

Abstract
This paper describes three protocols for identifying interacting surfaces on 15N-labeled target proteins of known structure by using in-cell NMR spectroscopy. The first protocol describes how to identify protein quinary structure interaction surfaces in prokaryotes by using cross-relaxation-induced polarization transfer, CRIPT, based in-cell NMR. The second protocol describes how to introduce labeled protein into eukaryotic (HeLa) cells via electroporation for CRIPT-based in-cell studies. The third protocol describes how to quantitatively map protein interacting surfaces by utilizing singular value decomposition, SVD, analysis of STructural INTeractions by in-cell NMR, STINT-NMR, data.
Keywords: In-cell NMR spectroscopy, Quinary structure, Electrophoresis, Singular value decomposition (SVD), Isotopic labeling, Protein-protein interactions, HeLa cells
1 Introduction
The ultimate goal of structural and biochemical research is to understand how macromolecular interactions give rise to and regulate biological activity in living cells. The challenge is formidable due to the complexity that arises not only from the number of proteins (gene products) expressed by the organism, but also from the combinatorial interactions between them [1, 2]. Despite ongoing efforts to decipher the complex nature of protein interactions, new methods for structurally characterizing protein complexes are needed to fully understand molecular networks [3]. With the onset of in-cell NMR spectroscopy [4] molecular structures can be studied under physiological conditions shedding light on the structural underpinning of biological activity.
The interior of a living cell is an extremely crowded environment and NMR-active nuclei in biological macromolecules are extremely sensitive to this environment and changes in its composition. In a cell the concentrations of macromolecules can exceed 400 mg/mL [5], hydrophobic and hydrophilic interactions are perturbed, there is limited bulk water, cytosolic water forms solvent shells on protein surfaces [6], and the distances between macromolecules are less than the characteristic Debye screening length of ion charges [7]. In addition, specific and nonspecific binding interactions with ions, small effector ligands and other macromolecules, as well as changes due to biochemical modifications, alter the chemical environment. As a result, in-cell NMR spectra are inherently noisier than spectra acquired in vitro.
The low signal-to-noise ratio arises from the myriad of interactions between the target protein and components of the cytosol. While these interactions do not result in large differences in the chemical shift between spectra acquired in-cell versus in vitro, they do result in slower tumbling times, a dramatic increase in the apparent molecular weight of the target, and extensive broadening of many or all of the spectral peaks [8–11]. For example, Thioredoxin, a 12 kDa protein, exhibits an apparent molecular weight of ~1 MDa in-cell. In particular, due to the high intracellular concentration, ribosomal and messenger RNA interact extensively with target proteins, altering the structure and activity in unpredictable ways [11–13]. These inherently transient interactions constitute a fifth level of protein structural organization, quinary structure [14].
Protein quinary interactions are refractory to biochemical analyses [15]. Studying these interactions requires in-cell methodologies because isolation techniques will disrupt protein complexes with limited thermodynamic stability [16]. Two-dimensional in-cell NMR spectra acquired by using 15N heteronuclear single quantum coherence, 1H-15N HSQC, experiments [17] do not resolve signals from target proteins engaged in quinary interactions due to broadened peaks. Transverse proton relaxation of the in-cell NMR signal, T2, is so short for high molecular weight complexes that only proteins that are unstructured, such as the prokaryotic ubiquitin-like protein, Pup, and thus have a longer T2 due to internal flexibility or that interact extremely weakly with the cytosol, have been studied by using 1H-15N HSQC.
To overcome the inability to detect high molecular weight species in-cell, relaxation optimized 15N-edited cross-relaxation enhanced polarization transfer, CRINEPT, heteronuclear multiple quantum coherence, HMQC, transverse relaxation optimized spectroscopy, TROSY, 1H-15N CRINEPT-HMQC-TROSY experiments are attractive for in-cell NMR studies due to superior sensitivity to NMR signals and relative insensitivity to unavoidable magnetic field inhomogeneity [18]. In vitro solution NMR performed on extensively deuterated supramolecular structures successfully used CRINEPT NMR spectroscopy to study complexes with molecular weights greater than 100 kDa [19]. For in-cell experiments, REDuced PROton density (REDPRO) labeling is used to exchange α and β protons of amino acids for deuterons to minimize proton relaxation [20, 21], and the optimal CRINEPT transfer time is calibrated for the target protein in-cell to maximize the NMR peak intensities [11] (Fig. 1). The resulting in-cell 1H-15N CRINEPT-HMQC-TROSY spectrum thus reveals the structure of the protein engaged in quinary interactions (Fig. 2a).
Fig. 1.
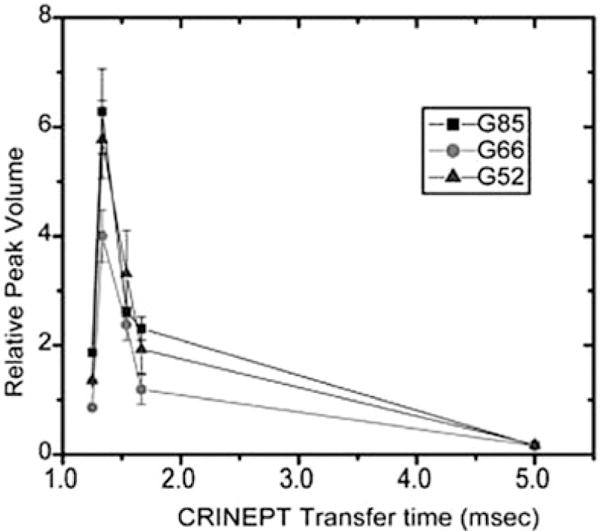
The relative volumes of the G52, G66, and G85 crosspeaks in the in-cell 1H-15N CRINEPT-HMQC-TROSY spectra of Trx are plotted against the CRINEPT transfer delay times. An endogenous tryptophan indole amide peak in the in-cell spectra is used as a reference. The optimum CRINEPT transfer delay for Trx is ~1.3 ms, which corresponds to an apparent molecular weight of 1.1 MDa. Reproduced from [11, 37] with permission from the American Chemical Society
Fig. 2.
