

Abstract
Adenosine-to-inosine (A-to-I) RNA editing regulates miRNA biogenesis and function. To date, fewer than 160 miRNA editing sites have been identified. Here, we present a quantitative atlas of miRNA A-to-I editing through the profiling of 201 pri-miRNA samples and 4694 mature miRNA samples in human, mouse, and Drosophila. We identified 4162 sites present in ∼80% of the pri-miRNAs and 574 sites in mature miRNAs. miRNA editing is prevalent in many tissue types in human. However, high-level editing is mostly found in neuronal tissues in mouse and Drosophila. Interestingly, the edited miRNAs in neuronal and non-neuronal tissues in human gain two distinct sets of new targets, which are significantly associated with cognitive and organ developmental functions, respectively. Furthermore, we reveal that miRNA editing profoundly affects asymmetric strand selection. Altogether, these data provide insight into the impact of RNA editing on miRNA biology and suggest that miRNA editing has recently gained non-neuronal functions in human.
MicroRNAs (miRNAs) are short (∼22 nt), endogenous, noncoding RNAs that regulate gene expression (Bartel 2004). miRNAs target most protein-coding genes via complementary base-pairing and play crucial roles in development and metabolism (Bushati and Cohen 2007; Bartel 2009). miRNA genes are first transcribed into primary miRNAs (pri-miRNAs), each of which contains a short (∼70 nt) double-stranded RNA (dsRNA) named precursor miRNA (pre-miRNA). Canonically, pri-miRNAs are cleaved by DROSHA to generate pre-miRNAs, which are further processed into mature miRNAs by DICER1 (Kim et al. 2009). Alternatively, a subset of intronic miRNAs utilize splicing to generate pre-miRNA hairpin mimics, termed mirtrons, which bypass DROSHA cleavage and are further processed by DICER1 (Westholm and Lai 2011).
Adenosine deaminases acting on RNA (ADARs) are known to bind dsRNA regions of protein-coding genes and noncoding sequences, particularly pri-miRNAs, and deaminate adenosine to inosine (Nishikura 2010, 2016). miRNA editing plays an important role in miRNA regulation and function (Kawahara et al. 2008; Wulff and Nishikura 2015). Editing in pri-miRNA regulates the processing of pre-miRNA (Yang et al. 2006). Editing in mature miRNA leads to the selection of new target genes (Kawahara et al. 2007b; Kume et al. 2014). The prevalence of RNA editing in protein-coding genes has been elucidated via extensive examination of RNA sequencing data (Ramaswami and Li 2016). However, in-depth study of pri-miRNA editing has been impeded by the infeasibility of detecting miRNA editing using traditional RNA-seq, since pri-miRNAs are rapidly processed and therefore maintained at much lower levels compared with protein-coding genes. Although several laboratories have examined RNA editing events in pri-miRNAs or mature miRNAs (Blow et al. 2006; Kawahara et al. 2008; de Hoon et al. 2010; Alon et al. 2012), systematical identification, quantification, and comparison of miRNA editing in multiple tissues or developmental stages have not been performed in human or animal model organisms.
In this study, we present the first quantitative atlas of miRNA A-to-I editing in animals. We selected human and two animal models (mouse and Drosophila melanogaster [D.mel], in which the ADAR mutant phenotypes were extensively studied) for RNA editing profiling. A human–mouse comparison will shed light on the miRNA editing dynamics and evolution in mammals. Additionally, the miRNA editing profiles in mouse and D.mel will be helpful for a better understanding of their phenotypic defects. A targeted RNA sequencing method that couples microfluidics-based multiplex PCR and deep sequencing (mmPCR-seq) (Zhang et al. 2014) was optimized to survey all pri-miRNAs in 201 human, mouse, and D.mel samples.
Results
Development of miR-mmPCR-seq method
Animal genomes typically encode several hundred to one thousand bona fide miRNA genes (Kozomara and Griffiths-Jones 2014). Because pri-miRNAs are poorly detected by RNA-seq (Chang et al. 2015), we reasoned that targeted RNA-seq, particularly a PCR-based approach that allows individualized and saturated amplification of different loci, is required to amplify all pri-miRNA loci and examine their editing profiles. We recently developed the mmPCR-seq method (Zhang et al. 2014), which can amplify and measure allelic ratios at roughly 1000 mRNA loci. Here, we have made substantial improvements to the mmPCR-seq to enable uniform amplification of pri-miRNA loci for miRNA editing profiling (Fig. 1A). This optimized method is hereafter referred to as miR-mmPCR-seq.
Figure 1.

The development and performance of miR-mmPCR-seq. (A) Schematic diagram of miR-mmPCR-seq. First, multiplex primers covering the pre-miRNA hairpin and flanking 70–150 bp regions were designed. Forty-eight preamplified cDNA samples and 48 pools of primers were loaded to the Fluidigm microfluidic chip. The PCR products from the same cDNA sample were automatically pooled. For each sample, the PCR products were barcoded and pooled together, and subject to 150-bp paired-end sequencing: (RT) reverse transcription; (GS) gene-specific. (B) Comparison of the cumulative distribution of amplicon coverage between unamplified and preamplified cDNA samples. The y-axis shows the number of reads in log2 scale. Read numbers are normalized to 0.25 million mapped reads per sample. (C,D) The reproducibility of RNA editing levels measured using preamplified and unamplified cDNA samples for sites with at least 50 reads in both samples. The color codes are the same as in B.
Compared with mRNA, the low expression level of pri-miRNA and the competition between cDNA extension and pre-miRNA hairpin reformation during reverse transcription (Schmittgen et al. 2004) make it more challenging to amplify pri-miRNA amplicons. We therefore tested a series of parameters to achieve uniform amplification across pri-miRNA loci. We selected 555 mouse miRNAs (Methods), designed 48 pools of 9- to 12-plex multiplex PCR primers (Supplemental Table S1) that target the pri-miRNA hairpin (pre-miRNA) and flanking sequences, and used mouse brain RNA for parameter optimization. In order to determine the effectiveness of pri-miRNA amplification, all tested samples were barcoded and deep sequenced. We reasoned that preamplification may improve the sensitivity and uniformity of the amplification because of the low-expression levels of pri-miRNAs. To assess this, we performed miR-mmPCR-seq with or without preamplification. We found that, unlike amplification of protein-coding gene loci (Zhang et al. 2014), preamplification substantially increased the number of pri-miRNAs amplified and led to a more uniform distribution of amplicons (Fig. 1B; Supplemental Fig. S1A). Most importantly, the reproducibility of measurements was largely increased with preamplification (Fig. 1C,D; Supplemental Fig. S1B–F). Concomitantly, at least two rounds of DNase I treatment are needed to completely remove the genomic DNA contamination (Supplemental Fig. S1G), which may otherwise interfere with RNA editing identification and quantification. Since the yield of highly structured RNA, such as pri-miRNA, varies when using different transcriptases with different reaction temperatures, we tested five reverse transcriptase kits working at temperatures from 42°C to 65°C. Additionally, to assess the performance of random primers or gene-specific primers, RNA was primed using either gene-specific primers or random hexamer primers. Unexpectedly, we found that more pri-miRNA amplicons, as well as a more uniform amplification, were obtained using cDNA generated with random primers (Supplemental Fig. S1H–L; Supplemental Note 1). SuperScript III with a reaction temperature at 50°C showed the best performance (Supplemental Fig. S1M), so this reaction condition was used for all further experiments.
