

Abstract
Background
Previous studies on the Burkholderia pseudomallei genetic diversity among clinical isolates from melioidosis-endemic areas have identified genetic factors contributing to differential virulence. Although it has been ruled out in Australian and Thai B. pseudomallei populations, it remains unclear whether B. pseudomallei sequence types (STs) correlate with disease in Malaysian patients with melioidosis.
Methods
In this study, multi-locus sequence typing (MLST) was performed on clinical B. pseudomallei isolates collected from Kelantan state of Malaysia, patients’ clinical data were reviewed and then genotype-risk correlations were investigated.
Results
Genotyping of 83 B. pseudomallei isolates revealed 32 different STs, of which 13(40%) were novel. The frequencies of the STs among the 83 isolates ranged from 1 to 12 observations, and ST54, ST371 and ST289 were predominant. All non-novel STs reported in this study have also been identified in other Asian countries. Based on the MLST data analysis, the phylogenetic tree showed clustering of the STs with each other, as well as with the STs from Southeast Asia and China. No evidence for associations between any of B. pseudomallei STs and clinical melioidosis presentation was detected. In addition, the bacterial genotype clusters in relation with each clinical outcome were statistically insignificant, and no risk estimate was reported. This study has expanded the data for B. pseudomallei on MLST database map and provided insights into the molecular epidemiology of melioidosis in Peninsular Malaysia.
Conclusion
This study concurs with previous reports concluding that infecting strain type plays no role in determining disease presentation.
Keywords: Burkholderia pseudomallei, Melioidosis, MLST, Sequence type, Risk
Background
Burkholderia pseudomallei (agent of melioidosis) is acquired by inoculation, inhalation and ingestion routes. It causes wide spectrum clinical presentations; particularly in patients with diabetes mellitus [1]. Marked heterogeneity is observed in the clinical presentation and disease severity among patients. The most severe manifestations of melioidosis are pneumonia and severe sepsis [2]. Melioidosis predominates in Southeast Asia and northern Australia [3, 4]. Regional variations in melioidosis signs and symptoms have been reported and prostatic abscess and encephalomyelitis are common in Australians. Parotid abscesses and hepatosplenic suppuration presentations have been described frequently in Thailand [5–7]. There is good evidence that certain B. pseudomallei genes contribute to different clinical presentations between Asia and Australia; in particular, the bimABm gene, which has been strongly associated with neurological melioidosis [8]. The reason behind this diversity remains unclear, but it may be due to host, bacterial, or environmental factors [2].
The study of molecular epidemiology has provided additional details regarding bacterial diversity and distribution [3]. Commonly applied B. pseudomallei molecular epidemiology procedures include pulsed-field gel electrophoresis (PFGE) [9, 10], random amplification of polymorphic DNA (RAPD) [11], ribotyping [12] and whole genome sequencing [13]. Multi-locus sequence typing (MLST) is another molecular approach that simplifies the exchange of local and global inter-laboratory genotyping data [14]. The discriminating ability of MLST between different B. pseudomallei genotypes was evaluated previously by comparison with PFGE and similar results were reported [2].
Typing of B. pseudomallei using MLST scheme is useful to explore sequence types (STs) in particular populations [15], predict the distribution of bacterial STs in a given geographical area [16], track the source of melioidosis outbreaks [17] and define whether recurrent melioidosis is due to a relapse of the same bacterial ST or reinfection with a different ST [18].
The B. pseudomallei STs must be studied in Peninsular Malaysia to understand the population genetics in this region and to determine the distribution and frequency of genotype associations in melioidosis cases. MLST was applied for this purpose. According to a literature database search, no national or local project has applied MLST to B. pseudomallei isolates collected from Peninsular Malaysia. However, some genotyping studies have used pooled isolates from different regions of Southeast Asia, including Malaysia [19]. Thus, this is the first study to compare STs of clinical isolates from Peninsular Malaysia and to determine whether particular STs are associated with particular clinical outcomes.
Methods
B. pseudomallei isolates source
Clinical samples were collected, bacteria were isolated and B. pseudomallei was identified and archived as part of routine diagnostics in accordance to the standard protocol at the Medical Microbiology & Parasitology Laboratory at the Hospital Universiti Sains Malaysia (HUSM). Only a single clinical isolate from each patient was obtained to preserve the assumption of independence of observations and to avoid repetition.
Multi-locus sequence typing
MLST was performed as described previously by Godoy et al. [19]. New allelic profiles were confirmed by a repeated MLST procedure. Novel STs were assigned new allelic profile numbers and were submitted, with the isolate information to the Burkholderia MLST database (http://pubmlst.org/bpseudomallei/). The submission process was completed from November 2012 to April 2015.
Phylogenetic analysis
Basic statistical quantities such as number of alleles, number of variable sites per allele, number and frequency of single nucleotides polymorphism (SNPs) in each locus and the nucleotide sequence diversity rate were calculated and displayed using functional options in molecular evolutionary genetics analysis version-6 (MEGA 6) software [20]. Relatedness among isolates was estimated based on two principles: differences in allelic profiles using eBURST v7 [21, 22]. and differences in the concatenated sequence of alleles at all loci using MEGA 6 software.
All STs were uploaded into eBURST v7 software to display the relatedness among the isolates obtained in this study, as well as among B. pseudomallei of the historical collection from different regions in Malaysia. Three population snapshot diagrams were generated: the first diagram displayed the relatedness of the novel and existing STs reported in this study. The second and third diagrams were made for STs of the MLST database for Malaysia before and after the addition of STs obtained from this study to display the significant changes on the full-size Malaysian MLST database population snapshot.
