

Abstract
Purpose
This study examines the role of PI3K/Akt pathway in δ-opioid receptor agonist (SNC-121)-induced RGC neuroprotection in a chronic glaucoma rat model.
Methods
Injecting hypertonic saline into the limbal veins of Brown Norway rats elevated IOP. Rats were treated either with 1 mg/kg SNC-121 or 3 mg/kg 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride (LY-294002; PI3K/Akt inhibitor) plus SNC-121 once daily for 7 days. Pattern ERGs were recorded in response to contrast reversal of patterned visual stimuli. Retinal ganglion cells (RGC) were visualized by Fluorogold retrograde labeling. Optic nerve head (ONH) astrocytes were pretreated with PI3K/Akt inhibitors for 30 minutes followed by 1-μM SNC-121 treatment. Changes in matrix metalloproteinases (MMP-1, -2, and -3) production and PI3K/Akt activation in optic nerve and TNF-α treated ONH astrocytes were measured by immunohistochemistry and Western blotting.
Results
SNC-121 activates the PI3K/Akt pathway in ONH astrocytes and the retina. In ONH astrocytes, SNC-121–induced Akt activation was fully inhibited by PI3K/Akt inhibitors. A sustained decline (7–42 days post injury) in Akt activation was seen in the ocular-hypertensive retina and optic nerve. This decline is reversed to normal levels by 1-mg/kg intraperitoneally (i.p.) SNC-121 treatment. Both pattern ERG amplitudes and RGC numbers were reduced in ocular hypertensive eyes, which were significantly increased in SNC-121–treated animals. Interestingly, SNC-121–induced increase in pattern-ERG amplitudes and RGC numbers were inhibited in LY-294002 pretreated animals. Additionally, SNC-121 treatment inhibited MMP-1, -2, and -3 production from the optic nerve of ocular hypertensive rats and TNF-α–treated ONH astrocytes.
Conclusions
PI3K/Akt pathway plays a crucial role in SNC-121–mediated RGC neuroprotection against glaucomatous injury.
Keywords: δ-opioid receptor, retina, glaucoma, retinal ganglion cells, PI3K/Akt, phosphorylation
Glaucoma is characterized by “cupping” of the optic nerve head (ONH), progressive loss of retinal ganglion cells (RGCs) whose axons course throughout the optic nerve head to synapse in the lateral geniculate body, and a decline in visual field. Accumulating literature suggests that multiple mechanisms and pathways are involved in RGC death in glaucoma including elevated IOP,1 inflammation,2–5 apoptosis,6–8 and neurotrophic factor deprivation.9,10 During the progression of glaucoma, activation of prodeath pathways cause RGC death; but, due to intrinsic neuroprotective capabilities of the retina, adaptive prosurvival signaling pathways are also simultaneously activated to counteract the injury. However, prodeath pathways predominate in RGCs when the intrinsic cell survival machinery loses the capacity to resist extrinsic injury. The current therapeutic strategy to cure glaucoma is to halt or slow down the disease progression by reducing elevated IOP. The lowering of IOP can help to retard the disease progression in many glaucoma patients. Unfortunately, it is not always successful, and many patients continue to experience the progressive loss of RGCs.11,12 Due to this limited therapy for glaucoma, there is a need to identify novel and unique targets to develop neuroprotective treatment strategies to cure glaucoma.
Glial cells (e.g., ONH astrocytes) provide cellular support to the axons. Normal ONH astrocytes are considered to be quiescent,13 but they become reactive in response to glaucomatous injury.14–16 Activated astrocytes have been shown to release a host of molecules ranging from proinflammatory cytokines, chemokines, immune mediators, adhesion molecules, nitric oxide, and reactive oxygen species, which establish a hostile microenvironment that is detrimental for RGCs.2,4,17,18 In spite of extensive research to understand glial-neuron interactions and behavior changes in response to injury, the actual sequence of events in glial cells that regulates the death/survival process for RGCs remains largely unclear. Hence, understanding the molecular mechanisms in ONH astrocytes that play a crucial role in the regulation of RGC death is critical for the development of better therapeutic agents for glaucoma treatment.
Earlier, we showed the neuroprotective roles of δ-opioid receptor agonists against ocular hypertension-induced injury in a chronic glaucoma rat model.2,19,20 However, the underlying molecular and cellular mechanisms in δ-opioid receptor–mediated neuroprotection remain poorly defined. In other systems, studies have shown that δ-opioid receptor–mediated neuroprotection involves multiple signaling cascades. These pathways include protein kinase C (PKC), mitogen-activated protein kinase, and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt).21–23 The PI3K/Akt signaling pathway protects neurons against various forms of injury in the brain.24 Mechanistically, phosphorylated and activated Akt inhibits downstream targets, such as proapoptotic proteins and caspases.25–28 The role of the PI3K/Akt pathway in δ-opioid receptor–mediated RGC neuroprotection remains unexplained. Therefore, the focus of the current study is to determine the potential neuroprotective role of the PI3K/Akt (e.g., prosurvival) pathway in δ-opioid receptor agonist–mediated neuroprotection in chronic glaucoma rat model.
Methods
Animals
Equal numbers of adult male and female Brown Norway rats (2–4 months of age; 125–200 g; Harlan Laboratories, Inc., Indianapolis, IN, USA) were used in this study. Animals were kept under a cycle of 12-hours light and 12-hours dark for all the studies. Prior to each animal procedure, rats were anesthetized with ketamine (75 mg/kg) and xylazine (8 mg/kg). The body temperature of each animal maintained at 37°C with a heating pad. All the animals were handled as per the guidelines of ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The Animal Care and Use Committee at the Medical University of South Carolina approved this study. SNC-121 (1 mg/kg) and 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride (LY-294002; 3 mg/kg) were injected intraperitoneally (i.p.) in rats once a day for 7 days.
