

Abstract
Many factors during the transplantation process influence posttransplant graft function and survival, including donor type and age, graft preservation methods (cold storage, machine perfusion), and ischemia-reperfusion injury. Successively, they will lead to cellular and molecular alterations that determine cell and ultimately organ fate. Oxidative stress and autophagy are implicated in posttransplant outcome since they are both affected by the stress responses triggered in each step (donor, preservation, and recipient) of the transplantation process. Furthermore, oxidative stress influences autophagy and vice versa. Interestingly, both processes have positive as well as negative effects on graft outcome, suggesting they are tightly linked during the transplantation process. In this review, we discuss the importance, regulation and crosstalk of oxidative signals, and autophagy in the field of transplantation medicine.
1. Introduction
For patients with end-stage organ disease, organ transplantation has become the treatment of choice. However, the success of transplantation is limited by a global shortage of suitable organs as well as loss of grafts following transplantation due to primary nonfunction or rejection. The gap between supply and demand has steadily increased over the years and, as a result, so has the number of patients on the waiting list [1, 2]. This is an alarming increase, as organ transplantation significantly improves a patient's quality of life as well as survival rate when compared to patients who remain on the waiting list [3–5]. These problems can be addressed by increasing the use of older and higher risk donors, while simultaneously improving graft longevity.
Oxidative stress levels correlate with graft survival in all steps of the transplantation process including in the donor [6–16], during preservation [17, 18], and reperfusion in the recipient. In donation after brain death (DBD) donors, brain death pathophysiology leads to increased renal oxidative damage markers which correlate with acute rejection, delayed graft function (DGF), and allograft function [6–16]. In donation after cardiac death (DCD) donors, cardiac arrest causes warm ischemia which is associated with impaired graft function and higher mortality rates [6–8]. Increased oxidative stress markers are evident in organs from DCD donors, but no evidence is available correlating them with the outcome. Clinically proven donor treatments that benefit graft survival include dopamine administration and hypothermic cooling of DBD donors, of which the effects could be related to modulation of oxidative stress [19, 20].
Organ grafts suffer additional ischemic injury during preservation. Prolonged duration of cold ischemia is considered an independent risk factor for a nonfunctioning or dysfunctioning transplant, particularly in marginal or extended criteria donation (ECD) donors [21]. These side effects have recently led to the implementation of hypothermic machine perfusion (HMP). HMP has clear benefits over static cold storage, as evidenced by improved graft function and survival rates in kidney transplantation [22, 23], as well as reduced oxidative stress markers in experimental [24, 25] and clinical [26] liver preservation.
In the recipient, the mechanism of reperfusion injury, labeled ischemia-reperfusion (IR) injury (IRI), has been reported in most solid transplantable organs [27–31] and is mediated by reactive oxidative species (ROS) production most likely from donor-derived vascular cells [32, 33], followed by a second burst of ROS probably produced by the recipient's phagocytes [29, 34–40]. However, mitochondria are also implicated in ROS [29, 41–44] as well as nitric oxide (NO) production during IRI [45–47]. Despite overwhelming experimental evidence on the beneficial effects of attenuating ROS during this phase, clinical evidence remains limited.
Oxidative stress is a known inducer of autophagy. Even though autophagy regulation during the transplantation process is only starting to be understood, several autophagy modulators have already been implemented. In this review, we will first introduce autophagy in the context of the transplantation process and cover the current knowledge during each of these stages. Secondly, we will touch upon the complex, intertwined, and reciprocal relationship of oxidative stress and autophagy in the field of transplantation medicine while covering the therapeutic strategies that target each of these pathways.
2. Autophagy and Transplantation
2.1. Autophagy: Importance and Mechanisms
Several pathways of autophagy exist [48], of which macroautophagy is the best studied (and which will simply be referred to as “autophagy” for the remainder of this manuscript). It involves the formation of double-membranous vesicular “autophagosomes” that occlude and transport the soon-to-be degraded material to the lysosomes (Figure 1). This process is normally constitutively active in cells, albeit at a low basal level, thereby maintaining cellular homeostasis (Figure 1). However, autophagy is stimulated upon stress through several signaling pathways, of which the mammalian target of rapamycin (mTOR) pathway is the most notable in response to nutrient stress. Energy deprivation attenuates mTOR signaling through the activation of AMP-activated kinase (AMPK), which also directly stimulates autophagy by the phosphorylation of the autophagy-initiating ULK1 complex. Another initiation complex is constituted by Vps34 (class III PI3K) and Beclin 1, an autophagy-specific BH3-only domain-containing protein that is inhibited by several antiapoptotic Bcl-2 family proteins (Figure 1). As such, apoptotic signals can also trigger autophagy, in which the autophagic response often precedes apoptosis as the first attempt to survival [49]. However, when autophagy fails, cells will eventually activate apoptosis, which may even occur with the help of the still active autophagic machinery. This shows that autophagy may switch from a prosurvival to a prodeath pathway under certain conditions [50], although the exact mechanism, context, and details on this autophagy-dependent cell death remain currently elusive.
Figure 1.

Overview of the autophagy process. Autophagy is initiated by the ULK1 complex, which is negatively regulated by mTOR, but positively by AMPK. This way it responds to nutrient or energy deprivation. In addition, class III PI3K complex requires Beclin 1, which is inhibited by Bcl-2. During elongation, Atg5-Atg12 and LC3-II are required. The latter is attached to the autophagosomal membranes. LC3-II will be delipidated on the outer membrane by Atg4 (a process inhibited by H2O2) but remains on the inner membrane and will be degraded inside the lysosomes. Mitochondria can be also degraded (mitophagy), via recruitment of Sqstm1/p62. The latter protein also recruits Keap1 for degradation, thereby enabling Nrf-2-dependent antioxidant transcription. Eventually, autophagosomes fuse with lysosomes, the cellular structures in which degradation takes place. The levels of autophagy determine the outcome on cellular injury and need to stay balanced in order not to provoke death.