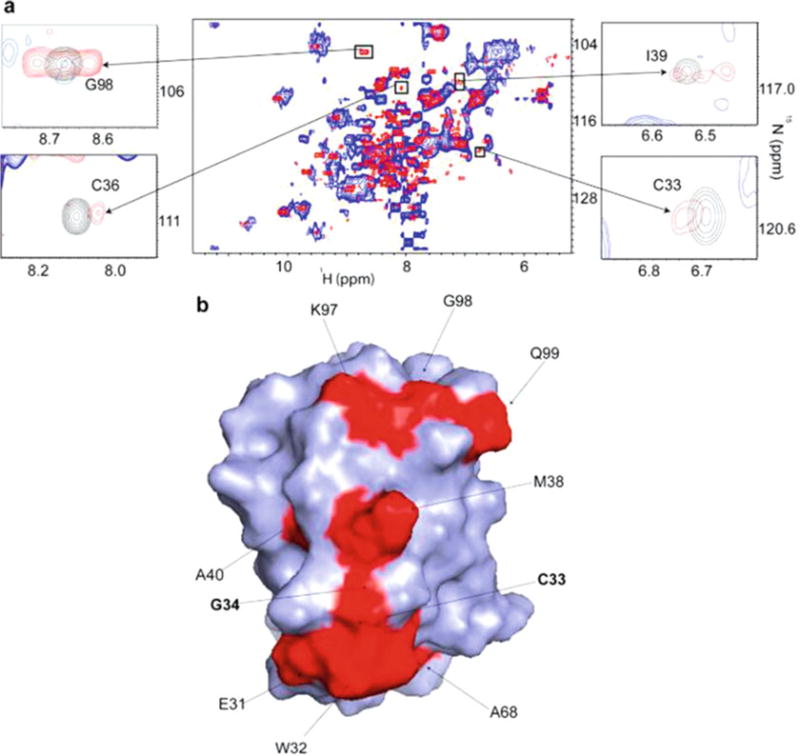
(a) Overlay of the in-cell 1H-15N CRINEPT-HMQC-TROSY spectrum of REDPRO labeled Trx (blue) and that of the cellular lysate (red). The intensities of C33, C36, I39, and G98 peaks, from the residues involved in the quinary interactions, are broadened out. The insets show overlays of the boxed regions of the in-cell spectrum (blue) and the corresponding regions of the 1H-15N CRINEPT-HMQC-TROSY spectrum of lysate (red), and 1H-15N HSQC spectrum of purified Trx in 10 mM potassium phosphate buffer at pH 6.5 (black). (b) Residues involved in the quinary interactions (red) are mapped onto the molecular surface of Trx (PDB code 1X0B); active site residues, C33 and G34, are in bold. Reproduced from [11] with permission from the American Chemical Society
To acquire an in-cell NMR spectrum, the concentration of labeled nuclei must be high enough to provide well-resolved resonances for unambiguous identification. In prokaryotes and some eukaryotes overexpression of target protein in a labeled medium is used to generate an intracellular concentration of target protein sufficiently high to acquire NMR spectra [22–25]. The in-cell NMR spectrum of a fully expressed protein is compared with the spectrum of free protein either purified or from cell lysates. Residues exhibiting the greatest reduction in intensity are presumed to contribute to the quinary interacting surface.
In eukaryotic cells overexpression of target proteins is often too weak to produce an intracellular concentration of target protein sufficient to acquire in-cell NMR spectra. Several methods have been developed to introduce 15N labeled target proteins into both cells including cell-penetrating peptides [26], toxins [27], micro injections [28, 29], and electroporation [11, 30]. Each method has its advantages and disadvantages, but all have proven effective in delivering sufficient quantities of labeled target protein to acquire reproducible in-cell NMR spectra (Fig. 3a).
Fig. 3.
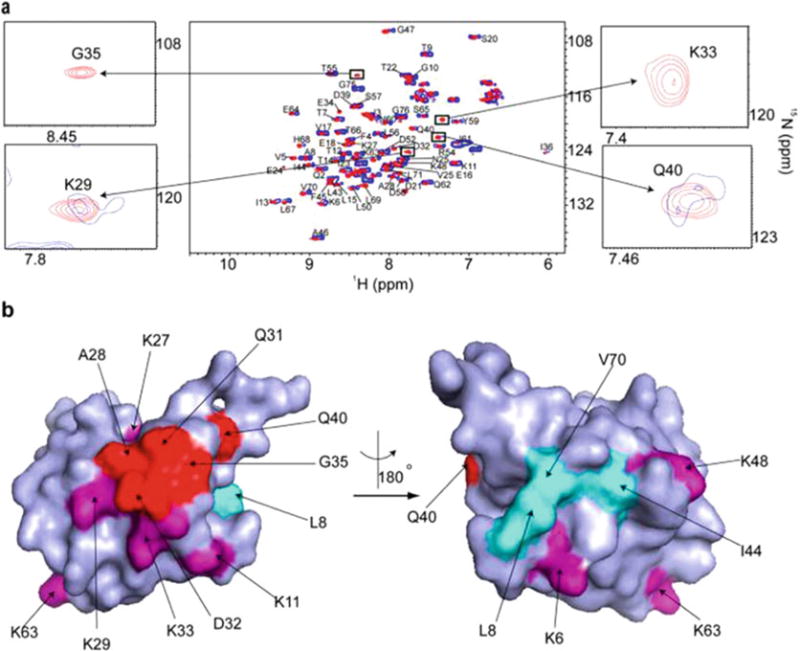
(a) Overlay of the in-cell 1H-15N CRINEPT-HMQC-TROSY spectrum of REDPRO labeled Ubiquitin electroporated into HeLa cells (blue) and the 1H-15N HSQC spectrum of the cell lysate. NMR peaks corresponding to K29, K33, G35, and Q40 (insets) are broadened in the in-cell spectrum, suggesting that Ubiquitin is involved intransient interactions with cellular components of the cytosol. (b) Residues (shown in red), whose NMR peaks are broadened out form a contiguous interaction surface involved in Ubiquitin quinary interactions (PDB code 1D3Z). The seven lysines of Ubiquitin, which are used for ubiquitylation, are in purple. K27, K29, and K33 are a part of the interaction surface and affected by the quinary interactions. The canonical I44 hydrophobic patch of Ubiquitin (shown in cyan) spanning L8, I44, and V70 is unperturbed by quinary interactions. Reproduced from [11] with permission from the American Chemical Society
Following the in-cell experiment, cells are lysed and the 1H-15N CRINEPT-HMQC-TROSY spectrum is re-acquired on free protein. The difference between the normalized peak intensities of the in-cell and lysate spectra is calculated for each residue (Fig. 4). Residues exhibiting the greatest change in intensity are mapped onto the surface of the target protein (Figs. 2b and 3b). Such analyses can provide insights into the effect of protein quinary structure on the regulation of biological activity in living cells [11, 31].
Fig. 4.

Bar plot showing the relative changes in in-cell 1H-15N CRINEPT-HMQC-TROSY peak intensities of Trx residues due to quinary interactions. Residues E31, W32, C33, C36, M38, A40, A68, and Q99, annotated with (*), are also affected in total RNA bound Trx. The horizontal line differentiates residues whose NMR peaks undergo significant broadening. Reproduced from [11] with permission from the American Chemical Society
Unlike quinary interactions, which are of comparatively low (micromolar or weaker) affinity, specific high (sub-micromolar) affinity interactions between a target and interactor protein are studied by using STINT-NMR [32, 33], which elucidates STructural INTeractions between proteins within their native environment by using in-cell NMR [22–25, 33]. In its simplest form, STINT-NMR can identify the interacting surface of a target protein when a single interactor protein (ligand) binds to it. This is accomplished in prokaryotes by overexpressing the target protein on the [U-15N] medium, then sequentially overexpressing interactor protein on the unlabeled medium. Samples are collected at various times and the in-cell NMR spectrum of the target protein is acquired. As the concentration of the interactor increases, the spectrum of the target changes to reflect the different chemical environment of the residues that have been affected by the binding interaction. The resulting NMR data provide a complete titration of the interaction and define structural details of the interacting surfaces at atomic resolution.
