

Abstract
Ammonia is a ubiquitous by-product of cellular metabolism, however the biological consequences of ammonia production are not fully understood, especially in cancer. We find that ammonia is not merely a toxic waste product, but is recycled into central amino acid metabolism to maximize nitrogen utilization. Cancer cells primarily assimilated ammonia through reductive amination catalyzed by glutamate dehydrogenase (GDH), and secondary reactions enabled other amino acids, such as proline and aspartate, to directly acquire this nitrogen. Metabolic recycling of ammonia accelerated proliferation of breast cancer. In mice, ammonia accumulated in the tumor microenvironment, and was used directly to generate amino acids through GDH activity. These data show that ammonia not only is a secreted waste product, but a fundamental nitrogen source that can support tumor biomass.
Increased nutrient consumption can supply carbon, nitrogen, oxygen, and sulfur to accommodate the extensive bioenergetic, biosynthetic, and pro-survival requirements of rapidly proliferating cells (1–3). As a consequence, such cells generate an excess of metabolic waste which are cleared in mammals through the excretory system. However, in the tumor microenvironment, metabolic waste such as lactate and ammonia accumulate (4, 5). Although lactate is well studied in cancer, little is known about the mechanisms by which cancer cells manage increased amounts of ammonia (NH3) generated by glutamine and asparagine catabolism, de novo cysteine synthesis through the transsulfuration pathway, and salvage nucleotide metabolism (6). Ammonia has been considered a toxic by-product that must be exported from cells and is subsequently cleared through urea cycle activity in the liver (7–9).
Glutamine has been called a “nitrogen reservoir” for cancer cells because of its anabolic role in nucleotide synthesis (6, 10). However, the role of glutamine as a nitrogen reservoir is contradicted in catabolic glutamine metabolism, because nitrogen is liberated as the by-product ammonia (11). The fate of ammonia in metabolism of proliferating cells and tumors remains unclear. We hypothesized that ammonia might be re-assimilated into central metabolism to maximize the efficiency of nitrogen utilization. In this study we sought to clarify roles of ammonia as 1) a toxic waste product or 2) a biosynthetic metabolite (Fig. 1a).
Figure 1. Glutamine-Derived ammonia is recycled.
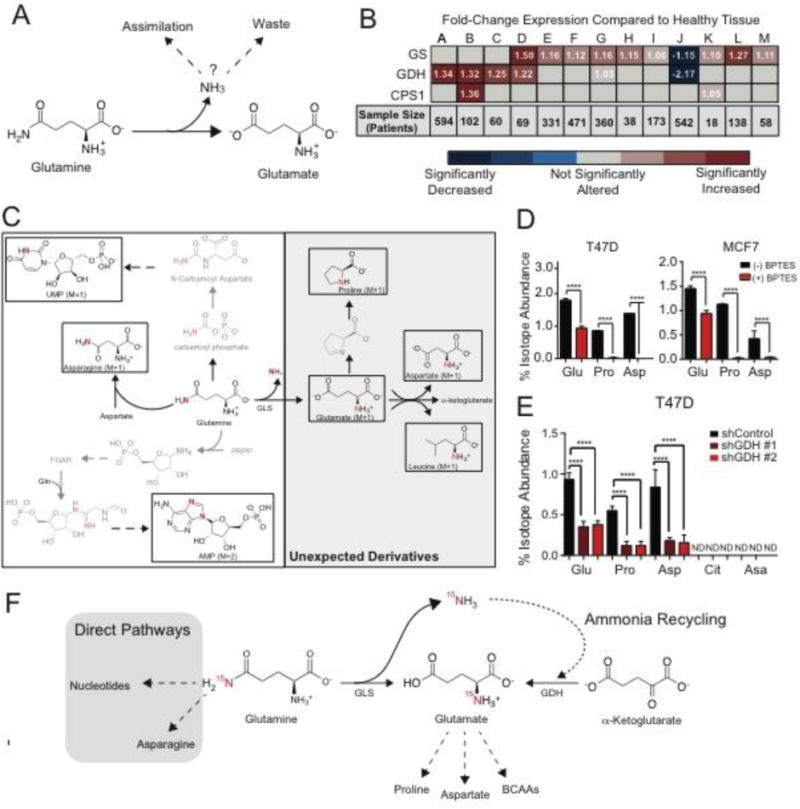
A. Schematic of fates of ammonia in cancer. B. mRNA expression of ammonia-assimilating enzymes from The Cancer Genome Atlas in cancer compared to its normal tissue. Fold-change (cancer/normal) for GS (Glutamine Synthetase), GDH1 (Glutamate Dehydrogenase), and CPS1 (Carbamoyl Phosphate Synthetase 1) RNA levels were assessed using Oncomine.org. Values are the mean of fold change (cancer/normal) measured across the number of patients listed. (A) Ovarian Serous Cystadenocarcinoma, (B) Colon Adenocarcinoma, (C) Rectal Adenocarcinoma, (D) Lobular & Ductal Breast Carcinoma, (E) Lung Adenocarcinoma, (F) Squamous Lung Cell Carcinoma, (G) Endometrial Adenocarcinoma, (H) Bladder Urothelial Carcinoma, (I) Gastric Adenocarcinoma, (J) Glioblastoma, (K) Pancreatic Adenocarcinoma, (L) Hepatocellular Carcinoma, (M) Cutaneous Melanoma. C. Schematic of 15N-isotopologues after treatment with 15N-(amide)glutamine. D. Isotopologue abundance of unexpected 15N-(amide)glutamine derivatives +/− 1 uM BPTES in T47D and MCF7 cell lines. Values represent mean +/− SEM, n=4 per condition. E. Isotope abundance of 15N-(amide)glutamine-derived metabolites in control cells and cells depleted of GDH (shGDH#1 & shGHD#2). ND= 15N-Isotopologue not detected. Glu = Glutamate M+1, Pro = Proline M+1, Asp = Aspartate M+1, Cit = Citrulline M+1, Asa = Argininosuccinate M+1. Values represent mean +/− SEM, n=4 per condition. F. Schematic of ammonia recycling. For all comparisons two-tailed t test was used. *P < 0.05, **P < 0.01, ***P<0.001, ****P<0.0001.