Identification and quantification of RNA editing in pri-miRNAs
To construct a comprehensive atlas of miRNA A-to-I editing in animals, we sequenced miR-mmPCR libraries constructed from 30 human adult and fetal tissues, 21 inbred mouse adult and embryonic tissues, and 31 D.mel developmental stages (Methods; Supplemental Tables S1, S2). RNA variants were identified as previously described with some modification (Methods; Ramaswami et al. 2012, 2013). With a minimum variant frequency cutoff at 5% (Methods), the average fractions of all RNA variants that are A-to-G/T-to-C type (indicative of A-to-I editing) are 97.6% (human Alu-derived miRNAs), 94.5% (human repetitive non-Alu-derived miRNAs), 89.2% (human nonrepetitive miRNAs), 91.3% (mouse repetitive miRNAs [Supplemental Fig. S2; Supplemental Note 2]), 87.3% (mouse nonrepetitive miRNAs), and 89.3% (D.mel miRNAs) of all variants. This indicates that the estimated false discovery rate (FDR) is <5% for each category (Ramaswami et al. 2012, 2013). The proportion of A-to-G/T-to-C and the number of A-to-I sites in each sample were shown in Supplemental Figure S3–S5. In total, we identified 2711, 959, and 492 A-to-G sites in human, mouse, and D.mel, respectively (Supplemental Table S3). Even with a minimum variant frequency cutoff at 2%, the FDR rates are considerably low (Supplemental Fig. S6A). At this cutoff, 7033, 5950, and 3075 A-to-G variants were identified (Supplemental Table S4), and >80% of the pre-miRNAs is edited in at least one sample for each species (Supplemental Fig. S6B), suggesting that ADARs may bind and edit a majority of the miRNAs. Because sites with relatively low editing levels (<5%) may have no biological significance, in the following analysis, we only used sites with ≥5% editing level.
We found that the A-to-G sites were associated with known features of the ADAR-binding sequence motif (Supplemental Fig. S6C; Supplemental Note 3). We validated 202 of 221 (91%) selected A-to-G sites in 36 miRNAs (with ≥15% editing level) in all three species using PCR and Sanger sequencing (Supplemental Fig. S7; Supplemental Table S5), including 75 of 84 (89%) sites in human non-neuronal tissues. The high accuracy and reproducibility of miR-mmPCR-seq were further verified using nascent RNA-seq of the D.mel Adar null mutant and editing level comparison between technical or biological replicates (Supplemental Fig. S8; Supplemental Note 4), consistent with our previous study (Zhang et al. 2014).
We also applied Porath's method (Porath et al. 2014) to identify hyperediting sites in our data set. We identified 114, 42, and 52 miRNAs with hyperediting events in human, mouse, and D.mel (Supplemental Fig. S9–S11; Supplemental Table S6), respectively. The hyperediting levels of most of the miRNAs are <2%. We therefore did not include them in the following analysis.
Global characterization of RNA editing in miRNAs
Our comprehensive list provided the basis for the inference of global observations regarding the miRNA editing in animals. We first examined the editing levels of both known and novel sites and found that both spanned a wide spectrum of editing levels (Fig. 2A). More than 1000 novel sites were edited at levels ≥20%, which vastly expanded the total number of moderately or highly edited sites in miRNAs. We next examined the editing site distributions in different species. We found that editing tends to occur in the stem region, whereas the end loop appears to be devoid of editing (Fig. 2B; Supplemental Figs. S12, S13A). Human nonrepetitive or repetitive non-Alu-derived miRNAs have fewer editing sites compared with Alu-derived miRNAs, which often form longer double-stranded stem loops. Moreover, miRNAs were grouped into mirtrons and non-mirtrons, because these two groups have different structural features (Westholm and Lai 2011). Interestingly, editing sites in mirtrons are highly selected and edited at a much higher level than that in non-mirtron miRNAs (Supplemental Fig. S13B). Within the highly edited stem regions, a large fraction of editing events occur in the mature miRNA region, which could be processed to edited mature miRNAs. When performing cross-species comparison, we found human has a significantly higher pre-miRNA editing density compared with mouse and D.mel (Fig. 2C). We found that sequences around pre-miRNAs are edited, albeit less frequently than pre-miRNAs (Supplemental Fig. S13C). When examining the secondary structure of flanking sequences, we found that they are significantly more structured than shuffled sequences (Supplemental Fig. S13D). Therefore, the pre-miRNA hairpin structure and highly structured flanking regions together may facilitate the binding of ADARs and subsequent ADAR-mediated editing.
Figure 2.

Identification, characterization, and regulation of miRNA editing sites. (A) The distributions of RNA editing levels for known and novel sites in different species. For this analysis, we used the representative editing level of each editing site, which is the maximum editing level across all samples we profiled. (B) Metagene profiles depicting the editing site distribution across pri-miRNAs. The structure predicted using dme-mir-2492 precursor and the flanking 150-nt sequence is used as the representative pri-miRNA secondary structure. Percentage of As which were edited at each position is indicated by color. (C) Box plot showing the number of editing sites per pre-miRNA in human, mouse, and D.mel. P-values were calculated using Wilcoxon test: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (D) The proportion and mean representative editing level comparison between editing sites with different complementary nucleotides. Schematic diagram was used to explain how we categorized the editing sites. Edited As are highlighted in red. Others, editing sites that cannot be categorized into the previous 5 types. The mean editing levels for each type were shown as the line plot. The color codes for species are the same as in C.
Next, we investigated the triplet motif and editing complementary sequence requirements for pri-miRNA editing. We confirmed the previously reported high-frequency editing at the adenosine residue within the UAG triplet in human (Kawahara et al. 2008), particularly for the sites with high editing levels (Supplemental Fig. S13E). We also found that AAG and AAA triplets are frequently edited in human miRNAs (Supplemental Fig. S13F). Additionally, the triplet preference is different between mammals and D.mel, likely because there are two functional ADARs in mammals with different motif preferences but only one ADAR in D.mel. For the editing site complementary sequences, we found that the majority of the editing occurs within A-U (50%) and A-C (8%) pairs (Fig. 2D). Furthermore, A-C pairs have the highest editing levels (Fig. 2D). These data reveal the genome-wide pattern of complementary sequence preference in miRNA editing.
To compare the conservation of miRNA editing among animal species, we analyzed both the miRNA and the editing site conservation. We found that within about 270 miRNAs conserved between human and mouse, 94 sites were edited in both species, whereas 920 and 363 were edited only in human or mouse, respectively. Additionally, 742 and 188 sites are located at human- or mouse-specific miRNAs. No conserved editing was observed between mammals and D.mel. Conserved editing sites have significantly higher editing levels compared with nonconserved sites (Supplemental Fig. S13G), suggesting more stringent functional constraint. Notably, a large number of lineage-specific sites are highly edited and may perform lineage-specific functions.