Sequences of every allelic profile were joined in the order of loci used to define the allelic profile to achieve a concatenated sequence of 3399 bp. The topology and grouping of all STs retrieved from this study were displayed on the constructed bootstrapped phylogenetic trees using Unweighted Pair Group Method with Arithmetic average (UPGMA) method in MEG 6 software. STs obtained from this study were analyzed with selected 88 STs representing Malaysia and regional endemic countries including India, China, Singapore, Indonesia, Laos, Vietnam, Philippines, Bangladesh and Thailand.
Genotype-disease associations
Patient records were reviewed for specific clinical manifestations and disease outcomes, including types of melioidosis (bacteremic, nonbacteremic, disseminated or localized), organs involved (lungs, liver, spleen, bone, soft tissues, brain and genitourinary) and death. All clinical definitions and classifications were categorized as mentioned by Zueter et al. [23]. Strain tropism and virulence were studied by displaying clinical outcomes throughout the phylogenetic tree topology prepared from the STs. On the other hand, all closely-related STs were gathered into groups and analyzed as independent variables (predictors) against clinical outcomes that were identified as dependent variables. Statistical analyses were performed to analyze each genotype cluster with every clinical outcome using Pearson’s chi-square or Fisher exact tests.
Ethics statement
Ethical approval was obtained from the Universiti Sains Malaysia Research Ethics Committee (Human) (USM/JEPeM/15110495) and data were analyzed anonymously.
Results
Of the 83 clinical B. pseudomallei isolates obtained in this study, 32 STs were identified. The frequencies of STs among the 83 isolates were 1–12 observations with a predominance of ST54 (n = 12), ST371 (n = 7) and ST289 (n = 7).
Among the obtained STs, the number of alleles per locus varied from 3 to 6. SNPs were observed at all seven loci, with the number of SNPs ranging from 2 to 21, while the number of polymorphic (variable) sites within the different alleles at the seven loci varied between 2 and 15. The levels of locus sequence diversity among all 32 STs were 2.5 to 5.3% (Table 1). All STs identified in this study were deposited in the MLST database with complete reference annotation (Table 2).
Table 1.
Properties of the MLST loci in the clinical B. pseudomallei isolates from Peninsular Malaysia
| Locus | No. of nucleotides analyzed | No. of alleles | No. of SNP | SNP Frequencya | No. of variable sites | Sequence diversity rateb |
|---|---|---|---|---|---|---|
| Ace | 519 | 4 | 3 | 0.6% | 3 | 4.1% |
| gltB | 522 | 5 | 8 | 1.5% | 3 | 3.1% |
| gmhD | 468 | 5 | 12 | 2.5% | 5 | 4.0% |
| lepA | 486 | 6 | 21 | 4.3% | 15 | 5.3% |
| lipA | 402 | 5 | 7 | 1.7% | 4 | 2.9% |
| narK | 561 | 4 | 9 | 1.6% | 5 | 3.3% |
| Ndh | 443 | 3 | 2 | 0.5% | 2 | 2.5% |
aRate of SNPs diversity in relation with locus length (no. of SNP/locus length)
bRate of allele diversity in relation with the number of total referenced database alleles
Table 2.
Properties of B. pseudomallei sequence types in this study
| Isolate code | Origin (specimen) | Sequence type | ||
|---|---|---|---|---|
| Strain name | MLST database ID | |||
| 2 | Blood | 54 | USM2 | 3668 |
| 3 | Blood | 54 | USM3 | 3669 |
| 7 | Pus | 54 | USM7 | 3670 |
| 15 | Body fluid | 54 | USM15 | 3671 |
| 69 | Blood | 54 | USM69 | 4066 |
| 47 | Pus | 54 | USM47 | 3672 |
| 48 | Blood | 54 | USM48 | 3673 |
| 50 | Blood | 54 | USM50 | 3674 |
| 43 | Urine | 54 | USM43 | 3675 |
| 22 | Body fluid | 54 | USM22 | 3676 |
| 27 | Body fluid | 54 | USM27 | 3677 |
| 40 | Blood | 54 | USM40 | 3678 |
| 8 | Blood | 371 | USM8 | 3679 |
| 12 | Blood | 371 | USM12 | 3718 |
| 14 | Blood | 371 | USM14 | 3680 |
| 24 | Blood | 371 | USM24 | 3681 |
| 33 | Blood | 371 | USM33 | 3682 |
| 35 | Blood | 371 | USM35 | 3683 |
| 71 | Blood | 371 | USM71 | 4018 |
| 6 | Pus | 46 | USM6 | 3684 |
| 45 | Sputum | 46 | USM45 | 3685 |
| 20 | Blood | 46 | USM20 | 3686 |
| 57 | Blood | 46 | USM57 | 3687 |
| 32 | Blood | 46 | USM32 | 3688 |
| 61 | Pus | 46 | USM61 | 3689 |
| 39 | Blood | 84 | USM39 | 3690 |
| 9 | Pus | 84 | USM9 | 3691 |
| 28 | Body fluid | 84 | USM28 | 3692 |
| 64 | Blood | 84 | USM64 | 3693 |
| 42 | Blood | 289 | USM42 | 3694 |
| 44 | Blood | 289 | USM44 | 3695 |
