

Case
A 31 years old woman referred to the outpatients with lower extremity and low back pain. Proximal muscle weakness was also a major complaint. She had lower extremity tenderness or antalgic gait on examination and osteomalacia. Radiological findings revealed atraumatic fractures of the long bones and vertebrae (T9-T12 and L1-L2) and bone demineralization (Figure 1, A-D). DEXA showed that bone mineral density (BMD) was 0.64 g/cm2 (lumbar spine). T-score was -3.7 SD and Z-score was -3.7 SD. Laboratory showed metabolic acidosis (pH:7.3 and HCO3-: 22 mmol/L) and a decrease in Phosphate, 25(ΟΗ)VitD blood levels, whereas ALP and PTH were increased. A 24-hour urine collection revealed increased Phosphate, and Potassium levels (Table 1). Anthropometric parameters were also influenced (Height:143 cm, Weight: 37.7 kg, BMI:18.4 g/m2). She reported that her symptoms started ten years ago, with atraumatic hip and rib fractures. Since then, she was taking active vitamin D 1 µg/Day and Oral phosphorus, 1.5 g/Day.
Figure 1.
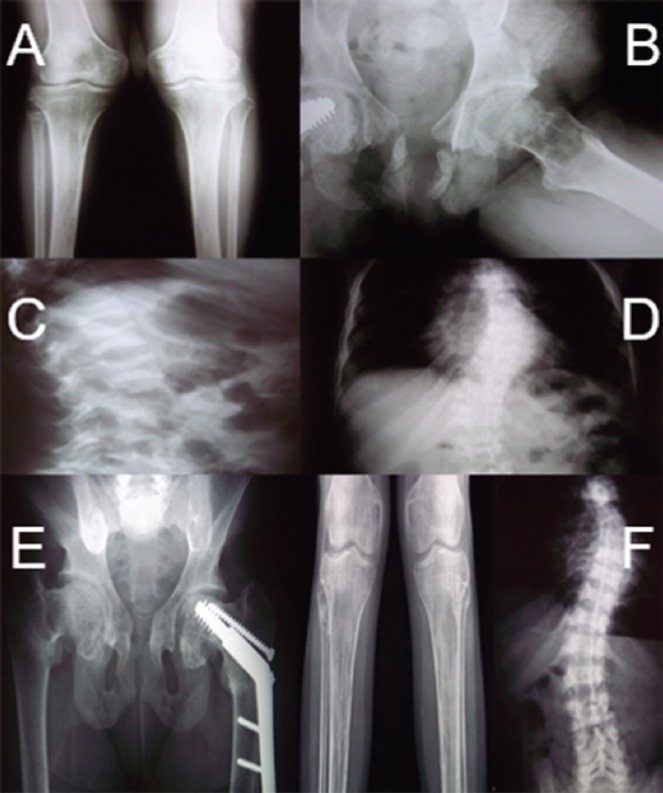
Patient’s imaging before therapy: (A) left proximal tibia fracture; (B) subtrochanteric and femoral neck intracapsular hip fractures; (C,D) vertebral compression; (E,F) Patient’s imaging after therapy: subtrochanteric and femoral neck intracapsular hip fractures and left proximal tibia fracture healed; vertebral compression stability.
Table 1.
Patient’s laboratory before and after treatment.
| Before treatment (Blood) | After treatment (Blood) | Before treatment (Urine) | After treatment (Urine) | |
|---|---|---|---|---|
| Na K Ca |
139 (136-146 mmol/l) 5.5 (3.5-5.3 mmol/l) 10 (8.8-10.4 mg/dl) |
139 4.2 10.2 |
4.2 (3-6 g/24h) 2.7 (2-3.5 g/24h) 1.4 (0.01-0.3 g/24h) |
3.9 2.9 0.96 |
| P | 2.0 (2.5-4.5 mg/dl) | 3.4 | 2128 (400-1300 mg/24h) | 954 |
| Mg | 1.9 (1.8-2.6 mg/dl) | 2.0 | ||
| ALP | 1266 <120 IU/L | 193 | ||
| PTH | 551 (15-65 pg/ml) | 281 | ||
| 25(ΟΗ) | 8.1 (20-32 ng/ml) | 18 | ||
| β-Cross | 3748 <584 pg/ml | 395 |
The diagnosis of adult acquired idiopathic form Fanconi’s syndrome was made, following the criteria of: (1) urinary hyperexcretion of amino acids of all classes (2) decreased serum phosphate on unrestricted dietary intake, (3) evidence of type 2 (proximal) renal tubular acidosis, including metabolic acidosis[1]. Her parents and her sister subsequently tested negative for Fanconi’s syndrome with a urinary amino acid screen.
