |
| Mucin‐type O‐glycan biosynthesis | 9.70 × 10−6 | 14 |
| Transcriptional misregulation in cancerc | 2.95 × 10−5 | 21 |
Diseases and molecular functions associated with the 29 deregulated miRNAs identified by IPA.
KEGG pathways associated with the 29 deregulated miRNAs identified by DIANA miRPath.v3.
Association with cancer development and progression.
Alterations in miRNA expression may have an extensive effect on gene expression, as one miRNA can directly or indirectly regulate more than 100 target genes by binding to their 3′UTR. Genes targeted by at least one of the 29 differentially expressed miRNAs were therefore identified using miRWalk2.0 analyzing only experimentally validated target genes. Genome‐wide RNA expression changes in OS were established by analyzing the 23 OS samples from the discovery cohort with RNA expression microarrays containing 27958 Entrez Gene RNAs (Andersen et al., in preparation). The identified target genes were correlated with genes identified as differentially expressed in the OS samples compared to OB cell lines (> 2‐fold change and P‐value < 0.05). This identified 301 genes with upregulated expression in OS samples, targeted by one or more of the identified downregulated miRNAs, as well as 60 genes with decreased expression in the OS samples targeted by one or more of the miRNAs with increased expression in the OS samples (Table S5).
The possible influence of the deregulation of miRNA expression on OS development, associated with the affected target genes, was investigated using IPA and KEGG pathway analysis. The most significant pathways, as well as biological functions associated with disease and cellular functions for validated target genes, are reported in Table 4. For IPA, six of the top ten biological functions and three of the top five pathways were associated with cancer development and progression. Furthermore, one of the biological functions, for which deregulated genes were enriched, was ‘Skeletal Disorders’ and one of the top canonical pathways was ‘Role of Osteoblasts, Osteoclasts, and Chondrocytes in Rheumatoid Arthritis’. For the KEGG analysis, nine of the ten highest‐ranking pathways were related to cancer development and progression. The 27th pathway for the KEGG analysis was ‘Osteoclast Differentiation’, thereby linking bone cell biology and cancer development and progression for the target genes with altered gene expression.
Table 4.
Pathways and biological functions associated with genes targeted by deregulated miRNAs
| IPA canonical pathways | P‐value | |
|---|---|---|
| Atherosclerosis signaling | 5.60 × 10−7 | |
| Role of NANOG in mammalian embryonic stem cell pluripotencya | 2.53 × 10−6 | |
| Role of osteoblasts, osteoclasts, and chondrocytes in rheumatoid arthritisb | 5.59 × 10−6 | |
| Role of tissue factor in cancera | 1.41 × 10−5 | |
| Natural killer cell signalinga | 1.53 × 10−5 |
| IPA biological functions | P‐value | No. of genes associated |
|---|---|---|
| Diseases and disorders | ||
| Connective tissue disorders | 2.86 × 10−13 | 77 |
| Inflammatory disease | 2.86 × 10−13 | 91 |
| Organismal injury and abnormalities | 2.86 × 10−13 | 348 |
| Skeletal and muscular disordersb | 2.86 × 10−13 | 104 |
| Cancera | 4.66 × 10−11 | 343 |
| Molecular and cellular functions | ||
| Cellular growth and proliferationa | 1.28 × 10−11 | 121 |
| Cellular movementa | 1.70 × 10−10 | 80 |
| Cellular Developmenta | 1.05 × 10−9 | 86 |
| Cell‐to‐cell signaling and interactiona | 5.07 × 10−8 | 60 |
| Cell death and survivala | 6.10 × 10−7 | 102 |
| KEGG pathways | No. of genes associated | |
|---|---|---|
| Metabolic pathwaysa | 22 | |
| MAPK signaling pathwaya | 17 | |
| Pathways in cancera | 16 | |
| Cytokine/cytokine receptor interactiona | 14 | |
| PI3K‐Akt signaling pathwaya | 14 | |
| Ras signaling pathwaya | 14 | |
| Proteoglycans in cancera | 13 | |
| Regulation of actin cytoskeletona | 12 | |
| Rap1 signaling pathway | 12 | |
| MicroRNAs in cancera | 11 |
IPA and KEGG analysis of the 301 upregulated and 60 downregulated genes identified to be targeted by the 29 deregulated miRNAs.
Association with cancer development and progression.
Association with bone cell biology.
An analysis of the biological relations between the target genes identified a network of 21 upregulated and eight downregulated genes significantly associated with ‘Cell Death and Survival, Cancer, Hematological System Development and Function’ (P‐value = 10−42). The intimate relationship of the 29 genes and the seven downregulated and four upregulated miRNAs validated to target these genes is illustrated in Fig. 4. The identified network comprised five genes (CCNG1, MDM2, TNFSF10, TP53, and UCHL1) directly associated with the TP53 pathway. Three additional target genes (ABL1, FASLG, and UBD) not part of the identified network were associated with the TP53 pathway. Four target genes (ABL1, MDM2, STMN1, and TP53) were associated with the RB pathway.
Figure 4.