| 49 | Blood | 289 | USM49 | 3696 |
| 13 | Blood | 289 | USM13 | 3697 |
| 5 | Blood | 289 | USM5 | 3698 |
| 66 | Pus | 289 | USM66 | 4016 |
| 63 | Blood | 289 | USM63 | 3699 |
| 29 | Blood | 271 | AMON29 | 3714 |
| 74 | Blood | 271 | USM74 | 4025 |
| 78 | Blood | 271 | USM78 | 4026 |
| 79 | Blood | 271 | USM79 | 4027 |
| 36 | Blood | 306 | USM36 | 3700 |
| 53 | Pus | 306 | USM306 | 3701 |
| 58 | Blood | 306 | USM58 | 3702 |
| 37 | Pus | 306 | USM37 | 3703 |
| 10 | Blood | 55 | USM10 | 3708 |
| 23 | Blood | 55 | USM23 | 3709 |
| 18 | Pus | 50 | USM18 | 3704 |
| 51 | Sputum | 50 | USM51 | 3705 |
| 54 | Blood | 50 | USM54 | 3706 |
| 41 | Blood | 50 | USM41 | 3707 |
| 38 | Blood | 376 | USM38 | 3710 |
| 17 | Pus | 376 | USM17 | 3711 |
| 31 | Pus | 507 | HANA31 | 3713 |
| 46 | Blood | 51 | ZED46 | 3712 |
| 60 | Body fluid | 10 | USM60 | 4015 |
| 67 | Blood | 164 | USM67 | 4022 |
| 73 | Blood | 164 | USM73 | 4023 |
| 80 | Blood | 164 | USM80 | 4024 |
| 68 | Blood | 369 | USM68 | 4017 |
| 72 | Blood | 402 | USM72 | 4019 |
| 82 | Blood | 368 | USM82 | 4021 |
| 75 | Blood | 47 | USM75 | 4028 |
| 77 | Blood | 47 | USM77 | 4029 |
| 81 | Blood | 47 | USM81 | 4030 |
| 83 | Pus | 47 | USM83 | 4031 |
| 76 | Blood | 168 | USM76 | 4020 |
| 11 | Blood | 1319 | 11 | 3659 |
| 65 | Blood | 1319 | USM65 | 4067 |
| 1 | Blood | 1317 | 1 | 3657 |
| 4 | Blood | 1318 | 4 | 3658 |
| 19 | Blood | 1320 | 19 | 3660 |
| 21 | Blood | 1321 | 21 | 3661 |
| 25 | Body fluid | 1322 | 25 | 3662 |
| 26 | Body fluid | 1322 | AMAR26 | 3715 |
| 30 | Pus | 1323 | 30 | 3663 |
| 16 | Blood | 1323 | USM16 | 4014 |
| 34 | Blood | 1324 | 34 | 3664 |
| 52 | Body fluid | 1325 | 52 | 3665 |
| 55 | Blood | 1326 | 55 | 3666 |
| 56 | Blood | 1326 | HAMZ56 | 3716 |
| 59 | Blood | 1327 | 59 | 3667 |
| 62 | Pus | 1358 | ABD12 | 4032 |
| 70 | Body fluid | 1359 | NOR13 | 4033 |
Genetic relatedness among studied B. Pseudomallei sequence types
Half of the STs were clustered into a single group of 16 STs, of which four were novel (Fig. 1). The STs were presented in 44 isolates clustered into a major group and emerged from ST271 representing the predicted founder. An additional three subgroup founders branched from ST271 were also identified including ST50, ST369 and ST1317. ST84 was the predicted as ancestor to another smaller population group consisting of six STs, and most were novel. The remaining STs were singletons.
Fig. 1.
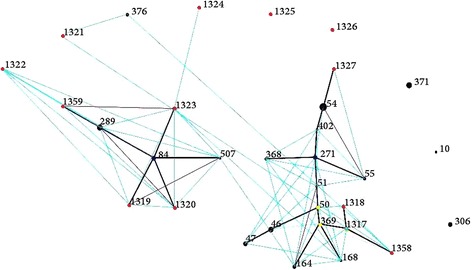
eBURST diagram representing the relatedness between 32 STs identified in 83 isolates. Black dot: existing ST. Red dot: novel ST. Blue dot: predicted group ancestor. Yellow dot: predicted subgroup ancestor. Green dot: novel and subgroup ancestor ST. Black and purple lines: single locus variants (SLVs). Blue line: double locus variant (DLV). Re-samplings for bootstrapping = 10,000; minimum number of identical loci for group definition =6; minimum number of SLV for subgroup definition =3. The size of the dot reflects the individual ST frequency among the 83 strains
Genetic relatedness among B. Pseudomallei sequence types in Malaysia
Thirteen STs identified in this study were novel, including ST1317, ST1318, ST1319, ST1320, ST1321, ST1322, ST1323, ST1324, ST1325, ST1326, ST1327, ST1358 and ST1359. On the other hand, the other STs (n = 19) reported in this study were also characterized elsewhere in the Indian subcontinent, China and Southeast Asia.
Total of 264 B. pseudomallei isolates and 59 STs were already registered in the database (MLST.net) until April 2015, all of which were from Malaysia. The present study uploaded additional 83 B. pseudomallei isolates and 32 STs from the same country. Before the present study, almost half of Malaysian STs were clustered into a single group with ST50 as the predicted founder. The remaining STs were singletons. No sub-groups were reported (Fig. 2). The present study has expanded the former Malaysian clonal cluster by adding more branching STs. In addition, new clonal expansion has emerged from ST84 to create another group in the Malaysian database (Fig. 3). This expansion was characterized by conversion of ST84 from an existing ST into a new ancestral group founder from which other single and double locus variant STs have emerged. In addition, another sub-clonal expansion was created from ST51, ST271, ST46, ST369 and ST1317.
Fig. 2.
