

Abstract
Stargardt disease is caused by variants in the ABCA4 gene, a significant part of which are noncanonical splice site (NCSS) variants. In case a gene of interest is not expressed in available somatic cells, small genomic fragments carrying potential disease-associated variants are tested for splice abnormalities using in vitro splice assays. We recently discovered that when using small minigenes lacking the proper genomic context, in vitro results do not correlate with splice defects observed in patient cells. We therefore devised a novel strategy in which a bacterial artificial chromosome was employed to generate midigenes, splice vectors of varying lengths (up to 11.7 kb) covering almost the entire ABCA4 gene. These midigenes were used to analyze the effect of all 44 reported and three novel NCSS variants on ABCA4 pre-mRNA splicing. Intriguingly, multi-exon skipping events were observed, as well as exon elongation and intron retention. The analysis of all reported NCSS variants in ABCA4 allowed us to reveal the nature of aberrant splicing events and to classify the severity of these mutations based on the residual fraction of wild-type mRNA. Our strategy to generate large overlapping splice vectors carrying multiple exons, creating a toolbox for robust and high-throughput analysis of splice variants, can be applied to all human genes.
Technological advancements have enabled the cost-effective detection of sequence variations in human genomes (Carss et al. 2017). Millions of single-nucleotide variants (SNVs) have been reported per individual, many thousands of which are rare and can be involved in rare monogenic disorders. Unfortunately, the enormous progress made in SNV detection has not been accompanied by the development of functional assays to investigate the effects of SNVs in a robust manner. Although it is very challenging to predict the effect of missense variants on protein function, characterizing putative splice variants is feasible. In the absence of patient cells, or if the gene of interest is not expressed in available somatic cells, in vitro splice assays are widely used. In these assays, small genomic fragments carrying potential disease-associated variants are tested for splice abnormalities. However, with the increasing detection of noncoding variants located within large introns, it is apparent that minigene assays are inadequate for testing the effects of deep-intronic variants without an intact genomic context, i.e., flanking exons and cis-acting elements that influence exon recognition.
The ATP-binding cassette transporter type A4 (ABCA4) is a 128-kb gene consisting of 50 exons and characterized by retina-specific expression (Allikmets et al. 1997). It encodes a multidomain transmembrane protein localized at the outer segments of photoreceptor cells (Sun and Nathans 1997; Molday et al. 2000). The estimated incidence of individuals with ABCA4-associated inherited retinal diseases is 1/10,000 (Blacharski 1988). ABCA4 variants have been observed in the vast majority (95%) of patients with autosomal recessive (ar) Stargardt disease (STGD1), ∼30% of patients with ar cone-rod dystrophy (CRD) and ∼5% of patients with ar panretinal dystrophy (Cremers et al. 1998; Maugeri et al. 1999, 2000). The phenotypic heterogeneity observed in individuals with ABCA4-associated retinal dystrophies can be explained by a genotype-phenotype correlation model, in which the residual activity of the mutant ABCA4 protein determines the clinical outcome. Typical STGD1 patients are usually characterized by a combination of a severe and a mild variant or two moderately severe variants, while patients affected by more severe phenotypes such as CRD and panretinal dystrophy carry a severe and a moderately severe variant or two severe variants, respectively (Cremers et al. 1998; Maugeri et al. 1999, 2000).
In order to provide an accurate prognosis for people with ABCA4-associated disease, it is essential to assess the functional consequences of all variants. Thus far, functional studies have been limited to selected missense variants that were studied at the protein level (Sun et al. 2000; Suarez et al. 2002; Quazi and Molday 2013), and only a small number of splice site variants have been examined (Rivera et al. 2000; Shroyer et al. 2001; Wiszniewski et al. 2005; Schulz et al. 2017). While the high impact of protein-truncating ABCA4 variants (nonsense, frameshift, canonical splice site variants) is clear, the effect of noncanonical splice site (NCSS) variants still remains largely unknown. These are either exonic or intronic variants located at the splice donor sites and splice acceptor sites. Intronic NCSS variants at splice donor sites are mostly situated at positions +3 to +6 downstream from exons, while intronic NCSS variants at splice acceptor sites are located at positions −14 to −3 upstream of exons. Exonic ABCA4 NCSS variants can only be found within the first or the last two nucleotides of an exon (Cornelis et al. 2017), which agrees with in silico predictions (Shapiro and Senapathy 1987).
Very recently, all published ABCA4 variants from 3928 retinal dystrophy cases were collected in a Leiden Open Variation Database (www.LOVD.nl/ABCA4), and an in silico pathogenicity assessment was provided for the 913 unique variants (Cornelis et al. 2017). Forty-four NCSS variants were identified that together account for 10% of all published ABCA4 alleles associated with STGD1 (Cornelis et al. 2017), and 41% of all ABCA4 alleles in patients with STGD1 from the Radboudumc in Nijmegen (N Bax, S Lambertus, FPM Cremers, and CB Hoyng, unpubl.). In silico programs assessing the pathogenicity of NCSS variants cannot always predict the effect of pre-mRNA splicing. For instance, the second most frequent ABCA4 variant, c.5461−10T>C, was predicted to be a mild variant, but it led to skipping exon 39, or exons 39 and 40, when tested in patient-derived photoreceptor precursor cells and a minigene assay (Sangermano et al. 2016), and in fibroblasts (Aukrust et al. 2017). It thereby could be classified as a severe ABCA4 variant.
In view of the large number of NCSS variants in ABCA4 with unknown functional effect, we designed a protocol to prepare sizeable multiple-exon ‘midigenes’ in order to test the consequences of all ABCA4 NCSS variants in their ‘natural context,’ resulting in a robust in vitro splice assay system. We generated a nearly complete library of overlapping wild-type midigenes from the ABCA4 gene, which allowed us to systematically assess the effect of 47 NCSS variants on RNA splicing.
Results
A large minigene enables accurate RNA analysis of the noncanonical splice site variant c.5714+5G>A
Previous genotype-phenotype studies in a large family with individuals showing pseudodominant ar retinitis pigmentosa (RP) due to a homozygous c.4539+1G>T ABCA4 splice variant, or ar CRD due to compound heterozygous c.4539+1G>T and c.5714+5G>A ABCA4 variants, led to the hypothesis that the NCSS variant c.5714+5G>A had a moderately severe effect (Cremers et al. 1998). Intriguingly, HEK293T cells transfected with small wild-type and mutant minigenes that contained only exon 40 showed partial skipping of exon 40 for the wild-type construct and full exon 40 skipping due to the c.5714+5G>A variant (Fig. 1A). Since our previous work (Cremers et al. 1998) and recent statistical analysis (Cornelis et al. 2017) suggested this variant had a moderate or mild effect on ABCA4 function, respectively, it seemed unlikely that the c.5714+5G>A variant would result in full exon 40 skipping. We noticed a similar artifact while studying the effect of another NCSS variant, c.5461−10T>C (Sangermano et al. 2016), so we hypothesized that the rhodopsin (RHO) exon 3 splice donor site and RHO exon 5 splice acceptor site are relatively strong (Human Splicing Finder scores of 87.5 and 90.7, respectively), resulting in erroneous exon 40 skipping. When we used larger minigenes containing exons 39 through 41, the erroneous exon 40 skipping in the wild-type construct was not observed anymore, and the mutant construct showed normal RNA product in addition to the exon 40 skipping product (Fig. 1B), as expected for a moderately severe variant.