Trans-regulation explains tissue-specific miRNA editing and cross-species editing profile differences
We next analyzed the spatial distribution of RNA editing from our data set (Fig. 3). We found frequent miRNA editing across different types of tissues in human (Fig. 3A,D). In sharp contrast, high-level editing events were mostly present in neuronal tissues (brain and spinal cord) in mouse (Fig. 3B,E). To have a fair comparison, we further examined conserved editing sites in 16 tissue types shared between human and mouse. The same pattern was consistently observed (Supplemental Fig. S14A). In D.mel, editing typically begins in pupal stages. The level of editing increases throughout development, and adult heads have the highest editing levels (Fig. 3C,F).
Figure 3.

Editing level profiles across different species. (A–C) Heatmap of editing levels in human (A), mouse (B), and D.mel (C). For each species, only sites with editing levels ≥10% in at least one sample were selected for analysis. Human: (ACG) anterior cingulate gyrus. Tissues (bone marrow and liver) that have more than 50 missing editing values were removed from analysis. (D.mel: em) embryo; (L) larvae; (WPP) White prepupae; (AdF_Ecl) adult female eclosion; (AdM_Ecl) adult male eclosion. Samples (L1, L3 PS3-6, and WPP4) that have more than 50 missing editing values were removed from analysis. (D–F), Pearson correlations for the editing levels of all sites in human (D), mouse (E), and D.mel (F). Gray means not available.
Next, we asked to what extent the editing pattern difference between human and mouse may be attributed to the expression of ADAR enzymes. We found that in mouse, both Adar and Adarb1 have much higher expression levels in neuronal tissues than in non-neuronal tissues (Fig. 4A). However, in human, the expression levels of ADAR and ADARB1 varied among tissues, and no such difference between neuronal and non-neuronal tissues was observed (Fig. 4A). We also used linear regression analysis to measure the contribution of ADAR expression to overall editing difference across different samples (Supplemental Fig. S14B). In mouse, we found a positive correlation between the editing level and the expression level of ADARs. However, in human, we did not observe such an obvious correlation. Taken together, the observed difference in global editing between human and mouse can be largely explained by the difference in ADAR expression. This result also suggests a recent gain of non-neuronal functions of miRNA editing in human.
Figure 4.

The trans- and cis-regulation of miRNA editing between human and mouse. (A) ADAR and ADARB1 expression levels for the 16 human and mouse tissues. The experiments were done in technical (Human) or biological (Mouse) replicates, and the mean expression levels were shown. (B) Dot plot showing editing level difference of conserved sites between human and mouse. Editing levels measured from the whole brain sample were used for analysis. Editing sites that are edited in at least one species and genomically encoded as “A” in both species were used for analysis. Differentially edited sites were determined by using Fisher's exact test. P-values were corrected using the Benjamini-Hochberg (BH) method (Benjamini and Hochberg 1995), and a confidence level of 0.05 was used as the cutoff. (C, left) Free energies comparison between miRNAs with “human-high” and “similar” sites. Free energy difference (y-axis) is defined as the free energy of a pre-miRNA in human minus that of its orthologous miRNA in mouse. (Right) Free energies comparison between miRNAs with “mouse-high” and “similar” sites. Free energy difference (y-axis) is defined as the free energy of a pre-miRNA in mouse minus that of its orthologous miRNA in human. The P-value was calculated using Wilcoxon test. (D) Comparison of the distance from nearest Alu element between miRNAs with “human-high” and “similar” sites. The P-value was calculated using one-tailed Wilcoxon test: (*) P < 0.05. (E) Comparison of the distance from nearest B1 repeat element between miRNAs with “mouse-high” and “similar” sites. The P-value was calculated using one-tailed Wilcoxon test. The color codes for species in C–E are the same as in B.
Cis-elements tune site-specific miRNA editing and account for individual site cross-species variation
Besides the difference in global tissue-specific miRNA editing between human and mouse, the editing level of orthologous sites between these two species in neuronal tissues are also varied (Supplemental Fig. S14A). To investigate whether this is caused by lineage-specific cis-element changes, we characterized the RNA secondary structures of these sites. We separated the sites into three groups: (1) “human-high” and (2) “mouse-high” groups that contain sites differentially edited between the two species in the brain (human-high: higher in human; mouse-high: higher in mouse); and (3) “similar” group that contains sites similarly edited in both species in the brain (Fig. 4B). We found that, compared with those containing sites from the “similar” group, the secondary structures of miRNAs with mouse-high sites tend to be more stable in mouse than in human (Fig. 4C). However, no such difference is found for miRNAs with human-high sites (Fig. 4C). Furthermore, human-high sites are significantly closer to Alu elements, which are known to affect the editing levels of many nonrepetitive sites (Fig. 4D; Ramaswami et al. 2012), whereas no significant effect is observed for mouse B1 repeats (Fig. 4E), despite that both B1 and Alu are derived from the 7SL RNA. These results indicate that the editing level changes of the individual site in human are mainly due to the Alu proximity.
Identification of edited mature miRNAs
Identification of edited mature miRNAs using small RNA-seq is especially challenging, due to the difficulty of accurately mapping very short reads (∼22 nt) with mismatches to the genome (de Hoon et al. 2010; Alon et al. 2012). Our comprehensive list of pre-miRNA editing provides a unique opportunity to systematically identify edited mature miRNAs. We constructed a custom pre-miRNA sequence database with the A replaced by G in edited positions. This allowed us to map the edited small RNA-seq reads into the custom database without any mismatch, thereby facilitating the accurate identification and quantification of edited mature miRNAs. We streamlined the analysis process (Methods; Supplemental Fig. S15A), curated, and analyzed a collection of 4694 small RNA-seq data sets (Supplemental Table S7). In total, we identified 367, 161, and 46 editing sites in mature miRNAs in human, mouse, and D.mel, respectively, most of which are newly reported (Supplemental Fig. S15B). Editing occurs in all positions in the mature miRNAs and is significantly enriched in miRNA seed region, which is critical for target recognition (Bartel 2009), in human and mouse, but not in D.mel (Fig. 5A). All detailed information related to each editing site, including the number of samples presented, the editing levels of the tissues in which it was observed, the number of A and G reads in the sample that shows the most convincing editing signal, and the averaged editing level for all samples with a given editing site were shown in Supplemental Table S8. We further analyzed the distribution of the editing sites shared by multiple samples (Supplemental Fig. S15C–E). Sites identified in multiple samples were defined as high-confidence sites (Supplemental Table S8).
Figure 5.

Edited mature miRNAs alter miRNA targeting. (A) The editing site density in mature miRNAs. The significant difference between the seed region of the miRNA (bases 2–8; region 1) and the rest of the sites (bases 1 and 9–24; region 2) was calculated using χ2 test: (human) P = 1.0 × 10−12; (mouse) P = 6.5 × 10−6; (D.mel) P = 0.85. (B, top) For each miRNA that was edited in the seed region, we grouped its targets into three categories: targeted by unedited version only (loss), by both unedited and edited versions (common), and by edited version only (gain). (Bottom) The distribution of the number of “gain” group target genes shared by multiple neuronal edited miRNAs in human was shown as an example. (C) Functional enrichment for “gain” group genes targeted by neuronal or non-neuronal edited miRNAs in human. Human edited miRNAs identified in neuronal or non-neuronal tissues (Supplemental Table S7) were used for analysis. “Gain” group genes targeted by at least six neuronal or four non-neuronal edited miRNAs were used for analysis (Methods). The sets of transcripts coexpressed with the miRNAs were used as background. P-values were corrected for multiple hypotheses testing using the BH method. (D) Functional enrichment for “gain” group genes targeted by neuronal edited miRNAs in mouse. Mouse edited miRNAs identified in neuronal tissues (Supplemental Table S7) were used for analysis. “Gain” group genes targeted by at least six miRNAs (Methods) were used for analysis. The sets of transcripts coexpressed with the miRNAs were used as background. The P-values were corrected for multiple hypotheses testing using the BH method. The top 20 significant GO terms were shown. (E) The comparison of edited neuronal miRNA seeds between human and mouse. A conserved edited seed means that both the miRNA seed and the editing position are conserved in human and mouse.