Following initiation, the formation of autophagosomes is mediated by the Atg5-Atg12 complex and the formation of phosphatidylethanolamine-conjugated LC3 (“LC3-PE” or “LC3-II”) (Figure 1). The latter is generally used as a marker for autophagy, as it is distinguishable from its precursor LC3-I on Western blot and its fluorescent labeling allows visualization of autophagosomes as GFP-LC3 punctae. Despite delipidation of LC3-II on the outer membrane by Atg4, LC3-II remains attached to the inner autophagosomal membranes even after fusion of autophagosomes with lysosomes. Therefore, an increase of LC3-II or GFP-LC3 punctae can signify stimulation of autophagy or attenuation of the final steps in autophagy (e.g., inhibition of fusion), leading to an accumulation of autophagosomes without any true upregulation of autophagy. Therefore, prudency is advised when interpreting autophagy data because the dynamic character of the “autophagic flux” should be taken into consideration [51].
As the graft endures several types of stress during transplantation, it is evident that the protective properties of autophagy might be important in restoring cellular homeostasis and function in the organ grafts. Interestingly, DGF increases with donor (and recipient) age as aging leads to increased susceptibility towards cellular stress [52, 53]. An underlying mechanism for this increased vulnerability of aged organs is the age-related reduction in autophagy [52]. It has therefore been suggested that pharmacological stimulation of autophagy could reduce graft injury and promote function [54]. However, as excessive autophagy may detrimentally impact cellular fate through autophagy-dependent cell death (Figure 1), it is important to first understand the dynamics and role of autophagy to determine whether autophagy stimulation or inhibition is the best option in transplantation.
2.2. Autophagy in the Donor
The autophagic response in the donor is likely the result of donor-related characteristics including donor age, gender, comorbidities, and donor type of death. Of course, ischemic time is strongly prolonged in DCD compared to DBD grafts, which could partially explain the stronger injury in DCD grafts. This is important, since autophagy's dynamics and role (protective versus detrimental) during ischemic stress are likely dependent on the extent of ischemic injury, at least in the kidney [55, 56] and heart [57]. As DCD donors suffer more extensive anoxic injury compared to DBD donors, this could trigger autophagy-dependent cell death (Figure 2) and suggests that therapeutic strategies involving autophagy modulation are strongly donor type-dependent.
Figure 2.

Regulation of oxidative signals and autophagy by transplantation-related factors. Excessive autophagy and/or oxidative stress can lead to increased graft injury and tilt the balance to the right. To tilt the balance towards survival, excessive signals need to be reduced towards protective levels of oxidative signals and autophagy.
Additionally, the higher posttransplant injury in ECD (e.g., older) kidneys may be attributed to a decline in autophagic activity with age [58–61], justifying autophagy stimulation as the preferred strategy in these donors. Compared to young mice, old mice showed a decreased autophagic response in terms of vacuole formation and elimination after stimulation of these processes with vinblastine and Triton X-100 [60]. In line with this, hypoxia-induced injury was reduced by starvation-induced autophagy in older kidneys [62]. Besides donor age, a less studied but seemingly equally important feature is the gender of the donor. In a cardiac IRI study in mice, males and females showed different autophagic activities: male mice show a decrease, and female mice an increase in LC3-II levels [61]. We have also observed gender differences in the kidney, with a decrease in male, but unchanged LC3-II levels in female Sprague-Dawley rats subjected to 45 min of ischemia followed by 3 h of reperfusion (Figure 3). As such, therapeutic strategies involving autophagy modulation may differ in organs coming from male and female donors (Figure 2).
Figure 3.

Gender differences in autophagy activation in response to ischemia-reperfusion injury. Western blot expression of autophagy-related proteins LC3-I, LC3-II, and Sqstm1/p62 in female and male Sprague-Dawley rat kidneys subjected to 45 min of warm ischemia (WI) followed by 3 h of reperfusion (3 h R). Each lane represents an independent experiment (N = 6). Quantification of p62 or LC3-II over GAPDH levels, compared to the mean of the corresponding Sham group. LC3-II and p62 are clearly lower in the WI group of females compared to the male WI group. Results are presented as mean ± SD (N = 6 per group) (∗p < 0.05; ∗∗∗p < 0.001).
2.3. Autophagy during Organ Preservation
Mixed reports exist on the effects of cold ischemia on autophagy. In mouse kidneys, cold ischemia resulted in increased autophagy markers [63]. Interestingly, repression of autophagic flux by means of lysosomal inhibitor bafilomycin A1 resulted in less apoptosis, suggesting that autophagy could trigger cell death during renal cold ischemia. Similar findings were observed in cold-preserved rat lungs [61], where prolonged preservation resulted in increased autophagy associated with cell death. Alternatively, decreased markers of autophagy were found following cold ischemia in marginal, steatotic rat livers [64–66]. Induction of autophagy in these marginal livers by means of melatonin and trimetazidine addition to the cold preservation medium improved organ quality as evidenced by lower levels of injury markers ALT and GLDH [66]. These benefits were attenuated when autophagy was suppressed with bafilomycin A1 [66]. Furthermore, oxygen insufflation of the cold preservation medium of marginal livers reversed the suppression of autophagy and functional impairment [65], suggesting beneficial effects of autophagy induction. These studies suggest that the differential effects of autophagy activation might be organ-dependent (Figure 2; see also Autophagy in the Recipient) and closely associated with organ function and cell death.
2.4. Autophagy in the Recipient
The role of autophagy in IRI is best studied in the context of the transplantation process. However, its dynamics and roles remain elusive and are likely dependent on different factors. Firstly, when studying the nature of autophagy dynamics, that is, whether autophagy is stimulated or attenuated during IRI, research suggests that autophagy is mostly upregulated in the heart [67] and the kidney [55], while findings in the liver are conflicting [64]. These discrepancies might be explained by several reasons (described in more detail in [55]), including the difficulty of measuring the dynamic process of autophagic flux in static samples. Indeed, an increase in LC3-II could equally well indicate stimulated autophagy as well as inhibited autophagic flux. In vivo experiments with chloroquine (an inhibitor of autophagosome-lysosome fusion, mimicking inhibited autophagic flux) suggest that autophagy inhibition might occur in the heart [68] and liver [69]. Moreover, as autophagy often fluctuates during prolonged stress, multiple time point postreperfusion should be investigated [55].