Conventional analyses of interacting proteins tend to incorrectly estimate the number of residues involved in the interaction because of the widespread signal broadening associated with the formation of a stoichiometric complex. The process of distinguishing which spectral changes are due to specific binding generally considers only the difference between the spectrum of free target protein and the final in-cell target spectrum following full over-expression of the interactor to assess the change in intensity of a given peak resonance [34]. This absolute difference is used to infer whether or not the corresponding amino acid contributes to the principal binding mode of the target. Time-dependent degradation of the target protein inside the cell or differences in sample preparations can lead to changes in the NMR spectra. A rigorous objective analysis of spectral changes is needed to unambiguously differentiate between signals that result from concentration-dependent and concentration-independent processes. This problem can be overcome by analyzing in-cell NMR data by using Singular Value Decomposition, SVD.
SVD is a mathematical technique used to identify the principal components of an arbitrary matrix that contribute maximally to the variance of its elements [35]. Over the course of a STINT-NMR titration, a series of in-cell NMR spectra are collected, and a matrix, M, is created that contains the changes in target protein peak intensities versus the expression time of the unlabeled binding partner (Fig. 5a). SVD analysis of matrix M discriminates between changes in the in-cell NMR spectrum of a target protein due to specific and nonspecific interactions and changes due to the presence of the complex cellular environment over the time course of interactor overexpression (Fig. 5b). A Scree plot (Fig. 5c) shows the distribution of singular values that defines the relative contribution of each binding mode to the change in chemical shift or intensity. An abrupt drop in the singular values following the first binding mode indicates the presence of a single principal binding mode; a gradual decrease in singular values corresponds to random binding of target protein to components of the cytosol. The analysis identifies the amino acid residues involved in the principal binding mode of a target protein with its interactor (Figs. 5d and 6).
Fig. 5.
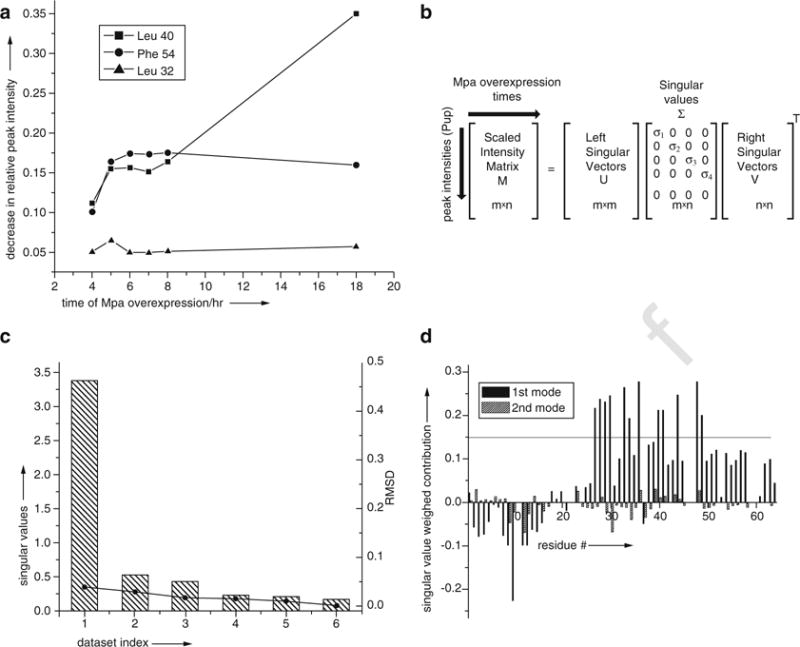
(a) The intensity of individual cross peaks of Pup change at different rates during interactor protein, Mpa, overexpression. SVD analysis evaluates the magnitude of the contribution of an intensity change to the NMR data over the experimental time course to identify concentration-dependent interactions. (b) SVD of the experimental data matrix M of size m × n yields the matrices U, Σ, and VT, with sizes m × m, m × n, and n × n, respectively, where m is the number of target protein amino acid residues used in the NMR analysis, n is the number of time-course NMR data sets, and VT is the transpose of matrix V. (c) The Scree plot shows the distribution of singular values for each data-set index (binding mode) from 1 to 6. The root mean square deviation, RMSD, values between respective components, and the complete dataset are indicated by solid circles. (d) The weighted contribution of each Pup amino acid residue to the Mpa principal binding modes, calculated as a product of a corresponding singular value and left singular vector, is shown for the 1st (black) and 2nd (hatched) binding modes. The threshold of 0.14 is chosen to highlight the 12 amino acids that exhibit the largest singular value weighted. Negative values are due to spectral overlap between the target protein and cellular metabolites. Reproduced from [37] with permission from Wiley and Sons
Fig. 6.
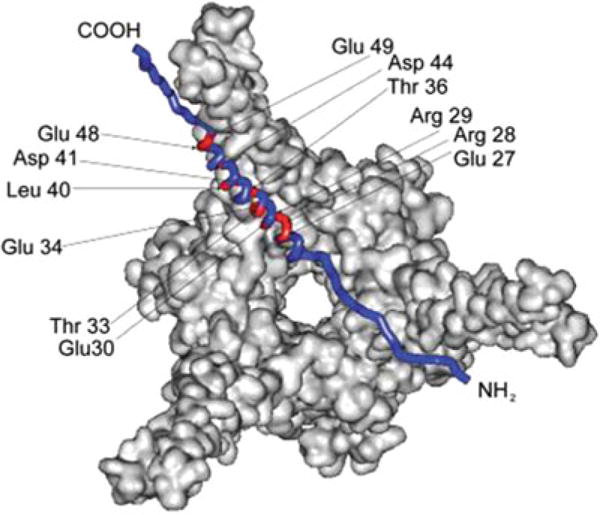
Residues comprising the principle-binding mode (red) between Pup and Mpa are mapped onto a Pup-Mpa complex (PDB code 3M9D) [38]. Reproduced from [37] with permission from Wiley and Sons
This paper describes three protocols for acquiring in-cell NMR spectra on 15N-labeled target proteins. The first protocol describes how to identify protein quinary structure interaction surfaces in prokaryotes by using CRIPT-based in-cell NMR. The second protocol describes how to introduce labeled protein into eukaryotic (HeLa) cells via electroporation for CRIPT-based in-cell. The third protocol describes how to quantitatively map protein interacting surfaces by utilizing SVD analysis of STINT-NMR data.
2 Materials
All the NMR experiments were performed at room temperature using a 700 MHz Bruker Avance spectrometer equipped with a TXI 2-gradient cryoprobe. All data were processed with Topspin 2.1 and analyzed by using CARA software. Interacting residues were mapped onto surface models of proteins using SWISS-PDB Viewer.