Mammals have three enzymes that can overcome the thermodynamic hurdles of ammonia assimilation: carbamoyl phosphate synthetase I (CPS1), the ATP-dependent, rate-limiting step of the urea cycle; glutamate dehydrogenase (GDH), an NAD(P)H-dependent enzyme that catalyzes reductive amination of α-ketoglutarate; and glutamine synthetase (GS), which catalyzes the ATP-dependent amination of glutamate to generate glutamine (12, 13) (fig. S1A). Analysis of transcriptomic data from The Cancer Genome Atlas for the ammonia-assimilating enzymes in healthy and cancerous tissues revealed that expression of GS and GDH mRNA was significantly increased across many cancer subtypes whereas CPS1 was only increased in colon (Fig. 1b). Among healthy tissues, GS and GDH are ubiquitously expressed and CPS1 is only expressed in the liver (fig. S1B). Breast cancers displayed increased expression of both GS and GDH. Specifically, Estrogen Receptor (ER) positive breast cancers have increased expression of GS and GDH compared to that in other subtypes (14). Therefore, we used ER positive breast cancer as a representative model to probe for ammonia assimilation.
To investigate the fate of glutamine-derived ammonia, we performed a metabolic tracing analysis with hydrophilic interaction liquid chromatography tandem mass spectrometry (HILIC-MS) and assessed the fate of 15N(amide)-glutamine, which liberates 15NH3 through glutaminase activity (15). To identify the metabolic derivatives of 15N(amide)-glutamine in an unbiased manner, we developed a method to screen the nitrogen metabolome, which contained 211 15N-isotopologues (table S1). The majority of the nitrogen metabolome did not acquire 15N-labeling; of 211 15N-isotopologues, only 33 metabolites were labeled (fig. S2). Consistent with previous studies, 15N-(amide)-glutamine was incorporated into asparagine and nucleotides (10) (Fig. 1C & fig. S3A). We also identified 15N-isotopologues of proline, aspartate, branched chain amino acids (BCAA), and glutamate, which have no previous biosynthetic connection to the amide-nitrogen on glutamine (Fig. 1C & fig. S3B). The labeled nitrogen was liberated as ammonia before production of these metabolites, suggesting that an ammonia recycling pathway may synthesize the other glutamine derivatives detected.
To test whether ammonia released during glutaminolysis was necessary for production of these unanticipated amide-nitrogen glutamine derivatives, cells were treated with the glutaminase inhibitor Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), which at 1 μM is not cytotoxic or cytostatic in T47D and MCF7 cell lines (16) (fig. S4A-C). BPTES treatment significantly decreased 15N-isotopologues of glutamate, proline, and aspartate, whereas metabolites involved in direct glutamine metabolism such as nucleotides and asparagine remained labeled (Fig. 1D & fig. S4D). Addition of ammonia to BPTES-treated cells restored metabolites depleted by glutaminase inhibition, demonstrating the specific contribution of ammonia (fig. S4E). This is consistent with findings that ammonia partially rescues proliferative defects in glutamine-deprived breast cancer cells(17).
We examined the potential mechanisms underlying assimilation of ammonia liberated during glutaminolysis. Because 15N-(amide)-glutamine did not elicit any isotopes of the urea cycle intermediates ornithine, citrulline, argininosuccinate and arginine, activity of CPS1 was ruled out as a mechanism for ammonia assimilation (fig. S2). Instead, our data indicated that GDH was the primary point of ammonia assimilation because glutamate is upstream of proline, aspartate, glutamine, and BCAA synthesis. However, the Km of GDH for ammonia is high (sub-mM) and GDH reportedly favors oxidative deamination over reductive amination in cancer cells (18–21). By contrast, GDH-catalyzed reductive amination is prevalent in the liver, where there is a sufficient concentration of ammonia to enable catalysis in this direction (22). We wondered if increased concentrations ammonia in the tumor microenvironment might also permit GDH-catalyzed reductive amination.
To determine whether GDH assimilates ammonia generated by glutamine catabolism, we used shRNA to deplete cells of GDH, cultured them with 15N-(amide)-glutamine and then subjected them to nitrogen metabolome scanning (fig. S4F). MCF7 and T47D cell lines express both GDH1 and GDH2 isoforms, and shRNAs targeted both (fig. S4G). Abundance of 15N-isotopologues of glutamate and downstream metabolites (proline, aspartate) was significantly decreased in cells depleted of GDH (Fig. 1E). Urea cycle intermediates (citrulline, argininosuccinate) remained unlabeled in cells lacking GDH, underscoring the lack of CPS1-mediated ammonia assimilation in breast cancer cells (Fig. 1E). Re-expression of shRNA-insensitive GDH1 rescued labeling onto glutamate and downstream metabolites (Fig. S5).