Development of Glaucoma Model by Hypertonic Saline Injection
Glaucomatous injury was induced in rat eyes by injecting hypertonic saline. A stable baseline IOP was documented prior to hypertonic saline injection. Before saline injections, proparacaine (0.5%) was applied topically to anesthetize the cornea. Approximately 50 μL of 2-M hypertonic saline was injected into the limbal venous system as previously described.2,20,29,30 To control the bacterial infection, antibacterial ointment (neomycin) was applied at the injection site. IOP was recorded as the average of six to eight consecutive measurements prior to surgery (baseline IOP; 18–20 mm Hg) followed by IOP measurement on a weekly basis after treatment, using a calibrated Tonolab tonometer (Colonial Medical Supply Company, Franconia, NH, USA), as described earlier.2,20,29 IOP exposure for each animal was calculated, as described earlier.29
Pattern Electroretinogram Recordings
Pattern ERGs were measured in nondark adapted animals (sequentially) 3 days prior to IOP elevation by hypertonic saline injection, and then biweekly postsurgery, as described earlier.2,20,29 The pattern ERG electrode was placed on the corneal surface using micromanipulator on undilated pupil as described earlier.2,20,29 A visual stimulus generated by black and white alternating contrast reversing bars (mean luminance, 50 cd/m2; spatial frequency, 0.033 cycle/deg; contrast, 99%; and temporal frequency, 1 Hz) was aligned with the projection of the pupil using the UTAS-2000 (LKC Technologies, Gaithersburg, MD, USA). The pattern ERGs were measured between a peak and adjacent trough of the waveform.
Retrograde Labeling of Retinal Ganglion Cells
RGCs were visualized by retrograde labeling as described earlier.2,20,29 Briefly, 3 μL of a 5% solution of Fluorogold in PBS was injected into the superior colliculus of anesthetized ocular-hypertensive animals immobilized in a stereotaxic apparatus. A small hole 1/8″ drilled in the skull 6 mm from bregma and 2 mm from lambda and Fluorogold was injected at 4-mm downward using a Hamilton syringe (Hamilton Company, Reno, NV, USA). Bone wax 903 (Lukens Cat # 2007-05; West Columbia, SC, USA) was used to fill the skull hole. After surgery, an antibacterial ointment (erythromycin) was applied to the wound to prevent infection. Animals were euthanized 7 days post Fluorogold injection. The eyes were enucleated and fixed in 4% paraformaldehyde (PFA) for 24 hours at 4°C. RGCs were counted in the inner and peripheral retina at a distance of 1.5 to 2.00 mm and 3.5 to 4.00 mm from the optic disc, respectively. The RGCs numbers averaged using eight microscopic fields of identical size (150 μm2; ×20 magnification) per retina using ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry
Eyes were removed at day 7 and 42 post-hypertonic saline injections and immunohistochemistry was performed as described previously.2,20,29 Eyes were fixed in 4% PFA for 4 hours followed by incubation in 30% sucrose solution overnight at 4°C. The eyecups were washed in ice cold PBS and frozen in optimal cutting temperature-embedding medium over dry ice. Cryosections were cut at −28°C, fixed in cold methanol for 10 minutes, and rinsed in 1× Tris-buffered saline (TBS), pH 7.5. Tissue permeabilization was performed using 0.2% Triton X-100 in TBS and washed again with TBS. Tissues were then blocked with 5% BSA in TBS for 1 hour at room temperature, followed by overnight incubation (4°C) with primary antibodies (e.g., anti–phospho-Akt antibody, 1:100 dilution; anti–glial fibrillary acidic protein [GFAP], 1:200; or 0.5% BSA). Tissues were then washed with TBS and incubated with fluorescein-conjugated secondary antibody (1:400 dilution; Alexa Fluor 488 or Rhodaminel Jackson Immuno Research Laboratories, Inc., West Grove, PA, USA) for 1 hour at 20°C. Primary antibody omitted from negative control slides. The slides were imaged under a bright-field microscope equipped with epifluorescence, and digitized images were captured.
Optic Nerve Head Astrocyte Cultures
Human eyes were obtained from NDRI (Philadelphia, PA, USA) to isolate ONH astrocytes as described previously.31 Briefly, primary ONH astrocytes were isolated from the ONH tissue of human eyes. The optic nerve freed of sclera tissue and the central retinal vessels were cut into four pieces. These pieces were then plated onto a collagen-I–coated cell culture plate and allowed to grow 2 to 3 weeks in Dulbecco's modified Eagle's medium F12 (DMEM F12; GE Healthcare Life Sciences, Logan, UT, USA) containing 10% fetal bovine serum (FBS) and antibiotics. ONH astrocytes were purified and immunopanned from the mixed cell culture as described earlier.31 Purified astrocytes were grown in astrocytes growth medium called “AGM SingleQuots kit” (catalog # CC4123; LONZA, Inc., Allendale, NJ, USA) which contains 3% FBS, 0.1% Ascorbic acid, 0.1% mEGF, 0.25% insulin, and 1% L-Glutamine. In this study, ONH astrocytes of passages two to five were used. ONH astrocytes were starved for 16 hours prior to SNC-121 and PI3K and Akt inhibitor treatment. Cells were starved in Astrocyte Basal Medium (ABM catalog # CC3187; LONZA, Inc.), which was devoid of serum, Ascorbic acid, 0.1% mEGF, and 0.25% insulin.
Western Blotting
Equivalent amounts of retina, optic nerve extract, or cell lysate of human ONH astrocytes (15-μg protein/lane) were loaded onto 10% SDS-PAGE, proteins separated and transferred to nitrocellulose membranes as described earlier.2,20,29,30 Membranes were incubated with appropriate primary antibodies (e.g., anti–phospho-PI3K, anti–phospho-Akt, anti-Akt, anti–MMP-1, anti–MMP-2, anti–MMP-3 at 1:1000 dilution or anti-β-actin at 1:3000 dilution) for 12 hours at 4°C. After washing, membranes were incubated for 1 hour at 20°C with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The chemiluminescent signal was captured using enhanced chemiluminescent reagent by a Biorad Versadoc imaging system (Biorad, Hercules, CA, USA).
Statistical Analysis
Statistical analysis was performed by Student's t-test for paired data. ANOVA Bonferroni posttest was used for multiple comparisons (GraphPad Software, Inc., San Diego, CA, USA). P ≤ 0.05 was considered significant.