2.1 Determining Protein Quinary Interaction Surfaces in Prokaryotes
pRSF-Trx: Plasmid used to overexpress bacterial Thioredoxin [11].
BL21(DE3) codon + (Novagen): E. coli strain used to overexpress plasmids.
Luria Broth (LB medium): Dissolve 20 g of LB Broth Lennox in 1 L of distilled water and autoclave.
Kanamycin (Kn) 1000× stock: Dissolve 0.35 g of kanamycin sulfate in 10 mL of distilled water, sterile filter and store at −20 °C.
LB-Kn: LB medium supplemented with 35 μg/mL of kanamycin.
LB-Kn agar plates: Dissolve 20 g of LB Broth Lennox in 1 L of distilled water, add 15 g of agar and autoclave. Monitor the temperature as it cools. When the temperature reaches 50–55 °C, add 1 mL of kanamycin stock solution. Pour 20–30 mL per plate into 100 × 10 mm Petri dishes. Let the agar solidify and the plates dry on the benchtop for several days. Store dried plates in a plastic bag at 4 °C.
1 M CaCl2: Dissolve 5.55 g of CaCl2 in 50 mL of distilled water and sterile filter.
M9 salts: Dissolve 6 g of Na2HPO4, 3 g of KH2PO4, and 0.5 g of NaCl in 1 L of distilled water. Adjust the pH to 7–7.4 and add 10 μL of 1 M CaCl2. Autoclave and store for up to 1 month at 4 °C. Autoclaving after adding CaCl2 will ensure that any calcium phosphate precipitates will be solubilized.
1 M Mg2SO4: Dissolve 6.02 g of MgSO4 in 50 mL of distilled water and sterile filter.
Solid NH4Cl and 15NH4Cl.
1 mg/mL thiamine HCl.
20% D-glucose: Dissolve 20 g of D-glucose in 80 mL of distilled water, sterile filter and store at 4 °C.
Glycerol.
M9 labeling medium: Add 1 g [U-15N]-NH4Cl, 2 mL of 1 M Mg2SO4, 1 mL of 1 mg/mL thiamine HCL and 20 mL of 20% (w/v) D-glucose, or 4 mL of glycerol to 1 L of M9 salts.
D2O.
Deuterated M9-Kn labeling medium: M9 labeling medium prepared using D2O and supplemented with 35 μg/mL of kanamycin.
1 M IPTG: Dissolve 2.383 g IPTG in 10 mL distilled water and store in −20 °C freezer.
NMR buffer: 10 mM potassium phosphate, pH 6.5.
5 mm standard sample tubes.
2.2 Determining Protein Quinary Interaction Surfaces in Eukaryotes
150 cm2 cell culture flasks.
Low glucose Dulbeccos’s Modified Eagle Medium (DMEM).
Fetal bovine serum (FBS).
Complete medium: DMEM supplemented with 10% FBS.
10× 0.5% Trypsin/5.3 mM EDTA stock (we use Gibco).
Balanced salt solution without calcium and magnesium: 5 mM KCl, 440 μM KH2PO4, 4.2 mM NaHCO3, 137 mM NaCl, 340 μM Na2PO4·7H2O.
Phosphate-buffered saline (PBS): Dissolve 8 g NaCl, 0.2 g of KCl, 1.44 g of Na2HPO4, and 0.24 g of KH2PO4 in 1 L of distilled water, adjust the pH to 7.4 and. Store PBS at room temperature.
Electroporation 2× salts stock: 100 mM sodium phosphate pH 7.2, 5 mM KCl, 15 mM MgCl2, 15 mM Hepes, 5 mM ATP, 5 mM reduced glutathione.
Electroporation buffer: 50% electroporation 2× salts stock and 50% Amaxa Nucleofector Solution R (Lonza).
We use a Amaxa™ Nucleofector 2b electroporator (Bio-rad). Any suitable electroporator may be used.
Electroporation cuvettes.
Culture dishes (Falcon).
NMR buffer (see Subheading 2.1).
D2O.
5 mm Shigemi sample tubes.
2.3 Using SVD Analysis to Determine Protein-Protein Structural Interactions
pASK-Pup-GGQ: Plasmid used to overexpress Pup-GGQ [36].
pRSF-Msm Mpa: Plasmid used to overexpress Mycobacterium smegmatis, (Msm) mycobacterial proteasome ATPase, Mpa [36].
BL21(DE3) codon + (see Subheading 2.1).
Kanamycin (Kn) 1000× stock (see Subheading 2.1).
Ampicillin (Ap) 1000× stock: Dissolve 1.00 g of kanamycin sulfate in 10 mL of distilled water, sterile filter and store at −20 °C.
LB-Ap-Kn: LB medium (see Subheading 2.1) supplemented with 35 μg/mL of kanamycin and 100 μg/mL of ampicillin.
LB-Ap-Kn agar plates: Dissolve 20 g of LB Broth Lennox in 1 L of distilled water, add 15 g of agar and autoclave. Monitor the temperature as it cools. When the temperature reaches 50–55 °C, add 1 mL of kanamycin and 1 mL of ampicillin stock solution. Pour 20–30 mL per plate into 100 × 10 mm Petri dishes. Let the agar solidify and the plates dry on the benchtop for several days. Store dried plates in a plastic bag at 4 °C.
M9 salts (see Subheading 2.1).
M9 labeling medium (see Subheading 2.1).
M9-Ap-Kn labeling medium: M9 labeling medium supplemented with 35 μg/mL of kanamycin and 100 μg/mL of ampicillin.
Dimethylformamide.
2 mg/mL anhydrotetracycline stock: dissolve 20 mg of anhydrotetracycline in dimethylformamide, store at −20 °C.
NMR buffer (see Subheading 2.1).
Glycerol.
1 M IPTG (see Subheading 2.1).
D2O.
Matlab 2009b or Octave 4.0.1 software.
3 Methods
3.1 Determining Protein Quinary Interaction Surfaces in Prokaryotes
This protocol requires an inducible plasmid that overexpresses the target protein and NMR peak assignments for the target protein. We will use Thioredoxin (Trx) as a specific example of the target protein, although any appropriately cloned target should work. Trx was cloned into expression vector pRSF-1b to yield pRSF-Trx [11]. pRSF-Trx confers kanamycin resistance, codes for lac repressor, and expresses N-terminal His-tagged bacterial Trx from the T7 promoter/lac operon, which is induced by IPTG. E. coli strain BL21 (DE3) codon + was transformed with pRSF-Trx using standard techniques and the transformed cells were selected on LB-Kn agar plates. NMR peak assignments for the target protein are required to complete the analysis.
Control experiments are performed to monitor the comparative viability of the transformed cells before and after acquisition of the in-cell spectrum by using cell growth plate assays. Additional controls test for cell leakage by acquiring a spectrum on the sample supernatant.