Fluids and electrolytes were administered. Metabolic acidosis was corrected with the i.v administration of bicarbonate solution (3 to 15 mEq/kg/day). Initially, Phosphates at the dose of 1.5 gr/day were given in 5 divided doses. Optimal serum phosphate levels were reached titrating the dose up to 2.4 gr/day. Active vitamin D at the dose of 1.25 gr/day and calcium carbonate starting at 500 mg/day and then titrated as needed up to 1500mg/day were also administered.
After 1 year of treatment, the fractures were healed, she gained muscle strength and she complained no more for back or lower extremities pain (Figure 1, E,F). DEXA showed that bone mineral density (BMD) was 0.96 g/cm2 (lumbar spine). T-score was -0.7 SD and Z-score was -0.7 SD. The quality of life of the patient was significantly improved after treatment.
Commentary
Fanconi syndrome is associated with renal proximal tubular dysfunction. The main pathogenic feature is a generalized reabsorption defect, leading to the wasting of phosphate, amino acids, glucose, and bicarbonate in erratic degrees. Clinical indices vary from direct or indirect turbulences of the tubule system, with bone diseases frequently presenting as sequelae of the syndrome[2]. Therefore, clinical manifestations of the disease differ and morbidity is secondary to the produced metabolic abnormalities. Most of these abnormalities, such as acidosis, calciuria, and phosphaturia, affect bone deposit and, thus, growth. Some forms of Fanconi syndrome, such as cystinosis, may lead to renal failure[2].
The onset of the disease varies; if it is acquired (medications, etc), it can present itself at any age. If it is inherited (autosomal recessive pattern), the onset is usually evident in early development.
Numerous mechanisms can result in reduced reabsorption of solutes by the proximal tubule. Functional modifications in the carriers that transport ingredients across the luminal membrane, disorders in cellular energy metabolism, and changes in the permeability of the tubular membranes interchange in several scenarios trying to elucidate the pathogenesis of the disease. Currently, there is not a well-established apparatus in the understanding of the pathophysiology of the disease[2].
The variety of transport abnormalities observed in Fanconi syndrome suggests that the alterations in the solute carriers at the proximal tubule is not likely to be the main pathophysiological mechanism A defect in cellular energy metabolism appears to be the prevailing scenario. The sodiumpotassium (Na+/K+)-adenosine triphosphatase (ATPase) pump, which is located at the basolateral membrane, provides the energy required for the function of these carriers. A defective cellular energy metabolism and decreased levels of ATP can lead to dysfunction of secondary active transport mechanisms, such as those of glucose, phosphate, or amino acids[3].
Evidence supporting this hypothesis can be found in various experimental models and clinical forms of Fanconi syndrome. In the most extensively studied model, glucosuria, phosphaturia, aminoaciduria, bicarbonaturia, and proteinuria, associated with decreases in Na+/K+ ATPase and ATP levels were developed, after maleic acid was injected in rats and dogs[3]. Similar changes were reported in animals injected with other substances, such as heavy metals (i.e, cadmium, lead, and mercury)[3].
Other studies showed that increased oxidative stress may contribute to tubular dysfunction in cystinosis. Cystinosis, caused by the accumulation of cystine in renal tubule cells, is the most common feature of Fanconi syndrome in children. In experimental animal models, when injecting dimethylester, increased deposition of cystine in the renal tubules was produced. Additionally, low rates of transport, reduced levels of ATP, oxygen consumption, and mitochondrial respiration were implicated. When ATP was added to the incubation media partially corrected these abnormalities.
Drugs that induce mitochondrial dysfunction, are candidates of causing Fanconi syndrome[4]. The underlying pathophysiological mechanisms for drug-induced Fanconi syndrome vary. Tetracycline metabolites can cause renal tubular disease with electrolyte imbalance and induce tubular impairment. Aminoglycosides irreversibly bind to the cellular membranes causing lysosomal swelling, whereas, Cisplatin is directly intoxicated for the proximal tubular cells, resulting in an increase in b2-microglobulin and/or aminoaciduria and/or proteinuria.
Clinical presentations of Fanconi syndrome vary, depending on the pathogenesis of the disease. Inherited Fanconi syndrome is often presented with proximal tubular renal acidosis, hypophosphatemic rickets, hypokalemia, polyuria, and polydipsia. Furthermore, Fanconi is associated with cystinosis, growth retardation, depigmentation of the retina, interstitial nephritis, and progressive renal failure. Acquired Fanconi syndrome features slightly different abnormalities, such as renal tubular acidosis, hypophosphatemia, hypokalemia, osteomalacia, and muscle weakness[3].
The idiopathic Fanconi syndrome occurs in the absence of any perceptible cause, and most cases are sporadic. Some cases are inherited, with no specific inheritance pattern (autosomal dominant, autosomal recessive, X-linked). Most often it features recurrent episodes of dehydration, rickets, and failure to thrive[3].