Functional significance of edited mature miRNAs in mammals
To reveal the possible functional significance of edited mature miRNAs, we compared the target genes of the unedited and edited versions of each miRNA with editing events at seed region in mammals. We found that edited and unedited versions of the miRNAs only shared 10%–35% of their target genes; thus, editing significantly changed the binding specificity of the miRNAs (Supplemental Fig. S15F). In order to deduce the new functions of edited miRNAs, we examined genes specifically targeted by the edited version of miRNAs (Fig. 5B). Since miRNA editing is more likely to play an important regulatory role for genes with more edited miRNA target sites, we examined Gene Ontology (GO) for genes regulated by multiple edited miRNAs (Fig. 5B). Edited miRNAs in neuronal and non-neuronal tissues were analyzed separately since they are likely involved in different functions. Interestingly, we found that in human, these targets of edited miRNAs in neuronal tissues are enriched in learning or memory and cognition (Fig. 5C), whereas targets of edited miRNAs in non-neuronal tissues are mainly enriched in organ development functions (Fig. 5C). Intriguingly, we found in mouse these targets of edited miRNAs in neuronal tissues are also enriched in learning or memory and cognition (Fig. 5D), although human and mouse have different pools of edited mature miRNA seeds (Fig. 5E). We further analyzed adult brain samples from wild-type and Adarb1−/− mice (Vesely et al. 2014) to test if edited miRNAs could alter miRNA targeting and affect target gene expression. No significant changes of target gene expression were observed, which may be because ADARB1 also regulates gene expression via functions that are unrelated to miRNA editing (Supplemental Note 5; Supplemental Fig. S16). Edited miRNAs in non-neuronal tissues in mouse were barely detectable, therefore were excluded from this analysis. Unexpectedly, these data suggest that RNA editing in miRNAs may be implicated in the neuronal function of ADARs in mammals. In concert with the observed prevalent miRNA editing in human non-neuronal tissues, these data also provide evidence that miRNA editing has recently gained non-neuronal functions in human.
RNA editing regulates asymmetric strand selection
miRNA biogenesis includes three important steps: DROSHA cleavage, DICER1 cleavage, and asymmetric strand selection. After being cleaved by DROSHA and DICER1, one miRNA strand in a given miRNA duplex is incorporated as the guide strand into Argonaute proteins during the formation of the RNA-induced silencing complex, whereas the other strand is discarded. The selection of the guide strand is an asymmetric, nonrandom process; in many cases, both strands function as mature miRNAs (Yang et al. 2011; Suzuki et al. 2015). RNA editing has been shown to affect DROSHA and DICER1 cleavage in several miRNAs, mostly by interfering with the miRNA processing (Yang et al. 2006; Kawahara et al. 2007a, 2008). To evaluate the impact of miRNA editing on miRNA biogenesis, we generated small RNA-seq data for human and mouse samples with a significant amount of pri-miRNA editing (Supplemental Table S7) and calculated the editing level difference between pri- and mature miRNAs, termed the “pri- versus mature miRNA difference” (PMD). We found that there exists a difference in editing levels between a substantial proportion of pri-miRNAs and their mature counterparts (Supplemental Fig. S17A–D). miRNAs with both positive PMD values and low editing levels in mature miRNAs have more editing sites (Supplemental Fig. S17B,D), suggesting that promiscuous miRNA editing may amplify such difference. Since miRNA editing is prevalent in different types of tissues in human, we next assessed the PMD values in multiple human tissues. We observed that miRNAs frequently have different PMD values in different tissues (Supplemental Fig. S17E), suggesting an unexpected dynamic tissue-specific PMD difference. The existence of the difference in editing levels between pri-miRNAs and their mature counterparts might be due to the interference of editing with the miRNA processing, which needs further experimental verification.
Asymmetric strand selection of single-stranded guide RNAs from miRNA duplex is the last and a crucial step in miRNA biogenesis; however, whether RNA editing plays a role in this process is unknown. The ample editing events identified in our study provide us a unique opportunity to address this question. We hypothesized that RNA editing occurring in miRNA duplex may reprogram asymmetric selection. To examine to what extent RNA editing affects strand selection, we integrated 4694 small RNA-seq data to calculate the correlation between editing level of an individual site and the 5p/3p ratio of the corresponding miRNA across all samples (Methods). We found that most of the editing sites in the 5p arm have negative correlations, whereas most of the sites in the 3p arm have positive correlations (Fig. 6A). This result suggests that, in many cases, RNA editing in one strand may promote the selection of the other strand. The correlation coefficients vary across different miRNAs and depend on the position of an editing event (Fig. 6A).
Figure 6.
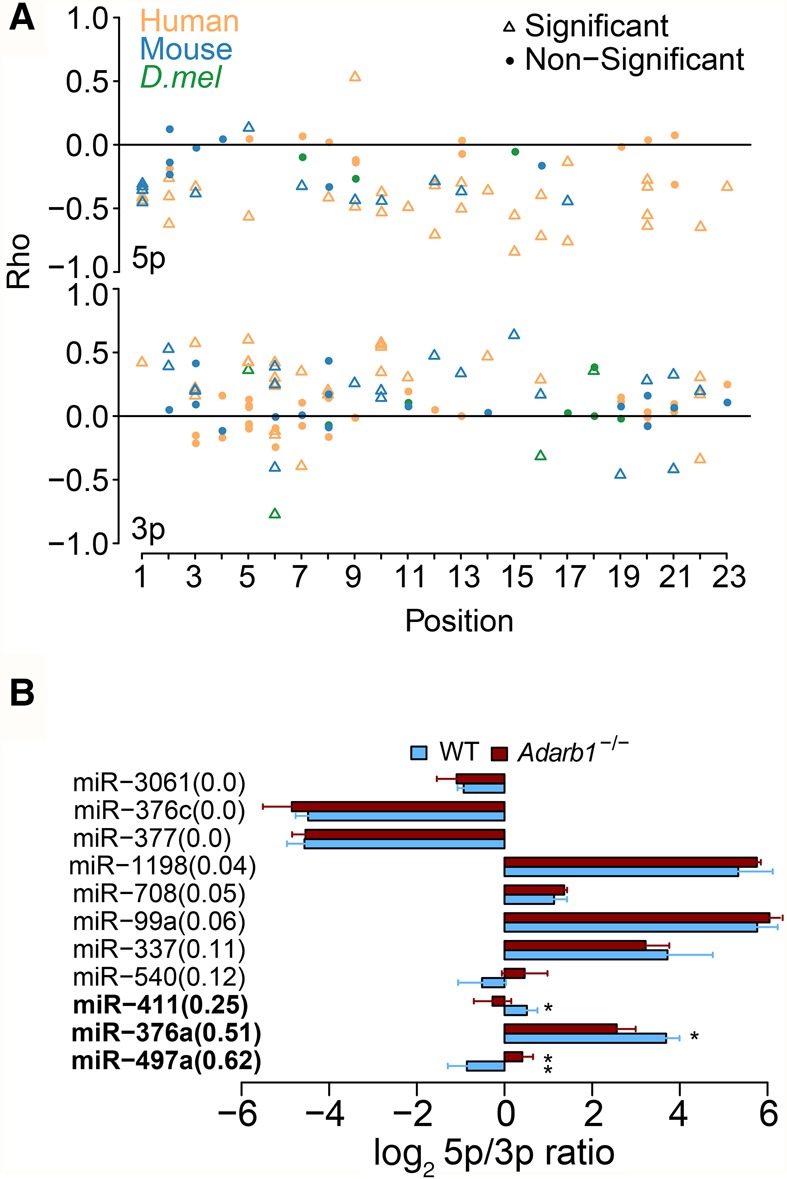
RNA editing modulates miRNA strand selection. (A) Spearman's correlation coefficient (rho) between the 5p/3p ratio and editing level for editing sites in 5p and 3p arms of the mature miRNAs were shown separately. (B) The comparison of the 5p/3p ratio of edited miRNAs between wild-type and Adarb1 knockout mice. We required that both 5p and 3p were covered by at least 30 reads and the 5p/3p ratio is between 0.01 and 100. The editing level difference between wild-type and Adarb1 knockout mice was shown in parentheses. (Error bars) SD. P-values were calculated using student t-test: (*) P < 0.05; (**) P < 0.01.