Next, we measured the amount of 15NH3 generated after 8 hour treatment with 15N-(amide)-glutamine using an LC-MS method for detection of 15NH3 from 15N-(amide)-glutamine (23). In MCF7 and T47D cells, only 3.5% of the total ammonia pool derived from glutaminolysis (fig. S6A-C). Since ~2% of the glutamate pool acquired this label in a GLS-dependent manner (fig S3B), we hypothesized that ammonia recycling from glutaminolysis is highly efficient. We quantified the efficiency of ammonia recycling from glutamine catabolism by incubating MCF7 cells with 15N2–13C5 glutamine. Glutaminolysis generated 15N-13C5-glutamate (glutamate M+6), and ammonia recycling was measured by detection of 15N-glutamate (glutamate M+1) (fig. S6D). We calculated the ratio of total amount of glutamate (glutamate M+6 & glutamate M+1) to glutamate directly generated in glutaminolysis (glutamate M+6) (fig. S6E). In total, 1.57 molecules of glutamate (by nitrogen) were generated from a single reaction of glutaminolysis, indicating a 57% efficiency of ammonia recycling (fig. S6F). Because both processes are mitochondrial, localization may support this high efficiency (24). Since GDH is a bi-directional enzyme, we also tested whether the catalytic activity of oxidative deamination or reductive amination was prevalent. In GDH-depleted cells, ammonia recycling (glutamate M+1) was decreased, but α-ketoglutarate (M+5), was unchanged, suggesting a net activity of reductive amination in this system (fig. S6G-H). In sum, these data indicate that ammonia derived from glutaminolysis is recycled by reductive amination catalyzed by GDH to support the synthesis of glutamate and downstream metabolites (Fig. 1F).
Because numerous reactions generate ammonia in addition to glutaminolysis, we investigated whether free ammonia could be assimilated into metabolic pathways. To optimize NH4Cl for tracing studies, we investigated whether exposure to increased concentrations of NH4Cl was toxic to tumor cells. Physiological concentrations of ammonia in plasma range between 0–50 μM in healthy human adults, 50–150 μM in newborns, and up to 1.0 mM in patients with hyperammonemia (25). Supraphysiological concentrations of ammonia are toxic to neurons, and sometimes assumed to also be toxic to tumor cells (7, 26, 27). However, NH4Cl was not toxic to tumor cells, even at concentrations that were toxic to primary human astrocytes (Fig. 2A, fig.S7A). Previous reports have shown that high concentrations of ammonia induce autophagy in tumor cells (5). In MCF7 and T47D cell lines, LC3II lipidation is not induced until 10 mM is added to media, which is greater than levels of ammonia reported in the tumor microenvironment. (fig. S7B) Moreover, ammonia concentrations of 0 to 10 mM did not alter uptake of glucose or glutamine, or basal respiration (fig. S7C-E). The expression of ammonia assimilating enzymes GS, GDH and CPS1 was not affected by increasing ammonia concentration (fig. S7F-I), nor did 10 mM ammonia alter the pH of the culture media (fig. S7J). These data indicate that supra-physiological concentrations of ammonia did not induce toxicity or metabolic stress in breast cancer cells.
Figure 2. Ammonia is assimilated by GDH to generate amino acids.
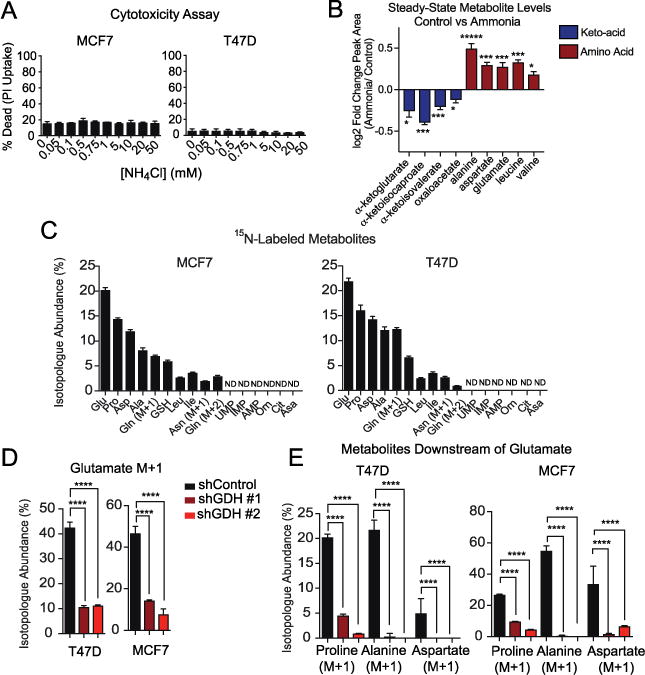
A. Propidium Iodide (PI) staining of cells treated with a dose of NH4Cl for 48 hours. Values represent mean +/− SEM, n=3. Representative experiment of three replicates. B. Heat map of fold-change in steady-state abundance of keto- and amino acids involved in transaminase reactions in T47D cells treated with 0.75 mM NH4Cl. Values represent mean +/− SEM, n=4. Representative experiment of two replicates. C. Abundance of 15N-isotopologues in MCF7 and T47D cells after 8 hours of treatment with 0.75 mM 15NH4Cl. (M+1) indicates a single nitrogen labeled and (M+2) indicates two nitrogen labeled. Values are scaled to account for total intracellular ammonia and represent mean +/− SEM, n=4. D. Isotopologue abundance of glutamate (M+1) in MCF7 and T47D cells treated for 8 hours with 0.75 mM 15NH4Cl in control and GDH-depleted cells. Values are scaled to account for total intracellular ammonia and represent mean +/− SEM, n=4. E. Abundance of 15N-isotopologues for metabolites downstream of glutamate treated for 8 hours with 0.75 mM 15NH4Cl in control and GDH depleted cells. Values are scaled to account for total intracellular ammonia and represent mean +/− SEM, n=4. For all comparisons two-tailed t test was used. *P < 0.05, **P < 0.01, ***P<0.001, ****P<0.0001.
