

SUMMARY
Deubiquitinating enzymes (DUBs) have garnered significant attention as drug targets in the last 5–10 years. The excitement stems in large part from the powerful ability of DUB inhibitors to promote degradation of oncogenic proteins, especially proteins that are challenging to directly target but which are stabilized by DUB family members. Highly-optimized and well-characterized DUB inhibitors have thus become highly sought after tools. Most reported DUB inhibitors, however, are polypharmacological agents possessing weak (micromolar) potency toward their primary target, limiting their utility in target validation and mechanism studies. Due to a lack of high resolution DUB•small molecule ligand complex structures, no structure-guided optimization efforts have been reported for a mammalian DUB. Here, we report a small molecule•ubiquitin specific protease (USP) family DUB co-structure and rapid design of potent and selective inhibitors of USP7 guided by the structure. Interestingly, the compounds are noncovalent active site inhibitors.
eTOC blurb
Lamberto, et al. report the structure-guided development of inhibitors of the deubiquitinating enzyme (DUB) USP7. The studies provide optimized and well-characterized probes for studying USP7 in normal and disease biology and furthermore, lend validation to the notion that potent and selective active site inhibitors of DUBs can be achieved.
INTRODUCTION
Ubiquitin is a 76-amino acid protein attached to substrate proteins post-translationally via iso-peptide bond formation between ubiquitin’s C-terminal glycine and a substrate lysine sidechain;(Komander and Rape, 2012) linear and branched polyubiquitin chains are assembled via attachment of another molecule of ubiquitin to one of seven lysines or the N-terminal methionine of ubiquitin.(Pickart and Fushman, 2004) Ubiquitin is attached to substrate proteins by the coordinated action of ubiquitin activating (E1), conjugating (E2), and ligating (E3) enzymes and removed by a family of proteases known as deubiquitinating enzymes (DUBs). The first recognized role of the ubiquitin system was controlling protein turnover.(Ciechanover et al., 1980; Hershko et al., 1980) Ubiquitin tags are also responsible for signaling a wide range of non-degradative functions. Ubiquitination can affect protein activity by modulating conformational changes, complexation with other proteins,(Ea et al., 2006; Wu et al., 2006) susceptibility to addition of other post-translation modifications (PTM) including phosphorylation and acetylation,(Hunter, 2007; Zhang et al., 2008; Zhao et al., 2008) and cellular localization(Li et al., 2003). Through combined degradative and non-degradative functions, ubiquitination coordinates a wide range of cellular processes including proteolysis,(Ciechanover et al., 2000) DNA repair,(Jackson and Durocher, 2013) chromatin remodeling,(Weake and Workman, 2008) receptor signaling,(Haglund and Dikic, 2012) and immunity,(Malynn and Ma, 2010; Zinngrebe et al., 2014) among others. Not surprisingly, aberrant ubiquitin system activity is linked to disease, including cancer,(Hoeller and Dikic, 2009; Pinto-Fernandez and Kessler, 2016) infection,(Isaacson and Ploegh, 2009; Maculins et al., 2016) and neurodegeneration(Ciechanover and Brundin, 2003; Ciechanover and Kwon, 2015). The relationship between ubiquitin and cancer biology has been clinically validated by the FDA approval of the proteasome inhibitor bortezomib for multiple myeloma.(Kane et al., 2003)
There are approximately 100 human DUBs belonging to six distinct families, five of which [ubiquitin specific protease (USP), ubiquitin C-terminal hydrolase (UCH), Ovarian tumor protease (OTU), Josephin, and Mindy] are cysteine proteases, and the sixth [JAB/MPN/MOV34 (JAMM/MPN)] is comprised of zinc metalloproteases.(Abdul Rehman et al., 2016; Clague et al., 2013; Komander et al., 2009; Komander and Rape, 2012) Many DUBs have been linked to physiological and/or pathophysiological functions. For example, USP1 and USP4 are involved in DNA damage repair,(Kee and Huang, 2015) USP22 and BAP1 have a role in chromatin function,(Atanassov et al., 2011) and USP2 and USP8 are reported to stabilize oncogenic proteins cyclin D1(Shan et al., 2009) and mutant EGFR,(Byun et al., 2013) respectively. While dozens of apo- and ubiquitin- bound structures have been solved,(Hu et al., 2002; Johnston et al., 1997; Komander et al., 2009) very few have been achieved with non-ubiquitin-based compounds.(Davies et al., 2012; Ratia et al., 2008; Schlierf et al., 2016) Notably, small molecule•DUB complex structures are lacking for the largest 56-member mammalian USP family.
The first DUB inhibitor, the dual USP14/UCHL5 inhibitor VLX1570, entered clinical trials in 2015.(Wang et al., 2016b) Overall though, DUB inhibitor development is still in early stages. Approximately 40 DUB inhibitors have been reported, although most are weak, multi-targeted agents.(D’Arcy et al., 2015; Ndubaku and Tsui, 2015) Given the current dearth of potent and selective inhibitors, skepticism remains as to whether or not this enzyme class will be druggable in a manner analogous to protein kinases, for example. A significant hindrance to the generation of potent and selective DUB inhibitors is a lack of structure-guided optimization efforts. One example of structure-guided development of a DUB inhibitor, which targeted the SARs DUB PLPro,(Baez-Santos et al., 2015), generated compounds with IC50s below 500 nM and exhibiting a high degree of selectivity relative to mammalian DUBs. In this case, selectivity was explained by significant structural differences between viral and mammalian DUBs. Breakthroughs in X-ray crystallography of small molecule DUB inhibitor complexes has the potential to enable rapid development of potent and selective inhibitors of mammalian DUBs.
The DUB USP7 has been shown to be involved in regulation of a myriad of cellular processes, including epigenetics, cell cycle, DNA repair, immunity, viral infection and tumorigenesis. USP7, also known as herpes virus-associated ubiquitin specific protease (HAUSP), was first discovered as a protein that plays a role in viral lytic growth.(Everett et al., 1997) Interest in the enzyme intensified when USP7 was implicated in regulating degradation of the tumor suppressor p53(Li et al., 2002) by stabilizing the major E3 ligase for p53, MDM2.(Cummins et al., 2004; Li et al., 2004) Recently, several epigenetic modifiers, including the methytransferase PHF8,(Wang et al., 2016a) demethylase DNMT1(Du et al., 2010; Felle et al., 2011; Qin et al., 2011) and acetyltransferase Tip60,(Dar et al., 2013) as well as H2B itself,(van der Knaap et al., 2005) have been identified as direct targets of USP7. Other notable targets of USP7 include the transcription factors FOXP3, which in Treg cells links this DUB enzyme to immune response(van Loosdregt et al., 2013), and N-Myc, which is stabilized in neuroblastoma cells.(Tavana et al., 2016) Consistent with its regulation of diverse substrates and biological processes, USP7 has emerged as a drug target in a wide range of malignancies including multiple myeloma,(Chauhan et al., 2012) breast cancer,(Wang et al., 2016a) neuroblastoma,(Tavana et al., 2016) glioma,(Cheng et al., 2013) and ovarian cancer.(Zhang et al., 2016)
P22077 and its close analog P5091 are the inhibitors most frequently utilized to probe USP7 functions (structures in Figure S1A). P22077 exhibits modest potency against USP7 (IC50= 8.0 μM) and equipotent inhibition of two additional DUBs, USP10 and USP47.(Altun et al., 2011; Ritorto et al., 2014) In addition to modest potency and selectivity, reported drawbacks of these nitro-thiophene-based compounds include poor solubility and general toxicity.(Chen et al., 2017) Additional USP7 inhibitors (shown in Figure S1B) have been identified although none possess features superior to P5091/P22077 and significant optimization efforts have not been undertaken.(Aleo et al., 2006; Colland et al., 2009; El-Desoky et al., 2017; Nicholson et al., 2008; Reverdy et al., 2012; Tanokashira et al., 2016; Yamaguchi et al., 2013)
Here we report the structure-guided development of next generation small molecule probes of USP7. High-resolution USP7•small molecule crystal structures enabled us to rapidly develop XL188, a highly selective 90 nM inhibitor of USP7, from a 7.2 μM lead, as a probe of USP7. Furthermore, we show that XL203C, the enantiomer of XL188, is over 80-fold less potent against USP7, and thus serves as an inactive control compound. In contrast to P22077/P5091, which target the invariant catalytic cysteine of USP DUBs, XL188 is a noncovalent active site inhibitor. We demonstrate that the XL188/XL203C active/inactive inhibitor pair is a powerful combination for studying USP7 function in cellular models.
RESULTS
XL188 is a potent and selective inhibitor of USP7
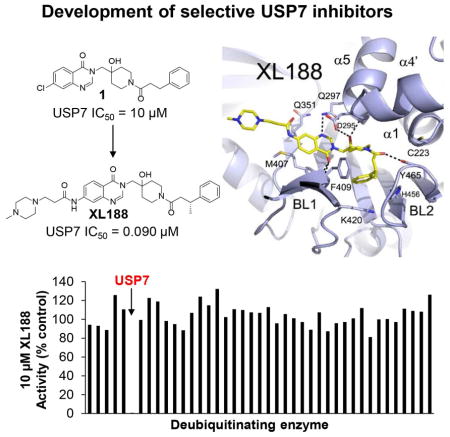
As part of an effort to identify chemical starting points for development of DUB inhibitors by profiling the inhibitory activity of compounds reported in peer-reviewed and patent literature for activity against large panels of DUBs,(Ritorto et al., 2014) we identified a highly selective inhibitor of USP7 (1, structure in Figure 1A) reported in a 2013 patent from Hybrigenics.(Colland, 2013; Kessler, 2014) When screened for inhibitory activity across a panel of 38 purified DUBs at a concentration of 100 μM, USP7 was the only DUB substantially inhibited (Figure S1C, Table S1). Dose-response analysis using USP7 catalytic domain (amino acids 208–560) or full-length enzyme (1–1102) and ubiquitin-aminomethylcoumarin (Ub-AMC) as substrate confirmed USP7 inhibitory activity, although potency was weak with IC50s in the double digit micromolar range (Figure 1B, S1D). Isothermal titration calorimetry (ITC), using catalytic domain, confirmed binding with a KD of 8 μM (Figure S1E, Table S2). We solved the structure of USP7 bound by 1, which enabled rapid structure-guided development of XL188 (Figure 1A), a highly potent and selective inhibitor of USP7. XL188 inhibited USP7 catalytic domain and full-length enzyme with IC50 values of 193 and 90 nM, respectively (Figure 1B). The interaction of XL188 with USP7 was confirmed using ITC and differential scanning fluorimetry (DSF) (Figure 1C, S1F, Table S2). Consistent with the 100-fold improvement in biochemical inhibition of USP7 by XL188 compared to 1, a KD of 104 nM was measured for USP7 catalytic domain using ITC (Figure 1C, Table S2). The selectivity of XL188 was assessed against a panel of 41 purified DUBs, using ubiquitin-rhodamine (Ub-Rho) as substrate. XL188 retained the excellent selectivity for USP7 observed with 1; at a concentration of 10 μM, XL188 exhibited little to no inhibition of any DUBs other than USP7 (Figure 1D, Table S1). In contrast, the enantiomer of XL188, XL203C (Figure 1A), showed 80-fold less potent inhibition of USP7 (IC50 = 7.18 μM, Figure 1B) and no significant inhibition of other DUBs (Figure S1G, Table S1).
Figure 1. Structure and selectivity of XL188.

A) Structure-guided optimization of 1 led to USP7 inhibitor XL188. The enantiomer of XL188, XL203C, is 80-fold less active.
B) Dose-dependent inhibition of USP7 catalytic domain (amino acids 208–560) and full-length USP7 (amino acids 1–1102) by 1, XL188 and XL203C using Ub-AMC as substrate.