Figure 1.
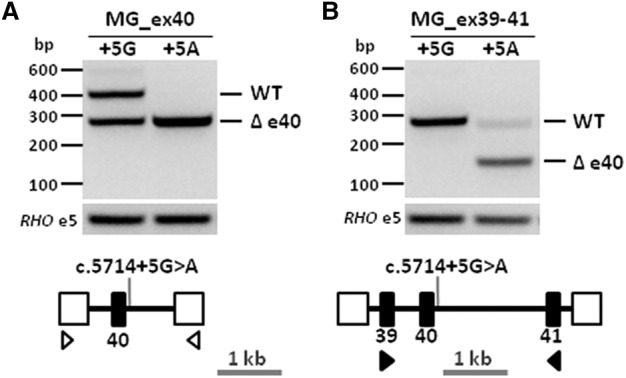
Large minigene enables accurate RNA analysis of the ABCA4 noncanonical splice site variant c.5714+5G>A. (A) Minigene containing the genomic region encompassing ABCA4 exon 40 (MG_ex40, black rectangle) and flanked by rhodopsin (RHO) exons 3 and 5 (open boxes) was designed to investigate the effect of the noncanonical splice site variant c.5714+5G>A. Reverse-transcription polymerase chain reaction (RT-PCR) performed using primers (open arrowheads) targeting RHO exons 3 and 5 showed an expected 404-bp wild-type fragment and a 274-bp fragment corresponding to exon 40 skipping (Δ) in control minigene (+5G) and full exon 40 skipping in mutant minigene (+5A). (B) Larger minigene containing the genomic region encompassing ABCA4 exons 39–41 (MG_ex39–41) was designed to investigate the same variant. RT-PCR using primers (black arrowheads) targeting exons 39 and 41 showed a single 260-bp wild-type fragment in the control minigene, while in the mutant minigene fragments of 260 and 130 bp corresponding to wild-type and exon 40 skipping were found.
Generation of ABCA4 wild-type midigene library
In view of these results and our goal to robustly test all reported NCSS variants identified in ABCA4, we generated multi-exon splice vectors using DNA from a bacterial artificial chromosome (BAC) clone (CH17-325O16) spanning the entire ABCA4 gene. In this way, a library of 29 overlapping wild-type midigenes (BA1–BA29) was generated (Fig. 2) by PCR amplification of fragments that were cloned in a Gateway-adapted vector containing the RHO exons 3, 4, and 5 (pCI-NEO-RHO) with the recombination sequences located between RHO exons 3 and 4 and between exons 4 and 5. The insert sizes for these constructs ranged from 4.0 to 11.7 kb, with, on average, three ABCA4 exons per construct. The maximal capacity of the vector is 12 kb. Hereafter, the pCI-NEO-RHO BA clones will be denoted as BA# clones.
Figure 2.

Overview of the wild-type midigene splice constructs of ABCA4 and locations of all 47 noncanonical splice site variants. Exons are represented here as black rectangles. Employing bacterial artificial chromosome DNA and Gateway sequence-tagged primers, we amplified 29 genomic fragments and cloned these into the pCI-NEO-RHO Gateway-adapted vector (BA1–BA29). Genomic DNA from control persons (MG30 and MG31) and a patient carrying c.6729+5_6729+19del (MG30) were used to clone wild-type and mutant fragments.
ABCA4 exons 1 and 50 were not contained in these clones, as they lack a splice acceptor and donor site, respectively. Due to difficulties in long-range PCR amplification at the 3′ end of the gene, we were also unable to amplify a genomic fragment containing exons 46–49. Instead, regular minigenes were generated for exons 48 (MG30) and 49 (MG31) using control genomic DNA as template. The midi- and minigenes together covered ∼92% of the 128-kb ABCA4 gene and 48 out of 50 exons (Fig. 2).
In all constructs, each ABCA4 exon was flanked by at least one upstream and one downstream ABCA4 exon, with the exception of BA1, BA4, BA6, BA8, BA10, BA29, MG30, and MG31. For constructs BA4, BA6, BA8, and BA10, the limitation was due to the large sizes of intron 6 (15.4 kb) and 11 (14.4 kb). BA5 and BA9 did not contain any exon, as they were generated to cover the middle parts of introns 6 and 11, respectively.
Sequence analysis of the wild-type BA clones revealed many polymorphisms but no deleterious variants due to the PCR amplification of the insert when compared to the human genome reference GRCh37/hg19 assembly. Nevertheless, despite the use of a proofreading high-fidelity DNA polymerase, we found minor sequence variations (Supplemental Table S1).
Selection of ABCA4 noncanonical splice site variants
The list of the 47 analyzed NCSS variants, their allelic frequencies, and predicted effect on splicing can be found in Supplemental Table S2. The majority of these variants (70%, n = 33) are located in noncoding elements; 30% (n = 14) are located in coding regions. Highly mutated intronic positions at the splice donor site were at the +5 (n = 12) and +3 (n = 6) positions. We did not observe significant “hotspots” at the splice acceptor site, although positions −3 and −10 were most frequently mutated (n = 3 each).
Generation of mutant midigenes
Eighteen BA clones were used to insert 46 NCSS variants (Fig. 2; Supplemental Fig. S1). Fifteen clones carry at least two flanking ABCA4 exons surrounding the tested NCSS variant, thereby allowing RT-PCR using ABCA4 primers instead of RHO primers. For three variants, this was not possible. c.160+5G>C and c.5312+3A>T were analyzed using a combination of a RHO exon 3 forward primer with ABCA4 exons 4 or 40 reverse primers, respectively. As c.768G>T resided in BA4, RT-PCR could only be performed using RHO exons 3 and 5 primers.
Eleven BA clones and MG31 were not used for mutagenesis purposes in this study. For variant c.6729+5_+19del, we did not perform mutagenesis as the wild-type and mutant minigenes were made using genomic DNA from a patient carrying this variant in a heterozygous manner.
Splice defects due to noncanonical splice site variants
We provided experimental evidence of splice defects for 44 out of 47 NCSS variants. For the RT-PCR control, cDNA from 18 wild-type BA clones was used (Fig. 2). Upon RT-PCR, 15 wild-type BA clones showed a single band corresponding to the expected splicing pattern, whereas BA1, BA12, and BA26 showed extra bands corresponding to alternative splice products (Supplemental Fig. S2). A comprehensive overview of all the splice assays for the 47 NCSS variants described in this study, as well as their sequencing analysis, can be found in Supplemental Figures S2 and S3. Representative results for nine variants are described below (Fig. 3; Supplemental Fig. S3).
Figure 3.