3.1.1 Target Protein Labeling and Overexpression
Inoculate 5 mL of LB-Kn (35 μg/mL) with a single colony of BL21(DE3) codon + containing pRSF-Trx.
Grow the culture overnight at 37 °C with shaking at 250 rpm.
Dilute the overnight culture to OD600 ~0.07 in 50 mL of LB-Kn.
Grow the cells at 37 °C, 250 rpm to OD600 ~0.9–1.0.
Centrifuge the cells at 4500 × g for 15 min at room temperature.
Wash the cells twice with a total of 50 mL of M9 salts.
Recentrifuge the cells and resuspend in 50 mL of deuterated M9-Kn labeling medium.
Incubate the culture at 37 °C, 275 rpm for 20 min.
Induce overexpression of Trx by adding 0.1% culture volume of 1 M IPTG.
Overexpress REDPRO-labeled target protein for up to 36 h at 37 °C, 275 rpm.
3.1.2 Sample Preparation
Centrifuge 50 mL of culture at 4500 × g for 15 min at room temperature.
Wash the cells twice with 50 mL of NMR buffer.
Resuspend the cell pellet in 0.45 mL of NMR buffer and 0.05 mL of D2O. Remove 10 μL for cell viability assays. Transfer the remaining sample to a standard 5 mm NMR tube.
Prepare 10−4, 10−5, and 10−6 serial dilutions of the 10 μL cell sample. Plate 100 μL of each dilution onto LB-Kn plates in triplicate. Incubate the plates overnight at 37 °C and count the resulting colonies.
3.1.3 NMR Spectroscopy
All of the experiments are performed at room temperature (RT) using a standard 5 mm NMR tube in a 700 MHz Bruker Avance II NMR spectrometer equipped with TXI z-gradient cryoprobe.
Shim the sample manually using the z, z2, and z3 shims.
Select the [1H-15N]-CRINEPT-HMQC-[1H]-TROSY experiment as described by Riek et al. [19] (Fig. 3b in [19]).
Apply water selective pulses to align water magnetization along the +z axis to achieve water suppression.
Set the recycle delay between the transients to 300 ms.
Set the number of transients to 64.
Set the spectral widths to 12 ppm and 30 ppm in the 1H and 15N dimensions respectively.
Set 1024 and 1 points in the 1H and 15N dimensions, respectively to optimize the CRINEPT transfer delay time. Increase the CRINEPT transfer time by 0.2–0.3 ms starting from 1 ms. Monitor the backbone amide envelope and tryptophan indole proton peaks to determine the CRINEPT transfer time that produces maximum peak volume, i.e., Topt (Fig. 1c). Set Topt to 1.3 ms for Trx.
Set 1024 and 128 points in the 1H and 15N dimensions, respectively.
Collect the [1H-15N]-CRINEPT-HMQC-[1H]-TROSY spectrum of the sample.
When data collection is completed, recover the sample. Remove a 10 μL aliquot for cell viability assays. Prepare 10−4, 10−5, and 10−6 serial dilutions of the 10 μL cell sample. Plate 100 μL of each dilution onto LB-Kn plates in triplicate. Incubate the plates overnight at 37 °C and count the resulting colonies.
Centrifuge the remaining sample at 4500 × g for 10 min at RT.
Transfer the supernatant to a standard 5 mm NMR tube and acquire a [1H-15N]-HSQC spectrum to test for cell leakage.
Resuspend the cell pellet in 0.3 mL of NMR buffer.
Lyse the remaining cells by freeze-thawing (−80 °C-RT) five times.
Centrifuge the sample at 18,000 × g for 15 min at RT.
Collect a [1H-15N]-CRINEPT-HMQC-[1H]-TROSY spectrum on the clarified lysate using the optimized value for Topt.
3.1.4 Data Processing and Analysis
Process data with Topspin 2.1 and analyze by using CARA software. Alternative offline data processing software may also be used.
Disregard residues with chemical shifts that differ by >0.1 ppm between the lysate and purified protein.
Normalize peak intensities for the in-cell and lysate spectra using an amino proton peak from either a glutamine or an asparagine sidechain that does not change chemical shift between in-cell and lysate spectra.
- Calculate changes in protein peak intensities, ΔI, due to quinary complexation as
where Ilysate and Iin – cell are the normalized, integrated peak intensities from the lysate and in-cell spectra. Plot ΔI versus protein residue (bar graph). Positive values correspond to peak broadening.
Draw a threshold line that distinguishes peaks with extreme broadening. Corresponding residues exceeding the threshold contribute to the quinary interaction surface.
Map broadened residues onto the protein surface based on PDB file(s) using SWISS-PDB Viewer.
3.2 Determining Protein Quinary Interaction Surfaces in Eukaryotes
Overexpressing labeled target protein in eukaryotic cells frequently does not generate an intracellular concentration that is sufficiently high to provide interpretable in-cell spectra. To overcome this problem, purified labeled target protein is introduced into HeLa cells by using electroporation. The protocol is a modification of that presented in Theillet et al. [30]. We will use Ubiquitin (Ubq) as a specific example of the target protein. This protocol requires purified REDPRO-labeled [U-15N]-Ubq [11, 20], and NMR peak assignments for the target protein to complete the analysis.
3.2.1 Preparation of HeLa Cells
Seed ~2 × 106 cells/flask into six 150 cm2 cell culture flasks.
Culture the cells for 3 days in 15 mL of complete medium, until 80% confluence is reached (0.8–1 × 107 cells/flask).
Remove the growth medium by sterile aspiration.
Dilute 10× 0.5% Trypsin/5.3 mM EDTA stock to 1× using balanced salt solution without calcium and magnesium.
To harvest the cells add 3 mL of 0.05% Trypsin/530 μM EDTA and incubate at 37 °C for 10 min.
Add 12 mL of complete medium to neutralize the Trypsin.
Centrifuge the cells at 200 × g for 10 min at RT.
Wash the cells with 15 mL of PBS pre-warmed to 37 °C and re-centrifuge.
Resuspend the cells to 2 × 106 cells/mL using PBS.
Transfer 1 mL of resuspended cells into Eppendorf tubes.
Centrifuge the cells at 200 × g for 10 min at RT, discard the supernatant.
3.2.2 Electroporation of HeLa Cells
Dilute purified REDPRO labeled Ubq to a final concentration of 0.8–1 mM with sterile filtered, freshly prepared electroporation buffer.
Prepare a protein electroporation (EP) sample by adding 100 μL of Ubq to the cell pellet and gently mixing.
Transfer the EP sample to an electroporation cuvette.
Electroporate the cells at RT using an Amaxa™ Nucleofector 2b electroporator (or any other suitable electroporator), pulse program B-28.
Pulse cells two to three times, gently mixing the sample between pulses.
Immediately add 1 mL of complete medium, pre-warmed to 37 °C, to each cuvette, and transfer the contents to pre-warmed 15 cm diameter cell culture dishes. Apply three to four samples to each dish.
Incubate the culture dishes in 5% CO2 incubators at 37 °C for 2 h to recover.