Next, we sought to validate our observation using mouse models depleted of ADAR or ADARB1 editing. First, we analyzed the wild-type and Adar−/− mouse model at E11 and E11.5 (Ota et al. 2013) due to embryonic lethality (Wang et al. 2000). We did not identify any miRNA editing event with an editing level ≥10%, because editing is barely detectable in the early embryo. From this data set, the maximal editing difference of any miRNA editing site between wild-type and Adar mutant is <10%. Such difference is too small to be used to validate the effect of editing on strand selection; we therefore excluded it for further analysis. Second, we analyzed adult brain samples from wild-type and Adarb1−/− mice (Vesely et al. 2014), since neuronal tissues have the highest overall miRNA editing levels. We found that for the edited miRNAs whose editing levels were altered the most in the Adarb1 knockout, the 5p/3p ratios are significantly different between wild-type and knockout mice (Fig. 6B). No such difference was observed for edited miRNAs with similar editing levels between wild-type and knockout mice. Thus, we provide evidence that miRNA editing is correlated with strand selection in vivo. Collectively, our data uncover a previously unknown role of miRNA editing on the asymmetric selection.
Discussion
In this work, through the profiling of 201 pri-miRNA samples and 4694 small RNA-seq samples, we present the first comprehensive miRNA A-to-I editing profiles in animals. This resource can serve as a reference for future studies on the regulation and functions of miRNA editing. Additionally, we developed a framework to identify and quantify RNA editing from small RNA-seq data using the pri-miRNA editing site resource we generated, further facilitating future miRNA editing studies. Most importantly, we found that miRNA editing is unexpectedly widespread; thus, RNA editing might impact miRNA biology more than previously thought. All the data from our study have been compiled into miREDB (miRNA editing database), a user-friendly database (http://miredb.sysu.edu.cn).
Besides A-to-I RNA editing, cytidine to uridine (C-to-U) RNA editing is also observed in mammals (Blanc and Davidson 2003; Zipeto et al. 2015). C-to-U RNA editing is mediated by cytidine deaminase APOBEC1, which is mainly expressed in the gastrointestinal tract, particularly small intestine, colon, and stomach (Uhlén et al. 2015). From our data, there are about 16 and 11 C-to-U variants per sample in human and mouse, respectively. To ask if C-to-U editing is present in miRNAs, we first compared the number of C-to-U variants identified in the gastrointestinal tract with the number of C-to-U variants identified in other tissues (Supplemental Fig. S18). If C-to-U editing does exist, we are expected to observe a higher C-to-U site number in the gastrointestinal tract. However, no such difference was observed, suggesting that C-to-U sites identified in miRNAs are likely to be false positive. We also randomly selected six C-to-U sites in the gastrointestinal tract and used PCR and Sanger sequencing to validate them. None of them were validated. Our result is consistent with two previous studies: One reported that APOBEC1 complementation factor (ACF) has high affinity to single-stranded but not double-stranded RNA (Mehta and Driscoll 2002); the other found no evidence for C-to-U editing of miRNAs from WT small intestine (Blanc et al. 2014).
It was previously revealed that site-selected editing of protein-coding genes in primates is induced by adjacent inverted Alu repeats or intronic duplex, which function as the recruitment elements for the ADAR enzymes (Daniel et al. 2013, 2014). In this study we found additional editing in the vicinity of pre-miRNAs. This observation suggests that the pre-miRNA hairpins may also work as the recruitment elements to facilitate the binding of ADARs and subsequent ADAR-mediated editing of flanking regions.
Interestingly, we also identified many more weak editing sites that do not have strong ADAR motif signature (Supplemental Fig. S6C). The fact that many As without strong motifs were edited in the pri-miRNA regions further supports the idea that pre-miRNA may form a rather tight dsRNA stem-loop structure, recruit ADARs, and function as the editing inducer to facilitate the RNA editing of many As without strong ADAR motifs.
A-to-I RNA editing has long been suggested to be involved in neuronal function, given that mouse and Drosophila mutants lacking ADAR enzymes display predominantly neurological phenotypes (Higuchi et al. 2000; Palladino et al. 2000). However, the specific editing events contributing to the neurological phenotypes are still to be discovered. miRNAs play regulatory roles in brain development, neuronal plasticity, as well as learning and memory (Kosik 2006; Im and Kenny 2012). Our work demonstrates that, in mammals, multiple edited miRNAs convergently obtain new targets related to synaptic function, learning or memory, and cognition. This observation sheds new light on the long-standing mystery in the RNA editing field, suggesting that miRNA editing events may contribute to the crucial regulatory role of ADARs in animal nervous system and be a driving force in human brain evolution.
Last, we observed a large difference of editing levels between pri- and mature miRNAs; in most cases, the editing level of a pri-miRNA is higher than that of the mature miRNA. Our observation is consistent with previous studies, which found that RNA editing of pri-miRNAs may inhibit miRNA processing (Yang et al. 2006; Kawahara et al. 2007a). It should be noted that a difference in the editing levels between pri- and mature miRNA does not always reflect the editing-induced difference in miRNA biogenesis. Other processes such as editing-induced mature miRNA degradation could also lead to such a difference. Additionally, caution should be taken since pri- and mature miRNA editing were detected by two different techniques in our analysis. Furthermore, we reveal, for the first time, the effect of RNA editing on miRNA asymmetric strand selection. This finding also has a wide range of translational applications to the oligonucleotide research, such as the design of siRNAs.
In conclusion, beyond being an epitranscriptomic resource, our work has provided unexpected biological insights into miRNA editing. These results together open new doors to the study of miRNA biogenesis and regulation.