C) Assessment of XL188 binding to USP7 using isothermal calorimetry.
D) Inhibitory activity of XL188 across a panel of 41 purified DUBs using ubiquitin-rhodamine (Ub-Rho) as substrate.
See also Figure S1, Table S1, S2.
XL188 binds the S4–S5 pocket of USP7
We determined co-crystal structures of 1 and XL188 in complex with purified, recombinant USP7 catalytic domain (Figure 2A–B, S2A–B) to 1.9 and 2.2Å, respectively. These high resolution structures revealed that the catalytic cysteine and switching loop were in the unproductive conformation, as seen in apo structures,(Hu et al., 2002) but with significantly different unit cell dimensions. A notable interaction in apo structures (represented by PDB 1NF8) was the face-to-face (or active site-to-active site) contact, highlighted by the mutual and complete insertion of Leu288’s side chain within a hydrophobic pocket near the catalytic cysteine, formed by helix α4, and loops -α1 and α4-α5. This interaction was not present in our complex structures; instead, the active site in our complex structure was nearly free of crystal contacts. Our results therefore further confirm the relevance of the proposed USP7 autoinhibited conformation; namely, that it is not a crystal contact artifact. Importantly, our complex structures revealed unambiguous electron density (Figure S2A, C) for the inhibitors in the substrate binding cleft leading to the active site. Inhibitors occupied the S4 and S5 subsites, about 5 Å removed from the catalytic triad (Figure 2A, S2A), involving multiple hydrogen bonds with four inhibitor hetero-atoms (Figure 2B, S2B) common to both compounds. Specifically, the quinazolinone ketone formed hydrogen bonds with peptide backbone nitrogens of Arg408 and Phe409, and the quinazolinone cyclic nitrogen formed a hydrogen bond with the amide side chain of Gln297. The tertiary hydroxy group was stabilized by hydrogen bonds with the carboxylic group of Asp295 as well as the peptide backbone nitrogen of Val296. Asp295 is highly conserved among the ubiquitin-specific protease (USP) family of deubiquitinating enzymes,(Quesada et al., 2004) as it hydrogen bonds with ubiquitin’s backbone P4 position, an interaction presumed to be important for substrate stabilization.(Hu et al., 2002) And the oxygen atom of the piperidine amide was within 3 Å of the hydroxyl group of Tyr465, a strictly-conserved DUB-family side chain (Figure S4A). In addition, the phenyl ring of 1 and XL188 was buried in the S4 pocket, which was bounded by the aromatic rings of Tyr514, His456, Phe409 and the aliphatic chains of Lys420 and Arg408. Notably, the sidechain of Phe409 flips to reveal the hydrophobic pocket, a conformational rearrangement also observed upon binding of ubiquitin,(Hu et al., 2002). The additional methyl group of XL188 present at the carbon alpha to the phenyl ring was involved in multiple van der Waals interactions including with the backbone of Asn460 and the phenyl side chain of Phe409, and it was associated with lower B-factors in the BL2 loop. Nearly all atoms of 1 and XL188 were buried except the chlorine atom and N-methyl-piperazine sidechain, respectively (Figure S2D–E). Compared with 1, XL188 was associated with a rotation of the fingers by about 5 degrees counterclockwise around an axis through the piperazine group.
Figure 2. Characterization of XL188 binding to USP7.

A) Ribbon diagram of USP7 with XL188.
B) Stereoview of USP7 (light blue) bound to XL188 (yellow). Hydrogen bonds are indicated by dashed lines.
C) Molecular surface representation of the USP7•XL188 co-structure. Highlighted regions indicate regions of altered HDX in the presence of XL188. Darker colors correspond to significant changes whereas lighter colors correspond to regions with subtle changes.
See also Figure S2, S3, Table S3.
Crystallographic studies were complemented with hydrogen deuterium exchange mass spectrometry (HDX MS) to monitor changes in protein dynamics. Exchange of backbone amide hydrogens with bulk solvent can be accurately measured upon inhibitor binding.(Iacob et al., 2009; Wales and Engen, 2006) Online digestion of USP7 was performed and 85 peptic peptides covering 85% of USP7 catalytic domain were investigated with HDX MS in the free and bound states (Figure S3A–B). Both XL188 and 1 protected the BL1 and α-4/5 loops (Figure 2C), confirming that the observed crystal structure interactions are also relevant in solution. While the locations of the major changes were the same, XL188 protected USP7 from exchange more than 1, consistent with increased affinity (Figure S3B–C). Moreover, HDX MS results showed ligand-induced conformational changes and stabilizations distant from the active site. Inhibitors also stabilized/protected the palm region that is near the catalytic cysteine, including helices α-3/4, consistent with a previously proposed allosteric regulatory mechanism for USP7.(Faesen et al., 2011) Furthermore, a disordered loop between α8/β14 was protected from deuterium incorporation suggesting this region becomes ordered upon inhibitor binding.
Mutagenesis studies reveal determinants of selectivity
Given the high degree of selectivity of this chemical series for USP7, we were surprised to discover that nearly every residue directly interacting with inhibitor and several additional amino acids lining the ligand binding pocket are conserved among the USP family of DUBs. Figure 3A shows a detailed ligand interaction diagram of XL188 with USP7; in the diagram, residues that are conserved, defined as >80% of 52 other USPs possessing an equivalent residue according to MView classification (Figure S4A),(Brown et al., 1998) are indicated with a red box. Previous site-directed mutagenesis studies show that amino acid substitutions at several of these positions abrogate the ability of the enzyme to cleave DUB substrates.(Hu et al., 2002) Thus, with the goals of gaining insight into the observed compound selectivity and identifying the most productive ligand interactions in our crystal structures, we primarily focused our mutagenesis studies on non-conserved residues contained within the ligand binding pocket. Seven USP7 mutants with a single amino acid substitution and one with double substitution were generated (Figure 3B). Glu351, Met407 and Met410 were substituted with Ser, Lys, and Ser, respectively, selected based on the prevalence of the amino acid at equivalent positions in other USPs (Figure S4A); all other mutated amino acids were replaced with alanine. Six of the eight mutant proteins retained the ability to cleave the DUB substrate Ub-AMC although two with significantly reduced activity relative to wild-type (Figure 3B). The mutagenesis studies were carried out in parallel to compound optimization studies, thus 1 was utilized to assess the inhibitory activity of the chemical series toward the mutants. 1 inhibited four of the active mutants with IC50 values within several fold of its IC50 for wild-type enzyme (Figure 3B). However, USP7Q351S and USP7Y514A were highly resistant to 1, with 1 exhibiting no inhibitory effect at concentrations up to 100 μM compound (Figure 3B, Figure S4B). Gln351 is unique to USP7, with only one other USP DUB, USP14, containing this residue at the equivalent position and 80% of USPs containing a residue with a small sidechain (Figure S4A). Tyr514, on the other hand, is highly conserved among DUBs. Thus, Gln351 may be an important determinant of selectivity for the hydroxypiperidine-based inhibitors. Initial mutagenesis studies were carried out using catalytic domain; full-length USP7Q351S was confirmed to be resistant to both 1 and XL188 (Figure 3C–D, S4C). Gln351 hydrogen bonds with Gln297 only in the auto-inhibited apo form and may be required for stabilization of the conformation bound by these inhibitors. Although a complete understanding of the molecular level contribution of Gln351S to selectivity awaits further structural studies, these studies support that the binding mode observed in crystallographic studies accurately represents the binding mode in solution.
Figure 3. Analysis of USP7 mutant proteins.

A) Detailed ligand interaction diagram of XL188 with USP7. Residues for which >80% of other USPs contain an amino acid belonging to the same class are boxed red.
B) Summary of activity against Ub-AMC and inhibition by 1 for USP7 mutant catalytic domain proteins.
C) Dose-response inhibition of full length USP7WT and USP7Q351 (amino acids 1–1102) by 1.
D) Dose-response inhibition of full length USP7WT and USP7Q351 (amino acids 1–1102) by XL188.
See also Figure S4.
SAR Investigation
The high resolution structure of USP7 bound by 1 enabled rapid optimization of potency, solubility and pharmacological properties of the compound leading to XL188. As detailed above, four compound hetero-atoms were involved in hydrogen bonding interactions with USP7, the phenyl ring was buried in the S4 hydrophobic pocket normally filled by the Leu73 sidechain of substrate ubiquitin and the chloro atom was solvent exposed. Initial SAR investigations (Figure 4A) focused on establishing the importance of the hydrogen bonding and hydrophobic interactions observed in the structure. All biochemical IC50s were measured using USP7 catalytic domain and Ub-AMC as substrate. 1 inhibited isolated USP7 catalytic domain with an IC50 of 12.3 μM and full-length protein with an IC50 of 10.2 μM. Compounds 2 and 3, in which the hydroxypiperidine –OH group was removed or replaced with –CN, respectively, exhibited little inhibition of USP7 at concentrations up to 100 μM. Similarly, removal of the amide carbonyl, as in 4, abrogated USP7 activity as did contraction of the 6-membered piperidine to a 5-membered ring (5). Occupancy of the S4 hydrophobic pocket by the phenyl ring was confirmed to be required for activity as removal of the moiety (6) or shortening the linker between the hydroxypiperidine and phenyl (7) resulted in biochemical IC50s > 100 μM. In contrast, installation of a racemic methyl group on the methylene adjacent to the phenyl ring (8) improved biochemical potency approximately 10-fold, consistent with data reported in the patent exemplifying 1. In order to explore the importance of stereochemistry of the methyl group we prepared both enantiomers which revealed that the (R)-stereoisomer (9) was approximately 100-fold more potent than the (S)-stereoisomer (10). Unfortunately, investigation of 9’s ability to bind and inhibit USP7 in cells was hampered by poor solubility in aqueous buffer. In order to improve this property we focused on installation of polar moieties in place of the solvent exposed chloro atom (Figure 4B). Installation of different groups, including an N-methyl-piperazine, piperidine, dimethylamine, and imidazole, linked to the 7-position of the quinazolinone via a short carbon chain and amide bond linkage, improved potency 2–4 fold relative to the parent compound 9. With several analogs exhibiting sub-micromolar USP7 IC50s, we considered metabolic stability as an additional parameter for compound prioritization since a probe suitable for in vivo studies would be highly valuable for pharmacological validation of USP7 in animal disease models. XL188 exhibited the greatest stability in the presence of mouse liver microsomes (MLMs) with a half-life of 31 min (Figure 4B). Competitive activity-based protein profiling using the DUB targeting activity based probe HA-Ub-Vs(Hewings et al., 2017) confirmed that XL188 bound native USP7 (Figure 4C). Treatment of HEK293T lysates with XL188 significantly blocked labeling of USP7 by HA-Ub-Vs with an IC50 of approximately 0.9 μM, a value that represents a significant improvement compared to 1 (at a concentration of 500 μM, 1 competes for 50% of probe labeling (Figure S5A)). Because in this experiment an irreversible probe (HA-Ub-Vs) is competing for occupancy of USP7 with a reversible inhibitor, the measured cell-based IC50s may underestimate binding. An HA blot of the same treated lysates (Figure S5B) confirmed the high degree of selectivity for USP7 observed in the 41 member purified enzyme panel. As a measure of general toxicity, we treated peripheral bone mononuclear cells (PBMCs) with XL188 and observed no growth suppression at concentrations up to 10 μM following 72 hours of treatment (Figure S5C). In order to achieve an ideally matched negative control compound to use in conjunction with XL188, we prepared its enantiomer, XL203C. XL203C showed 80–100-fold less potent inhibition of USP7 relative to XL188 in both biochemical (Figure 1B, 4B) and cellular target engagement assays (Figure 4C).
Figure 4. Structure-activity relationship studies.