Secondly, conflicts arise when looking at the proposed role of autophagy during IRI. In hepatic IRI models, most studies indicate a protective role for autophagy [64], whereas both protective and detrimental roles are assigned to autophagy in the heart and kidney [55, 67]. Besides the difficulties in measuring autophagic flux, nonspecific chemical modulators of autophagy also have secondary effects on mTOR (e.g., rapamycin), PI3K (e.g., 3-methyladenine), or lysosomal and endocytic function (bafilomycin A1 and chloroquine) [55]. Even data in conditional autophagy knockout mice (e.g., atg5−/−) are important to interpret with caution. As autophagy is an important mechanism for basal cellular homeostasis in cells (Figure 1), any stress addition in these models will likely lead to more injury than in wild-type. In this context, proximal tubule-specific Atg5 KO mice displayed strange concentric membranous structures in targeted cells [70], suggesting unhealthy cells.
Thirdly, several reports propose a dual role for autophagy in IRI, dependent of the extent of the stress [55, 71]; that is, mild IR stress leads to protective autophagy stimulation, while severe stress could trigger a switch towards autophagy-dependent cell death (Figures 1 and 2). In this respect, the duration of ischemia prior to transplantation is an important factor to consider. The longer the ischemic period, the more severe the reperfusion injury, which seems associated mostly with a detrimental role for autophagy, at least in the kidneys [55]. Besides the extent of the stress, the type of stress that initiates autophagy is also important. This is evident in the heart, where autophagy-dependent cell death during reperfusion seems to be dependent on the levels of Beclin 1, in which initiation is likely determined by apoptotic factors. Protective autophagy on the other hand seems to be dependent on AMPK activation and would therefore be related to changes in energy status (Figure 2) [71].
Finally, autophagy during IRI is strongly determined by the degree of autophagy dependency of the organs and even of different cell types within an organ (Figure 2). In renal podocytes, for example, autophagy is much more important for cellular homeostasis (as these cells are postmitotic) than in tubular cells. Also, cardiomyocytes are strongly dependent on basal autophagy, while hepatocytes rely more on stress-induced autophagy. In this respect, liver cells might have different mechanisms controlling autophagy as they can tolerate more severe stress than other organs. This might explain the more consistent data regarding the protective role for autophagy in this organ compared to the kidney and the heart during IR.
3. Autophagy and Oxidative Stress in Transplantation
3.1. The Relationship between Autophagy and Oxidative Stress
Both oxidative stress and autophagy have been described as both protective and detrimental pathways in response to cellular stressors [34, 55]. Therefore, it is feasible that the fine balance of oxidative stress levels and autophagy activation plays an important role in the long-term function and survival of organ grafts. This makes modulation of these pathways interesting targets to predict or improve graft function and survival after transplantation.
Surprisingly, the only currently known direct redox-based regulation of autophagy is the inhibitory oxidation of Atg4 by H2O2, which suppresses the delipidation of LC3-II [72] (Figure 1). Furthermore, H2O2 is also proposed to stimulate autophagy initiation directly via regulation of AMPK [73] (Figure 1). In a slower, more indirect fashion, oxidative stress also regulates transcription of Beclin 1 and LC3 [54]. Together, these protective, proautophagic effects are achieved with subtle changes in ROS. Conversely, acute and persistent ROS production can oxidatively modify macromolecules in such a way that they are only partially degraded by the autophagic/lysosomal pathway. This produces an indestructible product known as lipofuscin, which accumulates within the lysosomes, hampers their function, and sustains or even exacerbates oxidative injury [74]. Together, this suggests that the amount of ROS produced will determine whether autophagy will be activated or inhibited and whether it prevents or amplifies further damage.
Besides the regulation of autophagy by oxidative molecules, autophagy reciprocally regulates oxidative signals. As it is a clearance mechanism, autophagy may remove oxidatively damaged macromolecules or even entire organelles. Upon excessive ROS production, mitochondria risk severe damage and need to be partially removed [75]. This occurs through the selective autophagic degradation of damaged mitochondrial fragments called “mitophagy” (Figure 1). This process involves the recruitment of PINK1 and Parkin to the outer mitochondrial membrane where they promote ubiquitination (Ub) of several mitochondrial proteins [76, 77]. This Ub signal serves as a recognition for Sqstm1/p62, which is an adapter protein for LC3-II (Figure 1). The link between ROS and mitophagy was confirmed in a study on heart failure in mice, describing that p53-induced impairment of mitophagy resulted in mitochondrial dysfunction and increased ROS production [76, 77].
In addition to mitophagy, autophagy is shown to regulate antioxidant responses through interactions of Sqstm1/p62 with Keap1. Sqstm1/p62 recruits Keap1 via interactions with ubiquitinated aggregates, after which Keap1 is degraded via autophagy. Degradation of Keap1 in turn prevents it from ubiquitinating and degrading Nrf2-dependent transcription factors, thereby enabling the antioxidant response [78] (Figure 1).
3.2. Autophagy and Oxidative Stress in the Donor
The accumulation of acute and chronic injuries in the donor leads to increased senescence and reduced organ quality. In ECD donors, this may be due to an increase in age-related oxidative damage (Figure 2) [16] as well as a reduction of autophagic activity and hence the inability to remove oxidatively damaged organelles. This link was shown in the kidneys of aged rats, where a high caloric diet exacerbated oxidative damage and aging, while autophagy was reduced [79]. The opposite was observed in the low caloric group [79]. The link between caloric intake and autophagy regulation is interesting, as it highlights how energy deprivation (i.e., glucose and amino acid stress) can initiate protection via autophagy-mediated pathways to restore cellular energy supplies (Figure 2) [73]. In response to starvation, increased activity of the mitochondrial apparatus in turn increases mitochondrial ROS production, which activates autophagy [73]. Interestingly, a proteomics study in brain-dead rodents indicated that both metabolic changes and mitochondrial dysfunction are the two major canonical pathways affected in the kidney of brain-dead donors [80]. Even though no direct line of evidence connects these alterations to autophagy, it is nonetheless feasible that autophagy is indeed modulated in DBD donors.