The primary therapy for Fanconi syndrome is to treat the underlying causes and replace substances wasted in the urine. Fluids and electrolytes are administered orally or parenterally, to prevent dehydration resulting from polyuria[5]. Metabolic acidosis is most often present, caused by the inability of the kidney to absorb normal levels of bicarbonate. In the acute face, small boluses of IV sodium bicarbonate may be utilized to correct blood pH[5] In chronic conditions, bicarbonate is administered orally, ranging from 3 to 15 mEq/kg/day[5]. Administering bicarbonate in either form, orally or parenterally, may cause cells to increase potassium uptake; thus, monitoring of serum electrolytes is imperative in patients already predisposed to hypokalemia due to Fanconi syndrome. To minimize volume expansion and excretion of bicarbonate, diuretics, such as hydrochlorothiazide are employed at a dose of 1 to 3 mg/kg/day. Notable, if diuretics are not potassium sparing, a potassium supplement should be prescribed.
Another consideration in patients with Fanconi syndrome is bone disease. Due to the significant loss of phosphate, supplementation plus vitamin D, should be administered. Optimal serum phosphate levels can be reached with 1 to 3 g/day of supplemental phosphate, which is dose titrated. The patient should start with the lowest dose to avoid GI irritation and then increase it accordingly. For prophylaxis against hypokalemia, 20 mEq/day of P.O should be administered and then titrated as needed up to 100 mEq, given five times/day in adults[1]. Children may receive 3 to 8 mEq/kg/day orally in divided doses one to five times daily[1]. Vitamin D supplementation should be administered in its active form, whether exercise should be limited only in severe forms of Fanconi syndrome.
Patient prognosis depends on the cause of the syndrome and the severity of renal and extrarenal manifestations. Genetic forms are difficult to manage. They are usually associated with disruptions in growth, and involve other organs[3].
The multimodality of Fanconi syndrome presentations challenge medical professionals. Early diagnosis and management is a major concern, since life threatening situations can occur. Symptomatic management is still the therapy of choice. Thus, further research regarding etiology and therapy is needed.
Footnotes
The authors have no conflict of interest.
Edited by: P. Makras
Questions
1. Fanconi syndrome is associated with dysfunction in renal:
A. Proximal tubules
B. Ascending limb of loop of Henle
C. Descending limb of loop of Henle
D. Distal convoluted tubule
E. All the above
Critique
Fanconi syndrome is associated with renal proximal tubular dysfunction, leading to the wasting of phosphate, amino acids, glucose, and bicarbonate in erratic degrees.
The correct answer is A.
2. Which factors have shown a clinically proven action in the pathogenesis of the Fanconi syndrome?
A. Functional modifications in the carriers that transport ingredients across the luminal membrane
B. Disorders in cellular energy metabolism
C. Changes in the permeability of the tubular membranes
D. Increased oxidative stress
E. All the above
Critique
Numerous mechanisms can result in reduced reabsorption of solutes by the proximal tubule. Functional modifications in the carriers that transport ingredients across the luminal membrane, disorders in cellular energy metabolism, increased oxidative stress and changes in the permeability of the tubular membranes interchange in several scenarios trying to elucidate the pathogenesis of the disease. Currently, there is not a well-established apparatus in the understanding of the pathophysiology of the disease.
The correct answer is E.
3. The primary therapy for Fanconi syndrome is:
A. Fluids and electrolytes
B. Bicarbonate
C. Phosphate
D. Vitamin D supplementation
E. All the above
Critique
The primary therapy for Fanconi syndrome is to treat the underlying causes and replace substances wasted in the urine.
The correct answer is E.
References
- 1.Clarke BL, Wynne AG, Wilson DM, Fitzpatrick LA. Osteomalacia associated with adult Fanconi's syndrome: clinical and diagnostic features. Clin Endocrinol (Oxf) 1995;43:479–490. doi: 10.1111/j.1365-2265.1995.tb02621.x. [DOI] [PubMed] [Google Scholar]
- 2.Klootwijk ED, Reichold M, Unwin RJ, Kleta R, Warth R, Bockenhauer D. Renal Fanconi syndrome: taking a proximal look at the nephron. Nephrol Dial Transplant. 2015;30:1456–1460. doi: 10.1093/ndt/gfu377. [DOI] [PubMed] [Google Scholar]
- 3.Sirac C, Bridoux F, Essig M, Devuyst O, Touchard G, Cogne M. Toward understanding renal Fanconi syndrome: step by step advances through experimental models. Contrib Nephrol. 2011;169:247–261. doi: 10.1159/000313962. [DOI] [PubMed] [Google Scholar]
- 4.Izzedine H, Launay-Vacher V, Isnard-Bagnis C, Deray G. Drug-induced Fanconi's syndrome. Am J Kidney Dis. 2003;41:292–309. doi: 10.1053/ajkd.2003.50037. [DOI] [PubMed] [Google Scholar]
- 5.Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. 2017;13:115–131. doi: 10.1038/nrneph.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]