A–B) Structures, USP7 inhibitory activity and mouse liver microsome (MLM) stability of synthesized compounds.
C) Analysis of the ability of XL188 and XL203C to bind native USP7 across multiple doses in HEK293T lysates using competitive activity based protein profiling. Data are represented as mean ± SEM.
See also Figure S5.
XL188 promotes USP7 dependent loss of HDM2 and increase of p53 and p21
To further examine XL188’s on-target effects, we evaluated its impact on the USP7-HDM2-p53-p21 axis. The role of USP7 in promoting degradation of HDM2 resulting in increased levels of p53 and p21 is documented in multiple models.(Chauhan et al., 2012; Kon et al., 2010) In MCF7 cells treated with XL188, levels of p53 and p21 increased following 16 hours of treatment with concentrations in line with the IC50 of inhibition of native USP7 (Figure 5A). Consistent with the effects being USP7-dependent, no observable change in levels of the same proteins across the same range of concentrations with the comparatively inactive enantiomer XL203C was observed (Figure 5A). Degradation of HDM2 by XL188 was observed in the same system when new protein synthesis was blocked by addition of cycloheximide (Figure 5B). The necessity to block protein synthesis to observe degradation of HDM2 is consistent with the established negative feedback loop involving these proteins in which p53 transcriptionally upregulates HDM2.(Wu et al., 1993) Similarly, treatment of multiple myeloma MM.1S cells with XL188, but not XL203C, led to loss of HDM2 accompanied by an increase in downstream tumor suppressors p21 and p53, as has previously been reported for the USP7 inhibitor P5091 (Figure 5C–D).(Chauhan et al., 2012)
Figure 5. The USP7 inhibitor XL188 promotes loss of HDM2 and accumulation of p53 and p21.

A) Analysis of HDM2, p53 and p21 protein levels in MCF7 cells treated with XL188 or XL203C at the indicated concentration for 16 hours.
B) Analysis of HDM2, p53 and p21 protein levels in MCF7 cells following 16 hours of treatment with XL188 or XL203C at the indicated concentration with addition of cycloheximide for the last 2 hours.
C) Analysis of HDM2, p53 and p21 protein levels in MM.1S cells treated with XL188 or XL203C at the indicated concentration for 6 hours.
D) Analysis of HDM2, p53 and p21 protein levels in MM.1S cells following 6 hours of treatment with XL188 or XL203C at the indicated concentration with addition of cycloheximide for the last 2 hours.
DISCUSSION
Through their role in reversing ubiquitination and coordinating a wide range of cellular processes including proteolysis, DNA repair, receptor signaling, transcription and immunity, DUBs are emerging as a new class of drug targets. Although efforts to develop small molecule probes of DUBs to study their role in basic and disease biology have increased in recent years, these efforts have produced limited numbers of well characterized and optimized inhibitors. The DUB enzyme USP7 is one of the most well-studied DUBs and is reported to regulate the abundance of proteins involved in DNA damage repair, cell cycle regulation, and epigenetic and transcription factor control of gene expression. Notably, USP7 has been implicated in a range of different cancer types, controlling some of the most important proliferative processes. In order to identify the most promising disease contexts in which to deploy USP7 drugs and to more accurately catalog the activities of USP7 as it pertains to both the promise and liabilities associated with blockade of USP7 activity, highly potent and well-characterized probe compounds have been sought after. Here, we present an inhibitor of USP7, XL188, with double digit nanomolar potency toward USP7 and exquisite selectivity relative to the largest available panel of purified DUBs. We combined XL188 with a negative control compound, XL203C, to present a set of tools for interrogating USP7 biology. Thus, we confirmed USP7 as a key regulator of HDM2-p53-p21 signaling by showing that XL188, but not XL203C, promoted degradation of HDM2 resulting in increased levels of p53 and p21.
Our observation that the IC50s for minimum and full-length USP7 are similar (190 vs 90 nM), in light of the fact that USP7 catalytic domain efficiency is augmented by the presence of the c-terminal ubl domains,(Faesen et al., 2011) may suggest that the kinetics of the equilibrium between active and inactive states of the full-length enzyme is rapid. Alternatively, if we propose an additional stable intermediate towards the activated state, represented by Phe409 flipping, our results suggest that the full-length construct stabilizes this intermediate to a greater extent compared with the minimal construct. Although premature, we might further suggest that the mechanism by which the c-terminal domains up-regulate the catalytic domains is by stabilizing Phe409 flipping.
XL188 represents one of only a small set of mammalian DUB inhibitors with low nanomolar potency and a high degree of selectivity relative to other DUBs.(Dexheimer et al., 2010; Ndubaku and Tsui, 2015) To compare the state of the DUB field to the established kinase field: the first publication on phosphorylation came 23 years prior to the first publication on ubiquitination, and the current state of modulating ubiquitylation via DUBs is roughly where drugging phosphorylation via kinases was 20 years ago.(Cohen and Tcherpakov, 2010) In this project, rapid optimization of XL188 from 1 was enabled largely by our success in achieving a high resolution crystal structure of the lead compound in complex with USP7. We established a series of biochemical, biophysical, and selectivity assays to identify promising compounds for crystallographic and medicinal chemistry studies. By applying similar triaging assays, we believe similar success in crystallography and small molecule probe development can be achieved for other DUB family members.
Overall, our studies provide optimized and well-characterized small molecule probes for use in studying the roles of USP7 in normal and disease biology and furthermore, lend considerable validation to the notion that DUBs are a class of enzymes for which potent and selective inhibitors can be achieved.
SIGNIFICANCE
DUBs are a 100-member family of proteases that control levels and activation of proteins through removal of post-translational ubiquitin moieties. Of particular interest to drug discovery, DUBs stabilize disease linked proteins, including many targets that remain undruggable. Thus, DUB-targeting drugs can promote degradation of disease causing proteins, representing a novel strategy for modulating the activity of challenging targets in cancer and other diseases. Progress in the development of potent and selective DUB inhibitors for use as chemical probes and translation to the clinic has been slow, however. Of note, structure-guided medicinal chemistry campaigns have been lacking for mammalian DUBs, suggesting that success in crystallography of small molecule DUB co-structures enabling such efforts could significantly impact the field. We succeeded in solving high resolution X-ray crystal structures of USP7 bound by a small molecule ligand series. The co-structures enabled us to rapidly improve the potency of the lead inhibitor 100-fold to 90 nM. Furthermore, we show that the newly-developed inhibitor exhibits exquisite selectivity for USP7 relative to broad panels of purified and cellular DUBs and that the compound, but not a negative control stereoisomer, promotes degradation of a known USP7 substrate. The developed compounds are thus validated as high quality probes for pharmacological interrogation of the diverse reported functions of USP7. Importantly, this work provides proof-of-concept that potent and selective DUB inhibitors can be achieved and that structural characterization of chemical leads with their cognate DUB target could accelerate probe and drug discovery programs.
METHODS
CONTACT FOR REAGENT SHARING AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sara Buhrlage (saraj_buhrlage@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cells were obtained from the James E. Bradner’s laboratory and were not further authenticated. MM.1S cells were obtained from Kenneth C. Anderson’s laboratory and were not further authenticated. MCF7 cells were obtained from Jean Zhao’s laboratory and were not further authenticated. 293T cells were grown in DMEM with high glucose (Gibco 11965-118) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco 16140-071), and penicillin/streptomycin (Gibco 15140-122) and maintained in a humidified 37 °C/5% CO 2 incubator. MCF7 and MM.1S cells were grown in RPMI (Gibco 11875-119) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco 16140-071), and penicillin/streptomycin (Gibco 15140-122) and maintained in a humidified 37 °C/5% CO 2 incubator.
Primary cell cultures
Primary cells were obtained through written consent under approval of the Dana-Farber Cancer Institute Institutional Review Board. Peripheral blood mononuclear cells (PBMCs) from normal individuals were isolated by density gradient centrifugation through Ficoll-Plaque Plus (Amersham Pharmacia Biotech AB, Uppsala, Sweden) at 400xg for 25 minutes, followed by two washes in PBS. Cells were then maintained in RPMI media, supplemented with 10% FBS, and maintained in a humidified 37 °C/5% CO 2 incubator.
METHOD DETAILS
Chemical synthesis
General Methods and Materials
All commercially available starting materials were purchased from Sigma Aldrich, Fisher Scientific, Oakwood Chemical and Combi Block. All reagents were used as received without further purification. Known compounds were synthesized according to published literature procedures and any modifications are noted. Anhydrous solvents, such as tetrahydrofuran (THF), diethyl ether, dichloromethane (DCM), dimethyl formamide (DMF), dimethylsulfoxide (DMSO), 1,4-dioxane, and toluene (PhMe) were purchased from Fisher Scientific, and used as received. If necessary, air or moisture sensitive reactions were carried out under an inert atomsphere of nitrogen.
Removal of solvents was accomplished on a Büchi R-300 rotary evaporator and further concentration was done under a Welch 1400B-01 vacuum line, and Labconco FreeZone 6 plus system. Purification of compounds was performed by normal phase column chromatography using Teledyne CombiFlash chromatography system, and/or reversed phase chromatography on Waters Micromass ZQ preparative system with SunFire® Prep C18 OBDTM 5μM column. The purity was analyzed on Waters Acquity UPLC system. Analytical thin layer chromatography (TLC) plates were purchased from Fisher Scientific (EMD Millipore TLC Silica Gel60 F254). Visualization was accomplished by irradiation under UV light (254 nm).
All 1H-NMR spectra were recorded at 298K on a Bruker ARX 500 (500 MHz) spectrometer. 13C-NMR spectra were recorded on a Bruker ARX 500 (126 MHz) spectrometer. Samples were dissolved in CDCl3, DMSO-d6, or CD3OD. The spectra were referenced to the residual solvent peak (chlorofrom-d: 7.26 ppm for 1H-NMR and 77.16 ppm for 13C-NMR; DMSO-d6: 2.50 ppm for 1H-NMR and 39.25 ppm for 13C-NMR, CD3OD: 3.31 ppm for 1H NMR and 49.00 ppm for 13C NMR or tetramethylsilane (TMS) as the internal standard. Chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, br=broad peak), coupling constants (Hz), and number of protons. Mass spectrometry (LCMS) data were obtained on Waters Acquity UPLC system in positive ESI mode.
Synthetic procedures
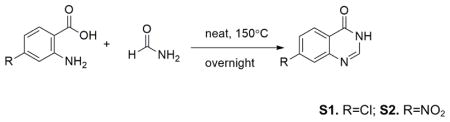
2-aminobenzoic acids (10.0mmol) and formamide (1.8g, 40.0mmol) were mixed in pressure tube, which was heated at 150°C overnight. Then the reaction was cooled to room temperature. The solid was suspended in cold water, then collected by vacuum filtration, and dried on high vacuum line. The products 1.6g (-Cl) and 1.8g (-NO2) were isolated as light brown solid in 88% (-Cl) and 95% (-NO2) yields with no further purification.
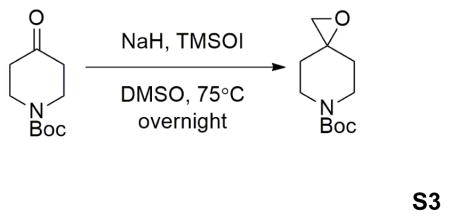
Sodium hydride (60% dispersion in mineral oil) (0.88g, 22.0mmol) was dissolved in 40mL anhydrous DMSO at 0°C under N2. Trimethylsulfoxonium iodide (4.84g, 22.0mmol) was added into the solution portionwisely. When addition completed, the mixture was warmed up to room temperature, and stirred for 40min. Then 1-Boc-4-piperidone (3.98g, 20.0mmol) was added portionwisely. The reaction mixture was then stirred at room temperature for 1 hour, then at 65°C for another hour. Then the mixture was poured on 100mL ice. Aqueous phase was extracted using EtOAc (50mL×2). Combined organic phase was washed with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. The crude material was purified by flash column chromatography (50% EtOAc in hexanes) to afford 2.98g product in 70% yield.