A recent study from our group showed that the treatment of brain-dead rats with thyroid hormone T3, both a metabolic regulator and a powerful inducer of autophagy, improved liver function, while reducing apoptosis and oxidative stress [81]. In prosecution of this work [81], the protective effects of T3 preconditioning in brain-dead rats were indeed accompanied by an induction of autophagy in the liver, as evidenced by increased formation of LC3-II together with decreased (albeit nonsignificantly) levels of the autophagic substrate SQSTM1/p62 (Figure 4). Interestingly, in the kidney, T3 neither altered autophagy nor attenuated injury or apoptosis (Figure 4). Therefore, this suggests that T3-induced autophagy is not evident in the kidney and consequently, neither a reduction in apoptosis nor injury. In contrast, the reduction in apoptosis and injury markers in the liver does appear to be associated with increased hepatic autophagy, suggesting an important role for autophagy stimulation in the livers from brain-dead animals. We hypothesize that T3 boosts a transient peak in mitochondrial activity and ROS production, resulting in autophagy induction and subsequent protective effects during BD in the liver (Figure 5). This is supported by a recent study by Sinha et al. that showed that T3 induces autophagy via ROS-related pathways in the liver both in vivo and ex vivo [82, 83]. This phenomenon might not be evident in the brain-dead kidney due to extensive levels of oxidative stress [11], which might push the balance from a protective to a detrimental role for autophagy. This work therefore suggests that triggering oxidative pathways while simultaneously increasing autophagy could be an interesting strategy to improve liver function in DBD donors. In this respect, modulation of (mitochondrial) metabolism by compounds such as T3 might be the key to stimulation of transient ROS and subsequently protective autophagy activation (Figure 2).
Figure 4.

Posttranscriptional reduction of apoptosis and induction of autophagy in the liver of brain-dead rats following T3 treatment, yet no effects in the kidney. Western blot expression of proapoptotic protein Bax and autophagy-related proteins LC3-II and SQSTM1/p62 in the liver (a, c) and kidney (b, d) and renal injury markers in plasma (e) of T3- or vehicle-pretreated brain-dead rats. Each lane represents an independent experiment. Results are presented as mean ± SD (N = 7 per group) (∗∗p < 0.01).
Figure 5.
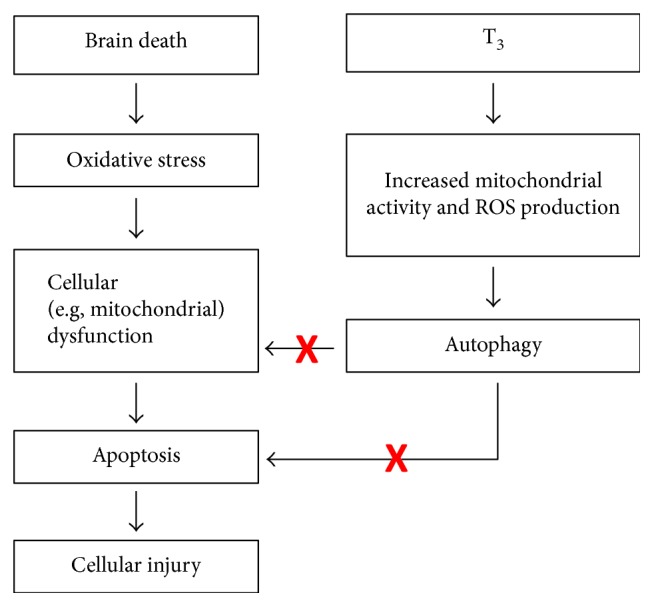
Proposed mechanism of T3 preconditioning during brain death in the liver.
3.3. Autophagy and Oxidative Stress during Preservation
Since most organs worldwide are still preserved on ice, several developments have been made to reduce ischemic injury during cold storage. The gold standard for preservation solutions in kidney, liver, pancreas and small bowel preservation is the University of Wisconsin (UW) storage solution [22, 84]. When comparing UW to HTK solution, the use of UW leads to decreased renal apoptosis and was linked to lower graft injury in human renal allografts [85]. UW contains, besides energy supplies (adenosine) and osmotic compounds, the antioxidants glutathione and allopurinol [84, 86] (Figure 2). However, the beneficial effects of these compounds are questionable considering their short life-spans and the fact that supplementation of glutathione to UW solution was unable to improve renal transplantation outcomes [84]. Nonetheless, a rodent study on kidney transplantation showed that treatment with hydrogen-rich saline (HRS), a novel antioxidant, immediately before ischemia attenuated transplantation-related renal injury and oxidative stress, while simultaneously inducing autophagic markers Beclin 1 and LC3-II [87]. Inhibition of autophagy with chloroquine attenuated these protective effects of HRS, suggesting that these effects were indeed autophagy-mediated and linked to oxidative stress [87]. Therefore, the addition of antioxidants to the preservation solution remains an interesting option to reduce graft injury (Figure 2). Conversely, prolonged cold ischemia of healthy kidneys in UW solution showed increased numbers of apoptotic and autophagic cells. In this case, the addition of bafilomycin A1 inhibited both autophagy and apoptosis [63]. These studies suggest that for the kidney the extent of cold ischemia injury could dictate whether either autophagy induction (in the case of shorter cold ischemia times) or inhibition (in the case of prolonged cold ischemia times) might be beneficial (Figure 2).