3.2.3 Sample Preparation
Add 15 mL of complete medium to each culture dish and incubate for 2–3 h.
Remove non-adherent cells by sterile aspiration.
Wash each culture dish three times with 15 mL of PBS.
Harvest the cells by treatment with Trypsin/EDTA (see Subheading 3.2.1, steps 4–7).
Wash the cells once with 1 mL of PBS and twice with 1 mL of NMR buffer.
Resuspend the cells in 250 μL of NMR buffer containing 10% D2O.
Transfer the cell suspensions into 5 mm diameter Shigemi tubes for NMR analysis.
3.2.4 NMR Spectroscopy
The same procedure is followed to collect in-cell NMR spectra from eukaryotic cells as is used for prokaryotic cells. The major difference being that due to the instability of transfected HeLa cells, the CRIPT transfer delay is not optimized. All of the experiments are performed at RT using a standard 5 mm NMR tube in a 700 MHz Bruker Avance II NMR spectrometer equipped with TXI z-gradient cryoprobe.
Shim the sample manually using the z, z2, and z3 shims.
Select the [1H-15N]-CRINEPT-HMQC-[1H]-TROSY experiment as described by Riek et al. [19] (Fig. 3b in [19]).
Apply water selective pulses to align water magnetization along the +z axis to achieve water suppression.
Set the recycle delay between the transients to 300 ms.
Set the number of transients to 64.
Set the spectral widths to 12 ppm and 30 ppm in the 1H and 15N dimensions, respectively.
Set Topt to 1.4 ms for Ubq.
Set 1024 and 128 points in the 1H and 15N dimensions, respectively.
Collect the [1H-15N]-CRINEPT-HMQC-[1H]-TROSY spectrum of the sample.
When data collection is completed, recover the sample. Centrifuge the cells at 200 × g for 10 min at RT.
Transfer the supernatant to a standard 5 mm NMR tube and acquire a [1H-15N]-HSQC spectrum to test for cell leakage.
Resuspend the cell pellet in 0.3 mL of NMR buffer.
Lyse the remaining cells by freeze-thawing (−80 °C-RT) five times.
Centrifuge the sample at 18,000 × g for 15 min at RT.
Collect a [1H-15N]-CRINEPT-HMQC-[1H]-TROSY spectrum on the clarified lysate using the optimized value for Topt.
3.2.5 Data Processing and Analysis
Process data with Topspin 2.1 and analyze by using CARA software.
Disregard residues with chemical shifts that differ by >0.1 ppm between the lysate and purified protein.
Normalize peak intensities for the in-cell and lysate spectra using an amino proton peak from either a glutamine or an asparagine sidechain that does not change chemical shift between in-cell and lysate spectra.
- Calculate changes in protein peak intensities, ΔI, due to quinary complexation as
where Ilysate and Iin–cell are the normalized, integrated peak intensities from the lysate and in-cell spectra. Plot ΔI versus protein residue (bar graph). Positive values correspond to peak broadening.
Draw a threshold line that distinguishes peaks with extreme broadening. Corresponding residues exceeding the threshold contribute to the quinary interaction surface.
Map the broadened residues onto the protein surface based on PDB file(s) using SWISS-PDB Viewer (or any other structure viewer).
3.3 Using SVD Analysis to Determine Protein-Protein Structural Interactions
All of the experiments are performed at RT using a standard 5 mm NMR tube in a 700 MHz Bruker Avance II NMR spectrometer equipped with TXI z-gradient cryoprobe. This protocol requires two expression plasmids that contain compatible origins of replication, different antibiotic resistance, and overexpress the target and interactor proteins from two distinct inducible promoters. NMR peak assignments for the target protein are also required to complete the analysis. We will use the prokaryotic Ubiquitin-like protein, Pup, as a specific example of the target protein, and mycobacterial proteasome ATPase, Mpa, from Mycobacterium smegmatis, Msm, as a specific example of the interactor protein.
Pup-GGQ, a precursor to Pup-GGE, was cloned into expression vector pASK3+ to yield pASK-Pup-GGQ [36]. pASK-Pup-GGQ contains an f1 origin, confers ampicillin resistance, codes for Tet repressor, and expresses Pup-GGQ from the tet promoter/operator, which is induced by tetracycline or anhydrotetracycline. Mpa was cloned into expression vector pRSF-1b (Novagen) to yield pRSF-Msm Mpa [36]. pRSF-Msm Mpa contains an RSF origin, confers kanamycin resistance, codes for lac repressor and expresses N-terminal His-tagged Msm Mpa from the T7 promoter/lac operon, which is induced by IPTG. E. coli strain BL21(DE3) codon + was co-transformed with pASK-Pup-GGQ and pRSF-Msm Mpa using standard techniques and the transformed cells were selected on LB-Ap-Kn agar plates.
Pup-GGQ does not engage in extensive quinary interactions, therefore unlike the previous protocols that utilized CRINEPT-HMQC-TROSY, Pup-GGQ is easily visualized in-cell by using 1H{15N}-HSQC.
3.3.1 Target Protein Labeling and Overexpression
Inoculate 50 mL of LB-Ap-Kn medium with a single colony of BL21(DE3) codon + containing pASK-Pup-GGQ and pRSF-Msm Mpa.
Grow the culture overnight at 37 °C with shaking at 250 rpm.
Centrifuge the cells at 4500 × g for 15 min at room temperature.
Wash the cells once with 50 mL of M9 salts.
Resuspend the cells to OD600 ~0.5 in 500 mL of M9-Ap-Kn labeling.
Incubate the culture at 37 °C, 250 rpm for 15 min.
Induce overexpression of Pup-GGQ by adding 2 mg/mL of anhydrotetracycline in dimethylformamide to a final concentration of 0.2 μg/mL.
Allow the induction to proceed for 4 h at 37 °C and 250 rpm.
Centrifuge 100 mL of culture at 4500 × g for 15 min at RT. Reserve the remaining culture for interactor overexpression (see Subheading 3.3.2).
Wash the cells twice with 50 mL of NMR buffer.
Resuspend the final cell pellet in 1 mL of NMR buffer containing 10% glycerol and store the sample at −80 °C for subsequent NMR analyses. This control sample provides the basis spectrum for the target protein in the absence of inter-actor protein.
3.3.2 Interactor Protein Overexpression
Centrifuge the remaining culture at 4500 × g for 15 min at RT.
Wash the cells once with 500 mL of M9 salts.
Resuspend the cells to OD600 ~0.5 in 1000 mL of LB-Ap-Kn.
Incubate the culture at 37 °C, 250 rpm for 15 min.
Induce overexpression of Msm Mpa by adding 0.1% culture volume of 1 M IPTG. Allow the induction to proceed at 37 °C and 250 rpm.
Remove 100 mL of culture at ~4 h, 5 h, 6 h, 7 h, 8 h, and 18 h post-induction.
Centrifuge each sample at 4500 × g for 15 min at RT.
Wash the cells twice with 50 mL of NMR buffer.