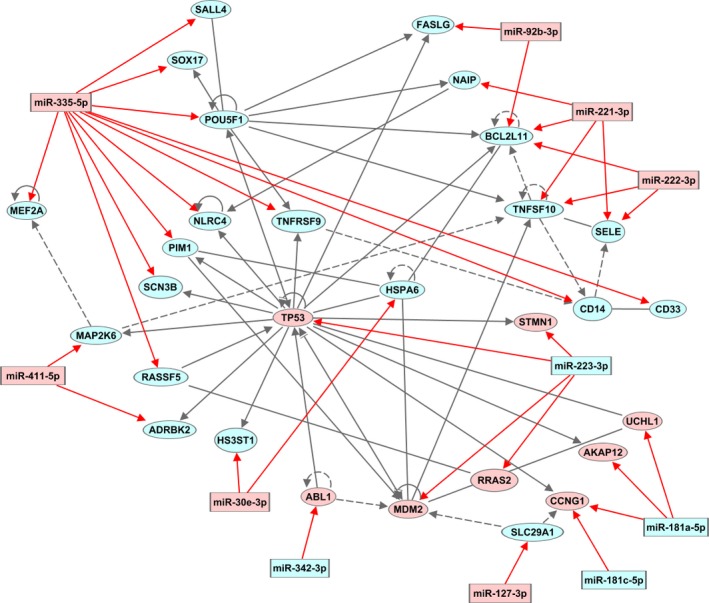
Network of validated target genes. Network of validated target genes associated with the terms ‘Cell Death and Survival, Cancer, Hematological System Development and Function’. The relation of 29 validated target genes (miRWalk2.0) assembled in a network associated with 11 miRNAs altered in OS in this study. Eight genes were downregulated and 21 upregulated. Red: downregulated; blue: upregulated. Gray lines: association between genes; Red lines: association between gene and miRNA; continuous line: direct association; dashed line: indirect association.
3.5. miRNAs associated with the metastatic status and response to chemotherapy
To identify miRNAs with an overall difference in expression based on the metastatic status, the discovery cohort was separated into metastatic and nonmetastatic primary OS (8 and 15 OS samples, respectively). This analysis revealed three miRNAs (miR‐29b‐3p, miR‐29c‐3p, and miR‐374a‐5p) significantly differentially expressed between metastatic and nonmetastatic OS samples (Table S9 and Fig. S3). The response to neoadjuvant chemotherapy was analyzed by subdividing the discovery cohort into poor responders (< 90% necrosis after chemotherapy) and good responders (> 90% necrosis after chemotherapy) (8 and 9 OS samples, respectively). This identified miR‐663b as significantly differentially expressed. Also, miR‐664a‐3p showed a strong trend toward differential expression (Table S9 and Fig. S3). However, when these miRNAs were analyzed in the samples from the validation cohort, none of the three miRNAs associated with the metastatic status nor the two miRNAs associated with the chemotherapeutic response displayed a significant difference (Table S9 and Fig. S3).
3.6. Prognostic value of the 29 differentially expressed miRNAs
The only prognostic markers for OS are currently absence of metastases at the time of diagnosis and response to chemotherapy. As 25–30% of OS patients develop distant metastases and the five‐year survival rate for patients with relapse is only 10–40%, it is critical to identify new prognostic biomarkers for OS.
We therefore investigated the prognostic value in function of survival and time to development of metastatic disease of the 29 differentially expressed miRNAs. Kaplan–Meier analysis was performed on the discovery cohort divided into two groups based on the median expression (high and low expression groups for each miRNA). For the overall survival analysis, two miRNAs (miR‐128‐3p and miR‐34a‐3p) were borderline significant in the discovery cohort (P = 0.0880 and 0.0760, respectively). No significant difference was, however, observed in the larger OS cohort (P = 0.5645 and 0.7507, respectively).
Analysis of the miRNA expression in function of time to metastasis, describing the aggressiveness of the OS tumor, identified miR‐222‐3p and miR‐221‐3p as significantly (P = 0.0359) and borderline significantly (P = 0.0863) associated with time to metastasis in the discovery cohort, respectively (Fig. S4A,C). High expression of miR‐221‐3p and low expression of miR‐222‐3p correlated with a poor prognostic value and earlier development of distant metastases. The association between expression level and poor prognosis in terms of earlier development of distant metastases was highly significant in the validation cohort (P = 0.0065 and 0.0074, respectively) (Fig. S4B,D) and when combining results from both the discovery and validation cohorts (P = 0.0007 and 0.0015, respectively) (Fig. 5A,B). When combining the expression level of the two miRNAs into one prognostic signature, there was a clear separation between OS patients with low‐miR‐221‐3p/high‐miR‐222‐3p expression and OS patients with high‐miR‐221‐3p/low‐miR‐222‐3p for both the discovery and validation cohorts and in combination (P = 2.4 × 10−8) (Fig. 5C and Fig. S4E,F).
Figure 5.