We also examined ammonia uptake by cells. When breast cancer cells were cultured in low concentrations of ammonia (0 to 1.0 mM) we observed a net output of ammonia, which reverted to net uptake as the extracellular concentration of NH4Cl increased above 1 mM (fig. S7K). At approximately 1.0 mM NH4Cl, ammonia was taken up from the medium, such that ammonia entry may be regulated by diffusion. In agreement, the characterized mechanism of ammonium (NH4+) import and export is through facilitated diffusion with Rhesus glycoproteins (RhC and RhG) (28). Also, ammonia (NH3) can diffuse across the plasma membrane.
We performed steady-state and tracing experiments in the presence of 0.75 mM NH4Cl because it is the inflection point of ammonia uptake and secretion and represents a low concentration of ammonia that is relevant to the tumor microenvironment. We used MetaboAnalyst 3.0 to perform an unbiased pathway analysis on the steady-state metabolites from cells cultured with or without ammonia (fig. S8A). The most significantly altered pathway was glutamate, aspartate, and alanine metabolism. Exposure to NH4Cl elicited a signature of increased transaminase activity, whereby abundance of ketoacids decreased and that of amino acids derived from them increased (Fig. 2B). Although amounts of non-essential amino acids increased, the abundance of other amino acids remain unchanged by ammonia, indicating ammonia did not affect universal amino acid metabolism (fig. S8B). Nor did ammonia alter the abundance of metabolites from the urea cycle and nucleotides (fig. S8C-D).
MCF7 and T47D cells were treated with 0.75 mM 15NH4Cl and scanned for 15N-isotopologues (fig. S9). Isotopologue abundances were scaled to represent total ammonia pools, since treatment with 0.75 mM enriched for ~35% of the intracellular ammonia pool (table S2). Consistent with tracing performed with glutamine-derived ammonia, we detected 15N-labeling of glutamate and downstream metabolites, such as proline and aspartate (Fig. 2C–D). Upon tracing with low levels of ammonia, a striking 20% of the glutamate pool was labeled, implying an important role for ammonia assimilation in glutamate metabolism in cancer, as glutamate exists near sub-millimolar levels (11). Tracing with high levels of ammonia that have been reported in the tumor microenvironment (3 mM), also elicited the same signature of ammonia assimilation (fig. S10A).
Consistent with steady-state data, all of the amino acids labeled were generated through glutamate-dependent transaminase reactions, except proline and glutathione, which are made in direct synthetic pathways from glutamate (fig. S10B). Other nitrogen-containing metabolites, particularly urea cycle intermediates, and essential amino acids were not labeled by ammonia (fig. S9). Furthermore, in spite of ammonia generating 15N-isotopologues of glutamine, we detected no 15N-isotopologues of any nucleotides, which is distinct from ammonia metabolism in LKB low tumors (30). We speculate that because labeled glutamine is generated in the mitochondria, this pool may not access the cytoplasm where de novo nucleotide synthesis occurs, rendering nucleotides unlabeled.
A time course of 15NH4Cl tracing revealed that ammonia was rapidly converted into glutamate, the first metabolite to reach steady-state (fig. S10C–F). Thus, ammonia appears to be primarily assimilated to generate glutamate and other labeled metabolites are produced in secondary reactions. Therefore, we investigated which metabolic derivatives of ammonia required activity of GDH. In cells depleted of GDH, 15NH4Cl labeling of glutamate was diminished, as was labeling of metabolites downstream of glutamate (Fig. 2E and 2F). Indeed this labeling was rescued when shRNA-insensitive GDH1 was overexpressed (fig. S10G-H). We did not observe adaptation through ammonia assimilating enzymes GS or CPS1 in cells lacking GDH. In both T47D and MCF7 cells, glutamine and asparagine labeling did not change in cells depleted of GDH (fig. S11). Metabolites of the urea cycle were unlabeled in cells depleted of GDH, indicating that adaptive reprogramming of ammonia assimilation into the urea cycle is not important in cultured breast cancer cells (fig. S9). Our data reveal a general mechanism by which free ammonia in the tumor microenvironment can be harnessed for biosynthetic pathways.