Overview of splice defects due to nine noncanonical splice site variants in ABCA4. All wild-type and mutant midigenes were transfected in HEK293T cells and their RNA subjected to RT-PCR. (A,B) RT-PCR for wild-type and c.160+5G>C mutant BA1 midigene showed complex splicing defects when using primers in RHO exon 3 and ABCA4 exon 4. Five defects (fragments 2 through 6) were observed next to the wild-type fragment 1. Asterisks denote fragments for which we obtained no sequence information. The RT-PCR products showed skipping of exon 2, exon 3, or exons 2 and 3. (C) Sequencing of pGEM-T-cloned fragment 4 revealed a 14-nt insertion due to the activation of a cryptic splice donor site in intron 2 and the absence of exon 3. The triangle points to the +5G>C variant. (D,E) RT-PCR products of wild-type and mutant BA1 containing NCSS variants residing at splice sites of exons 3 and 4. Primers in ABCA4 exons 2 and 4 reveal exon 3 skipping and/or exon 4 elongation. Note that the wild-type vector BA1 shows exon 3 skipping (fragment 5), which is more pronounced when using a forward RHO exon 3 primer (A) compared to using a forward ABCA4 exon 2 primer (D). (F) Sanger sequence of gel-purified fragment 3 shows a 2-nt elongation at the 3′ end of exon 4 due to the activation of a cryptic splice acceptor site in intron 3. Sanger sequencing results for all fragments without an asterisk are provided in Supplemental Figure S3. (G,H) RT-PCR and Sanger sequencing of RNA extracted from c.768G>T mutant BA4 midigene showed a 35-nt exon 6 elongation. (I) Human Splice Finder (HSF) splice site scores (blue arrowheads) for wild-type and mutant sites. (J,K) RT-PCR, gel analysis, and Sanger sequencing showed the wild-type product and a 12-nt exon 13 elongation due to c.1937+13T>G. M and WT denote mutant and wild-type splice products. (L) Details of the exon elongation defect. As represented by the HSF prediction software, the scores of the normal and intronic splice donor sites (SDSs) are not changed due to this variant. Most of the RT-PCR product consists of the exon elongation which introduces a premature stop-codon. (M–O) Variants at the penultimate position of exon 30 (c.4538A>G and c.4538A>C) resulted in a correct mRNA (fragment 1), a 30-nt exon 30 elongation due to the presence of a strong cryptic intronic splice donor site (fragment 2), a deletion of 73 nt of the 3′ end of exon 30 (fragment 3), and a 30-nt exon 30 elongation in combination with a 114-nt deletion of the 5′ end of exon 30 (fragment 4). Fragments 3 (227 bp) and 4 (216 bp) could not be resolved. The exon 30 deletions are the results of cryptic splice donor and acceptor sites that overlap at exonic positions c.4465–4468 (Supplemental Fig. S2, BA20). (P) RT-PCR of BA27 carrying the c.5898+5del variant resulted in at least two exon 42 elongation products of 717 and 780 bp, as well as intron 42 retention. (Q) The exon elongation products were due to the reduction of the strength of the exon 42 splice donor site, as shown with red numbers, in combination with the presence of nearby cryptic intronic splice donor sites with strong predicted scores. Only HSF and SpliceSiteFinder (SSF)-like scores are provided. Note that SSF-like is the only software that predicts the rare splice donor sites (positions c.5898+108 and c.5899−23) that contain the canonical GC sequence instead of GU. Intron 42 retention likely is due to the very strong splice donor site (HSF 98.0) flanking exon 43. The open arrowhead denotes the 1-bp deletion.
Variants c.160+5G>C, c.161G>T, c.302+4A>G, and c.303−3C>G were located in or near exons 2 and 4 and were introduced in BA1 (Figs. 2, 3A–F). Variant c.160+5G>C was predicted to significantly reduce the strength of the exon 2 splice donor site and likely cause exon skipping (Supplemental Table S2). RT-PCR using a primer at the 5′ side of exon 2 therefore was not useful. Hence, we used primers in RHO exon 3 and ABCA4 exon 4. The wild-type BA1 showed two main bands of 399 and 271 bp. Sanger sequence analysis showed that these were the expected mRNA fragment and a fragment in which exon 3 was skipped, respectively (Fig. 3A,B). c.160+5G>C showed a complex splicing pattern, i.e., correct and three main aberrant splice products, that after pGEM-T cloning and Sanger sequencing revealed bands of 305, 285, and 271 bp, which represented the skipping of exon 2, a 14-nt elongation of exon 2 along with skipping of exon 3, and skipping of exon 3, respectively (Fig. 3A–C). Exon 2 elongation can be explained by the presence of a cryptic splice donor site 14-nt downstream from the actual splice site. The cryptic donor site is not shown in Figure 3C as it can only be seen in the genomic DNA sequence. RT-PCR spanning ABCA4 exons 2–4 showed a wild-type 289-bp band for BA1. The introduction of variants c.161G>T and c.302+4A>C both showed a unique 147-bp band corresponding to skipping of exon 3, while c.303−3C>G showed two bands of 291 and 149 bp, corresponding to a dinucleotide elongation of exon 4 and to the same elongation along with the skipping of exon 3, respectively (Fig. 3D–F). The exon 4 elongation can be explained by the creation of a cryptic splice acceptor site due to the C>G change. The cryptic acceptor site is not shown in Figure 3F as it can only be seen in the genomic DNA sequence. The skipping of exon 3 in the wild-type BA1 clone probably was not an artifact, as we also observed exon 3 skipping in human retina RNA (Supplemental Fig. S2, BA1, panel C).
Variant c.768G>T is the second most frequent NCSS variant (allele frequency 0.106) in STGD1 cases in the Radboudumc, Nijmegen (N Bax, S Lambertus, FPM Cremers, and CB Hoyng, unpubl.). Splice predictions showed a significant weakening of the exon 6 splice donor site (Fig. 3I). For the wild-type and c.768G>T mutant BA4 clone, RT-PCR encompassing RHO exons 3 and 5 was performed, which yielded a wild-type band of 472 bp for BA4 and a 35-nt elongation of exon 6 for c.768G>T (Fig. 3G,H), explained by the presence of a cryptic splice donor site 36-nt downstream (Fig. 3I). The reading frame shift results in a predicted truncated protein, p.(Leu257Valfs*17). To validate this result ‘ex vivo,’ we generated photoreceptor precursor cells (PPCs) from somatic cells of a STGD1 patient carrying c.768G>T in a homozygous state. RT-PCR analysis of RNA from this patient and of normal human retina are shown in Supplemental Figure S4. The PPC results are identical to those observed in the in vitro splice assay in HEK293T cells.
The novel variant c.1937+13T>G was not predicted to result in a splicing defect (Fig. 3L), but surprisingly, in 86% of mRNA products, a 12-nt elongation of exon 13 was observed for BA11 carrying c.1937+13T>G (Fig. 3J,K). This led to a premature stop codon, resulting in p.(Phe647*) (Fig. 3L).
For exon 30, the penultimate nucleotide substitutions c.4538A>G and c.4538A>C were introduced into BA20, and HEK293T cell transfection resulted in four fragments after pGEM-T cloning (Fig. 3M). In addition to the correct mRNA (fragment 1), a 30-nt elongation due to the presence of a strong cryptic intronic splice donor site (fragment 2, Fig. 3M,O), a deletion of 73 nt from the 3′ end of exon 30 (fragment 3, Fig. 3N), and a 30-nt elongation in combination with a 114-nt deletion from the 5′ end of exon 30 (fragment 4, Supplemental Figs. S2, S3) were found. Fragments 3 (227 bp) and 4 (216 bp) could not be resolved. The exon 30 deletions were the result of partially overlapping cryptic splice donor and acceptor sites, the noncanonical sequences (AG/GT) of which reside at exonic positions c.4465–4468 (Supplemental Fig. S2, BA20, panel D).