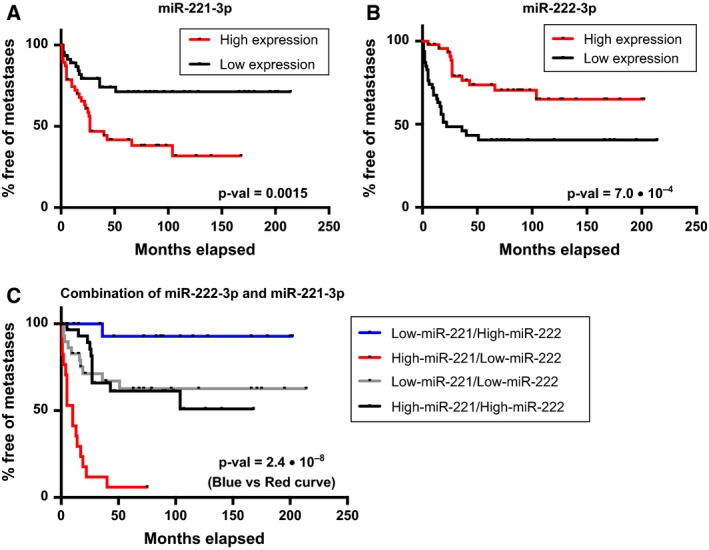
The prognostic value of the expression of miR‐221‐3p and miR‐222‐3p in function of time to metastasis. (A) Kaplan–Meier analysis of time to metastasis when subdividing the OS samples into high/low expression groups for miR‐221‐3p (based on the median expression). (B) Kaplan–Meier analysis of time to metastasis when subdividing the OS samples into high/low expression groups for miR‐222‐3p (based on the median expression). (C) Kaplan–Meier analysis of time to metastasis when combining the expression of both miR‐221‐3p and miR‐222‐3p for each patient. The discovery cohort and validation cohort have been combined for all three analyses. The Kaplan–Meier analysis of the high/low expression groups for the individual analyses of miR‐221‐3p and miR‐222‐3p contained 47 OS samples in each group. For the combined analysis of miR‐221‐3p/miR‐222‐3p expression, the number of OS samples in each group was as follows: 29 OS samples in high‐miR‐221‐3p/high‐miR‐222‐3p; 30 OS samples in low‐miR‐221‐3p/low‐miR‐222‐3p; 17 OS samples in high‐miR‐221‐3p/low‐miR‐222‐3p; and 18 OS samples in low‐miR‐221‐3p/high‐miR‐222‐3p. Data for the individual cohorts are shown in Fig. S4.
4. Discussion
OS is a complex disease exhibiting alterations in numerous pathways and with major cell‐to‐cell variation. The understanding of the etiology of OS is limited and no common molecular alteration has been identified. This designates the need for a more fundamental understanding of the underlying mechanisms leading to OS development and progression. Alterations in miRNA expression are one of the mechanisms associated with tumor development. As one miRNA potentially regulates the expression of more than 100 target genes, alterations in miRNA expression can have a vast impact on neoplastic initiation and progression. Hence, the focus of this study was the analysis of miRNA expression changes in OS. Profiling of miRNA expression in OS has been addressed in previous studies (Duan et al., 2011; Jones et al., 2012; Lulla et al., 2011; Maire et al., 2011; Namlos et al., 2012; Thayanithy et al., 2012; Won et al., 2013; J. Zhang et al., 2015). However, the limited number of samples of 2–18 OS samples in the discovery cohorts and 2–12 samples in the respective validation cohorts makes it challenging to draw reliable conclusions from the published data.
We have in this study investigated the expression of 752 miRNAs in 23 OS samples and validated significant findings in 78 additional OS samples. These are, to our knowledge, both the largest discovery and validation cohorts so far used for miRNA expression analysis in OS. The identified miRNA expression signature showed strong statistical significance in both the discovery and the validation cohorts comprising together a total of 101 OS samples. This provides robust evidence of the 29 miRNAs with an altered expression level between OS samples and OB cell lines to be truly deregulated in OS. A difference in miRNA expression between histological subtypes was not identified designating a universal role of the deregulated miRNAs in OS tumorigenesis.
A significant role of miRNAs in OS tumorigenesis was supported by the investigation of biological functions associated with the 29 deregulated miRNAs, revealing a strong correlation with cancer development and progression. This was further substantiated by the investigation of the validated miRNA/mRNA targets, identifying 361 genes with a significantly changed expression level in the OS samples. These genes were associated with multiple pathways and biological functions related to cancer development and progression. Furthermore, both a canonical pathway and biological functions were associated with bone cell pathogenesis linking the cancer‐related pathways with OS tumorigenesis.
A relation between the identified pathways associated with the target genes and the TP53 pathway, which is often altered in OS, also supported a correlation between the deregulated miRNAs and OS tumorigenesis. The network analysis and target genes comprised a total of eight genes directly associated with the TP53 pathway (ABL1, CCNG1, FASLG, MDM2, TNFSF10, TP53, UBD, and UCHL1) and four genes (ABL1, MDM2, STMN1, and TP53) associated with the RB pathway. Hence, the altered miRNAs identified in this study fit well with the current view of OS pathogenesis that the TP53 and RB pathways are the two major pathways involved in OS development and progression. Many potential functional miRNA/mRNA relationships were identified in this study. These need to be further investigated to determine their function and involvement in OS tumorigenesis.