Ammonia assimilation in yeast has a fundamental role in supporting growth and proliferation (31, 32). Because ammonia was not toxic to tumor cells (Fig. 2A), we tested whether ammonia might facilitate growth and proliferation of breast cancer cells. As in yeast, addition of NH4Cl to cell culture media increased proliferation in breast cancer cell lines (fig. S12A-B). Media was changed daily to minimize ammonia accumulation in culture media, which we measure to be approximately 0.3 mM per day from glutamine degradation and cellular metabolism (fig. S12C-F). Moreover, in 3D culture, addition of ammonia to media stimulated sphere growth and cell proliferation (Fig. 3A–B & fig. S12G). However, proliferation of primary human fibroblasts was not changed when ammonia was added to culture media (fig. S13A). Using 15NH4Cl tracing, we found that fibroblasts centrally assimilated ammonia to generate glutamine (fig. S13B), in line with their high expression of glutamine synthetase (33). Moreover, 15N-amide-glutamine tracing revealed that fibroblasts did not recycle glutamine-derived ammonia to generate glutamate, aspartate or proline (fig. S13C). Thus we hypothesized that, ammonia assimilation to generate glutamate through GDH may be important for its role in increased proliferation observed in breast cancer cells. Indeed, depletion of GDH prevented the accelerated growth of breast cancer cells treated with ammonia (Fig. 3C). Interestingly, the glutamate derivatives proline, aspartate and glutathione are associated with proliferation and tumorigenesis (34–38).
Figure 3. Ammonia stimulates breast cancer growth and proliferation.
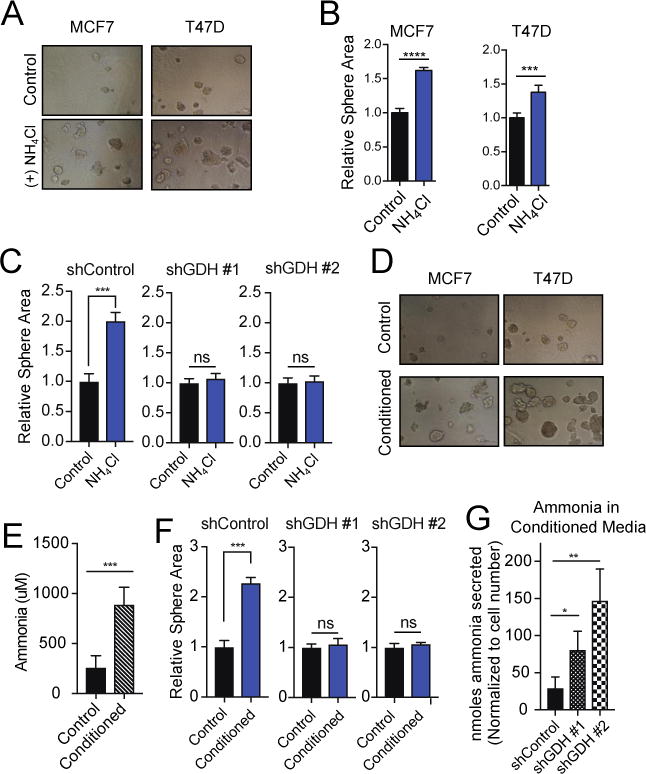
A. Representative images of 3D culture models of MCF7 and T47D cells treated with 0.5 mM NH4Cl compared to control conditions. B. Quantification of average sphere area of 100–200 spheres per well in 3D culture models of MCF7 and T47D cells treated with ammonia and control conditions for 7 days. Values represent mean area +/− SEM, n=4. Representative experiment of five replicates. C. Quantification of average sphere area of 200–250 spheres per well in 3D culture models of MCF7 cells harboring stable shRNA-mediated knockdown of GDH or control hairpin. Cells were treated for 8 days. Values represent mean area +/− SEM, n=4. Representative experiment of three replicates. D. Representative images of MCF7 and T47D cells in control conditions (daily media change) and conditioned media (media changed every 72 hours). Cells were treated for 8 days. E. Ammonia measurement in conditioned media compared to control after 8 days. F. Quantification of average sphere area of 200–250 spheres per well in 3D culture models of MCF7 control cells or cells depleted of GDH. Cells were treated in control or conditioned media for 8 days. Values represent mean area +/− SEM, n=4. Representative experiment of three replicates. G. Nmoles ammonia secreted per cell after 72 hours in control cells or cells depleted of GDH. Values represent mean +/− SEM, n=3. For all comparisons two-tailed t test was used. *P < 0.05, **P < 0.01, ***P<0.001, ****P<0.0001.
To assess the effect of tumor-generated ammonia on growth and proliferation, we compared the ability of cancer cells to grow in 3D cultures in which the medium was changed either daily or every 3 days, allowing ammonia to accumulate. The latter procedure provided a growth advantage for breast cancer cells, which correlated with ammonia accumulation in the media (Fig. 3D–E). Therefore, we tested whether ammonia recycling through GDH was a critical aspect that influenced proliferation. Cells depleted of GDH had no growth defect when the culture media was changed daily, but the growth advantage when media was changed after 3 days was abrogated (fig. S14A–C & Fig. 3F). Furthermore, cells depleted of GDH secreted more ammonia into the medium, consistent with impairment of ammonia recycling (Fig. 3G). In addition, treatment of MCF7 cells in 3D culture with high ammonia concentrations (3 mM NH4Cl) also stimulated proliferation (fig. S14D).