In the liver, cold storage of hepatic cells inhibited autophagic flux in both UW and Celsior solutions. However, only Celsior-stored cells reactivated autophagy upon reperfusion, while UW-stored cells displayed impaired lyso-autophagosomal fusion and elevated cell death [88]. Interestingly, pretreatment with simvastatin before cold ischemia led to the restoration of autophagy even in UW solution. This was accompanied by preserved cellular viability [88], suggesting possible benefits for hepatocytes when autophagy is induced during cold storage. These benefits might be linked to oxidative stress, as simvastatin-induced autophagy in cold-stored rat steatotic livers was accompanied by attenuation of oxidative stress [89], showing a link between ROS reduction and autophagy induction during cold liver preservation. In support of this, a study on liver transplantation in which the preservation medium (IGL-1) was enriched with trimetazidine, a fatty acid oxidation inhibitor, showed reduced oxidative stress yet activation of autophagy accompanied with reduced mitochondrial damage and hepatic injury [90]. These data therefore suggest that during cold storage of the liver, a strategy is preferred that simultaneously attenuates oxidative stress and stimulates autophagy.
The advantage of HMP over static cold storage has led to an increased implementation of this technique in the clinical setting. However, the lack of oxygen during HMP initiates high metabolic flux and subsequent high ROS production, particularly during the first minutes of reperfusion [91]. Therefore, benefits of oxygen addition to HMP solution are currently being investigated. Animal studies on liver and kidney HMP indicate that oxygenated HMP reduced mitochondrial flux and subsequent ROS production [91–93]. ROS production is further reduced when grafts are perfused at higher temperatures, such as subnormothermic (25°C) or normothermic (37°C) temperatures [94, 95]. Unfortunately, the effects of HMP on autophagy remain unexplored (Figure 2).
3.4. Autophagy and Oxidative Stress in the Recipient
As the extent of the oxidative response is dependent on ischemic duration, it is possible that the role of autophagy during IRI is dependent on the extent of oxidative stress. In this case, mild IR stress would trigger protective autophagy to counteract the oxidative damage experienced, for example, through autophagy-specific degradation of mitochondria (mitophagy). Regulation of mitophagy is thought to occur via oxidation of cardiolipin [29, 96–98], which plays a central role by mediating increased mitochondrial ROS release which serves as the signal for mitophagy initiation [99]. However, upon a very strong oxidative response (during severe reperfusion injury), an energy-dependent process as autophagy is not advised. In this case, autophagy stimulation could deprive much-needed energy, induce still elusive autophagy-dependent cell death mechanisms [100], or lead to the production of toxic lipofuscin (Figure 2) [74].
In the liver, IRI induced oxidative stress, autophagy, and apoptosis. These effects were attenuated by pretreatment with the antioxidant astaxanthin, potentially via modulation of the MAPK protein family [101]. In a study on renal IRI, increased oxidant production and autophagy markers were evident which suggests a tight and direct relationship between these two processes in which autophagy serves a protective role against renal oxidative damage [102]. In the heart, autophagic flux was analyzed during IRI in the presence of either H2O2 or the antioxidant N-2-mercaptopropionyl glycine (MPG). H2O2 increased, while MPG decreased autophagic flux, suggesting that autophagy is regulated by oxidative signals during cardiac IR. Interestingly, autophagy attenuation (by using heterozygous beclin 1+/− mice) improved injury in this experimental setting [103]. Altogether, these findings imply that oxidative signals influence autophagy during IR and might be responsible for the exact role (protective or detrimental) autophagy plays in IRI, depending on the type of organ.
Several modulators of oxidative stress and autophagy have been clinically tested or are commonly used following transplantation. One method to combat the oxidative burst in recipients is ischemic postconditioning (IPoC), a technique that involves the temporal cessation of blood flow to a remote tissue such as a limb or locally via constriction of a nearby afferent artery. Despite promising preclinical results of local [104] and remote IPoC [105], clinical trials investigating both techniques did not report improved renal function after transplantation [104, 106]. Interestingly, IPoC also stimulates autophagy in the heart [107, 108], but this seems to depend on the postreperfusion time [109]. A clinical study on the addition of the antioxidant human recombinant SOD showed decreased rejection rates and improved survival following cadaveric kidney transplantation, despite mixed preclinical results [17, 110]. These results might be autophagy-dependent, as SOD overexpression resulted in attenuation of starvation-induced autophagy and apoptosis [111].
Many of the compounds given to transplant recipients as part of their immunosuppressive regimen also modulate autophagy. These include calcineurin inhibitors cyclosporine and tacrolimus, mTOR inhibitors sirolimus (rapamycin) and everolimus, mycophenolate mofetil (MMF), and corticosteroids [55, 112]. Cyclosporine is a known autophagy inducer that has now largely been replaced by tacrolimus as the treatment of choice in most European and American transplantation centers, mostly because of its proposed nephrotoxic side effects [112]. Interestingly, these side effects are thought to be related to increased autophagic clearance and autophagosome formation, phenomena possibly mediated via oxidative stress and apoptosis [92, 113]. Tacrolimus, on the other hand, was recently identified as an autophagy modulator that acts via activation of transcription factor EB, which in turn increases the expression of both autophagy and lysosomal genes [114, 115]. However, it is likely that this is associated with a mild, beneficial induction of oxidative stress [116]. The use of rapamycin as well as second generation mTOR inhibitors such as everolimus and deferolimus has been used as part of immunosuppressive therapy mostly for their ability to limit T-cell proliferation, but these compounds also influence autophagy by means of mTOR inhibition. However, the use of rapamycin in animal models of renal IRI and transplantation has yielded questionable results. Some studies suggest that rapamycin improves mitochondrial homeostasis and, subsequently, reduces ROS production and cellular senescence [46]. On the contrary, a study on renal transplant recipients suffering from DGF shows that patients who received rapamycin had significantly lower chance to resolve DGF [117]. Interestingly, combined treatment with tacrolimus or mycophenolate mofetil (MMP), an immunosuppressive drug that is known to activate chaperone-mediated autophagy, enhanced the positive effects of rapamycin [55, 118]. The mixed effect of rapamycin treatment might be attributed to the amount of injury and subsequent extent of autophagy activation [55]. Finally, corticosteroids have been part of most postoperative and maintenance immunosuppressive regimens over the past years [119]. Interestingly, methylprednisolone (MP) both suppresses and stimulates autophagy in animal models [55]. This difference could be related to the extent of the preceding injury, or could be dose- and time-dependent, as MP treatment has opposite effects on oxidative injury when it is administered acutely (beneficial) or chronically (damaging) in rat lungs [120]. If and how MP affects autophagy modulation posttransplantation and whether this is influenced by oxidative signals remains to be elucidated.