The prognostic value of the 29 deregulated miRNAs was also investigated in function of the overall survival and time to metastasis by subdividing the OS samples into high/low expression groups. This identified two miRNAs, miR‐221‐3p and miR‐222‐3p, in which the expression level showed a significant association with time to metastasis. When combining the high/low expression scores for the two miRNAs for each OS patient, the separation became even more distinct and significant, signifying the robustness of the miR‐221/222 signature as a prognostic biomarker. Increased expression of miR‐221 and miR‐222 has also been shown to be significantly associated with lymphatic metastasis in gastric, colorectal, and breast cancer (Falkenberg et al., 2013; Fu et al., 2014; Liu et al., 2012b; Sun et al., 2011). Furthermore, these two miRNAs have been shown to be a promising biomarker in breast cancer in function of time to developing distant metastasis, although a high expression of both miRNAs was associated with poor prognosis in breast cancer (Falkenberg et al., 2013). The discrepancy of miR‐222 expression between these studies and the OS samples in this study may be explained by the ability of a miRNA to target several hundred genes, thereby promoting tumor growth in some malignancies and repressing it in others. This is further substantiated by the fact that miR‐222 was shown to target MMP1, which is associated with increased aggressiveness and metastatic risk in oral tongue squamous cell carcinoma (Liu et al., 2009) (MMP1 showed a 10‐fold increase in expression in the OS samples in this study). High expression of MMP1 has also been associated with the development of metastasis in colorectal and breast cancer (Liu et al., 2012a; Sunami et al., 2000). miR‐221 targets among other genes BCL2L11, which exhibits pro‐apoptotic properties, thereby suppressing tumor growth (Aichberger et al., 2009), as well as the gatekeeper PTEN (Li et al., 2016). In different cancers, high expression of miR‐221/222 has also been shown to facilitate epithelial/mesenchymal transition, a prerequisite for metastasis formation (Li et al., 2015; Stinson et al., 2011). Furthermore, modulation of miR‐221 levels in OS cell lines demonstrated its role in carcinogenesis with high levels leading to increasing invasiveness and migration of the cells (Zhu et al., 2015). Hence, both the deregulation itself and the target genes of miR‐221 and miR‐222 indicate a prominent role of these two miRNAs in the ability of a neoplasm to spread to other organs and in the aggressiveness of specific cancer types.
Of the 29 deregulated miRNAs identified in this study, expression changes in nine miRNAs have not previously been associated with OS in miRNA profiling studies, while 20 miRNAs previously associated with OS development and progression were uncovered (Duan et al., 2011; Jones et al., 2012; Lulla et al., 2011; Maire et al., 2011; Namlos et al., 2012; Thayanithy et al., 2012; Won et al., 2013; Zhang et al., 2015). However, as OS is characterized by many different alterations, confirmation of previous findings is important for this particular disease. Furthermore, many of the formerly reported miRNA changes in OS have been identified in cohorts comprising less than 10 OS samples. Previous studies profiling miRNA expression changes in OS revealed 14 miRNAs significantly changed in these studies, which were not among the 29 miRNAs identified in our study. This may be due to differences in the cohorts, but also due to low sample number in the published studies or the use of normal bone as the control tissue, comprising a lower number than the OS cohorts (comprising 2–12 normal bone samples) (Jones et al., 2012; Namlos et al., 2012; Thayanithy et al., 2012; Won et al., 2013; Zhang et al., 2015). The cell of origin for OS has not yet been completely clarified. Both mesenchymal stem cells (MSCs) and derived osteoprogenitors such as OBs have been analyzed for their ability to form OS. Several studies have shown that OB precursors presented a higher incidence of OS compared to early mesenchymal progenitors (Abarrategi et al., 2016), supporting the model of OS originating from cells with OB commitment rather than immature MSCs. OB cell lines have therefore been used as the normal controls in this study, as well as in several previous studies (comprising 1–3 OB cell lines) (Duan et al., 2011; Lulla et al., 2011; Maire et al., 2011). Only one study has so far been using both normal bone tissue and OB cell lines as controls, but analyzed only 15 miRNAs previously identified to be differentially expressed between normal bone and OS cell lines. Nine of these were validated in primary OS samples when compared to normal bone. However, for some of these miRNAs, the authors observed opposite expression changes in OS when compared to either OB cell lines or normal bone (Namlos et al., 2012). This may be due to OS being at an intermediate state between the undifferentiated OB cells and the fully differentiated bones comprising several different cell types. Furthermore, normal bone was obtained from cancer patients suggesting a different age distribution between OS samples and bone samples, which could lead to differential miRNA expression profiles, and raise the question of the possible presence of cancer‐related epigenetic alterations in these samples. These points, as well as the small number of miRNAs analyzed in primary OS, bone and OB cell lines, make it difficult to draw general conclusions from this study. Even though OB cells can be considered to be the cell of origin of OS, the use of cell lines as normal control holds the limitation of identifying miRNAs with altered expression due to differences between cell lines and human tissue and not due to the cell origin. Unfortunately, no normal bone tissue was available for the current study. Therefore, future studies should comprise pediatric OB cells as normal control as well as normal bone to obtain a more comprehensive description of the role of the identified miRNAs in OS tumorigenesis.
Several of the 24 significantly downregulated miRNAs identified in this study have previously been identified as tumor suppressor miRNAs in other malignancies, among others miR‐100‐5p, miR‐125b‐5p, miR‐127‐3p, miR‐370‐3p, miR‐335‐5p, and miR‐411‐5p (Gao et al., 2016; He et al., 2013; Jiang et al., 2014; Li et al., 2014; Luan et al., 2016; Zhang et al., 2016). Of these, miR‐100‐5p, miR‐127‐3p, and miR‐335‐5p have previously been identified as downregulated in OS studies analyzing 2–18 samples (Jones et al., 2012; Maire et al., 2011; Thayanithy et al., 2012; Zhang et al., 2015). However, miR‐127‐3p and miR‐370‐3p have also been detected upregulated in two OS samples in one study (Zhang et al., 2015). miR‐125b‐5p and miR‐411‐5p have not previously been identified having an altered expression in any OS miRNA profiling studies. miR‐125b has been shown to be an important regulator of both proliferation and differentiation in different cell types (Scott et al., 2007; Sempere et al., 2004). Moreover, miR‐125b has been shown to inhibit normal OB proliferation in mouse cells (Mizuno et al., 2008), indicating a role of this miRNA in bone development and thereby in OS tumorigenesis. miR‐335‐5p has also been identified to be important in the differentiation process of bone cells, as a positive regulator of Wnt signaling through the inhibition of Wnt antagonists (Zhang et al., 2011). Wnt signaling is elevated when osteoprecursor cells differentiate into OBs. However, Wnt signaling is inhibited when the OB cells differentiate further toward the terminal stage of bone‐forming cells, which may be a functional explanation for the downregulation of miR‐335‐5p in OS.