Results
Effects of SNC-121 on PI3K/Akt Activation in ONH Astrocytes
To determine the effects of serum on Akt phosphorylation and whether phosphorylation was impacted by SNC-121 treatment in the presence of serum, we treated ONH astrocytes with 3% FBS alone and in the presence of 1-μM/L SNC-121. As shown in Figure 1, serum (3%) increases the Akt phosphorylation by 111% (P < 0.05), which was not increased further when cells were treated with SNC-121 in the presence of serum. Considering this observation, we chose to starve the cells (serum deprivation) prior to SNC-121 treatment in all subsequent experiments. To determine if a selective δ-opioid receptor agonist, SNC-121, activated and phosphorylated the PI3K/Akt pathway, primary cultures of human ONH astrocytes were treated with 1-μM/L SNC-121 and changes in the phosphorylation state of PI3K and Akt were measured by Western blotting. As shown in Figures 2A and 2B, SNC-121 increased phosphorylation of Akt (60 kDa) and its upstream regulator, PI3K (p55), in a time-dependent manner, with maximum activation occurring at 15 to 30 minutes. Furthermore, 1-μM/L SNC-121–induced Akt phosphorylation was fully blocked by a PI3K/Akt inhibitor, LY-294002 (1 μM/L), and highly selective Akt inhibitors, perifosine (10 μM/L), and MK-2206 (100 nM/L, Figs. 3A–C).
Figure 1.

Effects of 3% FBS and SNC-121 on Akt phosphorylation in ONH astrocytes. Cells were treated with 3% FBS alone or 3% FBS + SNC-121 (1 μM) for 30 minutes. After incubation, cells were lysed using buffer (20 mM β-glycerophosphate, pH 7.5 containing 1% Triton X-100, 20-mM EGTA, 20-mM NaF, 15-mM MgCl2, 1-mM Sodium Orthovanadate, and 0.3% Mercaptoethanol). Cell lysates (15-μg proteins) were analyzed by Western blotting using selective anti–phospho-Akt and anti-Akt antibodies. The signal was captured using enhanced chemiluminescent reagent and the Biorad Versadoc imaging system. Data expressed as mean ± SE. *P < 0.05; n = 9.
Figure 2.

SNC-121–induced activation of PI3K (A) and Akt (B) in ONH astrocytes. ONH astrocytes were starved in serum-free medium for 16 hours. Cells were then treated with SNC-121 (1 μM) for the indicated time intervals. Cell lysates (15-μg proteins) were analyzed by Western blotting using selective anti–phospho-PI3K or anti-phospho-Akt antibodies. The band intensities were captured using enhanced chemiluminescent reagent and the Biorad Versadoc imaging system. Data are expressed as mean ± SE. *P < 0.05; n = 5–9 for phospho-PI3K and n = 7–13 for phospho-Akt.
Figure 3.

Effects of PI3K/Akt inhibitor LY-294002 (A), Akt inhibitor, perifosine (B), and Akt inhibitor, MK-2206 (C) on SNC-121-induced phosphorylation of Akt in ONH astrocytes. ONH astrocytes were starved in serum-free medium for 16 hours followed by SNC-121 (1 μM) treatment for 30 minutes. ONH astrocytes were preincubated with LY-294002 (1 μM/L), perifosine (10 μM/L), or MK-2206 (100 nM/L) to block Akt phosphorylation for 30 minutes before SNC-121 treatment. Cells lysates (15-μg proteins) were analyzed by Western blotting using selective anti–phospho-Akt and anti-Akt antibodies. The band intensities were captured using enhanced chemiluminescent reagent and the Biorad Versadoc imaging system. Data are expressed as mean ± SE. *P < 0.05; n = 5–9, ANOVA.
Effects of SNC-121 on Akt Activation in Ocular-Hypertensive Eyes
Next, we wanted to determine if ocular hypertension caused a decline in Akt phosphorylation and activation in the retina and optic nerve. The IOP was elevated by injecting hypertonic saline in limbal veins in Brown Norway rats, and a significant increase in IOP was observed as early as 7 days (Table), which remained significantly higher than the contralateral eyes for up to 6-weeks post injury (P < 0.05). Both retinas and optic nerves of contralateral and ocular-hypertensive eyes were analyzed for phospho-Akt at day 7 and 42, post injury. A sustained decline (7- to 42-days post injury) in Akt phosphorylation was seen in the ocular-hypertensive retina (Fig. 4) and optic nerve (Figs. 5, 6). To determine the effects of a selective δ-opioid receptor agonist (SNC-121), animals were treated with 1-mg/kg (i.p.) SNC-121 for 7 days, once daily. Interestingly, the decline in phospho-Akt in hypertensive optic nerve was reversed by SNC-121 treatment at day 7 (Figs. 5, 7) and 42 (Fig. 6), post injury. Furthermore, we analyzed the co-localization of p-Akt and GFAP (glial cell marker) in optic nerve of contralateral, ocular hypertensive, and SNC-121–treated ocular-hypertensive eyes. We noticed mild staining of p-Akt and GFAP in the contralateral eyes. As expected, p-Akt staining was diminished and GFAP staining was increase robustly in ocular hypertensive eyes. Subsequently, SNC-121 treatment increases the staining of p-Akt and decreases the staining of GFAP, respectively, in ocular-hypertensive eyes (Figs. 5, 6). We also noticed a co-localization of p-Akt and GFAP in optic nerve sections (shown by arrowhead), suggesting that glial cells are one of the targets where Akt phosphorylation is changing by SNC-121 treatment.
Table.
Measurements of IOP in Normal and Ocular Hypertensive Eyes
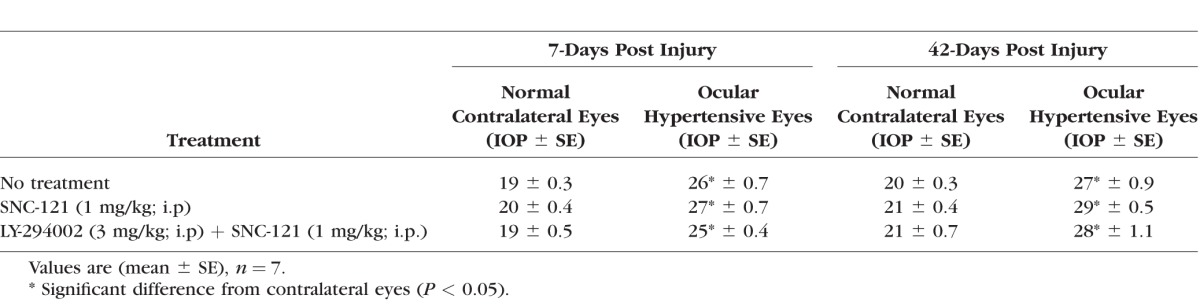
Figure 4.