Resuspend the final cell pellet in 1 mL of NMR buffer containing 10% glycerol and store each sample at −80 °C for subsequent NMR analyses.
3.3.3 Sample Preparation
Thaw the sample and centrifuge at 4500 × g for 15 min at RT.
Wash the cells once with 10 mL of NMR buffer.
Resuspend the cell pellet in 0.45 mL of NMR buffer and 0.05 mL of D2O. Remove 10 μL for a cell viability assays. Transfer the remaining sample to a standard 5 mm NMR tube.
Prepare 10−4, 10−5, and 10−6 serial dilutions of the 10 μL cell sample. Plate 100 μL of each dilution onto LB-Ap-Kn plates in triplicate. Incubate the plates overnight at 37 °C and count the resulting colonies.
3.3.4 NMR Spectroscopy
Shim the sample manually using the z, z2, and z3 shims.
Select the Watergate version of an 1H{15N}-edited HSQC experiment.
Set the number of transients to 32.
Set the spectral widths to 12 ppm and 30 ppm in the 1H and 15N dimensions respectively.
Set 512 and 64 points in the 1H and 15N dimensions, respectively, apodized with a squared cosine-bell window function and zero-filled to 1024 and 128 points, respectively, prior to Fourier transformation.
Collect the 1H{15N}-edited HSQC spectrum of the sample.
When data collection is completed, recover the sample. Remove a 10 μL aliquot for cell viability assays. Prepare 10−4, 10−5, and 10−6 serial dilutions of the 10 μL cell sample. Plate 100 μL of each dilution onto LB-Ap-Kn plates in triplicate. Incubate the plates overnight at 37 °C and count the resulting colonies.
Centrifuge the remaining sample at 4500 × g for 10 min at RT.
Transfer the supernatant to a standard 5 mm NMR tube and acquire a 1H{15N}-edited HSQC spectrum to test for cell leakage.
Resuspend the cell pellet in 0.3 mL of NMR buffer.
Lyse the remaining cells by freeze-thawing (−80 °C-RT) five times.
Centrifuge the sample at 18,000 × g for 15 min at RT.
Collect a 1H{15N}-edited HSQC spectrum on the clarified lysate using the optimized value for Topt.
3.3.5 Data Processing and Analysis
This section describes how to perform an SVD analysis on a hypothetical file named ChemShiftSVD.txt. MATLAB commands are indicated in bold Calibri font.
Process data with Topspin 2.1 and analyze by using CARA software.
- Calculate changes in protein peak intensities, ΔI, as
where (I/Iref)free is the scaled intensity of individual peaks in the in-cell spectrum of free Pup-GGQ and (I/Iref)bound is the scaled intensity of individual peaks in the in-cell spectrum of the Pup-Msm Mpa complex, and Iref is a glutamine peak at 7.45 ppm and 112.5 ppm in the proton and nitrogen dimensions respectively, that does not shift during titration. Assemble the data matrix in Excel in order of residue numbers (see Note 1).
Save the data as an ASCII text file, ChemShiftSVD.txt.
Start MATLAB (see Note 2) and enter clear all.
Use the file browser to locate the ChemShiftSVD.txt file.
Enter load ChemShiftSVD.txt to load the data for analysis.
Enter M = ChemShiftSVD; to name the matrix.
Enter [U,S,V] = svd(M) to perform SVD analysis (see Note 3).
Enter save (‘S.txt’, ‘S’,’-ascii’) to save the S matrix.
Enter B1 = S(1,1)*U(:,1) to determine the first binding mode (see Note 4).
Enter save (‘B1.txt’, ‘B1’,’-ascii’) to save the B1 matrix.
Enter B2 = S(2,2)*U(:,2) to determine the second binding mode (see Note 5).
Enter save (‘B2.txt’, ‘B2’,’-ascii’) to save the B2 matrix.
Divide the square of the singular value of the first binding mode by the sum of the squares of all of the singular values (the square of the Frobenius norm of S) and multiply by 100 to compute the percentage of change represented in the first binding mode (see Note 6).
Plot the singular values (bar plot) against the binding mode number (dataset index) to generate a Scree plot.
Plot (bar plot) the first and the second binding modes against the residue numbers.
Identify interacting residues by comparing the difference in the magnitude of the first vs the second binding modes. The highest value of the second binding mode is a threshold value that separates residues affected and not affected by binding.
Enter clear all before proceeding to analyze new data.
Map the interacting residues onto the protein surface based on PDB file(s) using SWISS-PDB Viewer or any other suitable structure viewer.
Footnotes
Enter either the chemical shift or the intensity changes for each residue measured at each time point in a single column. Do not enter residue numbers; i.e., a data set containing three chemical shift changes determined at three time points will have three columns of data in the matrix file.
Matlab 2009b or Octave 4.0.1 were used; however, later versions are acceptable.
Three main outputs are “U=”, left singular vectors, “S=”, singular values, and “V=”, right singular vectors. There are the same number of columns in the “S=” output as there are in the original data file. The singular values will appear as nonzero numbers on the diagonal of the matrix. E.g., for ChemShiftSVD, the “S=” output should appear as:
| 4.1582 | 0 | 0 |
| 0 | 0.3947 | 0 |
| 0 | 0 | 0.3023 |
where the first binding mode has a singular value of 4.1582, the second 0.3947, and the third 0.3023.
The larger absolute values represent greater contribution of the residue to the binding mode.
To calculate the values for other binding modes, use the command B# = S(#,#)*U(:,#) where # is the number of the binding mode. Do not exceed the dimensions of the matrix.
Using the hypothetical singular values for ChemShiftSVD, 98.59% of the changes occur in the first binding mode. Usually, true binding modes should represent more than 90% of the square of the Frobenius norm of S.