4. Conclusion
Oxidative stress is an important component of the transplantation process as well as a known inducer of autophagy. Although the exact mechanism behind the complex reciprocal relationship between oxidative stress and autophagy is only beginning to be understood, it seems to play a major role in the different roles autophagy and oxidative signals seem to play during transplantation. The potential protective properties of autophagy and low levels of oxidative stress, yet detrimental effects when excessive activation occurs (Figure 2), make these two processes interesting therapeutic or diagnostic targets in each step of the transplantation process, while their tight interaction supports the possibility to target both pathways with only one compound. However, how these processes are preferentially modulated depends on the specific step in the transplantation process, the type of organ, the age and gender of the donor, the ischemic time, and other contributing factors. Therefore, future research should try and decipher the complex, intertwined relationship of oxidative stress and autophagy during the transplantation process. Finding the optimal balance between autophagic and oxidative processes is crucial for the optimization of cellular longevity and thereby graft survival.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- 1.Eurotransplant. 2017. http://statistics.eurotransplant.org/index.php?search_type=transplants&search_period=by+year+chart.
- 2.National data: donors recovered in the U.S. by donor type. 2017. https://optn.transplant.hrsa.gov/data/view-data-reports/national-data/#.
- 3.Wolfe R. A., Ashby V. B., Milford E. L., et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. The New England Journal of Medicine. 1999;341(23):1725–1730. doi: 10.1056/NEJM199912023412303. [DOI] [PubMed] [Google Scholar]
- 4.Singer L. G. Cost-effectiveness and quality of life: benefits of lung transplantation. Respiratory Care Clinics of North America. 2004;10(4):449–457. doi: 10.1016/j.rcc.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Yang L. S., Shan L. L., Saxena A., Morris D. L. Liver transplantation: a systematic review of long-term quality of life. Liver International. 2014;34(9):1298–1313. doi: 10.1111/liv.12553. [DOI] [PubMed] [Google Scholar]
- 6.Tennankore K. K., Kim S. J., Alwayn I. P. J., Kiberd B. A. Prolonged warm ischemia time is associated with graft failure and mortality after kidney transplantation. Kidney International. 2016;89(3):648–658. doi: 10.1016/j.kint.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Marasco S. F., Kras A., Schulberg E., Vale M., Lee G. A. Impact of warm ischemia time on survival after heart transplantation. Transplantation Proceedings. 2012;44(5):1385–1389. doi: 10.1016/j.transproceed.2011.12.075. [DOI] [PubMed] [Google Scholar]
- 8.Morariu A. M., Schuurs T. A., Leuvenink H. G. D., van Oeveren W., Rakhorst G., Ploeg R. J. Early events in kidney donation: progression of endothelial activation, oxidative stress and tubular injury after brain death. American Journal of Transplantation. 2008;8(5):933–941. doi: 10.1111/j.1600-6143.2008.02166.x. [DOI] [PubMed] [Google Scholar]
- 9.Schuurs T. A., Morariu A. M., Ottens P. J., et al. Time-dependent changes in donor brain death related processes. American Journal of Transplantation. 2006;6(12):2903–2911. doi: 10.1111/j.1600-6143.2006.01547.x. [DOI] [PubMed] [Google Scholar]
- 10.Rebolledo R. A., Hoeksma D., Hottenrott C. M. V., et al. Slow induction of brain death leads to decreased renal function and increased hepatic apoptosis in rats. Journal of Translational Medicine. 2016;14(1):p. 141. doi: 10.1186/s12967-016-0890-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoeksma D., Rebolledo R. A., Hottenrott C. M. V., et al. Inadequate anti-oxidative responses in kidneys of brain-dead rats. Transplantation. 2017;101(4):746–753. doi: 10.1097/tp.0000000000001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosieradzki M., Kuczynska J., Piwowarska J., et al. Prognostic significance of free radicals: mediated injury occurring in the kidney donor. Transplantation. 2003;75(8):1221–1227. doi: 10.1097/01.TP.0000065282.46425.87. [DOI] [PubMed] [Google Scholar]
- 13.Stiegler P., Sereinigg M., Puntschart A., et al. Oxidative stress and apoptosis in a pig model of brain death (BD) and living donation (LD) Journal of Translational Medicine. 2013;11(1):p. 244. doi: 10.1186/1479-5876-11-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain S. K., Shohet S. B. Calcium potentiates the peroxidation of erythrocyte membrane lipids. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1981;642(1):46–54. doi: 10.1016/0005-2736(81)90136-x. [DOI] [PubMed] [Google Scholar]
- 15.Vladimirov Y. A., Olenev V. I., Suslova T. B., Cheremisina Z. P. Lipid peroxidation in mitochondrial membrane. Advances in Lipid Research. 1980;17:173–249. doi: 10.1016/b978-0-12-024917-6.50011-2. [DOI] [PubMed] [Google Scholar]
- 16.Balibrea J. M., Núñez-Peña J. R., García-Martín M. C., et al. The differential tissue expression of inflammatory, oxidative stress, and apoptosis markers in human uncontrolled non-heart-beating donors. Transplantation. 2013;95(11):1346–1353. doi: 10.1097/TP.0b013e31828ee151. [DOI] [PubMed] [Google Scholar]
- 17.Kosieradzki M., Rowiński W. Ischemia/reperfusion injury in kidney transplantation: mechanisms and prevention. Transplantation Proceedings. 2008;40(10):3279–3288. doi: 10.1016/j.transproceed.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Nagelschmidt M., Minor T., Gallinat A., et al. Lipid peroxidation products in machine perfusion of older donor kidneys. Journal of Surgical Research. 2013;180(2):337–342. doi: 10.1016/j.jss.2012.04.071. [DOI] [PubMed] [Google Scholar]