Both contralateral and ocular hypertensive eyes of Brown Norway rats were removed on day 7 and 42, post injury. Contralateral eyes were used as the control. Cryosections were immunostained by anti–phospho-Akt antibodies. We did not see any positive staining when primary antibodies were omitted (not shown). Data are a representation of at least four independent experiments. Comparable staining for phospho-Akt was seen in at least eight animals. NFL, nerve fiber layer; RGCL, retinal ganglion cells layer; IPL, inner plexiform layer.
Figure 5.

Changes in Akt phosphorylation in the optic nerve of contralateral, ocular hypertensive, and SNC-121–treated ocular hypertensive eyes. Optic nerves of untreated and SNC-121–treated animals were collected at day 7, post injury. Coronal sections of optic nerve were analyzed for phospho-Akt by immunohistochemistry using anti–phospho-Akt and anti-GFAP antibodies. Data are a representation of at least four independent experiments. Scale bar: 20 μm.
Figure 6.

Co-localization of p-Akt and GFAP and changes in Akt phosphorylation and GFAP expression in the optic nerve of contralateral, ocular hypertensive, and SNC-121–treated ocular-hypertensive eyes. Optic nerves of untreated and SNC-121–treated animals were collected at day 42, post injury and analyzed for phospho-Akt and GFAP by immunohistochemistry using anti–phospho-Akt (1:100) and anti-GFAP (1:200) antibodies. Co-localization of phosphor-Akt and GFAP is indicated by arrowhead. Data are a representation of at least four independent experiments. Scale bar: 20 μm.
Figure 7.
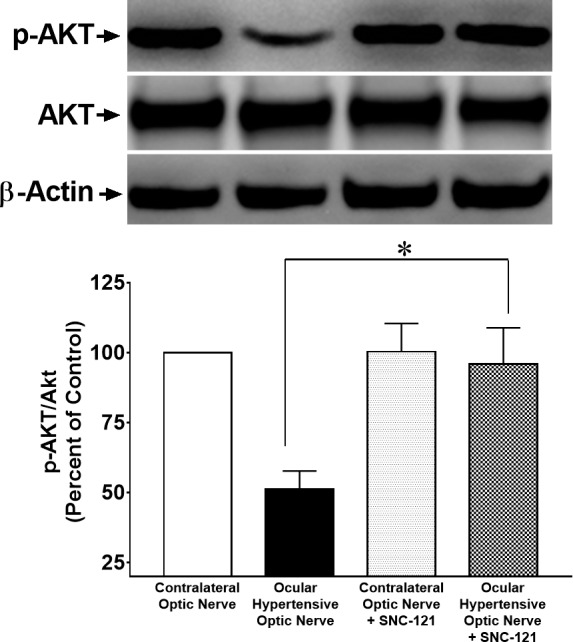
Changes in Akt phosphorylation in the optic nerve of ocular-hypertensive rats in response to the injury and SNC-121 treatment. Optic nerves of untreated and SNC-121–treated hypertensive eyes were collected at day 7, post injury and analyzed for phospho-Akt by Western blotting using anti–phospho-Akt antibodies. The quantitation of band intensities performed by densitometry. Data expressed as mean ± SE. *P < 0.05; n = 6.
Attenuation of SNC-121–Induced Neuroprotection by PI3K/Akt Inhibitor
Next, we assessed functional changes in RGCs by measuring the pattern ERGs as described earlier.2,20,29 Similar to our previous studies,2 SNC-121 treatment provided neuroprotection as determined by pattern ERGs at 2-, 4-, and 6-weeks post injury (Fig. 8). The SNC-121–induced RGC neuroprotection was blocked when animals were pretreated with a PI3K/Akt inhibitor (LY-294002; 3 mg/kg; i.p.) at all time points (e.g., 2-, 4-, and 6-weeks post injury; Fig. 8). We did not see any significant effects of LY-294002 and SNC-121 on pattern ERG amplitudes of normal contralateral eyes. However, we have seen a small decline in pattern ERG of ocular-hypertensive eyes that were treated with LY-294002. To further confirm that the declines in the pattern-ERG amplitudes were associated with RGC loss, RGCs were visualized by retrograde labeling with bilateral injections of Fluorogold into the superior colliculus. Representative micrographs of Fluorogold-labeled cells indicated a clear loss of RGCs in the ocular-hypertensive eye as compared with the contralateral eye. The loss of RGCs in ocular-hypertensive eyes was reduced when animals were treated with SNC-121 (1 mg/kg; i.p.) for 7 days (Figs. 9A, 9B). As expected, animals that received LY-294002 prior to SNC-121 treatment showed a significant reduction in RGC numbers. Quantification of RGCs in normal eyes (2150 ± 90), ocular-hypertensive eyes (1794 ± 105), SNC-121–treated eyes (2094 ± 76), and LY-294002 and SNC-121–treated ocular-hypertensive eyes (1584 ± 137) are presented in Figure 9B.
Figure 8.