Methods
Sample collection
Twenty-five human tissues were obtained. The total RNAs of the following 20 human tissues were purchased from Clontech: heart, bone marrow, cerebellum, brain (whole), fetal brain, liver, fetal liver, kidney, lung, placenta, prostate, skeletal muscle, spleen, testis, thymus, uterus, colon, small intestine, spinal cord, and stomach. An additional five human adult brain tissues dissected from specific brain regions from two individuals (donor N10 and N13) were obtained from the Chinese Brain Bank Center (Wuhan, China): anterior cingulate gyrus, callosum, cerebellar cortex, frontal gyrus, and substantia nigra. These human brain tissues were collected postmortem from individuals with no known medical history.
Twenty-one mouse tissues were obtained. C57BL/6J mice were purchased from the Laboratory Animal Center of Sun Yat-Sen University (SYSU). Adult tissues were obtained from 8- to 10-wk-old male and female mice. Fetal tissues were obtained from mice E18 to E20. All experiments were performed at the Animal Center of SYSU, in accordance with the Guide for the Care and Use of Laboratory Animals.
Thirty-one D.mel samples were obtained. Isogenic (w1118) embryos were collected at 2-h intervals for 24 h. Later-staged sample collections started with synchronized embryos and included resynchronizing with appropriate age indicators. Five larvae, six pupae, and two adult sexed stages (1- and 5-d old) were collected as previous described (Graveley et al. 2011).
RNA preparation
Total RNA was extracted with TRIzol and Direct-zol RNA Kits (Zymo Research).
miRNA selection
miRNA annotations were obtained from miRBase v20 (Human) or v21 (Mouse and D.mel). As pointed out by miRBase, many of the miRNAs deposited after 2007 were predicted from small RNA-seq experiments, and a substantial proportion of human and mouse miRBase miRNAs may not be real miRNA genes (Kozomara and Griffiths-Jones 2014). To obtain a set of bona fide human and mouse miRNAs for RNA editing identification, we selected miRNAs annotated as high-confidence miRNAs in miRBase. We also included additional low confidence miRNAs with considerable expression levels in small RNA-seq data analyzed by miRBase (more than 50 reads and greater than 50 RPM). Collectively, a refined list of 800 human miRNAs and 673 mouse miRNAs was used in our miR-mmPCR-seq experiments. For D.mel miRNAs, all 258 miRNAs from miRBase were used, since the Drosophila miRNA community has generally been conservative in their annotation of miRNAs, and the majority of the D.mel miRNAs are genuine (Kozomara and Griffiths-Jones 2014).
Mirtron annotations were obtained from previous studies (Chung et al. 2011; Ladewig et al. 2012).
Multiplex primer design
Multiplex PCR primers were designed as previously described (Zhang et al. 2014). Furthermore, to prevent primer design over variant sites, we masked any site that was polymorphic. For human miRNAs, we masked the genome using SNPs from dbSNP v138 and The 1000 Genomes Project (The 1000 Genomes Project Consortium 2012). Forty-eight pools of up to 16 primer pairs were designed (Supplemental Table S1), which cover a total of 712 miRNA loci (some loci contain two or three clustered miRNAs). For mouse miRNAs, we masked the genome using SNPs from dbSNP v138. Forty-eight pools of up to 12 primer pairs were designed (Supplemental Table S1), which cover a total of 555 loci. For D.mel miRNAs, we masked the genome using variants from the w1118 strain. Forty-three pools of up to eight primer pairs were designed (Supplemental Table S1), which cover a total of 247 loci. Most primers were designed to have amplicons between 160- and 300-bp long.
miR-mmPCR-seq
We previously developed mmPCR-seq (Zhang et al. 2014) to identify RNA editing sites and quantify their levels on protein-coding genes. To develop an optimized mmPCR-seq technology for pri-miRNA amplification, five reverse transcriptases were tested, including GoldScript (ThermoFisher, optimal reaction temperature 42°C), iScript (Bio-Rad, optimal reaction temperature 42°C), GoScript (Promega, optimal reaction temperature 42°C), SuperScript III (ThermoFisher, optimal reaction temperature 50°C–55°C), and ThermoScript (ThermoFisher, optimal reaction temperature 65°C).
Two rounds of DNase I treatment were performed to remove genomic DNA. Three micrograms of total RNA was used to synthesize the cDNA. cDNA was purified with Axygen AxyPrep Mag PCR clean-up beads (Axygen). Each cDNA library (300–500 ng) was preamplified using a primer pool (50 µM each) covering all sites. Preamplified product was purified using Axygen AxyPrep Mag PCR clean-up beads. Preamplified cDNAs and primer pools were loaded into the 48.48 Access Array IFC (Fluidigm), primed, mixed, amplified, and harvested. PCR products of each sample were subject to 15-cycle barcoding PCR and pooled together. All pools were combined at equal volumes and purified via Axygen AxyPrep Mag PCR clean-up beads. All libraries were sequenced on Illumina MiSeq or NextSeq 500 to produce 150-bp paired-end reads.
Identification of editing sites from miR-mmPCR-seq data
For each tissue or developmental stage, we merged all reads from biological and technical replicates into a single FASTQ file for variant calling. We adopted a pipeline that can accurately identify editing sites from RNA-seq data (Ramaswami et al. 2012). In brief, we first preprocessed the FASTQ files before mapping by trimming the adapters (cutadapt -e 0.1 --discard-trimmed -a AGATCGGAAGAGCACACGTC -a GACCTCGATAACGCTCGTGT), removing the first 20-bp primer sequences and last 10 bp of the reads (FASTX-Toolkit, fastx_trimmer -f 21 -l 140 -Q33), and filtering low-quality bases (cutadapt --trim-n -q 20,20 -m 90). We used BWA (Li and Durbin 2010) to align reads to the reference genome (bwa –n 6). We took variant positions in which the mismatch was supported by at least two mismatch reads with base quality score of 25 or higher and located at targeted loci. We further removed all known genomic variants: human, all SNPs present in dbSNP (except SNPs of molecular type “cDNA”; v138), The 1000 Genomes Project, and gDNA of donor N10 and N13; mouse, all SNPs present in dbSNP (except SNPs of molecular type “cDNA”; v138) and gDNA of C57BL/6J mouse; and D.mel, all SNPs present in gDNA of the w1118 strain. Additional filters were used to remove false positive sites and separate filtering criteria were used for variants occurring in Alu and non-Alu regions as we previously described (Ramaswami et al. 2012). We excluded sites with an extreme degree of variation (>97%), which are likely genomic SNPs. We removed sites in homopolymer runs of ≥5 bp. We removed sites in regions that were highly similar to other parts of the genome using BLAT (Kent 2002). Finally, we discarded samples with the proportion of A-to-G/T-to-C type <0.78 at the 5% editing level cutoff. We have provided the percentage of A-to-G/T-to-C of each sample (Supplemental Table S9) and a list of sites identified without the A-to-G/T-to-C proportion filter (Supplemental Table S4).