- 19.Schnuelle P., Gottmann U., Hoeger S., et al. Effects of donor pretreatment with dopamine on graft function after kidney transplantation: a randomized controlled trial. JAMA. 2009;302(10):1067–1075. doi: 10.1001/jama.2009.1310. [DOI] [PubMed] [Google Scholar]
- 20.Niemann C. U., Malinoski D. Therapeutic hypothermia in deceased organ donors and kidney-graft function. The New England Journal of Medicine. 2015;373(27):p. 2687. doi: 10.1056/NEJMc1511744. [DOI] [PubMed] [Google Scholar]
- 21.Guibert E. E., Petrenko A. Y., Balaban C. L., Somov A. Y., Rodriguez J. V., Fuller B. J. Organ preservation: current concepts and new strategies for the next decade. Transfusion Medicine and Hemotheraphy. 2011;38(2):125–142. doi: 10.1159/000327033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maathuis M.-H. J., Leuvenink H. G. D., Ploeg R. J. Perspectives in organ preservation. Transplantation. 2007;83(10):1289–1298. doi: 10.1097/01.tp.0000265586.66475.cc. [DOI] [PubMed] [Google Scholar]
- 23.Moers C., Pirenne J., Paul A., Ploeg R. J., Machine Preservation Trial Study Group Machine perfusion or cold storage in deceased-donor kidney transplantation. The New England Journal of Medicine. 2012;366(8):770–771. doi: 10.1056/NEJMc1111038. [DOI] [PubMed] [Google Scholar]
- 24.Zhao D.-F., Dong Q., Zhang T. Effects of static cold storage and hypothermic machine perfusion on oxidative stress factors, adhesion molecules, and zinc finger transcription factor proteins before and after liver transplantation. Annals of Transplantation. 2017;22:96–100. doi: 10.12659/aot.901897. [DOI] [PubMed] [Google Scholar]
- 25.Croce A. C., Ferrigno A., Bertone V., et al. Fatty liver oxidative events monitored by autofluorescence optical diagnosis: comparison between subnormothermic machine perfusion and conventional cold storage preservation. Hepatology Research. 2017;47(7):668–682. doi: 10.1111/hepr.12779. [DOI] [PubMed] [Google Scholar]
- 26.Henry S. D., Nachber E., Tulipan J., et al. Hypothermic machine preservation reduces molecular markers of ischemia/reperfusion injury in human liver transplantation. American Journal of Transplantation. 2012;12(9):2477–2486. doi: 10.1111/j.1600-6143.2012.04086.x. [DOI] [PubMed] [Google Scholar]
- 27.Downey J. M. Free radicals and their involvement during long-term myocardial ischemia and reperfusion. Annual Review of Physiology. 1990;52:487–504. doi: 10.1146/annurev.ph.52.030190.002415. [DOI] [PubMed] [Google Scholar]
- 28.Weyker P. D., Webb C. A. J., Kiamanesh D., Flynn B. C. Lung ischemia reperfusion injury: a bench-to-bedside review. Seminars in Cardiothoracic and Vascular Anesthesia. 2013;17(1):28–43. doi: 10.1177/1089253212458329. [DOI] [PubMed] [Google Scholar]
- 29.Granger D. N., Kvietys P. R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biology. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snoeijs M. G. J., van Heurn L. W. E., Buurman W. A. Biological modulation of renal ischemia-reperfusion injury. Current Opinion in Organ Transplantation. 2010;15(2):190–199. doi: 10.1097/MOT.0b013e32833593eb. [DOI] [PubMed] [Google Scholar]
- 31.Leindler L., Morschl E., László F., et al. Importance of cytokines, nitric oxide, and apoptosis in the pathological process of necrotizing pancreatitis in rats. Pancreas. 2004;29(2):157–161. doi: 10.1097/00006676-200408000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Jarasch E. D., Bruder G., Heid H. W. Significance of xanthine oxidase in capillary endothelial cells. Acta Physiologica Scandinavica. Supplementum. 1986;548:39–46. [PubMed] [Google Scholar]
- 33.Vickers S., Schiller H. J., Hildreth J. E., Bulkley G. B. Immunoaffinity localization of the enzyme xanthine oxidase on the outside surface of the endothelial cell plasma membrane. Surgery. 1998;124(3):551–560. doi: 10.1016/s0039-6060(98)70102-3. [DOI] [PubMed] [Google Scholar]
- 34.Land W. G. Emerging role of innate immunity in organ transplantation part III: the quest for transplant tolerance via prevention of oxidative allograft injury and its consequences. Transplantation Reviews. 2012;26(2):88–102. doi: 10.1016/j.trre.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez L. A., Grisham M. B., Twohig B., Arfors K. E., Harlan J. M., Granger D. N. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. American Journal of Physiology - Heart and Circulatory Physiology. 1987;253, 3, Part 2:H699–H703. doi: 10.1152/ajpheart.1987.253.3.H699. [DOI] [PubMed] [Google Scholar]
- 36.Romson J. L., Hook B. G., Kunkel S. L., Abrams G. D., Schork M. A., Lucchesi B. R. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67(5):1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 37.Kuzuya T., Hoshida S., Nishida M., et al. Role of free radicals and neutrophils in canine myocardial reperfusion injury: myocardial salvage by a novel free radical scavenger, 2-octadecylascorbic acid. Cardiovascular Research. 1989;23(4):323–330. doi: 10.1093/cvr/23.4.323. [DOI] [PubMed] [Google Scholar]
- 38.Jaeschke H., Farhood A., Smith C. W. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. The FASEB Journal. 1990;4(15):3355–3359. [PubMed] [Google Scholar]
- 39.Smith J. W., Matheson P. J., Morgan G., et al. Addition of direct peritoneal lavage to human cadaver organ donor resuscitation improves organ procurement. Journal of the American College of Surgeons. 2015;220(4):539–547. doi: 10.1016/j.jamcollsurg.2014.12.056. [DOI] [PubMed] [Google Scholar]