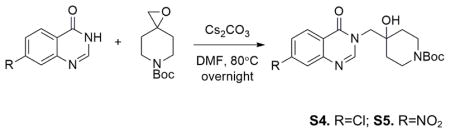
Into the solution of S1 (2.13g, 11.8mmol) in 50mL DMF was added S3 (2.78g, 13.0mmol) and Cs2CO3 (11.54g, 35.4mmol). The mixture was heated at 80°C overnight. Then the reaction was cooled to room temperature, and diluted with EtOAc. The solution was washed with saturated NH4Cl (50mL×2) Aqueous phase was extracted with more EtOAc. Combined organic phase was washed with brine, dried over MgSO4, followed by filtration and evaporation under reduced pressure. The crude material was purified by flash column chromatography (40% to 100% EtOAc in hexanes) to afford 3.94g product in 85% yield.
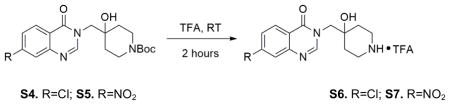
S4 was taken up in trifluoroacetic acid (TFA) as 1M solution, which was stirred at room temperature for 2 hours. The solution was concentrated under reduced pressure, further on high-vac overnight. S6 was directly used as starting material for the following synthesis without further purification.
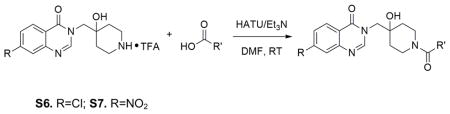
Amide formation by HATU-catalyzed coupling reaction: S6 was taken up in DMF as 1M solution, and 3 equivalence of Et3N was added. The carboxylic acid (1.2eq) was pre-mixed with HATU (2eq) and Et3N (5eq) in DMF at the same concentration, which was stirred at room temperature for 10min. Two solutions were then mixed together and further stirred at room temperature for 5 hours. The reaction was directly subjected to prep. HPLC purification. The isolated product was then further purified by normal phase flash chromatography to afford product with desired purity for following biological tests.
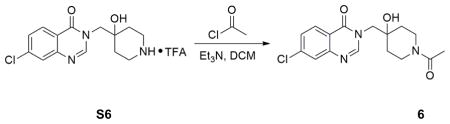
Amide formation by acylation using acid chloride: S6 (0.04g, 0.1mmol) was taken up in 2mL dichloromethane. Et3N (0.07mL, 0.5mmol) was added, followed by addition of acetyl chloride (0.015mL, 0.2mL). The reaction was kept at 0°C stirring for 2 hours. Then the reaction was quenched by adding drops of water, followed by immediate purification by flash column chromatography. The isolated product was further purified by HPLC to afford 19mg product in 57% yield.
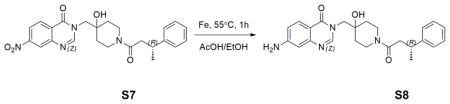
Reduction of aromatic nitro group: S7 (0.49g, 1.08mmol) was dissolved in 10mL AcOH/EtOH (1:1). Iron powder (0.25g, 4.39mmol) was added in one portion. The reaction was then stirred at 50°C for 1 hour. The iron powder was removed by filtration. Filtrate was concentrated under reduced pressure. The crude material was then purified by normal phase flash column chromatography (10% to 40% MeOH in EtOAc), followed by reverse phase HPLC to afford 0.22g product S8 in 53% yield
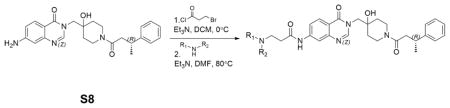
Installation of solubilizing groups: S8 (0.11g, 0.25mmol) was dissolved in 5mL dichloromethane. Et3N (0.035mL, 0.25mmol) was added at −20°C.3-bromopropionyl chloride (0.03mL, 0.25mmol) in 1mL DCM was added dropwisely. The reaction was stirred at 0°C for 3h. Then it was quenched by addition of drops of water, then concentrated under reduced pressure. The crude product was used for the next step without further purification.
Crude material from last step (0.06g, 0.1mmol) was dissolved in 1mL DMF. Into the solution was added N-methylpiperazine (0.016mL, 0.12mmol) and Et3N(0.028mL, 0.2mmol). The reaction was stirred at 80°C for 3 hours. The solution was directly subjected to reverse phase HPLC purification, followed by normal phase flash column chromatography (20% to 60% MeOH in EtOAc with 0.5% Et3N) to afford 0.043g product XL188 in 75% yield.
Compound characterization
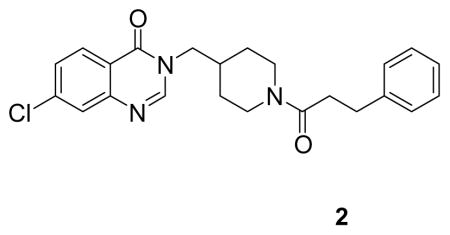
(7-chloro-3-((1-(3-phenylpropanoyl)piperidin-4-yl)methyl)quinazolin-4(3H)-one: white solid, 33% yield) 1H NMR (500 MHz, CDCl3) δ 8.21 (d, J = 8.6 Hz, 1H), 7.93 (s, 1H), 7.70 (d, J = 1.9 Hz, 1H), 7.46 (dd, J = 8.6, 2.0 Hz, 1H), 7.31 – 7.25 (m, 2H), 7.22 – 7.18 (m, 3H), 4.69 (d, J = 13.4 Hz, 1H), 3.88 – 3.73 (m, 3H), 3.00 – 2.92 (m, 2H), 2.91 – 2.82 (m, 1H), 2.60 (dp, J = 14.3, 7.3 Hz, 2H), 2.48 (td, J = 13.1, 2.4 Hz, 1H), 2.17 – 2.04 (m, 1H), 1.70 (d, J = 12.9 Hz, 1H), 1.62 (d, J = 12.8 Hz, 1H), 1.17 (qd, J = 12.5, 4.3 Hz, 1H), 0.97 (qd, J = 12.5, 4.2 Hz, 1H). 13C NMR (126 MHz, DMSO) δ 169.56, 159.76, 149.58, 148.95, 141.43, 138.88, 128.37, 128.18, 127.28, 126.30, 125.79, 120.37, 50.79, 44.55, 40.74, 38.22, 35.01, 33.98, 30.87, 29.54, 28.84. LCMS (ESI) m/z 410.29 [(M+H)+; calcd for C23H25ClN3O2+: 410.16].
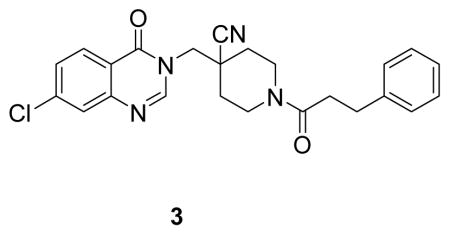
4-((7-chloro-4-oxoquinazolin-3(4H)-yl)methyl)-1-(3-phenylpropanoyl)piperidine-4-carbonitrile: white solid, commercial compound) 1H NMR (500 MHz, DMSO) δ 8.44 (s, 1H), 8.17 (d, J = 8.6 Hz, 1H), 7.79 (d, J = 2.0 Hz, 1H), 7.62 (dd, J = 8.6, 2.1 Hz, 1H), 7.25 (ddd, J = 13.3, 7.9, 4.0 Hz, 4H), 7.20 – 7.12 (m, 1H), 4.46 (d, J = 13.8 Hz, 1H), 4.36 – 4.24 (m, 2H), 3.97 (d, J = 14.3 Hz, 1H), 3.05 (t, J = 12.5 Hz, 1H), 2.80 (t, J = 7.7 Hz, 2H), 2.73 – 2.57 (m, 3H), 1.88 (t, J = 14.6 Hz, 2H), 1.61 (dtd, J = 17.1, 13.0, 3.9 Hz, 2H).13C NMR (126 MHz, DMSO) δ 170.41, 160.54, 149.90, 149.20, 141.81, 139.78, 128.99, 128.89, 128.70, 128.13, 126.94, 126.34, 121.06, 120.79, 50.64, 42.38, 40.40, 38.51, 34.30, 32.87, 32.28, 31.22. LCMS (ESI) m/z 435.29 [(M+H)+; calcd for C24H24ClN4O2+: 435.16].
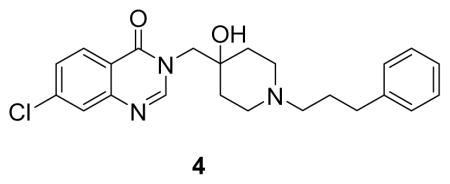
(7-chloro-3-((4-hydroxy-1-(3-phenylpropyl)piperidin-4-yl)methyl)quinazolin-4(3H)-one: white solid, 13% yield) 1H NMR (500 MHz, MeOD) δ 8.29 (s, 1H), 8.21 (d, J = 8.6 Hz, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.54 (dd, J = 8.6, 2.0 Hz, 1H), 7.26 (q, J = 7.1 Hz, 2H), 7.21 – 7.13 (m, 3H), 4.11 (s, 2H), 2.97 (d, J = 12.0 Hz, 2H), 2.76 – 2.59 (m, 6H), 1.98 – 1.81 (m, 4H), 1.63 (d, J = 13.4 Hz, 2H). 13C NMR (126 MHz, MeOD) δ 161.13, 149.97, 148.77, 140.38, 140.25, 128.24, 128.13, 128.00, 127.52, 126.00, 120.22, 67.60, 56.16, 53.81, 48.21, 32.27, 32.00, 25.90. LCMS (ESI) m/z 412.39 [(M+H)+; calcd for C23H27ClN3O2+: 412.18].
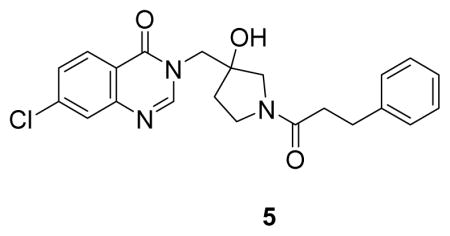
(7-chloro-3-((3-hydroxy-1-(3-phenylpropanoyl)pyrrolidin-3-yl)methyl)quinazolin-4(3H)-one: white solid, 8% yield) 1H NMR (500 MHz, DMSO) δ 8.29 (s, 1H), 8.17 (dd, J = 8.6, 4.3 Hz, 1H), 7.76 (t, J = 2.3 Hz, 1H), 7.59 (ddd, J = 8.7, 7.5, 2.1 Hz, 1H), 7.29 – 7.19 (m, 4H), 7.16 (t, J = 6.8 Hz, 1H), 5.27 (s, 1H), 4.17 (s, 1H), 4.14 (d, J = 4.0 Hz, 1H), 3.51 (s, 2H), 3.32 (dt, J = 23.6, 11.7 Hz, 3H), 2.84 – 2.74 (m, 2H), 2.59 – 2.51 (m, 2H), 2.47 – 2.40 (m, 1H), 1.95 (ddt, J = 39.5, 12.7, 9.3 Hz, 1H), 1.76 (ddd, J = 12.9, 11.0, 5.7 Hz, 1H). 13C NMR (126 MHz, DMSO) δ 169.93, 169.76, 160.22, 160.13, 150.33, 149.06, 149.04, 141.55, 141.51, 138.95, 128.45, 128.40, 128.35, 128.22, 127.24, 126.27, 125.81, 120.46, 78.33, 76.84, 55.97, 55.57, 51.10, 50.87, 44.46, 43.77, 36.06, 35.63, 35.06, 34.54, 30.29, 30.23. LCMS (ESI) m/z 412.29 [(M+H)+; calcd for C22H23ClN3O3+: 412.14].