Of the five significantly upregulated miRNAs identified in this study, miR‐181a‐5p, miR‐181c‐5p, miR‐223‐3p, and miR‐342‐3p have been described as oncomiRs in other cancer types (Mi et al., 2017; Tao et al., 2014; Walter et al., 2013; Wei et al., 2014). Of these, miR‐181a‐5p, miR‐181c‐5p, and miR‐223‐3p have previously been identified as upregulated in 4–18 OS samples (Jones et al., 2012; Lulla et al., 2011; Maire et al., 2011; Won et al., 2013). However, miR‐223‐3p has also been detected as downregulated in 18 OS samples in one study (Jones et al., 2012). miR‐342‐3p has not previously been associated with OS in miRNA profiling studies. miR‐181a‐5p has been shown to downregulate a Wnt antagonist, thereby activating the Wnt signaling, which is, as described above, important for the differentiation of osteoprecursor cells into OB cells (Lyu et al., 2017).
Future functional studies for each identified miRNA and their target genes in OS development and progression have a strong potential for discovering novel diagnostic and prognostic markers as well as developing new treatment strategies, thereby improving the long‐term survival rate for these young patients. Circulating miRNAs detected in serum and plasma of patients with cancer are being widely investigated for their potential as both diagnostic and prognostic markers of cancer, including OS (Ram Kumar et al., 2016). This might also provide a tool of diagnosing OS at an earlier time point when individuals complain repeatedly about ‘growing’ pains. Furthermore, miRNAs hold the promise of novel therapeutic targets, either by blocking the expression of an oncomiR or by substituting the loss of a tumor suppressor miRNA.
5. Conclusion
In conclusion, this study substantiates the importance of miRNA deregulation in OS and its association with OS tumorigenesis. We have identified and validated 29 deregulated miRNAs in, to our knowledge, the largest discovery and validation cohorts published, comprising a total of 101 OS samples. Both the miRNAs and their identified target genes are associated with multiple pathways and biological functions, related to cancer development and progression, as well as bone cell biology, thereby associating the deregulated miRNAs with OS tumorigenesis. Interestingly, a promising potential as prognostic biomarkers for the aggressiveness of OS was seen for two of these miRNAs (miR‐221‐3p and miR‐222‐3p).
Author contributions
GBA collected the patient samples, performed experiments, analyzed the data, performed statistical tests, and wrote the manuscript. AK purified RNA from FFPE samples and made cDNA. HH provided patient information and confirmed the presence of tumor cells from HE‐stained sections. LLH provided supervision and assistance for writing the manuscript and designing figures and tables. JT provided supervision and assistance for data analysis and interpretation and for writing the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig. S1. Principal component analyses of the 339 detected miRNAs.
Fig. S2. Boxplots of the expression level of 24 differentially expressed miRNAs.
Fig. S3. Boxplots of the expression level of three miRNAs associated with the metastatic potential of OS; two miRNAs associated with the chemotherapeutic response of OS.
Fig. S4. Prognostic value of the expression of two miRNAs in function of time to metastasis.
Table S1. Raw expression data (Cq values) for 752 miRNAs analyzed with the microRNA Ready‐to‐Use PCR, Human panel I and panel II v2 (Exiqon, Denmark) using the OS discovery cohort.
Table S2. Log2 normalized data for the 752 miRNAs analyzed for the OS discovery cohort.
Table S3. Raw expression data (Cq values) for 33 miRNAs to be validated and the five reference miRNAs analyzed with the Pick&Mix microRNA PCR Panel, 384 well Ready‐to‐Use (Exiqon, Denmark) using the OS validation cohort.
Table S4. Log2 normalized data for the 33 miRNAs analyzed in the OS validation cohort.
Table S5. Expression of 301 upregulated and 60 downregulated genes identified to be targeted by the 29 deregulated miRNAs.
Table S6. Expression of 76 miRNAs identified as differentially expressed in OS samples when compared to OB cell lines.
Table S7. Previous miRNA profiling studies describing the 33 miRNAs identified in the discovery cohort.
Table S8. Comparison of profiling studies investigating miRNA expression changes in OS and their expression in the OS investigation cohort in this study.
Table S9. Expression of three miRNAs associated with the metastatic potential of OS and two miRNAs associated with the chemotherapeutic response of OS.
Acknowledgements
The authors wish to thank Antoine Daunay and Tina Ellegaard Kjeldsen for excellent technical assistance and Tine Meyer for assisting with data information. The study was supported by grants from the Child Cancer Foundation, Arvid Nilssons Fond, Aase og Ejnar Danielsens fond, Neye Fonden, Ronald MacDonald Børnefond, and Krista og Viggo Petersens Fond.