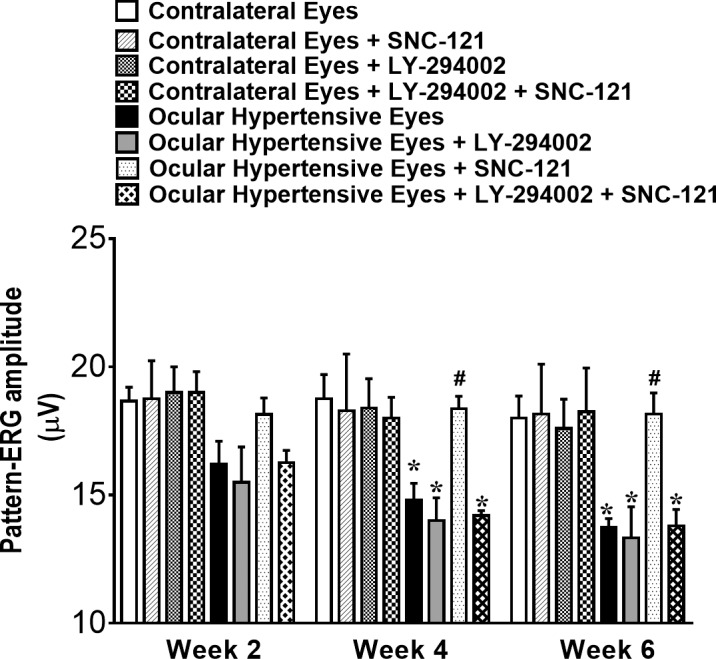
Pattern-ERG recordings in normal and ocular-hypertensive rat eyes. Each waveform is a mean of 300 individual waveforms taken at an interval of 1 second for each data point. One hour after hypertonic saline injections and subsequently once daily for 7 days, animals were treated with 1 mg/kg SNC-121 (i.p.). To block PI3K/Akt pathway, animals were pretreated with 3 mg/kg LY-294002 (i.p.) 1 hour prior to SNC-121 treatment for 7 days, once daily. The changes in pattern-ERG amplitudes of ocular-hypertensive eyes with and without drug treatment at each time point were analyzed using their respective values at day 0. Data are mean ± SE; *, #P < 0.05; n = 8. * Significant different from contralateral eyes and SNC-121–treated ocular-hypertensive eyes; # significantly different from ocular hypertensive eyes and LY-294002 + SNC-121–treated ocular-hypertensive eyes.
Figure 9.

(A) Fluorescence micrographs of flat-mounted retinas depicting Fluorogold-labeled RGCs in normal, ocular-hypertensive, SNC-121–treated, and LY-294002 + SNC-121–treated eyes. Briefly, 3 μL of a 5% solution of Fluorogold was injected into the superior colliculus of anesthetized animals. Seven days post Fluorogold injection, animals were euthanized, and retinas were prepared as flat-mounts, vitreous-side facing up. Fluorescent RGCs were visualized under Zeiss microscopy. Scale bar: 20 μm. (B) Rats were divided into three groups: ocular hypertensive (n = 8), SNC-121–treated ocular hypertensive (n = 8), and LY-294002 + SNC-121–treated ocular hypertensive (n = 8). In each retina, eight microscopic fields of identical size (150-μm2 area) were used to count RGCs by ImageJ software. *P < 0.05; n = 8 for each group.
Ocular Hypertension Induced Upregulation of MMPs in Optic Nerve
We also determined the upregulation of MMPs in optic nerve in response to ocular-hypertensive injury. The production of MMP-1 (151 ± 16; n = 6; P < 0.05), MMP-2 (332 ± 24; n = 4; P < 0.01), and MMP-3 (179 ± 26; n = 4; P < 0.05) was significantly increased in the ocular-hypertensive optic nerve as compared with the normal optic nerve at day 7, post ocular hypertensive injury (Figs. 10A–C). To determine if δ-opioid receptor activation in ocular-hypertensive animals opposes the production of MMPs, animals were treated with SNC-121 (1 mg/kg; i.p.) each day for 7 days. SNC-121 treatment reduced the production of MMP-1 (101 ± 6; n = 6; P < 0.02; Fig. 10A), MMP-2 (95 ± 14; n = 6; P < 0.007; Fig. 10B), and MMP-3 (105 ± 6; n = 4; P < 0.03; Fig. 10C) in ocular-hypertensive optic nerves. We have not seen any changes in MMP production in the presence of SNC-121 alone, in normal optic nerve (Figs. 10A–C).
Figure 10.

(A) MMP-1, (B) MMP-2, and (C) MMP-3 production in response to ocular hypertensive injury and its suppression by SNC-121 treatment in the optic nerve. Optic nerves of normal, ocular-hypertensive, and SNC-121–treated ocular-hypertensive eyes were collected at day 7, post injury and analyzed for MMP-1, MMP-2, and MMP-3 expression by Western blotting using selective antibodies for each MMP. Intensities of band were quantitated by densitometry. Data expressed as mean ± SE. *P < 0.05; n = 4–6.
TNF-α Induced Secretion of MMPs From ONH Astrocytes
To confirm a regulatory role of PI3K/Akt pathway in MMP production, we stimulated ONH astrocytes with TNF-α. Previous studies in our laboratory demonstrated that 25 ng/mL of TNF-α provided optimal secretion of MMPs, thus this concentration was chosen for this experiment. To determine if δ-opioid receptor activation by SNC-121 counteract the TNF-α–induced secretion of MMPs, ONH astrocytes were pretreated with SNC-121 prior to the TNF-α treatment. Treatment of the cells with SNC-121 significantly (P < 0.05) inhibited the TNF-α–induced secretion of MMP-1, MMP-2, and MMP-3 (Figs. 11A–C). To determine the roles of the PI3K/Akt pathway in TNF-α–induced MMP production, ONH astrocytes were treated with a PI3K inhibitor, LY-294002, prior to SNC-121 and TNF-α treatments. Interestingly, the SNC-121–mediated inhibition in MMP-1, MMP-2, and MMP-3 secretions were fully blocked when cells were pretreated with LY-294002 (Figs. 11A–C). However, TNF-α–induced increase in MMP-1, MMP-2, and MMP-3 secretion was not inhibited by LY-294002 treatment alone. In fact, we have noticed a trend that LY-294002 treatment slightly potentiated the TNF-α–induced MMP-1, MMP-2, and MMP-3 secretions (Figs. 11A–C).
Figure 11.

Effects of LY-294002 on MMP-1 (A), MMP-2 (B), and MMP-3 (C) secretion from ONH astrocytes at 6 hours. ONH astrocytes were starved in serum-free medium for 16 hours. Cells were then pretreated with LY-294002 (1 μM) for 30 minutes followed by SNC-121 (1 μM) and TNF-α (25 ng/mL) treatments for 6 hours. Secreted media was collected and concentrated 10-fold. An equal volume (40 μL) of conditioned media was analyzed by Western blotting using selective anti–MMP-1, anti–MMP-2, and anti–MMP-3 antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and the Biorad Versadoc imaging system. Data are expressed as mean ± SE. *P < 0.05; n = 4–7.