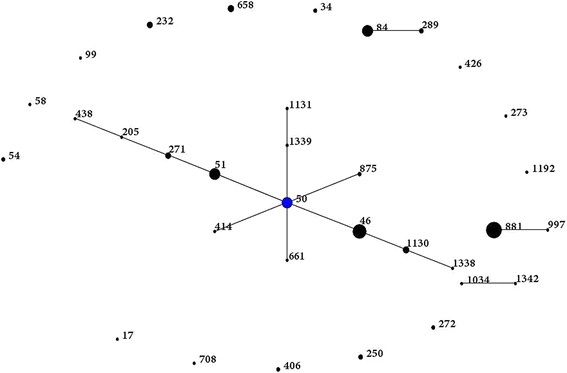
eBURST population snapshot for B. pseudomallei STs in Malaysia before conducting the present study. Blue dot refers to group founder. Each black dot represents single genotype. The size of the dot represents the ST frequency
Fig. 3.
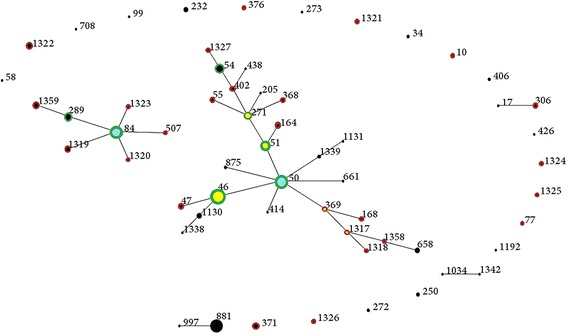
Overall B. pseudomallei STs in Malaysia showing STs added by this study. Black dot: ST only in Malaysian database query. Red hollow: ST only in this study. Green hollow: ST in both Malaysian query and present study. Yellow dot: subgroup founder. Blue dot: Group founder. Re-samplings for bootstrapping = 10,000; minimum number of identical loci for group definition =6; minimum number of SLV for subgroup definition =3. The size of the dot reflects the individual ST frequency among the 83 strains
Phylogenetic relationship among regional B. Pseudomallei sequence types
The majority of the STs formed unique sequences that differed by at least a single nucleotide and almost all were seen in all groups in the phylogenetic tree (Fig. 4). More than half of the group 1 STs were clustered with each other, as well as with STs from Malaysia, Thailand, Singapore, Cambodia, Vietnam, Laos and China. On the other hand, ST50 and the novel ST1327 were not grouped with any of our STs but were clustered with local STs and with narrower regional STs located in groups 2 and 8, respectively. The remaining STs were distributed among other groups with little distance between them. The STs in the lower sub-cluster of group 4 and in group 5 were clustered with STs that have been reported from Sarawak in West Malaysia. The majority of the novel STs were clustered with each other in any given group. Of the 13 novel STs, eight were located in group one. The only unique ST in this study was ST1326, which was novel and a singleton.
Fig. 4.
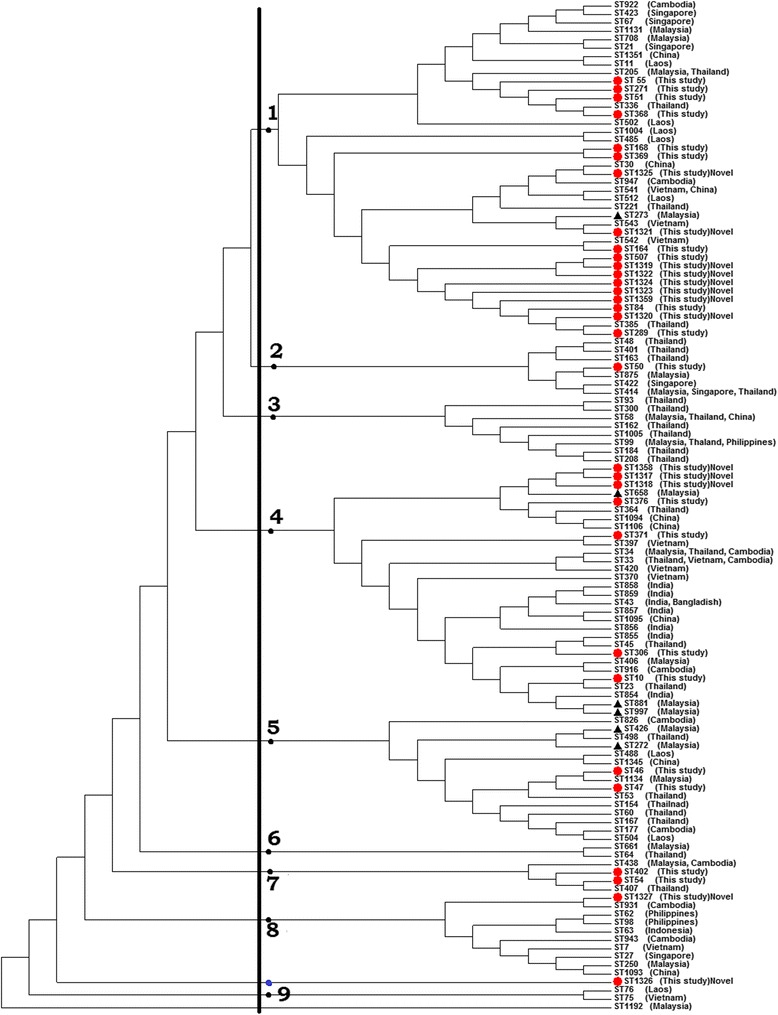
The evolutionary history inferred using the UPGMA method to analyze the studied 32 STs along with 88 historical STs represented India, China and Southeast Asian countries.▲: Sarawak ST
B. pseudomallei genotype - disease associations
The clinical histories of 70 subjects in whom bacterial genotypes were identified and archived were reviewed from 2007 to 2014. No evidence supporting an association between B. pseudomallei STs and any clinical presentation of melioidosis was observed on the phylogenetic tree; no clustering was noted for a given clinical outcome with a particular genotype (Fig. 5).
Fig. 5.