Six fragments were observed for c.5898+5del introduced in BA27, but we were only able to sequence three after pGEM-T cloning: Bands of 717 and 780 bp corresponded to 107-nt and 170-nt elongations of exon 42 due to the use of cryptic splice donor sites at positions c.5898+108 and +171, respectively, while a 1104-bp fragment corresponded to the entire retention of intron 42 (Fig. 3P,Q).
Classification of noncanonical splice site variants according to the splice defects
The percentage of correctly spliced ABCA4 RNA product for NCSS variants was calculated by using a capillary electrophoresis system (Fig. 4; Supplemental Table S3; Supplemental Fig. S5). Three variants, c.4634G>A, c.5196+3_5196+8del, and c.5585−10T>C, showed 100% correctly spliced ABCA4 mRNAs. We could rule out pathogenicity for the last two variants but not for c.4634G>A p.(Ser1545Asn), as this variant is significantly enriched in >3000 Caucasian STGD1 patients compared to the non-Finnish ExAC population (Cornelis et al. 2017). Sixteen variants showed between 4.3% and 79.6% normal RNA and were tentatively classified as severe, moderately severe, and mild, while the remaining 26 NCSS variants did not show any normal RNA and were thus deemed severe variants. For c.2382+5G>C and c.2588G>C, no quantification was performed as the wild-type BA12 clone also showed exon skipping. Based on the Sanger sequence validation, we were able to annotate all the variants at the RNA level and predict their effect at the protein level (Table 1).
Figure 4.
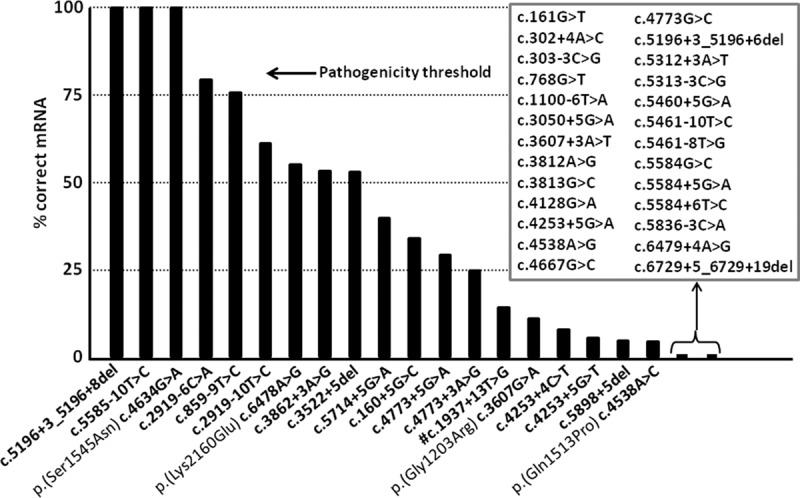
Percentages of remaining normal ABCA4 transcripts due to noncanonical splice site variants based on capillary electrophoresis system analyses. The percentages of normal ABCA4 transcript for 45 noncanonical splice site variants are represented by black bars. Nineteen variants showed varying fractions (4.3%–100%) of correct ABCA4 mRNA. Four of them also result in missense changes as depicted, but only p.(Ser1545Asn) and p.(Lys2160Glu) are likely to have an effect on protein function, as significant amounts of ABCA4 protein will be produced. For the remaining 26 variants (in the square box), no residual RNA was observed. (#) For this variant, densitometric scanning was performed.
Table 1.
In vitro tested noncanonical splice site variants and their observed RNA and predicted protein effects
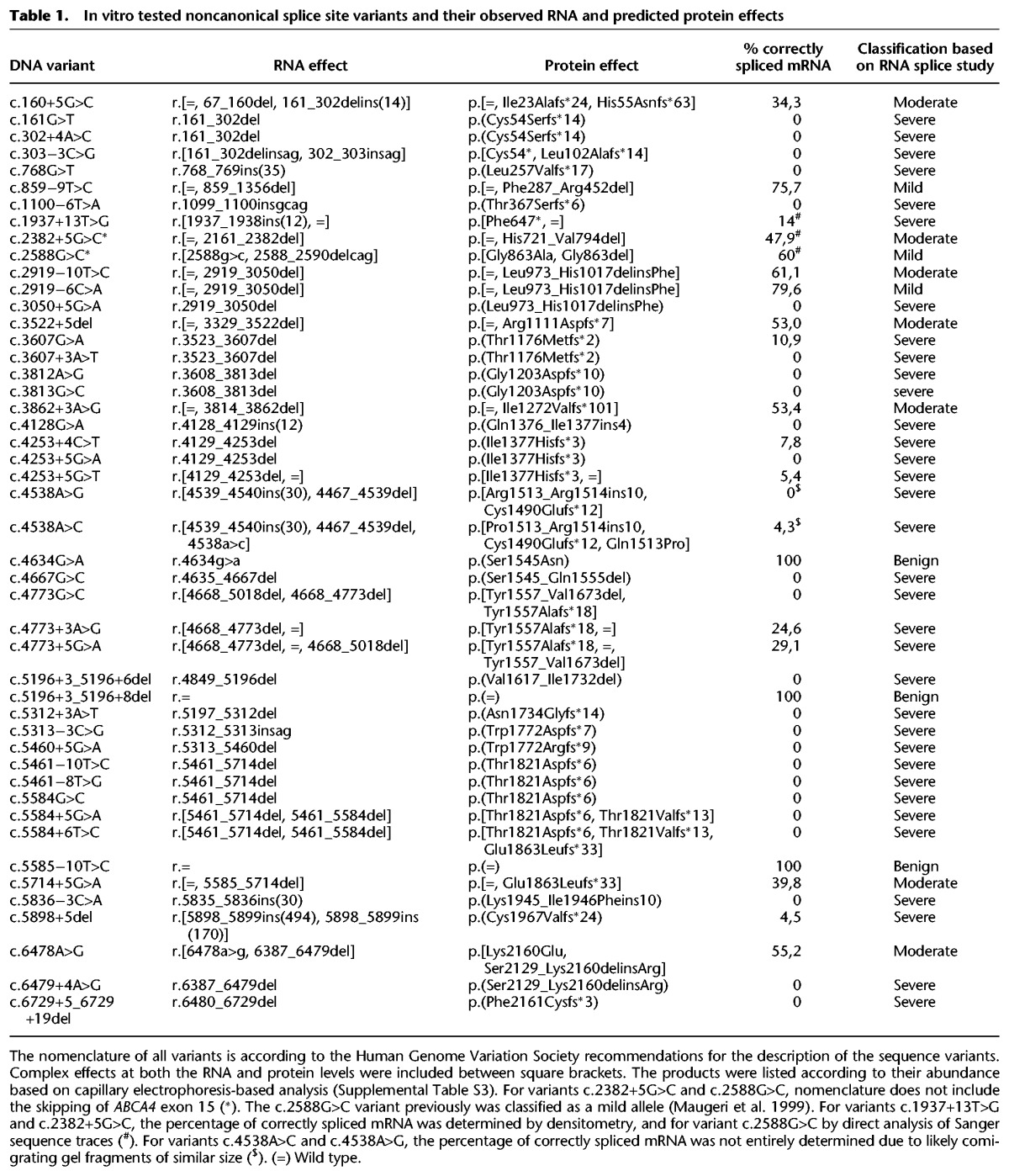
We correlated the predicted effect of the NCSS variants with the phenotypes in reported cases and, when the variants were not present in a homozygous state, with the severity of the second allele. For 25 of 47 variants, sufficient clinical data were available, and in all these cases, our predicted effect of the NCSS variants correlated with the observed phenotypes (Supplemental Table S4).