Discussion
Glaucoma is a progressive optic neuropathy characterized by optic disc cupping, RGC death, and vision loss. POAG is the most common type of glaucoma. Glaucoma pathogenesis is multifactorial; however, a major risk factor for the development of glaucoma is elevated IOP. The pathophysiological mechanisms by which elevated IOP leads to optic nerve atrophy and retinal degeneration are unknown. This manuscript provides novel findings that the PI3K/Akt pathway plays a key role in δ-opioid receptor–mediated RGC neuroprotection against ocular hypertension–induced injury, thus suggesting that PI3k/Akt pathway could be a potential therapeutic target for glaucoma therapy.
The protein kinase B (Akt) family is comprised of three highly homologous isoforms: Akt-1 (PKBα), Akt-2 (PKBβ), and Akt-3 (PKBγ). Upon binding to growth factor and to their specific membrane receptor, an upstream regulator, PI3 kinase, phosphorylates Akt at the serine 473 and threonine 308 residues.32 Phosphorylated and activated Akt then inhibits its downstream targets such as: proapoptotic proteins,25,26 caspases,27,28 and apoptosis signal regulating kinase-1.33 The PI3K/Akt pathway plays a central role in multiple cellular processes and exerts a prosurvival and antiapoptotic effect.34 In the eye, studies have provided limited evidence that the PI3K/Akt pathway protects the retina in nonglaucoma models35–39; however, none of the studies provide concrete evidence that direct activation of Akt provides RGC neuroprotection in glaucoma. This manuscript provides data that strengthens the idea that the PI3/Akt pathway plays a crucial role in the δ-opioid receptor–mediated neuroprotection against ocular hypertension–induced RGC injury.
Activation of δ-opioid receptors reduced infarct size in stroke and myocardial ischemia models.40,41 Activation of δ-opioid receptors is protective against hypoxia/excitotoxic injury in cortical neurons.42 DADLE, a δ-opioid receptor agonist, has been shown to improve hippocampal neuronal survival against ischemia in a rat model.43 Studies have also shown that δ-opioid receptor activation attenuates oxidative injuries in the brain exposed to ischemia/reperfusion by enhancing antioxidant ability and inhibition of caspase activity.44 Moreover, δ-opioid receptors have been involved in neuroprotective mechanisms of Alzheimer45 and Parkinson46 diseases. Earlier, we have shown an upregulation of δ-opioid receptors in response to glaucomatous injury, which has indicated their involvement in endogenous neuroprotective pathways.2 Additionally, we have shown that activation of δ-opioid receptors attenuates the production of TNF-α,2 nitric oxide, inducible nitric oxide synthase,20 and MMP-2 secretion from ONH astrocytes.19 Although beneficial effects of δ-opioid receptor agonists in nonocular systems such as chronic pain, emotional disorders, and neuroprotection against various forms of injuries have been defined, the usefulness of δ-opioid receptor agonists against glaucomatous injury requires additional studies. It would also be intriguing to understand how short-term (7 days) δ-opioid receptor agonist treatment provided long-term (up to 42 days) RGC neuroprotection. We know opioid-induced epigenetic mechanisms exert prolonged functional effects that often involve changes in acetylation.47–49 We speculate that δ-opioid receptor activation induces epigenetic changes that can provide long-term neuroprotection. The underlying mechanisms involved in δ-opioid receptor induced epigenetics remain largely unknown and require additional experiments that will be the focus of our future studies.
The current manuscript provides novel findings that the PI3K/Akt pathway is one of the potential targets of δ-opioid receptor agonist for RGC neuroprotection. We have shown that pretreatment of animals with a PI3/Akt inhibitor (LY-294002) blocked SNC-121–induced preservation of pattern ERGs and RGC integrity in a chronic glaucoma rat model. In this study, we have not determined axonal loss in ocular-hypertensive eyes. However, the reduction in Fluorogold labeling of RGCs in ocular-hypertensive eyes could be due to axonal loss. It is possible that axonal function also improved by SNC-121 treatment. Although studies have shown that opioid receptor agonists (e.g., bremazocine and morphine; 100 μg/2- to 4-kg body weight) have the ability to lower IOP in rabbits,50–52 it remain unknown if topical application of δ-opioid agonists can lower IOP in rodents. Numerous cell types including activated macrophages, astrocytes, microglia, Müller cells, and neuronal cells can undergo stress during the progression of glaucoma, which will produce various neurotoxic substances (e.g., proinflammatory cytokines, nitric oxide, and MMPs). Earlier, we have shown that SNC-121 treatment blocked the production of TNF-α and nitric oxide in an ocular hypertensive rat model.
In the current study, we have seen a reduction in p-Akt immunostaining in both retina and optic nerve of ocular hypertensive eyes, which were increased in SNC-121–treated ocular-hypertensive animals. We also noticed co-localization of p-Akt and GFAP in optic nerve of ocular hypertensive eyes. Additionally, MMP-1, MMP-2, and MMP-3 were produced from the optic nerve within a week in response to ocular hypertensive injury, which was also inhibited in SNC-121–treated ocular-hypertensive animals (Fig. 10). Furthermore, TNF-α induced MMPs production from ONH astrocytes is inhibited by SNC-121 treatment (Fig. 11). Both in vivo and in vitro data presented in this manuscript provide evidence that SNC-121 treatment activates and phosphorylate Akt both in the retina and optic nerve and reduces MMP-1, MMP-2, and MMP-3 production. These data provide initial clues that MMPs are primarily produced from ONH astrocytes. In this study, we have not tested MMPs secretion from other glial cells so the involvement of other glial cells in MMPs production in response to glaucomatous injury cannot be ruled out. At this point, it is not clear how SNC-121 treatment attenuates the production of MMPs; nor what would be the downstream signaling targets of the δ-opioid receptor agonist. To dissect out signaling mechanisms we determined the roles of the PI3K/Akt pathway in MMP production using ONH astrocytes. Data shown in Figures 11A to 11C demonstrated that TNF-α–induced MMP secretion from ONH astrocytes is marginally potentiated in the presence of PI3K inhibitor, LY-294002, suggesting that endogenous PI3K/Akt activity is required to regulate MMP secretion. Moreover, SNC-121–mediated inhibition of TNF-α–induced MMP secretion is fully blocked by LY-294002 (Figs. 11A–C). These data provide novel information that PI3K/Akt activity is augmented by the δ-opioid receptor agonist, SNC-121, which negatively regulates the production of MMPs. These findings provide novel leads that a δ-opioid agonist could directly mitigate the destabilization/remodeling of the optic nerve by blocking the excessive production of MMPs.