Quantification of editing levels from miR-mmPCR-seq data
For editing site identification, we mapped RNA-seq reads to the genome with BWA. For editing level quantification of known sites, we found that different aligners have very similar performance (Supplemental Fig. S19). Compared with BWA, gap-aware TopHat2 (Trapnell et al. 2009) can map spliced or unspliced RNA-seq reads to the genome at the same time. To simplify the pipeline, we used TopHat2 to quantify RNA editing level for both miR-mmPCR-seq and nascent RNA-seq data. For miR-mmPCR-seq data, we mapped the paired-end reads via TopHat2 (tophat2 --library-type fr-unstranded -N 6 --segment-mismatches 3 --read-edit-dist 10). Only sites covered by 50 or more reads were analyzed in all related analyses.
Validations by Sanger sequencing
To validate whether the newly identified editing sites are bona fide and to confirm the editing levels measured by miR-mmPCR-seq, we performed conventional Sanger sequencing on a set of randomly selected editing sites. A 25-μL PCR reaction was assembled with 2x rTaq Supermix (Genstar), ∼50 ng of gDNA (or ∼10 ng of cDNA) template, and 200 nM each of the forward and reverse primers. We used the following PCR program: 3 min at 94°C, 35 cycles for 15 sec at 94°C, 30 sec at 60°C, and 30 sec at 72°C. PCR amplicons were sequenced by BGI-Shenzhen. Primer sequences are listed in Supplemental Table S1.
Validation by nascent RNA-seq of the Adar null mutant
We obtained D.mel yellow white (yw) strain, Adar null mutant nascent RNA-seq from a recent study (Rodriguez et al. 2012). Reads were mapped via TopHat2 (Trapnell et al. 2009). Compared to BWA, gap-aware TopHat2 can map PE RNA-seq more accurately. We examined all identified A-to-G sites that are edited in the wild-type strain (defined as having more than two altered reads and editing levels >5%).
pri-miRNA editing analysis
To map each editing site to the so-called representative structure, we first apportioned each miRNA in the following regions: 5′ flanking region, pre-miRNA 5p region, central loop region, pre-miRNA 3p region, and 3′ flanking region. We further mapped each editing site to one of the regions and calculated the absolute distance between the editing site and the start/end of the region (Supplemental Fig. S13A). Next, the percentage of As that were edited at each position was calculated, using a selected miRNA as the representative secondary structure. Last, the miRNA editing density plot was drawn by SAVoR (Li et al. 2012). Free energy was calculated using the RNAfold program provided in the ViennaRNA package (Lorenz et al. 2011). To compare editing sites between human and mouse, we first identified miRNAs that are conserved between human and mouse. Conserved miRNAs were defined as two miRNA genes of reciprocal best hits. For conserved miRNAs, we further examined the editing site conservation. We converted the coordinates of sites between human and mouse using the LiftOver tool (http://genome.ucsc.edu). Since LiftOver does not provide strand information between two species, we obtained the strand information using pairwise alignment data from the UCSC Genome Browser. For positions that were successfully lifted over, we determined the nucleotide using the pairwise alignments in axt format from the UCSC Genome Browser.
mRNA quantification via real-time PCR (RT-PCR)
Total RNAs used for RT-PCR were pretreated with on-column DNase I in the purification step. To synthesize the cDNA with iScript Advanced cDNA Synthesis Kit (Bio-Rad), 2.5 µg of total RNA was used. PCR by SYBR green (Bio-Rad) for each cDNA was run in triplicates. The housekeeping genes (human, ACTB; mouse, Actb) were used as an internal control for mRNA RT-PCR. Primers are listed in Supplemental Table S10.
Small RNA-seq library construction
Small RNA-seq were performed as previously described (Vigneault et al. 2012) with the following modification: Instead of using a single RNA adapter for 5′ adapter ligation, we used pooled RNA adapters (rUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrN), which remove the sequence-specific biases of RNA ligases (Jayaprakash et al. 2011). All libraries were sequenced on HiSeq 2500 or NextSeq 500 (Illumina) to produce single-end 67-bp reads.
Identification of edited mature miRNAs
Custom edited pre-miRNA databases were generated by replacing the A with G for editing positions located at the mature miRNA region. For miRNAs with multiple editing sites in a mature miRNA region, edited pre-miRNAs with all possible combinations were generated.
Small RNA-seq data were obtained from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/). Only small RNA-seq data generated by Illumina platform were used. A total of 2721, 1552, and 421 samples in human, mouse, and D.mel were obtained (Supplemental Table S7). We removed low-quality reads, trimmed adapter sequences (cutadapt –q 20,20 --trim-n), collapsed identical reads, and retained reads with lengths ≥16 bp and ≤25 bp. As the 3′ end of animal miRNA sequences is subject to A or U addition (Burroughs et al. 2010), the last two bases of a read were trimmed if they are A or U. The filtered and trimmed reads were aligned using BLAST against the pre-miRNAs first (E-value <0.1; no mismatch allowed) (Altschul et al. 1990). The remaining unmapped reads were aligned using BLAST against custom edited pre-miRNA databases (E-value <0.1; no mismatch allowed). To ensure accurate RNA editing identification and measurement, only data sets in which the percentage of bases after quality trimming is ≥60% and the number of reads (with lengths ≥16 bp and ≤25 bp) mapped to miRNAs is ≥0.1 million were selected. Finally, for editing positions, we filtered Gs with low-quality scores (base quality score <30) (Alon et al. 2012); only positions supported by two or more G reads, ≥5% editing levels, and with significant modification (BH multiple testing correction P < 0.05) compared with the estimated sequencing error rate, as determined by binomial cumulative distribution, were recorded (Alon et al. 2012). Finally, the putative edited miRNAs were further mapped to the reference genome, and those perfectly mapped to the genome were removed.
miRNA target site analysis
We used the TargetScan algorithm (Agarwal et al. 2015) to predict miRNA target sites for the unedited and edited versions of miRNAs. Since I:C and G:C base pairs contribute similarly to the hybridization of miRNAs to their targets (Kawahara et al. 2007b), we replaced edited A with G when predicting targets of edited miRNAs. Only canonical 7–8 nt 3′ UTR sites were considered. For human and mouse miRNAs, we used the TargetScan7 context++ model and calculated context++ score of each target site (Agarwal et al. 2015), except that we did not account for the contribution of site conservation to achieve a fair comparison between unedited and edited miRNAs. For D.mel miRNAs, we used the TargetScan6 prediction since the TargetScan7 context++ model is not available and also did not account for the contribution of site conservation. All UTR sequences were downloaded from the TargetScan database (http://www.targetscan.org/). Human ORF sequences were downloaded from TargetScan database, and mouse ORF sequences were downloaded from UCSC Genome Browser. Putative targets with context++ score ≥0.8 were used for GO term analysis. GO term enrichment was analyzed in R (v3.3.1) using clusterProfiler packages (v3.0.4) (Yu et al. 2012) with the dependent packages of GO.db (v3.3.0) and GOSemSim (v1.30.3). We chose a miRNA number cutoff by which the genes targeted are about the top 15% of the “gain” group target genes. Using this cutoff, for human neuronal tissues, genes targeted by at least six edited miRNAs were used for GO term analysis; for human non-neuronal tissues, genes targeted by at least four edited miRNAs were used; for mouse neuronal tissues, genes targeted by at least six edited miRNAs were used. Mouse non-neuronal tissues were excluded from analysis due to the limited number of edited miRNAs identified. The lists of all genes expressed in corresponding tissues were used as background for enrichment analyses. The gene expression levels were obtained using Human BodyMap 2.0 (GSE30611) or Mouse Encode Project (http://chromosome.sdsc.edu/mouse/download.html). Genes with RPKM of 1 or more were selected. P-values were adjusted for multiple tests using the BH method.