Discussion
Splice site variants cause 15%–20% of human genetic disease (Matlin et al. 2005), and an increasing number of deep-intronic variants have been identified through targeted gene sequencing and whole-genome sequencing (www.dbass.soton.ac.uk) (Buratti et al. 2011; Carss et al. 2017). Contrary to canonical splice site variants, which in about two-thirds of cases result in exon skipping (Cheung et al. 2017), the effect of NCSS variants can vary tremendously, depending on the sequence alteration itself and its sequence environment. In recent studies, large-scale analyses of variants in or near consensus splice site sequences were performed using minigene constructs containing small exons flanked by minimal intronic sequences (Cheung et al. 2017). The maximum insert size of the tested genomic segments measured 170 bp, as the analysis of spliced sequences was performed by short-read next-generation sequencing. Although this type of study enabled high-throughput analysis of randomly designed splice site sequences (Rosenberg et al. 2015) or naturally occurring NCSS variants (Cheung et al. 2017), the size constraint of the employed constructs represents a major limitation to fully assess the effect of NCSS variants. As shown here, it is important to study NCSS variants in the context of their upstream or downstream introns and exons, as their effect often is influenced by other intronic cis-regulatory elements or affects exons located at considerable distances.
We have performed a systematic in vitro analysis of all reported and three novel NCSS variants in ABCA4 that are associated with STGD1 by generating midigenes: large, multi-exon splice vectors. Forty-four of 47 variants resulted in splicing defects. The most frequently observed defect, seen in 29 (62%) of the NCSS variants, was single or multi-exon skipping. For eight (17%) variants, we observed the simultaneous occurrence of different splice defects, e.g., exon skipping/elongation, skipping/retention, elongation/retention, and truncation/elongation. Six NCSS variants (13%) resulted in exon elongation.
The majority of NCSS variants (n = 35; 75%) affected the splice donor site. This concurs with the observation that the majority (65%) of pseudoexon mRNA insertions associated with human diseases also concern the activation of splice donor sites (www.dbass.soton.ac.uk) (Buratti et al. 2011). The longer intronic consensus sequences of splice acceptor sites (−3 to −14 for splice acceptor site vs. +3 to +6 for splice donor site) in which pyrimidine to pyrimidine substitutions have relatively little effect on the predicted splice strengths, may explain why variants that affect the splice acceptor site are generally less disruptive. The severe NCSS defect previously shown for c.5461−10T>C (exon 39 or exon 39 and 40 skipping) (Sangermano et al. 2016) prompted us to investigate the effect of two similar rare variants, i.e. c.2919−10T>C and c.5585−10T>C. We found that c.2919−10T>C resulted in exon skipping in 39% of the ABCA4 transcripts, while c.5585−10T>C showed no splice defects. The predicted reductions of the corresponding splice acceptor site splice scores all were minor. These contrasting results suggest that the effect of a NCSS variant also depends on the sequence context, e.g., the strengths of nearby splice acceptor and donor sites, the size of the flanking exons, and possibly exonic and intronic splice enhancer (ESE, ISE) and silencer motifs (ESS, ISS).
The effect of still unknown sequence motifs on splicing is also illustrated by our findings for c.1937+13T>G. This is not a typical NCSS variant as it is located downstream from the exon 13 splice donor site consensus sequence. As illustrated in Figure 3, J through L, this change does not lower the strength of the normal exon 13 splice donor site, nor does it create a stronger predicted downstream splice donor site. Nevertheless, it resulted in a 12-nt elongation of exon 13 and introduced a premature stop codon in 86% of the transcripts, based on Sanger sequencing. The Alamut ESEfinder did not predict this variant to create a splice enhancer (Cartegni et al. 2003; Smith et al. 2006). Follow-up studies are required to investigate the nature of this apparently unknown sequence motif that influences splicing. Importantly, this result also illustrates that the assessment of rare noncoding variants identified in diagnostic testing cannot fully rely on in silico predictions.
The generation of multiple-exon splice constructs in which most of the introduced NCSS variants were flanked by one or more upstream and downstream exons allowed us to investigate the full spectrum of splice defects. This is illustrated by two +5delG variants, c.3522+5del in Supplemental Figure S2 and c.5898+5del in Figure 3, P and Q, which not only showed multiple exon elongation but also retention of introns 23 and 42, respectively. The exon elongations could be explained by similarities shared by both introns, as they both carry multiple strong cryptic splice donor sites, small downstream introns (intron 23, 1081 bp; intron 42, 494 bp), and strong splice donor sites located at the 3′end of the following exon. The importance of the presence of the sequence context was also found for NF1 as the extent of exons 36 and 37 skipping due to a variant in exon 37 was determined by the presence of exons 34–38 in the splice construct (Baralle et al. 2006).
The observation that NCSS variants with an effect on RNA splicing resulted in a maximum of 79.6% of normal RNA leads us to believe that there may be a threshold for alleles to behave as ‘mild’ alleles. In our experiments, that threshold is ∼80% (Fig. 4). In cases where there is more than 80% normal RNA, the variants will likely be benign and will not contribute to the phenotype in patients with STGD1. Similarly, there likely are thresholds for moderately severe and severe alleles. However, as the clinical data of patients carrying NCSS variants were not described consistently, we could not accurately assign these thresholds. A similar threshold model was recently described for the remaining normal CEP290 protein in patients with Leber congenital amaurosis and various syndromic phenotypes, as a result of different combinations of coding and deep-intronic variants (Drivas et al. 2015).
The 29 BA constructs contain large (4.0–11.7 kb) segments of ABCA4. We could not clone genomic fragments larger than 11.7 kb due to the size constraint (12 kb) of the Gateway cloning system. We therefore cloned introns 6 and 11 in different overlapping fragments and performed transcript analysis using RHO primers. Constructs containing the first and last exon were not generated as they lack a splice acceptor and donor site, respectively. A difficult region to amplify was the 3′ part of the gene encompassing intron 48–exon 50. The absence of both flanking ABCA4 exons triggered splicing artifacts due to the presence of a strong RHO splice donor and acceptor site, as we observed erroneous exon skipping in MG_ex40 (Fig. 1).
Natural skipping of ABCA4 exons 3, 15, and 39/40 was observed by others (Braun et al. 2013) and by us (Supplemental Fig. S2), suggesting that the midigenes mimic the in vivo situation. An accurate comparison of the skipping events for exons 3 and 39/40 between wild-type midigene and human retina is not possible, as they result in frameshifts and the corresponding mRNAs are subject to nonsense-mediated decay in the human retina.