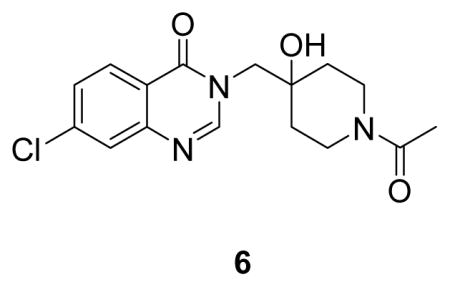
(3-((1-acetyl-4-hydroxypiperidin-4-yl)methyl)-7-chloroquinazolin-4(3H)-one: white solid, 57% yield) 1H NMR (500 MHz, DMSO) δ 8.40 (s, 1H), 8.15 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 8.6, 2.1 Hz, 1H), 4.09 – 3.96 (m, 4H), 3.58 (d, J = 13.4 Hz, 1H), 3.33 – 3.20 (m, 1H), 2.97 – 2.85 (m, 1H), 1.98 (s, 3H), 1.56 (td, J = 13.3, 4.3 Hz, 1H), 1.49 – 1.34 (m, 3H).13C NMR (126 MHz, DMSO) δ 167.66, 159.89, 150.25, 148.62, 138.65, 128.19, 126.92, 125.88, 120.05, 68.98, 53.39, 41.40, 36.44, 34.61, 33.93, 20.98. LCMS (ESI) m/z 336.18 [(M+H)+; calcd for C16H19ClN3O3+: 336.11].
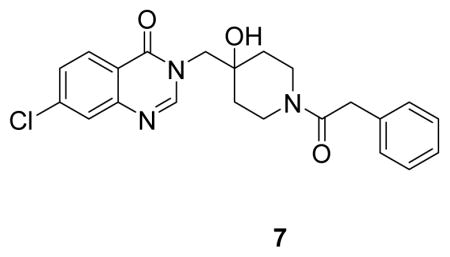
(7-chloro-3-((4-hydroxy-1-(2-phenylacetyl)piperidin-4-yl)methyl)quinazolin-4(3H)-one: white solid, commercial compound) 1H NMR (500 MHz, DMSO) δ 8.27 (s, 1H), 8.15 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 8.6, 2.1 Hz, 1H), 7.30 – 7.24 (m, 2H), 7.24 – 7.15 (m, 3H), 4.07 – 4.03 (m, 1H), 3.97 (dd, J = 38.6, 13.8 Hz, 2H), 3.74 – 3.64 (m, 3H), 3.29 – 3.20 (m, 1H), 3.03 – 2.92 (m, 1H), 1.51 – 1.27 (m, 4H).13C NMR (126 MHz, DMSO) δ 168.31, 159.88, 150.12, 148.71, 138.66, 135.76, 128.57, 128.19, 127.99, 126.93, 126.00, 125.95, 120.07, 68.97, 53.45, 41.15, 39.35, 36.86, 34.61, 33.99. LCMS (ESI) m/z 412.29 [(M+H)+; calcd for C22H23ClN3O3+: 412.14].
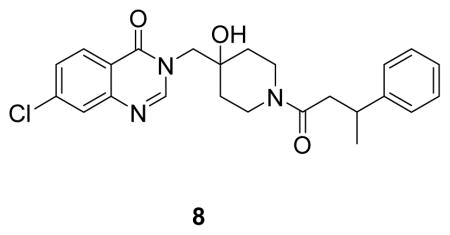
(7-chloro-3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)quinazolin-4(3H)-one: white solid, commercial compound) 1H NMR (500 MHz, DMSO) δ 8.29 (s, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.71 (d, J = 2.0 Hz, 1H), 7.56 (dd, J = 8.6, 2.1 Hz, 1H), 7.30 – 7.22 (m, 4H), 7.18 – 7.13 (m, 1H), 4.12 – 3.55 (m, 4H), 3.41 – 2.85 (m, 3H), 2.62 (dd, J = 14.9, 6.6 Hz, 1H), 2.54 (dd, J = 14.9, 7.6 Hz, 1H), 1.40 (t, J = 15.9 Hz, 4H), 1.25 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, DMSO) δ 168.89, 159.94, 159.88, 150.11, 148.74, 146.42, 146.30, 138.68, 128.20, 127.97, 127.94, 126.94, 126.66, 126.63, 125.98, 125.72, 125.67, 120.10, 69.04, 68.99, 53.57, 40.82, 40.71, 39.99, 36.67, 35.99, 35.77, 34.78, 34.64, 34.09, 33.95, 21.81, 21.61. LCMS (ESI) m/z 440.39 [(M+H)+; calcd for C24H27ClN3O3+: 440.17].
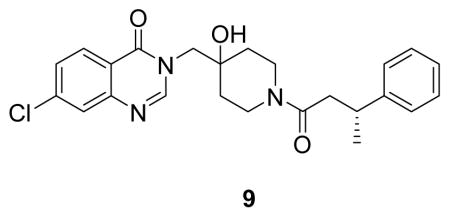
((R)-7-chloro-3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)quinazolin-4(3H)-one: white solid, 50% yield) 1H NMR (500 MHz, DMSO) δ 8.27 (d, J = 12.8 Hz, 1H), 8.16 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 1.7 Hz, 1H), 7.58 (dd, J = 8.6, 2.0 Hz, 1H), 7.26 (dd, J = 13.8, 6.7 Hz, 4H), 7.20 – 7.08 (m, 1H), 4.93 (d, J = 5.9 Hz, 1H), 4.04 (d, J = 13.6 Hz, 2H), 3.92 (q, J = 13.9 Hz, 1H), 3.65 (t, J = 12.5 Hz, 1H), 3.28 – 3.06 (m, 2H), 2.86 (ddd, J = 13.9, 8.6, 3.2 Hz, 1H), 2.68 – 2.50 (m, 2H), 1.62 – 1.26 (m, 3H), 1.26 – 1.12 (m, 3H).13C NMR (126 MHz, DMSO) δ 168.92, 159.90, 150.16, 148.76, 146.43, 146.31, 138.70, 128.22, 127.99, 127.96, 126.98, 126.68, 126.64, 125.99, 125.74, 125.69, 120.12, 69.05, 69.00, 53.58, 40.82, 40.71, 39.99, 36.68, 36.01, 35.78, 34.78, 34.65, 34.09, 33.95, 21.83, 21.63. LCMS (ESI) m/z 440.29 [(M+H)+; calcd for C24H27ClN3O3+: 440.17].
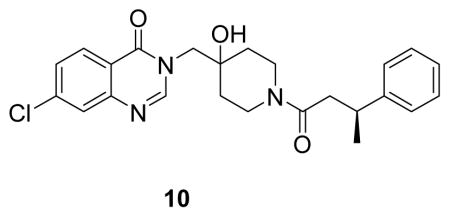
((S)-7-chloro-3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)quinazolin-4(3H)-one: white solid, 57% yield) 1H NMR (500 MHz, DMSO) δ 8.27 (d, J = 12.8 Hz, 1H), 8.16 (d, J = 8.6 Hz, 1H), 7.75 (d, J = 1.6 Hz, 1H), 7.58 (dd, J = 8.6, 2.0 Hz, 1H), 7.26 (dd, J = 13.7, 6.6 Hz, 4H), 7.21 – 7.09 (m, 1H), 4.93 (d, J = 5.8 Hz, 1H), 3.99 (dd, J = 43.9, 13.7 Hz, 2H), 3.91 (s, 1H), 3.65 (t, J = 12.2 Hz, 1H), 3.28 – 3.09 (m, 2H), 2.85 (d, J = 13.2 Hz, 1H), 2.68 – 2.55 (m, 2H), 1.60 – 1.25 (m, 3H), 1.20 (dd, J = 6.9, 1.5 Hz, 3H).13C NMR (126 MHz, DMSO) δ 168.89, 159.94, 159.88, 150.11, 148.74, 146.42, 146.30, 138.68, 128.20, 127.97, 127.94, 126.94, 126.66, 126.63, 125.98, 125.72, 125.67, 120.10, 69.04, 68.99, 53.57, 40.82, 40.71, 39.99, 36.67, 35.99, 35.77, 34.78, 34.64, 34.09, 33.95, 21.81, 21.61. LCMS (ESI) m/z 440.29 [(M+H)+; calcd for C24H27ClN3O3+: 440.17].
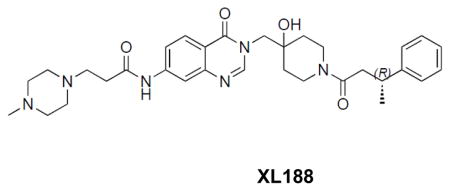
((R)-N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-(4-methylpiperazin-1-yl)propanamide: off-white solid, 75% yield) 1H NMR (500 MHz, DMSO) δ 10.57 (s, 1H), 8.20 (d, J = 13.1 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 8.02 (d, J = 1.6 Hz, 1H), 7.64 (d, J = 8.7 Hz, 1H), 7.25 (dd, J = 12.4, 6.2 Hz, 4H), 7.13 (dd, J = 18.7, 10.4 Hz, 1H), 4.96 (d, J = 5.8 Hz, 1H), 4.02 (d, J = 13.6 Hz, 1H), 3.90 (q, J = 14.0 Hz, 2H), 3.63 (dd, J = 29.0, 16.2 Hz, 1H), 3.27 – 3.12 (m, 2H), 2.86 (dd, J = 17.7, 14.8 Hz, 1H), 2.69 – 2.61 (m, 2H), 2.61 – 2.52 (m, 3H), 2.48 – 2.22 (m, 8H), 2.16 (s, 3H), 1.58 – 1.26 (m, 4H), 1.20 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 170.76, 168.83, 159.92, 159.87, 149.10, 148.73, 146.41, 146.28, 144.06, 127.93, 127.90, 126.97, 126.62, 126.60, 125.69, 125.63, 118.04, 116.26, 114.45, 69.03, 68.98, 54.37, 53.22, 51.96, 45.30, 40.81, 40.71, 39.97, 36.67, 35.95, 35.74, 34.77, 34.64, 34.09, 34.01, 33.95, 21.79, 21.61. LCMS (ESI) m/z 575.32 [(M+H)+; calcd for C32H43N6O4+: 575.33].
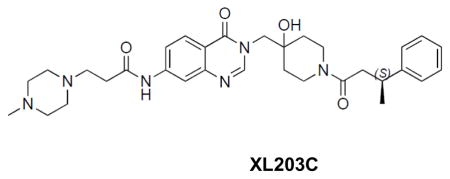
((S)-N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-(4-methylpiperazin-1-yl)propanamide: off-white solid, 26% yield) 1H NMR (500 MHz, DMSO) δ 10.52 (s, 1H), 8.19 (d, J = 13.1 Hz, 1H), 8.08 (d, J = 8.7 Hz, 1H), 8.02 (d, J = 2.0 Hz, 1H), 7.62 (dd, J = 8.8, 1.4 Hz, 1H), 7.25 (dd, J = 12.6, 6.3 Hz, 4H), 7.19 – 7.08 (m, 1H), 4.93 (s, 1H), 4.08 – 3.97 (m, 1H), 3.90 (q, J = 14.0 Hz, 2H), 3.64 (t, J = 12.9 Hz, 1H), 3.30 – 3.09 (m, 2H), 2.95 – 2.81 (m, 1H), 2.69 – 2.60 (m, 2H), 2.61 – 2.52 (m, 3H), 2.47 – 2.25 (m, 8H), 2.15 (s, 3H), 1.58 – 1.26 (m, 4H), 1.24 – 1.14 (m, 3H). 13C NMR (126 MHz, DMSO) δ 170.76, 168.82, 159.88, 149.11, 148.74, 146.41, 146.29, 144.04, 127.94, 127.91, 127.00, 126.63, 125.69, 125.64, 118.04, 116.28, 114.46, 68.99, 54.43, 53.24, 52.02, 45.38, 40.82, 40.71, 39.96, 36.68, 35.96, 35.75, 34.77, 34.64, 34.03, 21.80, 21.61. LCMS (ESI) m/z 575.32 [(M+H)+; calcd for C32H43N6O4+: 575.33].