To examine the physiological relevance of ammonia in the tumor microenvironment in vivo, we measured concentrations of ammonia that accumulated in the interstitial fluids of ER(+) xenograft tumors. ER(+) xenografts accumulated 0.8 to 3 mM ammonia in the interstitial fluids of the tumor microenvironment, compared with plasma ammonia concentrations approximately 300 μM (Fig. 4A). Importantly, this range of concentrations did not induce autophagy and was shown to accelerate growth and proliferation in vitro. Plasma ammonia concentrations in mice harboring tumors were not different than those in control mice (fig. S15A).
Figure 4. Contributions of systemic and tumor autonomous ammonia metabolism to amino acid synthesis.
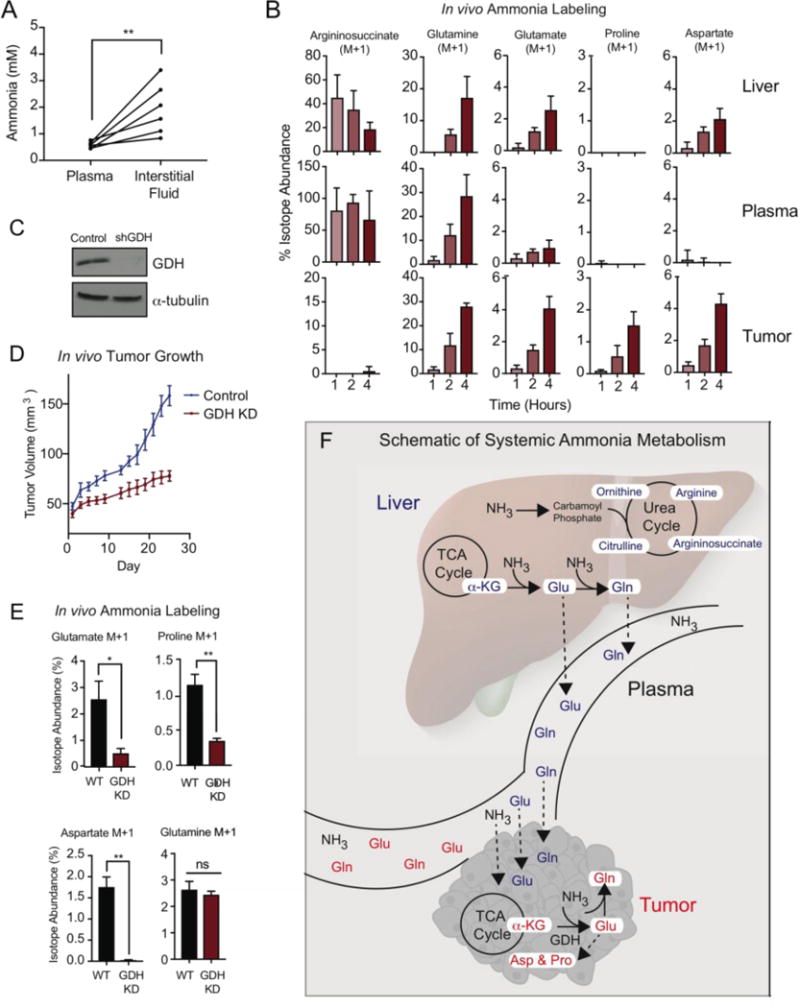
A. Measurement of ammonia in the interstitial fluids of the tumor microenvironment (TME) compared to plasma isolated from ER(+) breast cancer xenograft models. Lines connect values of ammonia in the plasma to that in the interstitial fluid of the TME. B. Isotope abundance of 15N-isotopologues isolated from the liver, plasma and tumor of mice IP injected with a bolus (9.0 mmol/kg) of 15NH4Cl. Tissues were harvested 1, 2, or 4 hours after injection. Values represent mean +/− SEM, n=4. 15N-isotopologues were corrected for natural abundance of tissues harvested from a control mouse treated with 9.0 mmol/kg NH4Cl for 4 hours. C. Western blot of GDH knockdown in T47D xenograft tumors. D. In vivo tumor growth of T47D control and GDH-depleted xenograft models (n=15 mice per group). Values represent mean tumor volume +/− SEM. E. In vivo tracing of 15NH4Cl in T47D control and GDH-depleted xenograft models. Values represent mean isotopologue abundance +/− SEM, n=4. F. Schematic of systemic and tumor autonomous ammonia metabolism. For all comparisons two-tailed t test was used. *P < 0.05, **P < 0.01, ***P<0.001, ****P<0.0001.
We tested whether accumulated ammonia in the tumor microenvironment was assimilated into metabolic pathways in vivo. Mice harboring subcutanenous T47D breast tumors were intraperitoneally injected with 15NH4Cl and the tumor, liver, and plasma were assessed for 15N-isotopologues over the next 1 to 4 hours (fig. S15B). The liver and tumor used distinct metabolic pathways for ammonia assimilation (Fig. 4B and fig. S16, S17A). In the liver, ammonia was sequestered into the urea cycle, leading to labeling of ornithine, citrulline, argininosuccinate and arginine (fig. S16). Although these labeled intermediates of the urea cycle were also detected in the plasma, they were undetectable in the tumor, indicating that these breast tumors did not engage the urea cycle for ammonia assimilation in vivo (Fig. 4B & fig. S16). Proline and aspartate, which were identified in vitro as metabolic derivatives of ammonia, were also labeled in the tumor in vivo. The metabolic pathways enabling proline and aspartate labeling were likely from tumor autonomous metabolism, as labeled proline and aspartate were not detected in the blood (Fig. 4B).