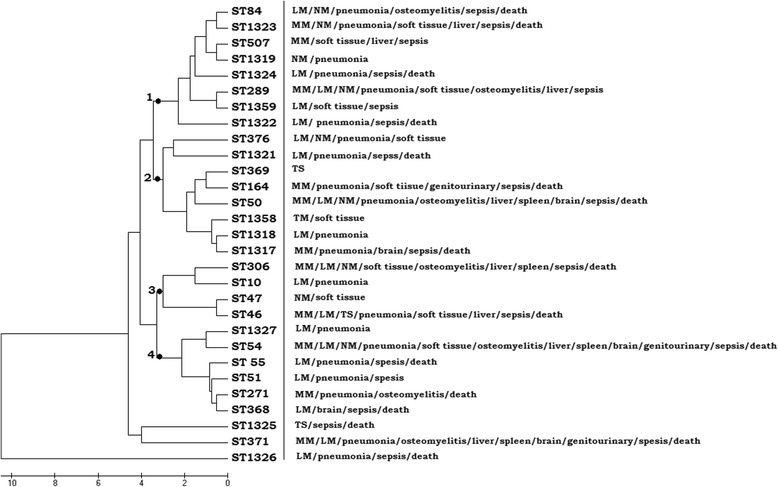
Topology of clinical outcomes on phylogenetic tree. MM: multifocal melioidosis, LM: localized melioidosis, NM: nonbacteremic melioidosis, TS: transient septicemia. 1: first genotype cluster; 2: second genotype cluster; 3: third genotype cluster; 4: fourth genotype cluster
In addition, no evidence of differential virulence or strain tropism was detected. For example, severe sepsis (n = 11) was caused by strains of seven different STs, whereas septic shock (n = 29) and abscess (n = 30) were caused by strains of 17 and 18 different STs, respectively.
The two-way tables for all bacterial genotype clusters in relation to clinical outcome variables were statistically non-significant (p > 0.05), with no reported risk estimate for any genotype cluster developing any of the clinical outcome (data not shown).
Discussion
Burkholderia pseudomallei is Gram negative saprophytic bacterium classified as Tier 1 Biological Select Agent [24]. Due to frequent recombination, the B. pseudomallei genome showed high plasticity that increases genetic divergence, and therefore strain-to-strain variation [25]. The spectrum of B. pseudomallei genetic diversity in Peninsular Malaysia and its association with clinical outcomes is not yet known. It is therefore important to determine ST genotypes to compare the molecular epidemiology of B. pseudomallei in Peninsular Malaysia with strains obtained from other regions, especially other countries within Asia, and to investigate genotype diversity as a possible explanation for differences in disease presentation, treatment response, prognosis and mortality [26].
In the present study, STs were identified with different frequencies, predominance, novelty, and allelic heterogeneity. The overall diversity of isolates found in the clinical specimens was 0.38 STs/isolate, compared with a diversity ratio of 0.65 STs/isolate reported in Australia.26 Several molecular studies that applied various genotyping methods to clinical B. pseudomallei isolates reported genotypic novelty and diversity with or without predominance of particular genotypes among single population communities of temperate endemic areas of Malaysia [10, 15, 27–29], Thailand [9, 30], India [31], and Australia [2, 32].
The presence of different genotypes with various frequencies reflects the historical introduction and dissemination of different B. pseudomallei genotypes into the study area or due to expansion of local STs that yielded new strains with novel STs [33]. Genotypic predominance might be attributed to localization of a particular genotype in the study area in which the contaminated environment became a rich source for infection by that genotype [34]. For example, the predominant STs found in this study were ST54, ST371, ST46 and ST84, which have been found in Malaysia and neighbouring countries. Moreover, some genotypes identified in this study such as ST402, ST55, ST271, ST376, ST47, and ST376, have been identified in soil and water sources in Malaysia and other neighboring countries [19, 33].
This genotypic picture for our clinical isolates might be linked to the endemic geographical distribution of B. pseudomallei in the environments our patients resided. This suggestion is supported by reports of melioidosis outbreaks caused by B. pseudomallei of the same genotypes as those of the suspected environmental sources [16, 35–38].
The presence of novel genotypes indicates local persistence of B. pseudomallei in the same geographical area and their ability to establish a new clone series producing novel offspring’s that carry new genotypes [39]. Several reports have documented the emergence of novel B. pseudomallei genotypes regardless of the number of the genotyped isolates [15, 31]. In this study, two of 10 strains isolated from patients residing in Bachok were novel genotypes, whereas 3/15 (20%), 1/10 (10%), 2/8 (25%) and 2/4 (50%) strains carried novel genotypes in Terengganu, Selangor, Pasir Puteh and Machang, respectively.
The characteristics of the alleles and loci were considerably diverse among the 32 STs. However, no new alleles have been reported. Previous studies suggested a high rate of recombination replacement relative to substitution mutations in B. pseudomallei that caused re-assortment of existing alleles, rather than emergence of new alleles, leading to a new generation of STs [32, 39].
The changes occurring in ST84 (as seen in the eBURST snapshots) before and after this study suggest the occurrence of clonal expansion of ST84. This conclusion was reached based on the presence of seven novel STs arising from ST84 and would be supported by confirming the evolutionary convergence of ST84 from a singleton ST to the group founder ST. In the same way, other sub-clonal expansions were created from ST51, ST271, ST46, ST369, and ST1317. Thus, the present study has markedly expanded the former Malaysian clonal cluster by adding more branching STs.