References
- Abarrategi A, Tornin J, Martinez‐Cruzado L, Hamilton A, Martinez‐Campos E, Rodrigo JP, Gonzalez MV, Baldini N, Garcia‐Castro J and Rodriguez R (2016) Osteosarcoma: cells‐of‐origin, cancer stem cells, and targeted therapies. Stem Cells Int 2016, 3631764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aichberger KJ, Gleixner KV, Mirkina I, Cerny‐Reiterer S, Peter B, Ferenc V, Kneidinger M, Baumgartner C, Mayerhofer M, Gruze A et al (2009) Identification of proapoptotic Bim as a tumor suppressor in neoplastic mast cells: role of KIT D816V and effects of various targeted drugs. Blood 114, 5342–5351. [DOI] [PubMed] [Google Scholar]
- Andersen CL, Jensen JL and Orntoft TF (2004) Normalization of real‐time quantitative reverse transcription‐PCR data: a model‐based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64, 5245–5250. [DOI] [PubMed] [Google Scholar]
- Bielack SS, Kempf‐Bielack B, Delling G, Exner GU, Flege S, Helmke K, Kotz R, Salzer‐Kuntschik M, Werner M, Winkelmann W et al (2002) Prognostic factors in high‐grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 20, 776–790. [DOI] [PubMed] [Google Scholar]
- Bishop MW, Janeway KA and Gorlick R (2016) Future directions in the treatment of osteosarcoma. Curr Opin Pediatr 28, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB and Cohen SM (2005) Principles of microRNA‐target recognition. PLoS Biol 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge JA, Nelson M, McComb E, McGuire MH, Rosenthal H, Vergara G, Maale GE, Spanier S and Neff JR (1997) Cytogenetic findings in 73 osteosarcoma specimens and a review of the literature. Cancer Genet Cytogenet 95, 74–87. [DOI] [PubMed] [Google Scholar]
- Calin GA and Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6, 857–866. [DOI] [PubMed] [Google Scholar]
- Duan Z, Choy E, Harmon D, Liu X, Susa M, Mankin H and Hornicek F (2011) MicroRNA‐199a‐3p is downregulated in human osteosarcoma and regulates cell proliferation and migration. Mol Cancer Ther 10, 1337–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweep H and Gretz N (2015) miRWalk2.0: a comprehensive atlas of microRNA‐target interactions. Nat Methods 12, 697. [DOI] [PubMed] [Google Scholar]
- Falkenberg N, Anastasov N, Rappl K, Braselmann H, Auer G, Walch A, Huber M, Hofig I, Schmitt M, Hofler H et al (2013) MiR‐221/‐222 differentiate prognostic groups in advanced breast cancers and influence cell invasion. Br J Cancer 109, 2714–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Qian F, Yang X, Jiang H, Chen Y and Liu S (2014) Circulating miR‐222 in plasma and its potential diagnostic and prognostic value in gastric cancer. Med Oncol 31, 164. [DOI] [PubMed] [Google Scholar]
- Gao YT, Chen XB and Liu HL (2016) Up‐regulation of miR‐370‐3p restores glioblastoma multiforme sensitivity to temozolomide by influencing MGMT expression. Sci Rep 6, 32972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- He J, Jing Y, Li W, Qian X, Xu Q, Li FS, Liu LZ, Jiang BH and Jiang Y (2013) Roles and mechanism of miR‐199a and miR‐125b in tumor angiogenesis. PLoS One 8, e56647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inose H, Ochi H, Kimura A, Fujita K, Xu R, Sato S, Iwasaki M, Sunamura S, Takeuchi Y, Fukumoto S et al (2009) A microRNA regulatory mechanism of osteoblast differentiation. Proc Natl Acad Sci U S A 106, 20794–20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Jin C, Liu J, Hua D, Zhou F, Lou X, Zhao N, Lan Q, Huang Q, Yoon JG et al (2014) Next generation sequencing analysis of miRNAs: MiR‐127‐3p inhibits glioblastoma proliferation and activates TGF‐beta signaling by targeting SKI. OMICS 18, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KB, Salah Z, Del Mare S, Galasso M, Gaudio E, Nuovo GJ, Lovat F, LeBlanc K, Palatini J, Randall RL et al (2012) miRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Res 72, 1865–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kager L, Zoubek A, Potschger U, Kastner U, Flege S, Kempf‐Bielack B, Branscheid D, Kotz R, Salzer‐Kuntschik M, Winkelmann W et al (2003) Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol 21, 2011–2018. [DOI] [PubMed] [Google Scholar]
- Kanehisa M and Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42(Database issue), 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansara M, Teng MW, Smyth MJ and Thomas DM (2014) Translational biology of osteosarcoma. Nat Rev Cancer 14, 722–735. [DOI] [PubMed] [Google Scholar]
- Li B, Lu Y, Wang H, Han X, Mao J, Li J, Yu L, Wang B, Fan S, Yu X et al (2016) miR‐221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed Pharmacother 79, 93–101. [DOI] [PubMed] [Google Scholar]
- Li H, Xie S, Liu M, Chen Z, Liu X, Wang L, Li D and Zhou Y (2014) The clinical significance of downregulation of mir‐124‐3p, mir‐146a‐5p, mir‐155‐5p and mir‐335‐5p in gastric cancer tumorigenesis. Int J Oncol 45, 197–208. [DOI] [PubMed] [Google Scholar]