In summary, this manuscript provides evidence that the δ-opioid receptor agonist, SNC-121, activates and phosphorylates Akt. Similar to our previously published studies, SNC-121 provided significant RGC neuroprotection up to 6-weeks post glaucomatous injury, and this neuroprotective effect of SNC-121 was blocked by LY-294002. The levels of phospho-Akt reduced in both the retina and optic nerves of ocular-hypertensive eyes, which was reversed to normal levels in SNC-121–treated animals; suggesting their potential role in neuroprotection. Overall, these studies provide novel insight that the PI3K/Akt signaling pathway plays a crucial neuroprotective role in δ-opioid receptor–mediated RGC neuroprotection. However, we cannot rule out the possibility of the involvement of other pathways/molecules in SNC-121–mediated neuroprotection.
Acknowledgments
Supported in part by National Institutes of Health (NIH)/National Eye Institute (NEI) Grants EY019081 and EY027355 (SH; Bethesda, MD, USA), NIH grant C06 RR015455 from the Extramural Research Facility Program of the National Center for Research Resources, and an unrestricted grant to MUSC-SEI from Research to Prevent Blindness (New York, NY, USA).
Disclosure: S. Husain, None; A. Ahmad, None; S. Singh, None; C. Peterseim, None; Y. Abdul, None; M.J. Nutaitis, None
References
- 1. Boland MV, Quigley HA. . Risk factors and open-angle glaucoma: classification and application. J Glaucoma. 2007; 16: 406– 418. [DOI] [PubMed] [Google Scholar]
- 2. Abdul Y, Akhter N, Husain S. . Delta-opioid agonist SNC-121 protects retinal ganglion cell function in a chronic ocular hypertensive rat model. Invest Ophthalmol Vis Sci. 2013; 54: 1816– 1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakazawa T, Nakazawa C, Matsubara A,et al. . Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006; 26: 12633– 12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tezel G, Li LY, Patil RV, Wax MB. . TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 2001; 42: 1787– 1794. [PubMed] [Google Scholar]
- 5. Zhou X, Li F, Kong L, Tomita H, Li C, Cao W. . Involvement of inflammation, degradation, and apoptosis in a mouse model of glaucoma. J Biol Chem. 2005; 280: 31240– 31248. [DOI] [PubMed] [Google Scholar]
- 6. Fuchs C, Forster V, Balse E, Sahel JA, Picaud S, Tessier LH. . Retinal-cell-conditioned medium prevents TNF-alpha-induced apoptosis of purified ganglion cells. Invest Ophthalmol Vis Sci. 2005; 46: 2983– 2991. [DOI] [PubMed] [Google Scholar]
- 7. McKinnon SJ, Lehman DM, Kerrigan-Baumrind LA,et al. . Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Invest Ophthalmol Vis Sci. 2002; 43: 1077– 1087. [PubMed] [Google Scholar]
- 8. Reichstein D, Ren L, Filippopoulos T, Mittag T, Danias J. . Apoptotic retinal ganglion cell death in the DBA/2 mouse model of glaucoma. Exp Eye Res. 2007; 84: 13– 21. [DOI] [PubMed] [Google Scholar]
- 9. Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. . Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000; 41: 764– 774. [PubMed] [Google Scholar]
- 10. Quigley HA, McKinnon SJ, Zack DJ,et al. . Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000; 41: 3460– 3466. [PubMed] [Google Scholar]
- 11. Kass MA, Heuer DK, Higginbotham EJ,et al. . The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002; 120: 701– 713; discussion 829–730. [DOI] [PubMed] [Google Scholar]
- 12. Lichter PR, Musch DC, Gillespie BW,et al. . Interim clinical outcomes in the Collaborative Initial Glaucoma Treatment Study comparing initial treatment randomized to medications or surgery. Ophthalmology. 2001; 108: 1943– 1953. [DOI] [PubMed] [Google Scholar]
- 13. Bachoo RM, Kim RS, Ligon KL,et al. . Molecular diversity of astrocytes with implications for neurological disorders. Proc Natl Acad Sci U S A. 2004; 101: 8384– 8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hernandez MR. . The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000; 19: 297– 321. [DOI] [PubMed] [Google Scholar]
- 15. Hernandez MR, Pena JD. . The optic nerve head in glaucomatous optic neuropathy. Arch Ophthalmol. 1997; 115: 389– 395. [DOI] [PubMed] [Google Scholar]
- 16. Morrison JC, Dorman-Pease ME, Dunkelberger GR, Quigley HA. . Optic nerve head extracellular matrix in primary optic atrophy and experimental glaucoma. Arch Ophthalmol. 1990; 108: 1020– 1024. [DOI] [PubMed] [Google Scholar]
- 17. Takai Y, Tanito M, Ohira A. . Multiplex cytokine analysis of aqueous humor in eyes with primary open-angle glaucoma, exfoliation glaucoma, and cataract. Invest Ophthalmol Vis Sci. 2012; 53: 241– 247. [DOI] [PubMed] [Google Scholar]
- 18. Ueda K, Nakahara T, Mori A, Sakamoto K, Ishii K. . Protective effects of TGF-beta inhibitors in a rat model of NMDA-induced retinal degeneration. Eur J Pharmacol. 2013; 699: 188– 193. [DOI] [PubMed] [Google Scholar]
- 19. Akhter N, Nix M, Abdul Y, Singh S, Husain S. . Delta-opioid receptors attenuate TNF-alpha-induced MMP-2 secretion from human ONH astrocytes. Invest Ophthalmol Vis Sci. 2013; 54: 6605– 6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Husain S, Abdul Y, Singh S, Ahmad A, Husain M. . Regulation of nitric oxide production by delta-opioid receptors during glaucomatous injury. PLoS One. 2014; 9: e110397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen YL, Law PY, Loh HH. . NGF/PI3K signaling-mediated epigenetic regulation of delta opioid receptor gene expression. Biochem Biophys Res Commun. 2008; 368: 755– 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Husain S, Abdul Y, Potter DE. . Non-analgesic effects of opioids: neuroprotection in the retina. Curr Pharm Des. 2012; 18: 6101– 6108. [DOI] [PubMed] [Google Scholar]