References
- 1.Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, Chemama Y, Labigne A, Legrain P. The protein-protein interaction map of helicobacter pylori. Nature. 2001;409(6817):211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 2.Sali A, Glaeser R, Earnest T, Baumeister W. From words to literature in structural proteomics. Nature. 2003;422(6928):216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]
- 3.Gerstein M, Lan N, Jansen R. Proteomics. Integrating interactomes. Science. 2002;295(5553):284–287. doi: 10.1126/science.1068664. [DOI] [PubMed] [Google Scholar]
- 4.Serber Z, Keatinge-Clay AT, Ledwidge R, Kelly AE, Miller SM, Dotsch V. High-resolution macromolecular NMR spectroscopy inside living cells. J Am Chem Soc. 2001;123(10):2446–2447. doi: 10.1021/ja0057528. [DOI] [PubMed] [Google Scholar]
- 5.Goodsell D. The machinery of life. Springer; New York: 2009. [Google Scholar]
- 6.Persson E, Halle B. Cell water dynamics on multiple time scales. Proc Natl Acad Sci U S A. 2008;105(17):6266–6271. doi: 10.1073/pnas.0709585105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benedek GB, Villars FMH. Physics: with illustrative examples from medicine and biology:electricity and magnetism. Springer-Verlag; New York: 2000. [Google Scholar]
- 8.Maldonado AY, Burz DS, Shekhtman A. In-cell NMR spectroscopy. Prog Nucl Magn Reson Spectrosc. 2011;59(3):197–212. doi: 10.1016/j.pnmrs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Wang GF, Wang Y, Creager-Allen R, Lutz EA, Scronce H, Slade KM, Ruf RA, Mehl RA, Pielak GJ. Protein (19)F NMR in Escherichia coli. J Am Chem Soc. 2010;132(1):321–327. doi: 10.1021/ja907966n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowley PB, Chow E, Papkovskaia T. Protein interactions in the Escherichia coli cytosol: an impediment to in-cell NMR spectroscopy. Chembiochem. 2011;12(7):1043–1048. doi: 10.1002/cbic.201100063. [DOI] [PubMed] [Google Scholar]
- 11.Majumder S, Xue J, DeMott CM, Reverdatto S, Burz DS, Shekhtman A. Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry. 2015;54(17):2727–2738. doi: 10.1021/acs.biochem.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbieri L, Luchinat E, Banci L. Protein interaction patterns in different cellular environments are revealed by in-cell NMR. Sci Rep. 2015;5:14456. doi: 10.1038/srep14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyne C, Ruhle B, Gautier VW, Crowley PB. Specific ion effects on macromolecular interactions in Escherichia coli extracts. Protein Sci. 2015;24:310–318. doi: 10.1002/pro.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McConkey EH. Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc Natl Acad Sci U S A. 1982;79(10):3236–3240. doi: 10.1073/pnas.79.10.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wirth AJ, Gruebele M. Quinary protein structure and the consequences of crowding in living cells: leaving the test-tube behind. Bioessays. 2013;35(11):984–993. doi: 10.1002/bies.201300080. [DOI] [PubMed] [Google Scholar]
- 16.Sarkar M, Smith AE, Pielak GJ. Impact of reconstituted cytosol on protein stability. Proc Natl Acad Sci U S A. 2013;110(48):19342–19347. doi: 10.1073/pnas.1312678110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR spectroscopy. Academic Press; New York: 2007. [Google Scholar]
- 18.Wider G. NMR techniques used with very large biological macromolecules in solution. Methods Enzymol. 2005;394:382–398. doi: 10.1016/S0076-6879(05)94015-9. [DOI] [PubMed] [Google Scholar]
- 19.Riek R, Wider G, Pervushin K, Wuthrich K. Polarization transfer by cross-correlated relaxation in solution NMR with very large molecules. Proc Natl Acad Sci U S A. 1999;96(9):4918–4923. doi: 10.1073/pnas.96.9.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shekhtman A, Ghose R, Goger M, Cowburn D. NMR structure determination and investigation using a reduced proton (RED-PRO) labeling strategy for proteins. FEBS Lett. 2002;524(1–3):177–182. doi: 10.1016/s0014-5793(02)03051-x. [DOI] [PubMed] [Google Scholar]
- 21.Ferrage F, Dutta K, Shekhtman A, Cowburn D. Structural determination of biomolecular interfaces by nuclear magnetic resonance of proteins with reduced proton density. J Biomol NMR. 2010;47(1):41–54. doi: 10.1007/s10858-010-9409-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serber Z, Dotsch V. In-cell NMR spectroscopy. Biochemistry. 2001;40(48):14317–14323. doi: 10.1021/bi011751w. [DOI] [PubMed] [Google Scholar]
- 23.Banci L, Barbieri L, Bertini I, Luchinat E, Secci E, Zhao Y, Aricescu AR. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat Chem Biol. 2013;9(5):297–299. doi: 10.1038/nchembio.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertrand K, Reverdatto S, Burz DS, Zitomer R, Shekhtman A. Structure of proteins in eukaryotic compartments. J Am Chem Soc. 2012;134(30):12798–12806. doi: 10.1021/ja304809s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamatsu J, O’Donovan D, Tanaka T, Shirai T, Hourai Y, Mikawa T, Ikeya T, Mishima M, Boucher W, Smith BO, Laue ED, Shirakawa M, Ito Y. High-resolution heteronuclear multidimensional NMR of proteins in living insect cells using a baculovirus protein expression system. J Am Chem Soc. 2013;135(5):1688–1691. doi: 10.1021/ja310928u. [DOI] [PubMed] [Google Scholar]
- 26.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, Shirakawa M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458(7234):106–109. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 27.Ogino S, Kubo S, Umemoto R, Huang S, Nishida N, Shimada I. Observation of NMR signals from proteins introduced into living mammalian cells by reversible membrane permeabilization using a pore-forming Toxin, Streptolysin O. J Am Chem Soc. 2009;131(31):10834–10835. doi: 10.1021/ja904407w. [DOI] [PubMed] [Google Scholar]
- 28.Selenko P, Serber Z, Gadea B, Ruderman J, Wagner G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc Natl Acad Sci U S A. 2006;103(32):11904–11909. doi: 10.1073/pnas.0604667103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakai T, Tochio H, Tenno T, Ito Y, Kokubo T, Hiroaki H, Shirakawa M. In-cell NMR spectroscopy of proteins inside Xenopus laevis oocytes. J Biomol NMR. 2006;36(3):179–188. doi: 10.1007/s10858-006-9079-9. [DOI] [PubMed] [Google Scholar]
- 30.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. 2016;530(7588):45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 31.Majumder S, DeMott CM, Reverdatto S, Burz DS, Shekhtman A. Total cellular RNA modulates protein activity. Biochemistry. 2016;55(32):4568–4573. doi: 10.1021/acs.biochem.6b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burz DS, Dutta K, Cowburn D, Shekhtman A. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR) Nat Methods. 2006;3(2):91–93. doi: 10.1038/nmeth851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burz DS, Shekhtman A. The STINT-NMR method for studying in-cell protein-protein interactions. Curr Protoc Protein Sci. 2010:11. doi: 10.1002/0471140864.ps1711s61. Chapter 17: Unit 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burz DS, Dutta K, Cowburn D, Shekhtman A. In-cell NMR for protein-protein interactions (STINT-NMR) Nat Protoc. 2006;1(1):146–152. doi: 10.1038/nprot.2006.23. [DOI] [PubMed] [Google Scholar]
- 35.Golub GH, Van Loan CF. Matrix computations. 4th. The Johns Hopkins University Press; Baltimore, USA: 2012. [Google Scholar]
- 36.Maldonado AY, Burz DS, Reverdatto S, Shekhtman A. Fate of pup inside the mycobacterium proteasome studied by in-cell NMR. PLoS One. 2013;8(9):e74576. doi: 10.1371/journal.pone.0074576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majumder S, DeMott CM, Burz DS, Shekhtman A. Using singular value decomposition to characterize protein-protein interactions by in-cell NMR spectroscopy. Chembiochem. 2014;15(7):929–933. doi: 10.1002/cbic.201400030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;17(11):1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]