Validating multiple splice products can be time-consuming as gel-purification and Sanger sequencing are hampered by contaminations from other fragments, thus requiring plasmid cloning and additional Sanger sequencing. A more general drawback of the in vitro splice assay is that, despite the observed splicing similarities between wild-type BA clones and human retina, splicing in HEK293T cells may not always mimic the splice defects as they might occur in the retina of patients. Other studies have indicated that indeed there are retina-specific splice factors (Murphy et al. 2016; Parfitt et al. 2016).
The use of bacterial artificial chromosome clones facilitated the generation of large, multi-exon wild-type splice vectors. In an alternative approach, a 5.6-kb BRCA1 minigene was constructed by cloning six consecutive fragments carrying exons 19–27 that, in the human genome, span 28.7 kb (Acedo et al. 2015). Our approach can readily be applied to generate midigenes from all human genes, as BAC clones are available for this purpose (https://bacpacresources.org/) (Osoegawa and de Jong 2004). These wild-type midigenes not only allow the accurate assessment of large splice defects due to NCSS variants but can also be used to test deep-intronic variants hypothesized to result in pseudo-exon insertions (R Sangermano, A Garanto, M Khan, RWJ Collin, and FPM Cremers, unpubl.).
We observed sizeable exon elongations in significantly occurring RNA products due to c.160+5G>C, c.768G>T, c.1937+13T>G, c.4538A>G, and c.4538A>C variants. Splice modulation using antisense oligonucleotides (AONs) may be an option to redirect the splicing back to the natural splice sites. AONs have already demonstrated their therapeutic potential for several eye disease-associated genes, such as CEP290 (Collin et al. 2012; Gerard et al. 2012; Garanto et al. 2016; Parfitt et al. 2016), USH2A (Slijkerman et al. 2016), or OPA1 (Bonifert et al. 2016), and have a great potential as therapeutic molecules for inherited retinal diseases.
In conclusion, we generated a complete set of sizeable wild-type ABCA4 splice vectors, which allowed us to gain insight into the severity of the poorly explored class of NCSS ABCA4 variants on a gene-wide scale. By assessing the functional consequences of putative splice defects, STGD1 patients can obtain a more accurate molecular diagnosis and thereby become eligible for novel therapies. Our approach can be used for all human genes to effectively test putative splice defects. Besides from NCSS variants, midigenes are also especially suited for testing rare deep-intronic variants and their effect on splicing in a reliable manner, as 90% of introns are <11 kb (Sakharkar et al. 2004) and thereby completely fit into midigenes, together with all cis-acting splice factor binding motifs and flanked by their natural exons.
Methods
STGD1 patients
Patient 1 is a 40-yr-old female who was diagnosed with typical STGD1 at the age of 16. Sanger sequence analysis of all 50 ABCA4 exons and a MLPA analysis (MRC Holland P151 and P152) revealed heterozygous variants identified as two noncanonical splice site variants, the previously described c.859−9T>C and the novel c.303−3C>G. Patient 2 is a 37-yr-old male with STGD1 who was diagnosed at the age of 35 but had been suffering from reduced visual acuity since the age of 22. Sanger sequence analysis of all 50 ABCA4 exons revealed the coding variant c.70C>T [p.(Arg24Cys)] and a novel intronic variant 13-bp downstream from exon 13 (c.1937+13T>G). Patient 3 is a 53-yr-old female with STGD1 who was diagnosed at the age of 49. Sanger sequence analysis of all 50 ABCA4 exons revealed a novel homozygous c.5461−8T>G variant. All three patients display extensive areas of retinal pigment epithelium atrophy throughout the posterior pole, extending beyond the vascular arcades. On electroretinography, the cone and rod responses were significantly and equally reduced in patients 2 and 3 at the age of diagnosis.
Generation of wild-type and mutant minigenes for exons 39–41, 40, 48, and 49
Wild-type minigenes for exons 39–41, 40, 48, and 49 were generated by amplifying genomic DNA encompassing these exons from genomic DNA of an anonymous control person. Mutant minigenes for c.5714+5G>A and c.6729+5_+19del variants were generated by amplifying these regions from the genomic DNA of two patients carrying these ABCA4 variants. Primer details can be found in Supplemental Tables S5 and S6. Minigenes were generated according to a previously described protocol (Sangermano et al. 2016).
Generation of ABCA4 wild-type midigene library
The BACPAC Resource Center (BPRC) at the Children's Hospital Oakland Research Institute (Oakland, CA) provided the bacterial artificial chromosome clone, CH17-325O16 (insert g.94,434, 639–94,670,492), containing the entire ABCA4 gene. Using NucleoBond Xtra Midi EF (cat. no. 740420.250, Macherey-Nagel), BAC DNA was isolated and used as a template to generate large wild-type ABCA4 fragments by PCR. PCR primers were designed using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0) and their specificity verified by using Human BLAT searches (https://genome.ucsc.edu/cgi-bin/hgBlat) (Kent 2002; Supplemental Table S6). PCR reaction mixtures (50 µL) contained 0.5 µM of each forward and reverse primer, 0.2 mM dNTPs, Phusion High-Fidelity DNA Polymerase (cat. no. M0530L, New England Biolabs), 1× Phusion GC Buffer, 3% DMSO, and 2.5 ng BAC DNA. PCR reaction was performed by using the Veriti Thermal Cycler (Thermo Fisher Scientific). All PCRs were performed using the following thermocycling conditions: initial denaturation at 98°C for 30 sec; 15–20 cycles of denaturation at 98°C for 10 sec each, annealing at 57°C, and extension at 72°C (1 min/kb); with a final extension at 72°C for 15 min. The PCR-amplified products were analyzed on a 0.6% (w/v) agarose gel. Amplified bands were excised, and the NucleoSpin Gel and PCR cleanup kit (cat. no. 740609.250, Macherey-Nagel) was used to purify the DNA fragments, according to the manufacturer's protocol.
Two hundred nanograms of each purified fragment were cloned into the pDONR201 vector (Invitrogen) by using the Gateway BP Clonase Enzyme Mix (cat. no. 11789021, Thermo Fisher Scientific). The generated wild-type entry clones (denoted Bacterial Artificial Chromosome ABCA4 [BA] clones) were digested with restriction enzymes and validated by Sanger sequencing (Supplemental Table S7), and each BA entry clone was transferred into the Gateway-adapted destination vector pCI-NEO-RHO as previously described (Sangermano et al. 2016).
Selection of candidate ABCA4 noncanonical splice site variants
Of the 47 ABCA4 NCSS variants investigated in this study, 43 were selected from the Leiden Open-source Variation Database (www.LOVD.nl/ABCA4) (Cornelis et al. 2017), and three were found in STGD1 patients (c.303−3C>G for patient 1, c.1937+13T>G for patient 2, c.5461−8T>G for patient 3) during routine diagnostic studies in the Radboudumc. c.2919−10T>C was a recently published novel NCSS variant (Schulz et al. 2017). Forty-six of the NCSS variants comply with the following three inclusion criteria: (1) located in intronic positions −14 to −3 (splice acceptor site), or +3 to +6 (splice donor site) or at the first or the last 2 nucleotides of exons; (2) a reduction in the strength of the mutant splice site compared to its wild-type reference when calculated by at least one of five algorithms (SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer, Human Splicing Finder) (Shapiro and Senapathy 1987; Reese et al. 1997; Pertea et al. 2001; Yeo and Burge 2004; Desmet et al. 2009) via Alamut Visual software version 2.7 (Interactive Biosoftware; www.interactive-biosoftware.com); and (3) an allele frequency of ≤0.005 in the gnomAD browser (accessed May 31, 2017) (Lek et al. 2016). Variant c.1937+13T>G is not located in the splice donor site consensus sequence, and the five splice prediction programs did not show any reductions in canonical and downstream splice sites. It was selected for a splice assay as it was the only rare intronic ABCA4 variant located outside the NCSS consensus sequences but within 20 bp of an exon junction that was identified in routine Sanger sequencing of ∼300 STGD1 cases.