We also observed labeling of glutamine and glutamate in the tumor. Because labeled glutamine and glutamate were also found in the liver and plasma, it is not clear whether these 15N-isotopologues were generated tumor autonomously. Furthermore, the kinetics of glutamine labeling in the tumor implied that a subset of the labeled glutamine pool in the tumor may be taken up from the plasma (Fig. 4B).
To distinguish systemic contributions of ammonia metabolism from tumor autonomous metabolic pathways we traced 15NH4Cl and 15N-(amide)-glutamine in tumors ex vivo (fig. S17B-D). With 15NH4Cl, labeling of glutamate, aspartate, proline and glutamine was recapitulated. Consistent with in vivo studies, the urea cycle intermediates, nucleotides, and other nitrogen-abundant metabolites were not labeled. These data underscore a fundamental role of ammonia for amino acid synthesis, particularly glutamate, aspartate and proline in vivo.
Consistent with in vitro experiments, tumors treated with 15N-(amide)-glutamine generated labeled glutamate and the downstream metabolites proline and aspartate, suggesting that glutamine-derived ammonia may be recycled in solid tumors (fig. S18A). In contrast to free 15NH4Cl, which did not label metabolites of the urea cycle in cells, in vivo or in solid tumors ex vivo, 15N-(amide)-glutamine, when added to the tumors ex vivo, elicited labeling of the urea cycle intermediate citrulline (fig. S17D). Thus, an alternative pathway that does not require ammonia may exist that connects the amide nitrogen on glutamine to citrulline production ex vivo.
We next generated a GDH—depleted xenograft model to further investigate the mechanism of ammonia assimilation in vivo (Fig. 4C). Tumor-specific depletion of GDH significantly decreases tumor growth in vivo, consistent with our findings that GDH-depleted cells grow slower in conditioned media and are insensitive to ammonia-induced growth in vitro (Fig. 4D, fig.S18A). Since GDH- catalyzed ammonia assimilation mediates growth and proliferation in vitro, we tested whether GDH also assimilates ammonia in vivo using IP injection of 15NH4Cl in control and GDH-depleted xenograft models. Glutamate, aspartate and proline labeling are significantly decreased in GDH depleted tumors compared to control tumors (Fig. 4E). Importantly, GDH-depletion did not abrogate glutamine labeling, underscoring the specificity of GDH for ammonia assimilation to generate glutamate and the downstream metabolites proline and aspartate. To further validate that GDH-mediated ammonia assimilation is tumor autonomous, ammonia was traced ex vivo in control and GDH-depleted tumors. GDH-depletion significantly decreases glutamate, aspartate and proline labeling (fig. S18B). Taken together, these data show that both tumor autonomous metabolism of ammonia, as well as systemic assimilation support tumor biomass, especially for glutamate-derived amino acids (Fig. 4F).
Ammonia accumulates in the tumor microenvironment because tumors are poorly vascularized, making this a unique niche for ammonia metabolism in the human body. Since ammonia transport is mediated by diffusion, elevated ammonia in the microenvironment leads to its accumulation inside of tumor cells (fig. S18C). Therefore, the ability to re-assimilate this ammonia into metabolic pathways is critical in this context. In contrast, the liver re-assimilates ammonia to generate urea, which is a sink for excess nitrogen and is excreted as metabolic waste to protect against toxicity associated with systemic ammonia accumulation. Tumor cells strictly recycle this nitrogen to generate amino acids downstream of glutamate and do not engage the urea cycle.
In sum, we identified that ammonia is an important nitrogen source for breast cancer metabolism. Ammonia is not simply a metabolic waste product, and it can be recycled to support the high demand for amino acid synthesis in rapidly proliferating cells. Although ammonia is sometimes considered a toxin, it stimulated growth and proliferation in breast cancer cells. This stimulatory effect appears to be mediated by GDH-catalyzed ammonia assimilation. Furthermore, ammonia accumulated in the tumor microenvironment, and was used by cancer cells for amino acid synthesis in vivo. These biosynthetic pathways are supported in both systemic and tumor autonomous metabolism. Thus, we showed that metabolic recycling of ammonia provides an important source of nitrogen for breast cancer biomass.
Supplementary Material
One Sentence Summary.
Ammonia is a fundamental nitrogen source that can support tumor biomass.
Acknowledgments
The authors acknowledge all members of the Haigis lab for insightful discussions. MCH is supported by the Ludwig Center at Harvard, Glenn Foundation for Medical Research and NIH grant R01CA213062. J.B.S is funded by the National Science Foundation Graduate Research Fellowship Program (Grant # DGE1144152). Harvard Medical School has filed a patent application on the data described in this manuscript.