- 23. Husain S, Potter DE. . The opioidergic system: potential roles and therapeutic indications in the eye. J Ocul Pharmacol Ther. 2008; 24: 117– 140. [DOI] [PubMed] [Google Scholar]
- 24. Kretz A, Happold CJ, Marticke JK, Isenmann S. . Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol Cell Neurosci. 2005; 29: 569– 579. [DOI] [PubMed] [Google Scholar]
- 25. Datta SR, Brunet A, Greenberg ME. . Cellular survival: a play in three Akts. Genes Dev. 1999; 13: 2905– 2927. [DOI] [PubMed] [Google Scholar]
- 26. Zhao H, Shimohata T, Wang JQ,et al. . Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005; 25: 9794– 9806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cardone MH, Roy N, Stennicke HR,et al. . Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998; 282: 1318– 1321. [DOI] [PubMed] [Google Scholar]
- 28. Zhou H, Li XM, Meinkoth J, Pittman RN. . Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000; 151: 483– 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Husain S, Abdul Y, Crosson CE. . Preservation of retina ganglion cell function by morphine in a chronic ocular-hypertensive rat model. Invest Ophthalmol Vis Sci. 2012; 53: 4289– 4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. . A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997; 64: 85– 96. [DOI] [PubMed] [Google Scholar]
- 31. Husain S, Liou GI, Crosson CE. . Opioid receptor activation: suppression of ischemia/reperfusion-induced production of TNF-alpha in the retina. Invest Ophthalmol Vis Sci. 2011; 52: 2577– 2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alessi DR, Andjelkovic M, Caudwell B,et al. . Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996; 15: 6541– 6551. [PMC free article] [PubMed] [Google Scholar]
- 33. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. . Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001; 21: 893– 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. . PI3K/Akt and apoptosis: size matters. Oncogene. 2003; 22: 8983– 8998. [DOI] [PubMed] [Google Scholar]
- 35. Dreixler JC, Hemmert JW, Shenoy SK,et al. . The role of Akt/protein kinase B subtypes in retinal ischemic preconditioning. Exp Eye Res. 2009; 88: 512– 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakazawa T, Shimura M, Tomita H,et al. . Intrinsic activation of PI3K/Akt signaling pathway and its neuroprotective effect against retinal injury. Curr Eye Res. 2003; 26: 55– 63. [DOI] [PubMed] [Google Scholar]
- 37. Weishaupt JH, Rohde G, Polking E, Siren AL, Ehrenreich H, Bahr M. . Effect of erythropoietin axotomy-induced apoptosis in rat retinal ganglion cells. Invest Ophthalmol Vis Sci. 2004; 45: 1514– 1522. [DOI] [PubMed] [Google Scholar]
- 38. Qi Y, Chen L, Zhang L, Liu WB, Chen XY, Yang XG. . Crocin prevents retinal ischaemia/reperfusion injury-induced apoptosis in retinal ganglion cells through the PI3K/AKT signalling pathway. Exp Eye Res. 2013; 107: 44– 51. [DOI] [PubMed] [Google Scholar]
- 39. Russo R, Cavaliere F, Berliocchi L,et al. . Modulation of pro-survival and death-associated pathways under retinal ischemia/reperfusion: effects of NMDA receptor blockade. J Neurochem. 2008; 107: 1347– 1357. [DOI] [PubMed] [Google Scholar]
- 40. Chen TY, Goyagi T, Toung TJ,et al. . Prolonged opportunity for ischemic neuroprotection with selective kappa-opioid receptor agonist in rats. Stroke. 2004; 35: 1180– 1185. [DOI] [PubMed] [Google Scholar]
- 41. Gross ER, Hsu AK, Gross GJ. . Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004; 94: 960– 966. [DOI] [PubMed] [Google Scholar]
- 42. Zhang J, Haddad GG, Xia Y. . delta-, but not mu- and kappa-, opioid receptor activation protects neocortical neurons from glutamate-induced excitotoxic injury. Brain Res. 2000; 885: 143– 153. [DOI] [PubMed] [Google Scholar]
- 43. Iwata M, Inoue S, Kawaguchi M, Nakamura M, Konishi N, Furuya H. . Effects of delta-opioid receptor stimulation and inhibition on hippocampal survival in a rat model of forebrain ischaemia. Br J Anaesth. 2007; 99: 538– 546. [DOI] [PubMed] [Google Scholar]
- 44. Yang Y, Xia X, Zhang Y,et al. . Delta-opioid receptor activation attenuates oxidative injury in the ischemic rat brain. BMC Biol. 2009; 7: 1– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thathiah A, De Strooper B. . The role of G protein-coupled receptors in the pathology of Alzheimer's disease. Nat Rev Neurosci. 2011; 12: 73– 87. [DOI] [PubMed] [Google Scholar]
- 46. Mabrouk OS, Marti M, Salvadori S, Morari M. . The novel delta opioid receptor agonist UFP-512 dually modulates motor activity in hemiparkinsonian rats via control of the nigro-thalamic pathway. Neuroscience. 2009; 164: 360– 369. [DOI] [PubMed] [Google Scholar]
- 47. Renthal W, Kumar A, Xiao G,et al. . Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009; 62: 335– 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang L, Lv Z, Hu Z,et al. . Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010; 35: 913– 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheng J, Lv Z, Wang L, Zhou Y, Hui B. . Histone H3 phosphoacetylation is critical for heroin-induced place preference. Neuroreport. 2011; 22: 575– 580. [DOI] [PubMed] [Google Scholar]
- 50. Dortch-Carnes J, Potter DE. . Bremazocine: a kappa-opioid agonist with potent analgesic and other pharmacologic properties. CNS Drug Rev. 2005; 11: 195– 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dortch-Carnes J, Russell KR. . Morphine-induced reduction of intraocular pressure and pupil diameter: role of nitric oxide. Pharmacology. 2006; 77: 17– 24. [DOI] [PubMed] [Google Scholar]
- 52. Stagni E, Bucolo C, Motterlini R, Drago F. . Morphine-induced ocular hypotension is modulated by nitric oxide and carbon monoxide: role of mu3 receptors. J Ocul Pharmacol Ther. 2010; 26: 31– 35. [DOI] [PubMed] [Google Scholar]