- Li J, Yao L, Li G, Ma D, Sun C, Gao S, Zhang P and Gao F (2015) miR‐221 promotes epithelial‐mesenchymal transition through targeting PTEN and forms a positive feedback loop with beta‐catenin/c‐Jun signaling pathway in extra‐hepatic cholangiocarcinoma. PLoS One 10, e0141168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian JB, Stein GS, van Wijnen AJ, Stein JL, Hassan MQ, Gaur T and Zhang Y (2012) MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol 8, 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kato Y, Erzinger SA, Kiriakova GM, Qian Y, Palmieri D, Steeg PS and Price JE (2012a) The role of MMP‐1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer 12, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Li G, Fan C, Diao Y, Wu B and Li J (2012b) Increased expression of microRNA‐221 in gastric cancer and its clinical significance. J Int Med Res 40, 467–474. [DOI] [PubMed] [Google Scholar]
- Liu X, Yu J, Jiang L, Wang A, Shi F, Ye H and Zhou X (2009) MicroRNA‐222 regulates cell invasion by targeting matrix metalloproteinase 1 (MMP1) and manganese superoxide dismutase 2 (SOD2) in tongue squamous cell carcinoma cell lines. Cancer Genomics Proteomics 6, 131–139. [PMC free article] [PubMed] [Google Scholar]
- Luan J, Wang J, Su Q, Chen X, Jiang G and Xu X (2016) Meta‐analysis of the differentially expressed microRNA profiles in nasopharyngeal carcinoma. Oncotarget 7, 10513–10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulla RR, Costa FF, Bischof JM, Chou PM, de F Bonaldo M, Vanin EF, Soares MB (2011) Identification of differentially expressed microRNAs in osteosarcoma. Sarcoma 2011, 732690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu X, Li J, Yun X, Huang R, Deng X, Wang Y, Chen Y and Xiao G (2017) miR‐181a‐5p, an inducer of Wnt‐signaling, facilitates cell proliferation in acute lymphoblastic leukemia. Oncol Rep 37, 1469–1476. [DOI] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K et al (2014) Epigenomic alterations define lethal CIMP‐positive ependymomas of infancy. Nature 506, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maire G, Martin JW, Yoshimoto M, Chilton‐MacNeill S, Zielenska M and Squire JA (2011) Analysis of miRNA‐gene expression‐genomic profiles reveals complex mechanisms of microRNA deregulation in osteosarcoma. Cancer Genet 204, 138–146. [DOI] [PubMed] [Google Scholar]
- McCarville MB 2009. The child with bone pain: malignancies and mimickers. Cancer Imaging 9 (Spec No A): S115–S121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers PA, Heller G, Healey JH, Huvos A, Applewhite A, Sun M and LaQuaglia M (1993) Osteogenic sarcoma with clinically detectable metastasis at initial presentation. J Clin Oncol 11, 449–453. [DOI] [PubMed] [Google Scholar]
- Mi Y, Zhang D, Jiang W, Weng J, Zhou C, Huang K, Tang H, Yu Y, Liu X, Cui W et al (2017) miR‐181a‐5p promotes the progression of gastric cancer via RASSF6‐mediated MAPK signalling activation. Cancer Lett 389, 11–22. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Yagi K, Tokuzawa Y, Kanesaki‐Yatsuka Y, Suda T, Katagiri T, Fukuda T, Maruyama M, Okuda A, Amemiya T et al (2008) miR‐125b inhibits osteoblastic differentiation by down‐regulation of cell proliferation. Biochem Biophys Res Commun 368, 267–272. [DOI] [PubMed] [Google Scholar]
- Namlos HM, Meza‐Zepeda LA, Baroy T, Ostensen IH, Kresse SH, Kuijjer ML, Serra M, Burger H, Cleton‐Jansen AM and Myklebost O (2012) Modulation of the osteosarcoma expression phenotype by microRNAs. PLoS One 7, e48086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini M, Nicoloso MS and Calin GA (2009) MicroRNAs and cancer – new paradigms in molecular oncology. Curr Opin Cell Biol 21, 470–479. [DOI] [PubMed] [Google Scholar]
- Ning ZQ, Lu HL, Chen C, Wang L, Cai W, Li Y, Cao TH, Zhu J, Shu YQ and Shen H (2016) MicroRNA‐30e reduces cell growth and enhances drug sensitivity to gefitinib in lung carcinoma. Oncotarget 8, 4572–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria: URL http://www.R-project.org/. [Google Scholar]
- Ram Kumar RM, Boro A and Fuchs B (2016) Involvement and clinical aspects of microRNA in osteosarcoma. Int J Mol Sci 17, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS and Benz CC (2007) Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro‐RNA miR‐125a or miR‐125b. J Biol Chem 282, 1479–1486. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha‐Rowe I, Moss E, Dmitrovsky E and Ambros V (2004) Expression profiling of mammalian microRNAs uncovers a subset of brain‐expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5, R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Pierron G, Klughammer J, Datlinger P, Schonegger A, Schuster M, Hadler J, Surdez D, Guillemot D, Lapouble E et al (2017) DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med 23, 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Russell RB and Cohen SM (2003) Identification of Drosophila microRNA targets. PLoS Biol 1, E60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O'Brien C Spoerke J, Jhunjhunwala S, Boyd Z, Januario T et al (2011) TRPS1 targeting by miR‐221/222 promotes the epithelial‐to‐mesenchymal transition in breast cancer. Sci Signal 4, ra41. [DOI] [PubMed] [Google Scholar]