Footnotes
Supplementary Materials
Materials and Methods
References (39–43)
Full Reference List
REFERENCES AND NOTES
- 1.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell metabolism. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coloff JL, et al. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell metabolism. 2016;23:867–880. doi: 10.1016/j.cmet.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Science signaling. 2010;3:ra31. doi: 10.1126/scisignal.2000911. [DOI] [PubMed] [Google Scholar]
- 6.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nature reviews Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kappler M, et al. Normoxic accumulation of HIF1alpha is associated with glutaminolysis. Clinical oral investigations. 2016 doi: 10.1007/s00784-016-1780-9. [DOI] [PubMed] [Google Scholar]
- 8.Tapper EB, Jiang ZG, Patwardhan VR. Refining the ammonia hypothesis: a physiology-driven approach to the treatment of hepatic encephalopathy. Mayo Clinic proceedings. 2015;90:646–658. doi: 10.1016/j.mayocp.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 9.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tardito S, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nature cell biology. 2015;17:1556–1568. doi: 10.1038/ncb3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeBerardinis RJ, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adeva MM, Souto G, Blanco N, Donapetry C. Ammonium metabolism in humans. Metabolism: clinical and experimental. 2012;61:1495–1511. doi: 10.1016/j.metabol.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Morris SM., Jr Regulation of enzymes of the urea cycle and arginine metabolism. Annual review of nutrition. 2002;22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- 14.Tyanova S, et al. Proteomic maps of breast cancer subtypes. Nature communications. 2016;7:10259. doi: 10.1038/ncomms10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.S AL, Roberts Lee D, Gerszten Robert E, Clish Clary B. Targeted Metabolomics. Current Protocols in Molecular Biology. 2012:30.32.31–30.32.24. doi: 10.1002/0471142727.mb3002s98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson MM, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) The Biochemical journal. 2007;406:407–414. doi: 10.1042/BJ20070039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng M, Chen S, Lao T, Liang D, Sang N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell cycle. 2014;9:3921–3932. doi: 10.4161/cc.9.19.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frigerio F, Casimir M, Carobbio S, Maechler P. Tissue specificity of mitochondrial glutamate pathways and the control of metabolic homeostasis. Biochimica et biophysica acta. 2008;1777:965–972. doi: 10.1016/j.bbabio.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 19.Treberg JR, Brosnan ME, Watford M, Brosnan JT. On the reversibility of glutamate dehydrogenase and the source of hyperammonemia in the hyperinsulinism/hyperammonemia syndrome. Advances in enzyme regulation. 2010;50:34–43. doi: 10.1016/j.advenzreg.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 20.B ME, Brosnan AJT. Hepatic glutamate metabolism: a tale of 2 hepatocytes. The american journal of clinical nutrition. 2009;90:857s–861s. doi: 10.3945/ajcn.2009.27462Z. [DOI] [PubMed] [Google Scholar]
- 21.Adam J, et al. A role for cytosolic fumarate hydratase in urea cycle metabolism and renal neoplasia. Cell reports. 2013;3:1440–1448. doi: 10.1016/j.celrep.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaganas I, et al. The effect of pH and ADP on ammonia affinity for human glutamate dehydrogenases. Metabolic brain disease. 2013;28:127–131. doi: 10.1007/s11011-013-9382-6. [DOI] [PubMed] [Google Scholar]
- 23.Spinelli JB, Kelley LP, Haigis MC. An LC-MS Approach to Quantitative Measurement of Ammonia Isotopologues. Sci Rep. 2017;7:10304. doi: 10.1038/s41598-017-09993-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.M B, Devlin ATM. Intramitochondrial localization of alanine aminotransferase in rat-liver mitochondria: comparison with glutaminase and aspartate aminotransferase. Amino acids. 1995:363–374. doi: 10.1007/BF00807273. [DOI] [PubMed] [Google Scholar]
- 25.Dasarathy S, et al. Ammonia toxicity: from head to toe? Metabolic brain disease. 2016 doi: 10.1007/s11011-016-9938-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braissant O, McLin VA, Cudalbu C. Ammonia toxicity to the brain. Journal of inherited metabolic disease. 2013;36:595–612. doi: 10.1007/s10545-012-9546-2. [DOI] [PubMed] [Google Scholar]
- 27.Hillmann P, Kose M, Sohl K, Muller CE. Ammonium-induced calcium mobilization in 1321N1 astrocytoma cells. Toxicology and applied pharmacology. 2008;227:36–47. doi: 10.1016/j.taap.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Gruswitz F, et al. Function of human Rh based on structure of RhCG at 2.1 A. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9638–9643. doi: 10.1073/pnas.1003587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vander Heiden MG, et al. Identification of small molecule inhibitors of pyruvate kinase M2. Biochemical pharmacology. 2010;79:1118–1124. doi: 10.1016/j.bcp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. 2017;546:168–172. doi: 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boer VM, Crutchfield CA, Bradley PH, Botstein D, Rabinowitz JD. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Molecular biology of the cell. 2010;21:198–211. doi: 10.1091/mbc.E09-07-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.S BM, Laxman Sunil, Shi Lei, Tu Benjamin P. Npr2 inhibits TORC1 to prevent inappropriate utilization of glutamine for biosynthesis of nitrogen-containing metabolites. Science signaling. 2014;7:ra120. doi: 10.1126/scisignal.2005948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermeulen T, et al. Glutamine synthetase is essential for proliferation of fetal skin fibroblasts. Archives of biochemistry and biophysics. 2008;478:96–102. doi: 10.1016/j.abb.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Phang JM, Liu W, Hancock CN, Fischer JW. Proline metabolism and cancer: emerging links to glutamine and collagen. Current opinion in clinical nutrition and metabolic care. 2015;18:71–77. doi: 10.1097/MCO.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahu N, et al. Proline Starvation Induces Unresolved ER Stress and Hinders mTORC1-Dependent Tumorigenesis. Cell metabolism. 2016;24:753–761. doi: 10.1016/j.cmet.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Jin L, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer cell. 2015;27:257–270. doi: 10.1016/j.ccell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birsoy K, et al. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sullivan LB, et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.