McCombie et al. [33] had studied the molecular epidemiology of B. pseudomallei using MLST of 207 historical isolates collected in Malaysia, Thailand and Vietnam. MLST revealed 80 STs and 56 were novel. When those STs were added to the B. pseudomallei MLST database and analyzed together, the historical-collection STs clustered significantly within the complex of the eBURST diagram in an ancestral pattern and expanded the B. pseudomallei population snapshot. In the same study, ST84 was likely a B. pseudomallei isolate characteristic of Southeast Asia rather than Australia based on abundance in several environmental isolates from Thailand and Malaysia.
Clustering of our STs in the phylogenetic tree with STs from Sarawak, Thailand, Singapore, Cambodia, Vietnam, Laos and China suggests their genetic relatedness with ST ancestors of these regions. In addition, all non-novel STs identified in this study were also identified in these countries at different frequencies, which suggesting that the Malaysian isolates may not be distinct from those of Southeast Asia. ST371, ST164, ST47, ST306, ST55, ST376, ST402, ST507, ST368, ST369, ST10 and ST168 were first identified in Malaysia. Nevertheless, these STs are not found exclusively in Malaysia only but also in other Southeast Asian countries. This topology explores the geographical expansion and spread of those STs among regional countries through environmental and human routes [32]. Such expansion was restricted to countries bordering with Malaysia but not other regions, such as Australia, Africa, or Latin America, due to the absence of shared STs with those regions, which concurs with previous findings of no shared STs among different continents. However, a few exceptions have been more recently reported; in one study, ST105 and ST849 were shared STs between Australia and Cambodia and both STs were isolated from patients from both countries [40]. Another study reported the isolation of ST562 from Australia and China [41].
Clinical outcome-genotyping association in human cases has not been clearly described in Malaysia and interpretative studies on the significance of genotyping results remain limited. In this study, tests to cluster clinical presentation on the phylogenetic tree, differential virulence tropism for an individual ST, and statistical associations between genotype clusters with clinical presentations did not detect any relationship between genotype and disease. Two Australian studies genotyped clinical isolates of B. pseudomallei using PFGE and MLST. The clinical history of each patient was reviewed and analyzed statistically in combination with the resulting genotypes. However, neither study found an association due to the high diversities of the genotypes and clinical presentations and low relative frequencies of each of them. In addition, no association was reported between a given genotype and a particular clinical presentation or site of infection [2, 26]. On the other hand, a study from Thailand reported partial and possible associations between B. pseudomallei ribotypes and clinical outcomes of melioidosis. However, that study was not conclusive due to low number of tested cases [11]. Our study concurs with the previous studies demonstrating a lack of an association between any ST and disease, but considers that host and environmental factors are reasons for the heterogenous nature of the clinical presentation of the disease.
Conclusion
The present study revealed the high diversity of B. pseudomallei in Malaysia, and several STs were discovered. Many of the non-novel STs found in this study were also reported from neighboring Asian countries. None of the STs were associated a specific disease presentation. Therefore, host and environmental factors play crucial roles in the diversity of clinical presentation and outcomes of the disease. Further studies on environmental samples (and a comparison with clinical isolates) may provide more extensive, representative data to elucidate the course and evolution of the B. pseudomallei population in this region. Expanding the clinical case review would provide more data for further understanding of specific genotype-disease association in melioidosis.
Acknowledgements
We thank Azlan Abdullah and Nurleem Mursheed from the Microbiology Laboratory USM for their help procuring the isolates and help with technical issues.
Funding
This project was funded by Malaysian Ministry of Education Exploratory Research Grant Scheme (ERGS) grant, no. 203/PPSP/6730024 awarded to Azian Harun. The funding body has a role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- MEGA
Molecular evolutionary genetics analysis
- MLST
Multi-locus sequence typing
- PCR
Polymerase chain reaction
- PFGE
Pulsed-field gel electrophoresis
- RAPD
Random amplification of polymorphic DNA
- SNP
Single nucleotides polymorphism
- ST
Sequence type
- UPGMA
Unweighted Pair Group Method with Arithmetic average
Authors’ contributions
AZ: did data collection, analyzed and drafted the article; ZAR: proofread and assisted data analysis, MAM: assisted in supervision of the clinical part and writing, AH: supervised and got the fund for whole project and assisted data collection, analysis and proofreading. All authors have read and approved the manuscript.
Ethics approval and consent to participate
Ethical approval was obtained from the Universiti Sains Malaysia Research Ethics Committee (Human) (USM/JEPeM/15110495) and data were analyzed anonymously. No consent, written or verbal, was not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Abdel Rahman Zueter, Phone: +962 798190685, Email: zeuterabdelrahman@gmail.com.
Zaidah Abdul Rahman, Email: drzaidah@usm.my.
Mahmoud Abumarzouq, Email: mahmoudabumarzouq@hotmail.com.
Azian Harun, Email: azian@usm.my.
References
- 1.Currie BJ. Melioidosis: evolving concepts in epidemiology, pathogenesis, and treatment. Semin Respir Crit Care Med. 2015;36(6):111–125. doi: 10.1055/s-0034-1398389. [DOI] [PubMed] [Google Scholar]
- 2.Cheng AC, Godoy D, Mayo M, Gal D, Spratt BG, Currie BJ. Isolates of Burkholderia pseudomallei from northern Australia are distinct by multilocus sequence typing, but strain types do not correlate with clinical presentation. J Clin Microbiol. 2004;42(12):5477–5483. doi: 10.1128/JCM.42.12.5477-5483.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Currie BJ, Dance DA, Cheng AC. The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans R Soc Trop Med Hyg. 2008;102(1,1);1–4. [DOI] [PubMed]
- 4.Puthucheary SD. Melioidosis in Malaysia. Med J Malaysia. 2009;64(4):266–274. [PubMed] [Google Scholar]
- 5.Currie BJ, Fisher DA, Howard DM, Burrow JN, Lo D, Selva-Nayagam S, Anstey NM, Huffam SE, Snelling PL, Marks PJ, Stephens DP, Lum GD, Jacups SP, Krause VL. Endemic melioidosis in tropical northern Australia: a 10-year prospective study and review of the literature. Clin Infect Dis. 2000;31(4,1): 981–986. [DOI] [PubMed]
- 6.White NJ. Melioidosis. Zentralbl Bakteriol. 1994;280:439–443. doi: 10.1016/S0934-8840(11)80502-5. [DOI] [PubMed] [Google Scholar]
- 7.Dance DAB. Melioidosis. In: Guerrant RL, Walker, D. H., Weller, P. F., editor. Tropical Infectious Diseases: principles, pathogens, & practice. 2 ed. Philadelphia: Elsevier/Churchill Livingston. 2004. p. 381–388.