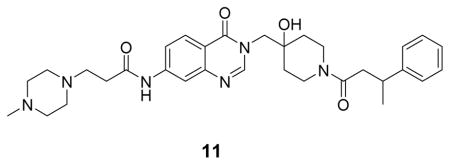
(N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-(4-methylpiperazin-1-yl)propenamide: white solid, 16% yield) 1H NMR (500 MHz, DMSO) δ 10.84 (s, 1H), 8.25 (d, J = 13.2 Hz, 1H), 8.15 – 7.99 (m, 2H), 7.69 (d, J = 8.7 Hz, 1H), 7.28 – 7.25 (m, 4H), 7.19 – 7.10 (m, 1H), 4.05 – 3.88 (m, 3H), 3.65 (t, J = 12.8 Hz, 1H), 3.28 – 3.14 (m, 10H), 2.92 – 2,85 (m, 3H), 2.79 (s, 3H), 2.64 – 2.53 (m, 4H), 1.53 – 1.28 (m, 4H), 1.20 (d, J = 6.0, 3H). 13C NMR (126 MHz, DMSO) δ 169.60, 160.66, 160.61, 158.82, 158.57, 149.95, 149.34, 147.15, 147.02, 144.63, 128.69, 128.66, 127.69, 127.38, 127.35, 126.44, 126.39, 118.90, 117.14, 115.37, 69.76, 69.71, 53.94, 41.57, 41.46, 40.88, 40.70, 37.42, 36.71, 36.49, 35.50, 35.37, 34.81, 34.66, 22.53, 22.34. LCMS (ESI) m/z 575.32 [(M+H)+; calcd for C32H43N6O4+: 575.33].
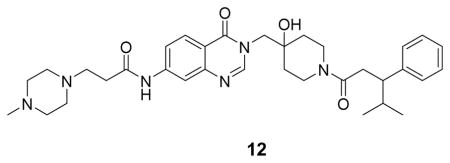
(N-(3-((4-hydroxy-1-(4-methyl-3-phenylpentanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-(4-methylpiperazin-1-yl)propenamide: off-white solid, 40% yield) 1H NMR (500 MHz, DMSO) δ 10.65 (s, 1H), 8.21 (d, J = 16.9 Hz, 1H), 8.11 (dd, J = 8.7, 1.9 Hz, 1H), 8.05 (s, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.23 (td, J = 7.6, 2.9 Hz, 2H), 7.15 (t, J = 6.5 Hz, 2H), 7.13 – 7.06 (m, 1H), 4.04 – 3.93 (m, 2H), 3.85 (s, 1H), 3.75 – 3.61 (m, 1H), 3.58 – 2.98 (m, 10H), 2.91 – 2.55 (m, 10H), 1.88 –1.80 (m, 1H), 1.53 – 1.25 (m, 3H), 1.12 – 1.06 (m, 1H), 0.89 (t, J = 7.2 Hz, 3H), 0.70 – 0.61 (m, 3H). 13C NMR (126 MHz, DMSO) δ 169.94, 169.86, 160.61, 160.53, 149.95, 149.29, 144.51, 144.09, 144.03, 128.82, 128.79, 128.24, 128.16, 127.81, 126.31, 118.89, 117.21, 115.33, 69.76, 69.66, 54.04, 50.83, 49.24, 49.08, 48.75, 42.48, 41.51, 37.43, 37.33, 36.01, 35.94, 35.55, 35.31, 34.72, 34.65, 32.83, 32.63, 32.39, 21.24, 21.19, 20.76, 20.56. LCMS (ESI) m/z 603.43 [(M+H)+; calcd for C34H47N6O4+: 603.37].
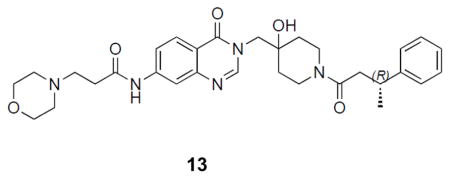
((R)-N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-morpholinopropanamide: off-white solid, 65% yield) 1H NMR (500 MHz, DMSO) δ 10.52 (s, 1H), 8.20 (d, J = 13.3 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 8.04 (d, J = 1.8 Hz, 1H), 4.96 (d, J = 6.4 Hz, 1H), 4.02 (d, J = 13.7 Hz, 1H), 3.90 (q, J = 14.0 Hz, 2H), 3.73 – 3.60 (m, 1H), 3.59 – 3.55 (m, 4H), 3.18 (ddd, J = 21.0, 18.3, 9.2 Hz, 2H), 2.93 – 2.80 (m, 1H), 2.65 (t, J = 6.9 Hz, 2H), 2.61 – 2.54 (m, 3H), 2.41 (m, 4H), 1.40 (dddd, J = 41.2, 26.0, 16.8, 9.5 Hz, 4H), 1.19 (dd, J = 6.9, 1.7 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 170.62, 168.81, 160.00, 149.15, 148.76, 146.44, 146.31, 144.09, 118.13, 116.29, 114.51, 69.06, 65.93, 53.73, 52.79, 40.85, 40.75, 39.98, 36.71, 36.00, 35.78, 34.79, 34.66, 34.11, 33.98, 33.81, 21.83, 21.64. LCMS (ESI) m/z 562.32 [(M+H)+; calcd for C31H40N5O5+: 562.30].
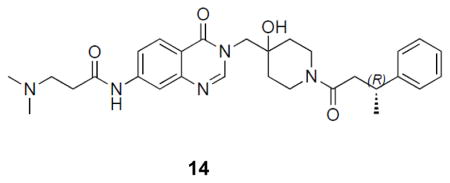
((R)-3-(dimethylamino)-N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)propanamide: off-white solid, 36% yield) 1H NMR (500 MHz, DMSO) δ 10.47 (s, 1H), 8.18 (dd, J = 13.0, 7.3 Hz, 1H), 8.06 (d, J = 8.7 Hz, 1H), 8.02 (d, J = 2.0 Hz, 1H), 7.63 – 7.59 (m, 1H), 7.24 (dd, J = 12.6, 6.3 Hz, 4H), 7.17 – 7.09 (m, 1H), 4.92 (s, 1H), 4.07 – 3.95 (m, 1H), 3.89 (q, J = 14.0 Hz, 2H), 3.63 (t, J = 13.0 Hz, 1H), 3.18 (ddd, J = 20.8, 18.0, 9.1 Hz, 2H), 2.86 (ddd, J = 13.8, 10.5, 5.5 Hz, 1H), 2.60 – 2.54 (m, 3H), 2.54 – 2.47 (m, 2H), 2.17 (s, 6H), 1.56 – 1.23 (m, 4H), 1.24 – 1.15 (m, 3H). 13C NMR (126 MHz, DMSO) δ 170.76, 168.83, 159.89, 149.09, 148.74, 146.42, 146.29, 144.07, 127.94, 127.92, 126.97, 126.61, 125.70, 125.64, 118.06, 116.26, 114.46, 68.99, 54.59, 53.29, 44.63, 40.82, 40.72, 39.98, 39.96, 36.68, 35.97, 35.75, 34.78, 34.65, 34.61, 34.11, 33.96, 21.80, 21.61. LCMS (ESI) m/z 520.31 [(M+H)+; calcd for C29H38N5O4+: 520.29].
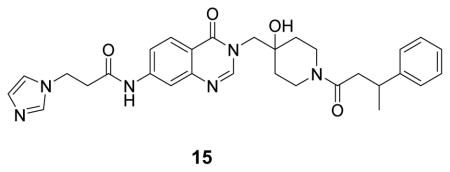
(N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-(1H-imidazol-1-yl)propenamide: white solid, 19% yield) 1H NMR (500 MHz, DMSO) δ 10.62 (s, 1H), 9.16 (s, 1H), 8.22 (d, J = 12.9 Hz, 1H), 8.09 (d, J = 8.7 Hz, 1H), 8.00 (s, 1H), 7.80 (s, 1H), 7.67 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.28 – 7.25 (m,4H), 7.15 (d, J = 3.6 Hz, 1H), 4.03 – 3.87 (m, 3H), 3.65 (t, J = 12.4 Hz, 1H), 3.26 – 3.14 (m, 2H), 3.08 (t, J = 6.3 Hz, 2H), 2.90 – 2.85 (m, 1H), 2.64 – 2.49 (m, 4H), 1.54 – 1.25 (m, 4H), 1.20 (d, J = 5.4 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 169.60, 169.43, 160.60, 160.55, 158.88, 158.60, 149.99, 149.25, 147.15, 147.03, 144.33, 136.33, 128.68, 128.66, 127.85, 127.37, 127.35, 126.44, 126.39, 122.58, 120.32, 118.86, 117.25, 115.33, 69.77, 69.71, 54.04, 44.94, 41.55, 41.44, 40.71, 37.41, 36.70, 36.57, 36.49, 35.52, 35.39, 34.80, 34.66, 22.53, 22.35. LCMS (ESI) m/z 543.22 [(M+H)+; calcd for C30H35N6O4+: 543.27].
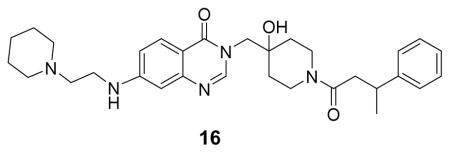
((N-(3-((4-hydroxy-1-(3-phenylbutanoyl)piperidin-4-yl)methyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-3-(piperidin-1-yl)propenamide: white solid, 18% yield) 1H NMR (500 MHz, DMSO) δ 10.69 (s, 1H), 9.32 (s, 1H), 8.23 (d, J = 12.8 Hz, 1H), 8.11 (dd, J = 8.7, 1.9 Hz, 1H), 8.05 (s, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.19 – 7.31 (m, 4H), 7.15 (s, 1H), 4.03 – 3.89 (m, 3H), 3.65 (t, J = 12.4 Hz, 1H), 3.48 – 3.40 (m, 4H), 3.26 – 3.15 (m, 2H), 2.98 – 2.89 (m, 5H), 2.64 – 2.51 (m, 2H), 1.84 (d, J = 13.3 Hz, 2H), 1.68 – 1.62 (m, 3H), 1.55 – 1.31 (m, 5H), 1.21 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 169.60, 169.22, 160.61, 160.56, 158.87, 158.59, 150.01, 149.29, 147.15, 147.03, 144.41, 128.68, 128.66, 127.87, 127.37, 127.35, 126.44, 126.39, 118.89, 117.27, 115.38, 69.77, 69.72, 54.05, 52.89, 52.05, 41.55, 41.45, 40.69, 37.41, 36.71, 36.49, 35.53, 35.40, 34.81, 34.67, 31.22, 23.00, 22.53, 22.35, 21.65. LCMS (ESI) m/z 560.22 [(M+H)+; calcd for C32H42N5O4+: 560.32]. NMR spectra are provided as Supplementary Materials.