- Sun K, Wang W, Zeng JJ, Wu CT, Lei ST and Li GX (2011) MicroRNA‐221 inhibits CDKN1C/p57 expression in human colorectal carcinoma. Acta Pharmacol Sin 32, 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami E, Tsuno N, Osada T, Saito S, Kitayama J, Tomozawa S, Tsuruo T, Shibata Y, Muto T and Nagawa H (2000) MMP‐1 is a prognostic marker for hematogenous metastasis of colorectal cancer. Oncologist 5, 108–114. [DOI] [PubMed] [Google Scholar]
- Tao K, Yang J, Guo Z, Hu Y, Sheng H, Gao H and Yu H (2014) Prognostic value of miR‐221‐3p, miR‐342‐3p and miR‐491‐5p expression in colon cancer. Am J Transl Res 6, 391–401. [PMC free article] [PubMed] [Google Scholar]
- Thayanithy V, Sarver AL, Kartha RV, Li L, Angstadt AY, Breen M, Steer CJ, Modiano JF and Subramanian S (2012) Perturbation of 14q32 miRNAs‐cMYC gene network in osteosarcoma. Bone 50, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T and Hatzigeorgiou AG (2015) DIANA‐miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res 43, W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter BA, Valera VA, Pinto PA and Merino MJ (2013) Comprehensive microRNA profiling of prostate cancer. J Cancer 4, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LL (2005) Biology of osteogenic sarcoma. Cancer J 11, 294–305. [DOI] [PubMed] [Google Scholar]
- Wei Y, Yang J, Yi L, Wang Y, Dong Z, Liu Z, Ou‐yang S, Wu H, Zhong Z, Yin Z et al (2014) MiR‐223‐3p targeting SEPT6 promotes the biological behavior of prostate cancer. Sci Rep 4, 7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won KY, Kim YW, Kim HS, Lee SK, Jung WW and Park YK (2013) MicroRNA‐199b‐5p is involved in the Notch signaling pathway in osteosarcoma. Hum Pathol 44, 1648–1655. [DOI] [PubMed] [Google Scholar]
- Zhang J, Tu Q, Bonewald LF, He X, Stein G, Lian J and Chen J (2011) Effects of miR‐335‐5p in modulating osteogenic differentiation by specifically downregulating Wnt antagonist DKK1. J Bone Miner Res 26, 1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang D, Xiong J, Chen L and Huang J (2015) MicroRNA‐33a‐5p suppresses growth of osteosarcoma cells and is downregulated in human osteosarcoma. Oncol Lett 10, 2135–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu G, Liu G, Ye Y, Zhang C, Fan C, Wang H, Cai H, Xiao R, Huang Z et al (2016) miR‐411‐5p inhibits proliferation and metastasis of breast cancer cell via targeting GRB2. Biochem Biophys Res Commun 476, 607–613. [DOI] [PubMed] [Google Scholar]
- Zhu J, Liu F, Wu Q and Liu X (2015) MiR‐221 increases osteosarcoma cell proliferation, invasion and migration partly through the downregulation of PTEN. Int J Mol Med 36, 1377–1383. [DOI] [PubMed] [Google Scholar]
- Zhu WY, Luo B, An JY, He JY, Chen DD, Xu LY, Huang YY, Liu XG, Le HB and Zhang YK (2014) Differential expression of miR‐125a‐5p and let‐7e predicts the progression and prognosis of non‐small cell lung cancer. Cancer Invest 32, 394–401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Principal component analyses of the 339 detected miRNAs.
Fig. S2. Boxplots of the expression level of 24 differentially expressed miRNAs.
Fig. S3. Boxplots of the expression level of three miRNAs associated with the metastatic potential of OS; two miRNAs associated with the chemotherapeutic response of OS.
Fig. S4. Prognostic value of the expression of two miRNAs in function of time to metastasis.
Table S1. Raw expression data (Cq values) for 752 miRNAs analyzed with the microRNA Ready‐to‐Use PCR, Human panel I and panel II v2 (Exiqon, Denmark) using the OS discovery cohort.
Table S2. Log2 normalized data for the 752 miRNAs analyzed for the OS discovery cohort.
Table S3. Raw expression data (Cq values) for 33 miRNAs to be validated and the five reference miRNAs analyzed with the Pick&Mix microRNA PCR Panel, 384 well Ready‐to‐Use (Exiqon, Denmark) using the OS validation cohort.
Table S4. Log2 normalized data for the 33 miRNAs analyzed in the OS validation cohort.
Table S5. Expression of 301 upregulated and 60 downregulated genes identified to be targeted by the 29 deregulated miRNAs.
Table S6. Expression of 76 miRNAs identified as differentially expressed in OS samples when compared to OB cell lines.
Table S7. Previous miRNA profiling studies describing the 33 miRNAs identified in the discovery cohort.
Table S8. Comparison of profiling studies investigating miRNA expression changes in OS and their expression in the OS investigation cohort in this study.
Table S9. Expression of three miRNAs associated with the metastatic potential of OS and two miRNAs associated with the chemotherapeutic response of OS.
Data Availability Statement
All miRNA data generated and analyzed during this study are included in this published article (Tables S1–S4 including both raw and log2‐normalized data). The normalized log2 expression data for the 361 genes analyzed in this study are provided in Table S5. The raw gene expression dataset used in the current study is part of another ongoing study and will be published and deposited in a public repository separately. Meanwhile, these data are available from the corresponding author upon request.