Generation of ABCA4 mutant constructs
Site-directed mutagenesis was performed on wild-type BA entry clones as depicted in Figure 2. Mutagenesis primers were designed by using the online tool QuikChange Primer Design (http://www.genomics.agilent.com/primerDesignProgram.jsp?toggle=upload Now&mutate=true&_requestid=1039517) and their GC content and temperature verified by using the online software Multiple Primer Analyzer (https://www.thermofisher.com/nl/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/multiple-primer-analyzer.html) (Supplemental Table S8). The mutagenesis PCR reaction mixture (50 µL) contained 0.5 µM of each forward and reverse primer, 0.2 mM dNTPs, 1 U Phusion High-Fidelity DNA Polymerase (cat. no. M0530L, New England Biolabs), 1× Phusion HF Buffer, 2× Q-solution (cat. no. 1005485, Qiagen), and 20 ng wild-type entry clone. All PCRs were performed using the following thermocycling conditions: initial denaturation at 94°C for 5 min; 15 cycles of denaturation at 94°C for 30 sec each, annealing between 55°C and 75°C, and extension at 72°C (1 min/kb), with a final extension at 72°C for 20 min. PCR-amplified products were incubated with DpnI restriction enzyme (cat. no. R0176S, New England Biolabs) for 4.5 hr, followed by heat inactivation at 65°C for 20 min to remove the wild-type plasmid DNA. Digested products were first run on a 0.6% (w/v) agarose gel to check for the presence of a unique linear fragment, corresponding to the mutant construct. Each of the NCSS variant mutants had their sequences validated (Supplemental Table S9), and finally, each validated mutant construct was cloned into the Gateway-adapted destination vector pCI-NEO-RHO as previously described (Sangermano et al. 2016).
In vitro splice assay and transcriptional analysis
To assess the effect of the NCSS variants on splicing, HEK293T cells (CRL-3216, ATCC) were transfected with wild-type and mutant constructs. Transfection, RNA isolation, and cDNA synthesis protocols were previously described (Sangermano et al. 2016). ABCA4 transcript analysis was generally carried out by performing RT-PCR analysis using ABCA4 exonic primers (Supplemental Table S10). To test the effect of NCSS variants located in or near exons that could not be flanked by other ABCA4 exons, e.g., due to the large size of some introns, primers were used from RHO exons 3 and 5. RT-PCR using primers targeting RHO exon 5 was used as a control for transfection efficiency (Supplemental Table S10).
All reaction mixtures (50 µL) contained 0.2 µM of each primer pair, 1 U Taq DNA Polymerase (cat. no. 11647679001, Roche), 1× PCR buffer with MgCl2, 1 mM MgCl2, 0.2 mM dNTPs, 1× Q-Solution (cat. no. 1005485, Qiagen), and 50 ng cDNA. PCR conditions were a first denaturation step of 94°C for 5 min, followed by 35 cycles of melting (94°C for 30 sec each), annealing (58°C for 30 sec), and extension (72°C for 5 min) steps, with a final elongation step of 72°C for 2 min. The RT-PCR products were run on a 2% (w/v) agarose gel and the resulting bands were excised and purified with the NucleoSpin Gel & PCR cleanup kit (cat. no. 740609.250, Macherey-Nagel) according to the manufacturer's protocol. Finally, 100 ng of the purified nucleic acid were analyzed by Sanger sequencing in a 3100 or 3730 DNA Analyzer (Thermo Fisher Scientific). For complex RT-PCR band patterns, 4 µL of the RT-PCR products, containing between 1 and 2 µg of DNA, were further cloned via the pGEM-T Easy Vector System I (cat. no. A1360 Promega) according to the manufacturer's protocol.
To determine the fraction of correctly spliced product, quantitative analysis of the RT-PCR bands was performed using the Fragment Analyzer Auto Capillary Electrophoresis System (Advanced Analytical Technologies, Inc.). RT-PCR of three constructs with no splice defects, 16 constructs with partial splice defects, and three constructs with no correctly spliced RNAs were performed in duplicate. Human retina cDNA was used as the control, as several ‘natural’ exon skipping events in human retina have been reported previously (Supplemental Tables S3, S11; Supplemental Figs. S5, S6; Braun et al. 2013). For variants c.1937+13G>T, c.2382+5G>C, and c.2588G>C, the fraction of correctly spliced product was assessed by densitometric analysis using Image J software (Schneider et al. 2012). The analysis was performed in duplicate and values were normalized for the size of the different fragments present in each RT-PCR product mixture (Supplemental Table S12).
ABCA4 transcript analysis of patient-derived photoreceptor precursor cells
Fibroblasts from a patient carrying the noncanonical splice site variant c.768G>T in a homozygous state were successfully reprogrammed in iPSCs and further differentiated into photoreceptor precursor cells as previously described (Sangermano et al. 2016). Photoreceptor precursor cells were harvested, total RNA extracted, and RT-PCR performed by using a forward primer located in ABCA4 exon 4 (5′-TCATGAATGCACCAGAGAGC-3′) and a reverse primer located in ABCA4 exon 7 (5′-ACGGCTGTCTAGGAGTGTGG-3′). Gel resolution, excision, and purification of the bands and Sanger sequencing were performed as previously described (Sangermano et al. 2016).
Data access
The raw sequencing data for all wild-type splicing constructs (WT_BA), mutant splicing BA constructs (BA_c.), and RT-PCR fragments (RT_BA) from splice assays in HEK293T cells from this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA417900.
Supplementary Material
Acknowledgments
We thank Erwin van Wijk for providing the Gateway-adapted minigene vector and Christian Gilissen for assistance in data processing. This work was supported by the FP7-PEOPLE-2012-ITN (Marie-Curie Actions) program EyeTN, agreement 317472 (to F.P.M.C.). This work was also supported by the Foundation Fighting Blindness USA, grant no. PPA-0517-0717-RAD (to F.P.M.C., R.W.J.C., S.A., and A.G.), RP Fighting Blindness, UK, grant no. GR591 (to F.P.M.C. and S.A.), the Macula Vision Research Foundation (to F.P.M.C.), the Rotterdamse Stichting Blindenbelangen, the Stichting Blindenhulp, the Stichting tot Verbetering van het Lot der Blinden (to F.P.M.C. and S.A.), and by the Landelijke Stichting voor Blinden en Slechtzienden, Macula Degeneratie fonds and the Stichting Blinden-Penning that contributed through Uitzicht (to F.P.M.C. and S.A.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226621.117.