- 8.Sarovich DS, Price EP, Webb JR, Ward LM, Voutsinos MY, Tuanyok A, Mayo M, Kaestli M, Currie BJ. Variable Virulence Factors in Burkholderia pseudomallei (Melioidosis) Associated with Human Disease. PLoS ONE. 2014;9(3):e91682. doi: 10.1371/journal.pone.0091682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koonpaew S, Ubol MN, Sirisinha S, White NJ, Chaiyaroj SC. Genome fingerprinting by pulsed-field gel electrophoresis of isolates of Burkholderia pseudomallei from patients with melioidosis in Thailand. Acta Trop. 2000;74(2):187–191. doi: 10.1016/S0001-706X(99)00069-8. [DOI] [PubMed] [Google Scholar]
- 10.Azura MN, Norazah A, Kamel AG, Zorin SADNA. Fingerprinting of septicemic and localized Burkholderia pseudomallei isolates from Malaysian patients. Southeast Asian J Trop Med Public Health. 2011;42(1):114–121. [PubMed] [Google Scholar]
- 11.Norton R, Roberts B, Freeman M, Wilson M, Ashhurst-Smith C, Lock W, Brookes D, La Brooy J. Characterisation and molecular typing of Burkholderia pseudomallei: are disease presentations of melioidosis clonally related? FEMS Immunol Med Microbiol. 1998;20(1):37–44. doi: 10.1016/S0928-8244(97)00104-1. [DOI] [PubMed] [Google Scholar]
- 12.Pitt TL, Trakulsomboon S, Dance DA. Molecular phylogeny of Burkholderia pseudomallei. Acta Trop. 2000;74(2):181–185. doi: 10.1016/S0001-706X(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 13.Price EP, Sarovich DS, Viberg L, Mayo M, Kaestli M, Tuanyok A, Foster JT, Keim P, Pearson T, Currie BJ. Whole-genome sequencing of Burkholderia pseudomallei isolates from an unusual melioidosis case identifies a polyclonal infection with the same multilocus sequence type. J Clin Microbiol. 2015;53(1):282–286. doi: 10.1128/JCM.02560-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95(6):3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zueter AM, Rahman ZA, Yean CY, Harun A. Brief communication genotyping of Burkholderia pseudomallei revealed high genetic variability among isolates from a single population group. Int J Mol Epidemiol Genet. 2015;6(1):41–47. [PMC free article] [PubMed] [Google Scholar]
- 16.Wuthiekanun V, Limmathurotsakul D, Chantratita N, Feil EJ, Day NP, Peacock SJ. Burkholderia Pseudomallei is genetically diverse in agricultural land in Northeast Thailand. PLoS Negl Trop Dis. 2009;3:e496. doi: 10.1371/journal.pntd.0000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inglis TJ, Garrow SC, Henderson M, Clair A, Sampson J, O'Reilly L, Cameron B. Burkholderia pseudomallei traced to water treatment plant in Australia. Emerg Infect Dis. 2000;6(1):56–59. doi: 10.3201/eid0601.000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maharjan B, Chantratita N, Vesaratchavest M, Cheng A, Wuthiekanun V, Chierakul W, Chaowagul W, Day NP, Peacock SJ. Recurrent melioidosis in patients in northeast Thailand is frequently due to reinfection rather than relapse. J Clin Microbiol. 2005;43(12):6032–6034. doi: 10.1128/JCM.43.12.6032-6034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godoy D, Randle G, Simpson AJ, Aanensen DM, Pitt TL, Kinoshita R, Spratt BG. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbiol. 2003;41(5):2068–2079. doi: 10.1128/JCM.41.5.2068-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013;30(12): 2725–2729. [DOI] [PMC free article] [PubMed]
- 21.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186(5):1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spratt BG, Hanage WP, Li B, Aanensen DM, Feil EJ. Displaying the relatedness among isolates of bacterial species-the eBURST approach. FEMS Microbiol Lett. 2004;241(2):129–134. doi: 10.1016/j.femsle.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 23.Zueter A, Yean YC, Abumarzouq M, Abdul Rahman Z, Deris ZZ, Harun A. The epidemiology and clinical spectrum of melioidosis in a teaching hospital in a north-eastern state of Malaysia: a fifteen-year review. BMC Infect Dis. 2016;16:333. doi: 10.1186/s12879-016-1583-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler D. Viral research faces clampdown. Nature. 2012;490:456. doi: 10.1038/490456a. [DOI] [PubMed] [Google Scholar]
- 25.Holden MT, Titball RW, Peacock SJ, Cerdeno-Tarraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, Sebaihia M, Thomson NR, Bason N, Beacham IR, Brooks K, Brown KA, Brown NF, Challis GL, Cherevach I, Chillingworth T, Cronin A, Crossett B, Davis P, DeShazer D, Feltwell T, Fraser A, Hance Z, Hauser H, Holroyd S, Jagels K, Keith KE, Maddison M, Moule S, Price C, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Simmonds M, Songsivilai S, Stevens K, Tumapa S, Vesaratchavest M, Whitehead S, Yeats C, Barrell BG, Oyston PC, Parkhill J. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci USA. 2004;101(39): 14240–14245. [DOI] [PMC free article] [PubMed]