USP7 cloning, expression, and purification
USP7 full length (amino acids 1-1102) and catalytic domain (208-560) were amplified using Addgene plasmid #16655(Cummins and Vogelstein, 2004) as a template and primers listed in Table S4 and cloned into pET28PP and pET28aLIC, respectively, using InFusion HD EcoDry Cloning Kit (Takara Bio Cat# 121416). Amino acid mutations of catalytic domain were introduced by PCR using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) following manufacturer’s protocol using primers listed in Table S4. All DNAs were handled in E.coli 10beta cells (New England Biolabs Cat# C3020K) and transformed in E.coli BL21(DE3) for protein expression.
A construct of human USP7 covering residues 208–560 in the pET28aLIC vector was overexpressed in E. coli BL21 (DE3) in terrific broth (TB) medium in the presence of 50 ug/ml of kanamycin. Cells were grown at 37°C to an OD of 0.8, cooled to 17°C, induced with 500 μM isopropyl-1-thio-D-galactopyranoside (IPTG), incubated overnight at 17°C, collected by centrifugation, and stored at −80°C. Cell pellets were sonicated in buffer A ( 50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 10 mM Imidazole, and 3 mM BME) and the resulting lysate was centrifuged at 30,000 xg for 40 min. Ni-NTA beads (Qiagen) were mixed with lysate supernatant for 30 min and washed with buffer A. Beads were transferred to an FPLC-compatible column and the bound protein was washed with 15% buffer B (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 300 mM imidazole, and 3 mM BME) and eluted with 100% buffer B. Thrombin was added to the eluted protein and incubated at 4°C overnight. The sample was then concentrated and passed through a Superdex 200 16/60 column (GE healthcare) in a buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol, and 1 mM TCEP. Fractions were pooled, concentrated and frozen at −80°C.
USP7 full length (amino acids 1-1102) in pET28aLIC was transformed in BL21(DE3) cells. An overnight culture was used to inoculate one liter of TB supplemented with 50 ug/ml kanamycin. Cells were grown at 37°C till they reached optical density (OD) ~0.6 at 600 nm. Protein expression was initiated by the addition of 0.4 mM IPTG. Cells were then grown for 16–20 hours at 17°C prior collection by centrifugation. Cell pellets were washed in PBS and resuspended in 25 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol and 1mM TCEP, 10 mM Imidazole, 0.1% IGEPAL sonicated and incubated with Ni-Nta beads (Quiagen) for 30 min at 4°C. Beads were washed with 10% buffer B (25 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol and 1mM TCEP, 250 mM Imidazole,) and eluted with 100% buffer B. Protein containing fractions were concentrated and loaded on Superdex 200 10/300 GL column in a buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol, and 1 mM TCEP. Fractions were pooled, concentrated and frozen at -80°C.
Ub-AMC Assay
USP7 and mutants were tested for their activity in Ubiquitin-AMC assay in presence or absence of inhibitors. For this assay USP7 catalytic domain WT or mutant was used at the following concentrations: 250nM USP7 WT, M407K, M407K/M410S or Q351S, 125 nM H461A, 600 nM Y514A and 10 nM M410S. For the same assay USP7 full length WT and Q351 mutant were used at 50 nM. USP7 variants were pre-incubated with different concentrations of inhibitors or DMSO as a control in 50 mM HEPES pH7.6, 0.5 mM EDTA, 11 uM ovalbumin, 5 mM DTT. The reaction was incubated 30 min at room temperature prior to the addition of 2 uM Ubiquitin-AMC (Boston Biochem) substrate. The initial rate of the reaction was measured by collecting fluorescence data at one minute interval over 30-minute period using a Clariostar fluorescence plate reader at excitation and emission wavelength of 345 and 445 nm respectively. The calculated initial rate values were plotted against inhibitor concentrations to determine IC50s. All the experimental data were plotted using Prism GraphPad.
Differential scanning fluorimetry
Differential Scanning Fluorimetry (DSF) experiments were carried out in a RTPCR 7500 Real-Time System (LifeTech) in 96 well plates, a total volume of 20 ul, and with an optimized SYPRO Orange dye concentration (5000x concentration in DMSO, Invitrogen). Compound dilutions in assay buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP) were first prepared by an NT8 liquid handler (Formulatrix) and then addition of dye was performed also by the same liquid handler. Sealed plates were heated at 1°C/min from 25°C to 95°C with fluorescence readings every 0.5°C. Tm values were determined as the minimum of the first derivative of the recorded fluorescence intensity versus temperature plot.
Isothermal titration calorimetry
The binding affinity of protein/ligand was measured by adding 0.02 mM protein in cell and titrating with 0.2 mM ligand in the syringe using an Auto-ITC200 microcalorimeter (Malvern) at 20°C. Proteins and ligands were prepared within ITC buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, and 2% DMSO. The data were fit using Origin 7.0 software. All ITC results were summarized in Table S2.
Selectivity profiling
Selectivity profiling (DUBProfiler) was performed by Ubiquigent using the manufacturer’s protocols.
Mouse liver microsome stability
Mouse liver microsome (MLM) half-life measurements were performed by Scripps Florida Pharmacology Core according their protocols.
Competitive activity based protein profiling
HEK 293T cells were pelleted, washed with PBS, lysed on ice (50 mM Tris pH 7.6, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 0.5% NP-40, 10% glycerol, 1 mM TCEP, phosphatase inhibitor cocktails (Sigma P5726 and Calbiochem 524624), and protease inhibitors (pepstatin, leupeptin, PMSF, and aprotinin), and clarified by centrifugation. Protein content was quantified by BCA, and 50 ug of lysate was diluted into 30 uL labeling buffer (50 mM Tris pH 7.6, 5 mM MgCl2, 0.5 mM EDTA, 250 mM sucrose, 1 mM TCEP) and incubated at room temperature with shaking with the indicated inhibitors for 30 minutes. Samples were then supplemented with 1 uM HA-Ub-VS and incubated at room temperature with shaking for 15 minutes. Reactions were quenched with 4x LDS sample buffer (Thermo Fisher B0007) supplemented with 10% BME, vortexed vigorously, and heated to 95°C for 5 minutes. Samples were resolved by SDS-PAGE and analyzed by Western blot with the indicated antibodies.
Cell treatments
Cells were treated with DMSO or different concentrations of XL-188, XL-203 for 6 (MM1S) or 16 (MCF7) hours in presence or absence of cycloheximide. For the experiments in which cycloheximide was used, cells were treated with compounds for 4 (MM1S) or 14 (MCF7) hours prior to the addition of 50 ug/ml of cycloheximide. At 6 or 16h time point cells were washed in PBS and lysed in modified RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 20 mM Tris, 150 mM NaCl, 1 mM EDTA) containing phosphatase inhibitor cocktails 1 and 2 (Sigma), and protease inhibitors. Protein concentrations were quantified using the BCA protein assay kit (Pierce) and samples were probed by immunoblotting using mdm2 (Santa cruz sc-965), p53 (Cell signaling 9282), p21 (Cell signaling 2947), GAPDH (Cell signaling 2118), USP7 (Cell signaling 4833) antibodies.
Peripheral blood mononuclear cell testing
Peripheral blood mononuclear cells (PBMCs) were generously provided by Dr. Steven Treon and Dr. Guang Yang. PBMCs from normal individuals were isolated by density gradient centrifugation through Ficoll-Plaque Plus (Amersham Pharmacia Biotech AB, Uppsala, Sweden) at 400xg for 25 minutes, followed by two washes in PBS. Cells were then maintained in RPMI media, supplemented with 10% FBS. Primary cells were obtained through written consent under approval of the Dana-Farber Cancer Institute Institutional Review Board. The trypan blue exclusion assay has been previously described(Weisberg et al., 2002) and was used for quantification of PBMCs prior to seeding for CellTiter-Glo Luminescent Cell Viability assays (Promega, Madison, WI). These assays were used for proliferation studies and carried out according to manufacturer instructions. Cell viability is reported as percentage of control (untreated) cells, and error bars represent the standard deviation for each data point.
Crystallization, data collection and structure determination
One equivalence of compound (from a 100 mM DMSO stock) was mixed with 1 mM protein and crystallized by sitting-drop vapor diffusion at 20 °C in the following crystallization buffer: 30% PEG 3350, 0.2 M NaFormate, 0.1 M NaCitrate-pH 5.5, and 10 mM DTT. Crystals were transferred briefly into crystallization buffer containing 25% glycerol prior to flash-freezing in liquid nitrogen. Diffraction data from complex crystals were collected at beamline 24ID-C of the NE-CAT at the Advanced Photon Source (Argonne National Laboratory). Data sets were integrated and scaled using XDS.(Kabsch, 2010) Structures were solved by molecular replacement using the program Phaser(McCoy et al., 2007) and the search model PDB entry 1NB8. The ligand was positioned and preliminarily refined using Buster and Rhofit.(Smart et al., 2012) Iterative manual model building and refinement using Phenix(Adams et al., 2010)and Coot(Emsley and Cowtan, 2004) led to a model with excellent statistics.
Hydrogen exchange experiments
Hydrogen exchange experiments were performed essentially as described in Iacob et al (Iacob et al., 2009). A stock solution of USP7 catalytic domain at 50 pmol/μL in 20mM Hepes (pH 7.5), 200 mM NaCl, 1 mM TCEP, 5% glycerol H2O was prepared. Deuterium exchange in USP7 alone was initiated by dilution with 15-fold D2O buffer (pD 7.5), at room temperature. At each deuterium exchange time point (from 10 s to 4 hours) an aliquot from the exchange reaction was removed and labeling was quenched by adjusting the pH to 2.5 with an equal volume of quench buffer (0.8% Formic Acid and 0.8M Guanidine Hydrochloride, H2O). Quenched samples were immediately injected into the LC/MS system.
For the HDX MS experiments of USP7 bound to compounds, each compound was individually incubated with USP7 as follows: XL188 was incubated at RT for 30 min with USP7 at a protein: compound ratio of 1:10, ensuring that 99.65 % was bound after dilution with D2O. 9 was mixed at a protein: compound ratio of 1:10, ensuring that 99.02 % was bound after dilution with D2O. 1 was mixed at a protein: compound ratio of 1:20, ensuring that 88.19% was bound after dilution with D2O. The same time course as for the protein alone was implemented for the compounds work (10sec–4h).
Mass analysis
Each sample was analyzed as previously described.(Wales et al., 2008) Briefly, the samples were digested online using a Poroszyme immobilized pepsin cartridge (2.1 mm x 30 mm, Applied Biosystems) at 15 °C for 30 s, then injected into a custom Waters nanoACQUITY UPLC HDX Manager™ and analyzed on a XEVO G2 mass spectrometer (Waters Corp., USA). The average amount of back-exchange using this experimental setup was 20–30%, based on analysis of highly deuterated peptide standards. Deuterium levels were not corrected for back-exchange and are therefore reported as relative.(Wales and Engen, 2006) All experiments were performed in duplicate. The error of measuring the mass of each peptide averaged ± 0.12 Da in this experimental setup (4). The HDX-MS data were processed using PLGS 3.0 and DynamX 3.0 (Waters Corp., USA).
QUANTIFICATION AND STATISTICAL ANALYSIS
All biochemical curves and statistical analyses were produced using Prism 6 (GraphPad Software).
DATA AND SOFTWARE AVAILABILITY
Structure files and coordinates have been deposited to PDB under these accession numbers: 5VSB for USP7/1, 5VSK for USP7/8 5VS6 for USP7/XL188.
Supplementary Material
Highlights.
Functional and structural characterization of USP7 inhibitors
Inhibitors bind the S4–S5 pocket of the enzyme
Inhibitors exhibit a high degree of selectivity for USP7 relative to 40 other DUBs
Acknowledgments
Crystallographic work was based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines (NIGMS P41 GM103403). Funding was also provided by NIH (R01 CA211681) (S.J.B, S.D., K.C.A), Dana-Farber/Northeastern University Joint Program in Cancer Drug Development (S.J.B., R.I., E.L.W.), Ellison Foundation (S.J.B.), Chleck Family Foundation (N.J.S.) and an award from TWD Kemtech J. Wang (S.J.B).