Asymmetric strand selection analysis
Editing levels and 5p/3p ratios were calculated from all filtered small RNA-seq data sets. Mature miRNAs that can be mapped to multiple positions in the genome were excluded from analysis. Editing levels of individual sites were quantified as the number of G reads divided by the total number of A and G reads mapped to an editing site when the latter was 20 or more reads. To calculate the 5p/3p ratio, we required that both the 5p and 3p arms are covered by 10 or more reads. To calculate the correlation between editing level of individual site and the 5p/3p ratio of the corresponding miRNA, several criteria were applied for preprocessing of data. We required that (1) the sample size is 15 or higher, (2) the editing level is ≥10% in at least one sample, and (3) the mean value of 5p/3p ratios of all samples is between 0.01 and 100. Spearman's rho and P-value were calculated using R (R Core Team 2014).
Data access
The sequence data generated for this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA398715. The Sanger traces have been submitted to NCBI Trace Archive (https://trace.ncbi.nlm.nih.gov/) under accession numbers 2344290146–2344290280.
Supplementary Material
Acknowledgments
We thank Jin Billy Li, Patricia Deng, Xin Hong, and members of the Zhang Laboratory for critical reading and discussion of the manuscript. We thank Guangzhou Xushen Intelligent Technology Co., Ltd. and SYSU Ecology and Evolutionary Biology Sequencing Core Facility for sequencing service. This study was supported by grants from the National Natural Science Foundation of China (31571341 and 91631108 to R.Z.), Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S638 to R.Z.), Guangdong Major Science and Technology Projects (2017B020226002 to R.Z.), and The Thousand Talents Plan–The Recruitment Program for Young Professionals (R.Z.).
Author contributions: R.Z. and N.G. conceived the project. L.L., N.G., W.C., Q.F., and R.Z. conducted the experiments. Y.S., X.S., J.L., S.X., Z.H., and R.Z. performed bioinformatics analysis. R.Z. wrote the manuscript with input from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.224386.117.
References
- The 1000 Genomes Project Consortium . 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam JW, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon S, Mor E, Vigneault F, Church GM, Locatelli F, Galeano F, Gallo A, Shomron N, Eisenberg E. 2012. Systematic identification of edited microRNAs in the human brain. Genome Res 22: 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc Ser B (Methodol) 57: 289–300. [Google Scholar]
- Blanc V, Davidson NO. 2003. C-to-U RNA editing: mechanisms leading to genetic diversity. J Biol Chem 278: 1395–1398. [DOI] [PubMed] [Google Scholar]
- Blanc V, Park E, Schaefer S, Miller M, Lin Y, Kennedy S, Billing AM, Ben Hamidane H, Graumann J, Mortazavi A, et al. 2014. Genome-wide identification and functional analysis of Apobec-1-mediated C-to-U RNA editing in mouse small intestine and liver. Genome Biol 15: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. 2006. RNA editing of human microRNAs. Genome Biol 7: R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y, et al. 2010. A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res 20: 1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. 2007. microRNA functions. Annu Rev Cell Dev Biol 23: 175–205. [DOI] [PubMed] [Google Scholar]
- Chang TC, Pertea M, Lee S, Salzberg SL, Mendell JT. 2015. Genome-wide annotation of microRNA primary transcript structures reveals novel regulatory mechanisms. Genome Res 25: 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Agius P, Westholm JO, Chen M, Okamura K, Robine N, Leslie CS, Lai EC. 2011. Computational and experimental identification of mirtrons in Drosophila melanogaster and Caenorhabditis elegans. Genome Res 21: 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Venø MT, Ekdahl Y, Kjems J, Öhman M. 2013. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res 40: 9876–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Silberberg G, Behm M, Öhman M. 2014. Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol 15: R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Taft RJ, Hashimoto T, Kanamori-Katayama M, Kawaji H, Kawano M, Kishima M, Lassmann T, Faulkner GJ, Mattick JS, et al. 2010. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res 20: 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81. [DOI] [PubMed] [Google Scholar]
- Im HI, Kenny PJ. 2012. MicroRNAs in neuronal function and dysfunction. Trends Neurosci 35: 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaprakash AD, Jabado O, Brown BD, Sachidanandam R. 2011. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res 39: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. 2007a. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer–TRBP complex. EMBO Rep 8: 763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. 2007b. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. 2008. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res 36: 5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139. [DOI] [PubMed] [Google Scholar]
- Kosik KS. 2006. The neuronal microRNA system. Nat Rev Neurosci 7: 911–920. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42(Database issue): D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume H, Hino K, Galipon J, Ui-Tei K. 2014. A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res 42: 10050–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. 2012. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res 22: 1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ryvkin P, Childress DM, Valladares O, Gregory BD, Wang LS. 2012. SAVoR: a server for sequencing annotation and visualization of RNA structures. Nucleic Acids Res 40(Web Server issue): W59–W64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Driscoll DM. 2002. Identification of domains in apobec-1 complementation factor required for RNA binding and apolipoprotein-B mRNA editing. RNA 8: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. 2013. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 153: 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. 2000. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102: 437–449. [DOI] [PubMed] [Google Scholar]
- Porath HT, Carmi S, Levanon EY. 2014. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5: 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Ramaswami G, Li JB. 2016. Identification of human RNA editing sites: a historical perspective. Methods 107: 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB. 2012. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 9: 579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Menet JS, Rosbash M. 2012. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 47: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Jiang J, Liu Q, Yang L. 2004. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res 32: e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Katsura A, Yasuda T, Ueno T, Mano H, Sugimoto K, Miyazono K. 2015. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat Struct Mol Biol 22: 512–521. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419. [DOI] [PubMed] [Google Scholar]
- Vesely C, Tauber S, Sedlazeck FJ, Tajaddod M, von Haeseler A, Jantsch MF. 2014. ADAR2 induces reproducible changes in sequence and abundance of mature microRNAs in the mouse brain. Nucleic Acids Res 42: 12155–12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneault F, Ter-Ovanesyan D, Alon S, Eminaga S, C Christodoulou D, Seidman JG, Eisenberg E, M Church G. 2012. High-throughput multiplex sequencing of miRNA. Curr Protoc Hum Genet Chapter 11: Unit 11.12.1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K. 2000. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290: 1765–1768. [DOI] [PubMed] [Google Scholar]
- Westholm JO, Lai EC. 2011. Mirtrons: microRNA biogenesis via splicing. Biochimie 93: 1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff BE, Nishikura K. 2015. Modulation of microRNA expression and function by ADARs. Curr Top Microbiol Immunol 353: 91–109. [DOI] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. 2006. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 13: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC. 2011. Widespread regulatory activity of vertebrate microRNA* species. RNA 17: 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Li X, Ramaswami G, Smith KS, Turecki G, Montgomery SB, Li JB. 2014. Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nat Methods 11: 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipeto MA, Jiang Q, Melese E, Jamieson CH. 2015. RNA rewriting, recoding, and rewiring in human disease. Trends Mol Med 21: 549–559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.