- 26.Cheng AC, Day NP, Mayo MJ, Gal D, Currie BJ. Burkholderia pseudomallei strain type, based on pulsed-field gel electrophoresis, does not determine disease presentation in melioidosis. Microbes Infect. 2005;7(1):104–109. doi: 10.1016/j.micinf.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 27.Vadivelu J, Puthucheary SD, Mifsud A, Drasar BS, Dance DA, Pitt TI. Ribotyping and DNA macrorestriction analysis of isolates of Burkholderia pseudomallei from cases of melioidosis in Malaysia. Trans R Soc Trop Med Hyg. 1997;91(3):358–360. doi: 10.1016/S0035-9203(97)90107-3. [DOI] [PubMed] [Google Scholar]
- 28.Radua S, Ling OW, Srimontree S, Lulitanond A, Hin WF, Yuherman LS, Rusul G, Mutalib AR. Characterization of Burkholderia pseudomallei isolated in Thailand and Malaysia. Diagn Microbiol Infect Dis. 2000;38(3):141–145. doi: 10.1016/S0732-8893(00)00189-9. [DOI] [PubMed] [Google Scholar]
- 29.Chua KH, See KH, Thong KL, Puthucheary SDDNA. Fingerprinting of human isolates of Burkholderia pseudomallei from different geographical regions of Malaysia. Trop Biomed. 2010;27(3):517–524. [PubMed] [Google Scholar]
- 30.Leelayuwat C, Romphruk A, Lulitanond A, Trakulsomboon S, Thamlikitkul V. Genotype analysis of Burkholderia pseudomallei using randomly amplified polymorphic DNA (RAPD): indicative of genetic differences amongst environmental and clinical isolates. Acta Trop. 2000;77(2):229–237. doi: 10.1016/S0001-706X(00)00137-6. [DOI] [PubMed] [Google Scholar]
- 31.Mukhopadhyay C, Kaestli M, Vandana KE, Sushma K, Mayo M, Richardson L, Tuanyok A, Keim P, Godoy D, Spratt BG, Currie BJ. Molecular characterization of clinical Burkholderia pseudomallei isolates from India. Am J Trop Med Hyg. 2011;85(1):121–123. doi: 10.4269/ajtmh.2011.11-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng AC, Ward L, Godoy D, Norton R, Mayo M, Gal D, Spratt BG, Currie BJ. Genetic diversity of Burkholderia pseudomallei isolates in Australia. J Clin Microbiol. 2008;46(1):249–254. doi: 10.1128/JCM.01725-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCombie RL, Finkelstein RA, Woods DE. Multilocus sequence typing of historical Burkholderia pseudomallei isolates collected in Southeast Asia from 1964 to 1967 provides insight into the epidemiology of melioidosis. J Clin Microbiol. 2006;44(8):2951–2962. doi: 10.1128/JCM.00725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Currie B, Smith-Vaughan H, Golledge C, Buller N, Sriprakash KS, Kemp DJ. Pseudomonas pseudomallei isolates collected over 25 years from a non-tropical endemic focus show clonality on the basis of ribotyping. Epidemiol Infect. 1994;113(2):307–312. doi: 10.1017/S0950268800051736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inglis TJ, Garrow SC, Adams C, Henderson M, Mayo M, Currie BJ. Acute melioidosis outbreak in Western Australia. Epidemiol Infect. 1999;123(3):437–443. doi: 10.1017/S0950268899002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Loh JP, Aw LT, Yap EP, Lee MA, Ooi EE. Rapid molecular typing of Burkholderia pseudomallei, isolated in an outbreak of melioidosis in Singapore in 2004, based on variable-number tandem repeats. Trans R Soc Trop Med Hyg. 2006;100(7):687–692. doi: 10.1016/j.trstmh.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Currie BJ, Haslem A, Pearson T, Hornstra H, Leadem B, Mayo M, Gal D, Ward L, Godoy D, Spratt BG, Keim P. Identification of melioidosis outbreak by multilocus variable number tandem repeat analysis. Emerg Infect Dis. 2009;15(2):169–174. doi: 10.3201/eid1502.081036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McRobb E, Sarovich DS, Price EP, Kaestli M, Mayo M, Keim P, Currie BJ. Tracing melioidosis back to the source: using whole-genome sequencing to investigate an outbreak originating from a contaminated domestic water supply. J Clin Microbiol. 2015;53(4):1144–1148. doi: 10.1128/JCM.03453-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spratt BG, Hanage WP, Feil EJ. The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr OpinMicrobiol. 2001;4(5):602–606. doi: 10.1016/s1369-5274(00)00257-5. [DOI] [PubMed] [Google Scholar]
- 40.De Smet B, Sarovich DS, Price EP, Mayo M, Theobald V, Kham C, Heng S, Thong P, Holden MT, Parkhill J, Peacock SJ, Spratt BG, Jacobs JA, Vandamme P, Currie BJ. Whole-genome sequencing confirms that Burkholderia pseudomallei multilocus sequence types common to both Cambodia and Australia are due to homoplasy. J Clin Microbiol. 2015;53(1):323–326. doi: 10.1128/JCM.02574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H, Xia L, Zhu X, Li W, Du X, Wu D, Hai R, Shen X, Liang Y, Cai H, Zheng X. Burkholderia pseudomallei sequence type 562 in China and Australia. Emerg Infect Dis. 2015;21(1):166–168. doi: 10.3201/eid2101.140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.