References
- Acedo A, Hernández-Moro C, Curiel-García Á, Díez-Gómez B, Velasco EA. 2015. Functional classification of BRCA2 DNA variants by splicing assays in a large minigene with 9 exons. Hum Mutat 36: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, et al. 1997. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15: 236–246. [DOI] [PubMed] [Google Scholar]
- Aukrust I, Jansson RW, Bredrup C, Rusaas HE, Berland S, Jorgensen A, Haug MG, Rodahl E, Houge G, Knappskog PM. 2017. The intronic ABCA4 c.5461–10T>C variant, frequently seen in patients with Stargardt disease, causes splice defects and reduced ABCA4 protein level. Acta Ophthalmol 95: 240–246. [DOI] [PubMed] [Google Scholar]
- Baralle M, Skoko N, Knezevich A, De Conti L, Motti D, Bhuvanagiri M, Baralle D, Buratti E, Baralle FE. 2006. NF1 mRNA biogenesis: effect of the genomic milieu in splicing regulation of the NF1 exon 37 region. FEBS Lett 580: 4449–4456. [DOI] [PubMed] [Google Scholar]
- Blacharski PA. 1988. Fundus flavimaculatus. In Retinal dystrophies and degenerations (ed. Newsome DA), pp. 135–159. Raven Press, New York. [Google Scholar]
- Bonifert T, Gonzalez Menendez I, Battke F, Theurer Y, Synofzik M, Schols L, Wissinger B. 2016. Antisense oligonucleotide mediated splice correction of a deep intronic mutation in OPA1. Mol Ther Nucleic Acids 5: e390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, Deluca AP, Fishman GA, Lam BL, Weleber RG, et al. 2013. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet 22: 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Chivers M, Hwang G, Vorechovsky I. 2011. DBASS3 and DBASS5: databases of aberrant 3′- and 5′-splice sites. Nucleic Acids Res 39: D86–D91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, Megy K, Grozeva D, Dewhurst E, Malka S, et al. 2017. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet 100: 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. 2003. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 31: 3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung R, Insigne KD, Yao D, Burghard CP, Jones EM, Goodman DB, Kosuri S. 2017. Large-scale screening of rare genetic variants in humans reveals frequent splicing disruptions. bioRxiv 10.1101/199927. [DOI]
- Collin RW, den Hollander AI, van der Velde-Visser SD, Bennicelli J, Bennett J, Cremers FP. 2012. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol Ther Nucleic Acids 1: e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis SS, Bax NM, Zernant J, Allikmets R, Fritsche LG, den Dunnen JT, Ajmal M, Hoyng CB, Cremers FP. 2017. In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum Mutat 38: 400–408. [DOI] [PubMed] [Google Scholar]
- Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, et al. 1998. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet 7: 355–362. [DOI] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. 2009. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37: e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drivas TG, Wojno AP, Tucker BA, Stone EM, Bennett J. 2015. Basal exon skipping and genetic pleiotropy: a predictive model of disease pathogenesis. Sci Transl Med 7: 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garanto A, Chung DC, Duijkers L, Corral-Serrano JC, Messchaert M, Xiao R, Bennett J, Vandenberghe LH, Collin RW. 2016. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet 25: 2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard X, Perrault I, Hanein S, Silva E, Bigot K, Defoort-Delhemmes S, Rio M, Munnich A, Scherman D, Kaplan J, et al. 2012. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common Leber congenital amaurosis CEP290 mutation. Mol Ther Nucleic Acids 1: e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—The BLAST-Like Alignment Tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CW. 2005. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol 6: 386–398. [DOI] [PubMed] [Google Scholar]
- Maugeri A, van Driel MA, van de Pol DJ, Klevering BJ, van Haren FJ, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, et al. 1999. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet 64: 1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri A, Klevering BJ, Rohrschneider K, Blankenagel A, Brunner HG, Deutman AF, Hoyng CB, Cremers FP. 2000. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 67: 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday LL, Rabin AR, Molday RS. 2000. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet 25: 257–258. [DOI] [PubMed] [Google Scholar]
- Murphy D, Cieply B, Carstens R, Ramamurthy V, Stoilov P. 2016. The Musashi 1 controls the splicing of photoreceptor-specific exons in the vertebrate retina. PLoS Genet 12: e1006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, de Jong PJ. 2004. BAC library construction. Methods Mol Biol 255: 1–46. [DOI] [PubMed] [Google Scholar]
- Parfitt DA, Lane A, Ramsden CM, Carr AJ, Munro PM, Jovanovic K, Schwarz N, Kanuga N, Muthiah MN, Hull S, et al. 2016. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell 18: 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Lin X, Salzberg SL. 2001. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res 29: 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Molday RS. 2013. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem 288: 34414–34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. 1997. Improved splice site detection in Genie. J Comput Biol 4: 311–323. [DOI] [PubMed] [Google Scholar]
- Rivera A, White K, Stohr H, Steiner K, Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP, Apfelstedt-Sylla E, et al. 2000. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet 67: 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Patwardhan RP, Shendure J, Seelig G. 2015. Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163: 698–711. [DOI] [PubMed] [Google Scholar]
- Sakharkar MK, Chow VT, Kangueane P. 2004. Distributions of exons and introns in the human genome. In Silico Biol 4: 387–393. [PubMed] [Google Scholar]
- Sangermano R, Bax NM, Bauwens M, van den Born LI, De Baere E, Garanto A, Collin RW, Goercharn-Ramlal AS, den Engelsman-van Dijk AH, Rohrschneider K, et al. 2016. Photoreceptor progenitor mRNA analysis reveals exon skipping resulting from the ABCA4 c.5461-10T→C mutation in Stargardt disease. Ophthalmology 123: 1375–1385. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz HL, Grassmann F, Kellner U, Spital G, Ruther K, Jagle H, Hufendiek K, Rating P, Huchzermeyer C, Baier MJ, et al. 2017. Mutation spectrum of the ABCA4 gene in 335 Stargardt disease patients from a multicenter German cohort—impact of selected deep intronic variants and common SNPs. Invest Ophthalmol Vis Sci 58: 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. 1987. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15: 7155–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR. 2001. Null missense ABCR (ABCA4) mutations in a family with Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci 42: 2757–2761. [PubMed] [Google Scholar]
- Slijkerman RW, Vache C, Dona M, Garcia-Garcia G, Claustres M, Hetterschijt L, Peters TA, Hartel BP, Pennings RJ, Millan JM, et al. 2016. Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol Ther Nucleic Acids 5: e381. [DOI] [PubMed] [Google Scholar]
- Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. 2006. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 15: 2490–2508. [DOI] [PubMed] [Google Scholar]
- Suarez T, Biswas SB, Biswas EE. 2002. Biochemical defects in retina-specific human ATP binding cassette transporter nucleotide binding domain 1 mutants associated with macular degeneration. J Biol Chem 277: 21759–21767. [DOI] [PubMed] [Google Scholar]
- Sun H, Nathans J. 1997. Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet 17: 15–16. [DOI] [PubMed] [Google Scholar]
- Sun H, Smallwood PM, Nathans J. 2000. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet 26: 242–246. [DOI] [PubMed] [Google Scholar]
- Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, Lupski JR. 2005. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet 14: 2769–2778. [DOI] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.