Footnotes
AUTHOR CONTRIBUTIONS
S.J.B, S.D, D.C., and K.C.A. initiated the project and S.J.B. and S.D. oversaw all aspects of the project. I.L. ran biochemical assays, designed and generated USP7 mutants and designed and performed cell experiments. X.L. designed and synthesized reported compounds. H-S.S. and S.D. carried out crystallographic studies and ITC and DSF experiments. T.M. generated USP7 mutants and performed biochemical assays. W. H. synthesized reported compounds. N.S. and D.D. performed cell experiments. R. I. and J. R. E. are responsible for the HDX MS studies. D.C. and K.C.A. oversaw cell studies. E. L. W. tested compounds on PBMCs. S.J.B. and S.D. wrote the manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul Rehman SA, Kristariyanto YA, Choi SY, Nkosi PJ, Weidlich S, Labib K, Hofmann K, Kulathu Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Molecular cell. 2016;63:146–155. doi: 10.1016/j.molcel.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer research. 2006;66:9235–9244. doi: 10.1158/0008-5472.CAN-06-0702. [DOI] [PubMed] [Google Scholar]
- Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, Kumar KG, Konietzny R, Fischer R, Kogan E, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chemistry & biology. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Atanassov BS, Koutelou E, Dent SY. The role of deubiquitinating enzymes in chromatin regulation. FEBS letters. 2011;585:2016–2023. doi: 10.1016/j.febslet.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos YM, St John SE, Mesecar AD. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral research. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NP, Leroy C, Sander C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics. 1998;14:380–381. doi: 10.1093/bioinformatics/14.4.380. [DOI] [PubMed] [Google Scholar]
- Byun S, Lee SY, Lee J, Jeong CH, Farrand L, Lim S, Reddy K, Kim JY, Lee MH, Lee HJ, et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3894–3904. doi: 10.1158/1078-0432.CCR-12-3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer cell. 2012;22:345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Song J, Wang J, Xu C, Chen C, Gu W, Sun H, Wen X. Synthesis and biological evaluation of thiazole derivatives as novel USP7 inhibitors. Bioorganic & medicinal chemistry letters. 2017;27:845–849. doi: 10.1016/j.bmcl.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Cheng C, Niu C, Yang Y, Wang Y, Lu M. Expression of HAUSP in gliomas correlates with disease progression and survival of patients. Oncology reports. 2013;29:1730–1736. doi: 10.3892/or.2013.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Heller H, Elias S, Haas AL, Hershko A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:1365–1368. doi: 10.1073/pnas.77.3.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Experimental & molecular medicine. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22:442–451. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbe S. Deubiquitylases from genes to organism. Physiological reviews. 2013;93:1289–1315. doi: 10.1152/physrev.00002.2013. [DOI] [PubMed] [Google Scholar]
- Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell. 2010;143:686–693. doi: 10.1016/j.cell.2010.11.016. [DOI] [PubMed] [Google Scholar]
- Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Molecular cancer therapeutics. 2009;8:2286–2295. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- Colland F, Marie-Edith G. W.I.P. Organization, editor . Selective and reversible inhibitors of ubiquitin specific protease 7. 2013. [Google Scholar]
- Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428:1p. doi: 10.1038/nature02501. following 486. [DOI] [PubMed] [Google Scholar]
- D’Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacology & therapeutics. 2015;147:32–54. doi: 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Dar A, Shibata E, Dutta A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Molecular and cellular biology. 2013;33:3309–3320. doi: 10.1128/MCB.00358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CW, Chaney J, Korbel G, Ringe D, Petsko GA, Ploegh H, Das C. The co-crystal structure of ubiquitin carboxy-terminal hydrolase L1 (UCHL1) with a tripeptide fluoromethyl ketone (Z-VAE(OMe)-FMK) Bioorganic & medicinal chemistry letters. 2012;22:3900–3904. doi: 10.1016/j.bmcl.2012.04.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer TS, Rosenthal AS, Liang Q, Chen J, Villamil MA, Kerns EH, Simeonov A, Jadhav A, Zhuang Z, Maloney DJ. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. Discovery of ML323 as a Novel Inhibitor of the USP1/UAF1 Deubiquitinase Complex. [PubMed] [Google Scholar]
- Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Science signaling. 2010;3:ra80. doi: 10.1126/scisignal.2001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Molecular cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- El-Desoky AH, Kato H, Tsukamoto S. Ceylonins G-I: spongian diterpenes from the marine sponge Spongia ceylonensis. Journal of natural medicines. 2017 doi: 10.1007/s11418-017-1087-4. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. The EMBO journal. 1997;16:566–577. doi: 10.1093/emboj/16.3.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faesen AC, Dirac AM, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Molecular cell. 2011;44:147–159. doi: 10.1016/j.molcel.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Felle M, Joppien S, Nemeth A, Diermeier S, Thalhammer V, Dobner T, Kremmer E, Kappler R, Langst G. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic acids research. 2011;39:8355–8365. doi: 10.1093/nar/gkr528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Dikic I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. Journal of cell science. 2012;125:265–275. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:1783–1786. doi: 10.1073/pnas.77.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings DS, Flygare JA, Bogyo M, Wertz IE. Activity-based probes for the ubiquitin conjugation-deconjugation machinery: new chemistries, new tools, and new insights. The FEBS journal. 2017;284:1555–1576. doi: 10.1111/febs.14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Molecular cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Iacob RE, Pene-Dumitrescu T, Zhang J, Gray NS, Smithgall TE, Engen JR. Conformational disturbance in Abl kinase upon mutation and deregulation. Proc Natl Acad Sci U S A. 2009;106:1386–1391. doi: 10.1073/pnas.0811912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson MK, Ploegh HL. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell host & microbe. 2009;5:559–570. doi: 10.1016/j.chom.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Molecular cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Johnston SC, Larsen CN, Cook WJ, Wilkinson KD, Hill CP. Crystal structure of a deubiquitinating enzyme (human UCH-L3) at 1.8 A resolution. The EMBO journal. 1997;16:3787–3796. doi: 10.1093/emboj/16.13.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta crystallographica Section D, Biological crystallography. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. The oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- Kee Y, Huang TT. Role of Deubiquitinating Enzymes in DNA Repair. Molecular and cellular biology. 2015;36:524–544. doi: 10.1128/MCB.00847-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler BM. Selective and reversible inhibitors of ubiquitin-specific protease 7: a patent evaluation (WO2013030218) Expert opinion on therapeutic patents. 2014;24:597–602. doi: 10.1517/13543776.2014.882320. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nature reviews Molecular cell biology. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annual review of biochemistry. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Kon N, Kobayashi Y, Li M, Brooks CL, Ludwig T, Gu W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene. 2010;29:1270–1279. doi: 10.1038/onc.2009.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Molecular cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- Maculins T, Fiskin E, Bhogaraju S, Dikic I. Bacteria-host relationship: ubiquitin ligases as weapons of invasion. Cell research. 2016;26:499–510. doi: 10.1038/cr.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malynn BA, Ma A. Ubiquitin makes its mark on immune regulation. Immunity. 2010;33:843–852. doi: 10.1016/j.immuni.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndubaku C, Tsui V. Inhibiting the deubiquitinating enzymes (DUBs) Journal of medicinal chemistry. 2015;58:1581–1595. doi: 10.1021/jm501061a. [DOI] [PubMed] [Google Scholar]
- Nicholson B, Leach CA, Goldenberg SJ, Francis DM, Kodrasov MP, Tian X, Shanks J, Sterner DE, Bernal A, Mattern MR, et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein science : a publication of the Protein Society. 2008;17:1035–1043. doi: 10.1110/ps.083450408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Current opinion in chemical biology. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Pinto-Fernandez A, Kessler BM. DUBbing Cancer: Deubiquitylating Enzymes Involved in Epigenetics, DNA Damage and the Cell Cycle As Therapeutic Targets. Frontiers in genetics. 2016;7:133. doi: 10.3389/fgene.2016.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Leonhardt H, Spada F. Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. Journal of cellular biochemistry. 2011;112:439–444. doi: 10.1002/jcb.22998. [DOI] [PubMed] [Google Scholar]
- Quesada V, Diaz-Perales A, Gutierrez-Fernandez A, Garabaya C, Cal S, Lopez-Otin C. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochemical and biophysical research communications. 2004;314:54–62. doi: 10.1016/j.bbrc.2003.12.050. [DOI] [PubMed] [Google Scholar]
- Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, Harpon J, Battaglia V, Vivat V, Sippl W, Colland F. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chemistry & biology. 2012;19:467–477. doi: 10.1016/j.chembiol.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Ritorto MS, Ewan R, Perez-Oliva AB, Knebel A, Buhrlage SJ, Wightman M, Kelly SM, Wood NT, Virdee S, Gray NS, et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nature communications. 2014;5:4763. doi: 10.1038/ncomms5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, Jones M, Hassiepen U, Schaefer M, Kiffe M, et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nature communications. 2016;7:13166. doi: 10.1038/ncomms13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Molecular cell. 2009;36:469–476. doi: 10.1016/j.molcel.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta crystallographica Section D, Biological crystallography. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanokashira N, Kukita S, Kato H, Nehira T, Angkouw ED, Mangindaan REP, de Voogd NJ, Tsukamoto S. Petroquinones: trimeric and dimeric xestoquinone derivatives isolated from the marine sponge Petrosia alfiani. Tetrahedron. 2016;72:5530–5540. [Google Scholar]
- Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, Chen C, Califano A, Yamashiro DJ, Sun H, Gu W. HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nature medicine. 2016;22:1180–1186. doi: 10.1038/nm.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap JA, Kumar BR, Moshkin YM, Langenberg K, Krijgsveld J, Heck AJ, Karch F, Verrijzer CP. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Molecular cell. 2005;17:695–707. doi: 10.1016/j.molcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, Meerding J, Berkers CR, Barbi J, Grone A, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–271. doi: 10.1016/j.immuni.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass spectrometry reviews. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Ma S, Song N, Li X, Liu L, Yang S, Ding X, Shan L, Zhou X, Su D, et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. The Journal of clinical investigation. 2016a;126:2205–2220. doi: 10.1172/JCI85747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Scientific reports. 2016b;6:26979. doi: 10.1038/srep26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Molecular cell. 2008;29:653–663. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, Gilliland DG, Griffin JD. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer cell. 2002;1:433–443. doi: 10.1016/s1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nature cell biology. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes & development. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Miyazaki M, Kodrasov MP, Rotinsulu H, Losung F, Mangindaan RE, de Voogd NJ, Yokosawa H, Nicholson B, Tsukamoto S. Spongiacidin C, a pyrrole alkaloid from the marine sponge Stylissa massa, functions as a USP7 inhibitor. Bioorganic & medicinal chemistry letters. 2013;23:3884–3886. doi: 10.1016/j.bmcl.2013.04.066. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang H, Tian L, Li H. Expression of USP7 and MARCH7 Is Correlated with Poor Prognosis in Epithelial Ovarian Cancer. The Tohoku journal of experimental medicine. 2016;239:165–175. doi: 10.1620/tjem.239.165. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, Wyce A, Thorne AW, Berger SL, McMahon SB. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Molecular cell. 2008;29:102–111. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S, Suzuki E, Le Guezennec X, Stunnenberg HG, Krasnov A, et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Molecular cell. 2008;29:92–101. doi: 10.1016/j.molcel.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Zinngrebe J, Montinaro A, Peltzer N, Walczak H. Ubiquitin in the immune system. EMBO reports. 2014;15:28–45. doi: 10.1002/embr.201338025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.