- 40.Victorino G. P., Ramirez R. M., Chong T. J., Curran B., Sadjadi J. Ischemia-reperfusion injury in rats affects hydraulic conductivity in two phases that are temporally and mechanistically separate. American Journal of Physiology - Heart and Circulatory Physiology. 2008;295(5):H2164–H2171. doi: 10.1152/ajpheart.00419.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanderson T. H., Reynolds C. A., Kumar R., Przyklenk K., Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Molecular Neurobiology. 2013;47(1):9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zorov D. B., Filburn C. R., Klotz L. O., Zweier J. L., Sollott S. J. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. The Journal of Experimental Medicine. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalogeris T., Bao Y., Korthuis R. J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biology. 2014;2(1):702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cardoso S., Correia S., Carvalho C., et al. Perspectives on mitochondrial uncoupling proteins-mediated neuroprotection. Journal of Bioenergetics and Biomembranes. 2015;47(1-2):119–131. doi: 10.1007/s10863-014-9580-x. [DOI] [PubMed] [Google Scholar]
- 45.Abu-Amara M., Yang S. Y., Seifalian A., Davidson B., Fuller B. The nitric oxide pathway - evidence and mechanisms for protection against liver ischaemia reperfusion injury. Liver International. 2012;32(4):531–543. doi: 10.1111/j.1478-3231.2012.02755.x. [DOI] [PubMed] [Google Scholar]
- 46.Pernow J., Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovascular Research. 2013;98(3):334–343. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- 47.Tratsiakovich Y., Yang J., Gonon A. T., Sjöquist P.-O., Pernow J. Arginase as a target for treatment of myocardial ischemia-reperfusion injury. European Journal of Pharmacology. 2013;720(1–3):121–123. doi: 10.1016/j.ejphar.2013.10.040. [DOI] [PubMed] [Google Scholar]
- 48.Hale A. N., Ledbetter D. J., Gawriluk T. R., Rucker E. B. Autophagy: regulation and role in development. Autophagy. 2013;9(7):951–972. doi: 10.4161/auto.24273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei Y., Pattingre S., Sinha S., Bassik M., Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Molecular Cell. 2008;30(6):678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y., Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death and Differentiation. 2015;22(3):367–376. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klionsky D. J., Emr S. D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slegtenhorst B. R., Dor F. J. M. F., Elkhal A., et al. Mechanisms and consequences of injury and repair in older organ transplants. Transplantation. 2014;97(11):1091–1099. doi: 10.1097/TP.0000000000000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kregel K. C., Zhang H. J. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2007;292(1):R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 54.Pallet N., Livingston M., Dong Z. Emerging functions of autophagy in kidney transplantation. American Journal of Transplantation. 2014;14(1):13–20. doi: 10.1111/ajt.12533. [DOI] [PubMed] [Google Scholar]
- 55.Decuypere J. P., Ceulemans L. J., Agostinis P., et al. Autophagy and the kidney: implications for ischemia-reperfusion injury and therapy. American Journal of Kidney Diseases. 2015;66(4):699–709. doi: 10.1053/j.ajkd.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 56.Decuypere J. P., Pirenne J., Jochmans I. Autophagy in renal ischemia-reperfusion injury: friend or foe? American Journal of Transplantation. 2014;14(6):1464–1465. doi: 10.1111/ajt.12717. [DOI] [PubMed] [Google Scholar]
- 57.Xu Q., Li X., Lu Y., et al. Pharmacological modulation of autophagy to protect cardiomyocytes according to the time windows of ischaemia/reperfusion. British Journal of Pharmacology. 2015;172(12):3072–3085. doi: 10.1111/bph.13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamaguchi O., Otsu K. Role of autophagy in aging. Journal of Cardiovascular Pharmacology. 2012;60(3):242–247. doi: 10.1097/FJC.0b013e31824cc31c. [DOI] [PubMed] [Google Scholar]
- 59.Schiavi A., Ventura N. The interplay between mitochondria and autophagy and its role in the aging process. Experimental Gerontology. 2014;56:147–153. doi: 10.1016/j.exger.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 60.Terman A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology. 1995;41(Supplement 2):319–326. doi: 10.1159/000213753. [DOI] [PubMed] [Google Scholar]
- 61.Chen C., Hu L.-X., Dong T., et al. Apoptosis and autophagy contribute to gender difference in cardiac ischemia-reperfusion induced injury in rats. Life Sciences. 2013;93(7):265–270. doi: 10.1016/j.lfs.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 62.Kume S., Uzu T., Horiike K., et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. The Journal of Clinical Investigation. 2010;120(4):1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Turkmen K., Martin J., Akcay A., et al. Apoptosis and autophagy in cold preservation ischemia. Transplantation. 2011;91(11):1192–1197. doi: 10.1097/TP.0b013e31821ab9c8. [DOI] [PubMed] [Google Scholar]
- 64.Cursio R., Colosetti P., Gugenheim J. Autophagy and liver ischemia-reperfusion injury. BioMed Research International. 2015;2015:16. doi: 10.1155/2015/417590.417590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minor T., Stegemann J., Hirner A., Koetting M. Impaired autophagic clearance after cold preservation of fatty livers correlates with tissue necrosis upon reperfusion and is reversed by hypothermic reconditioning. Liver Transplantation. 2009;15(7):798–805. doi: 10.1002/lt.21751. [DOI] [PubMed] [Google Scholar]
- 66.Zaouali M. A., Boncompagni E., Reiter R. J., et al. AMPK involvement in endoplasmic reticulum stress and autophagy modulation after fatty liver graft preservation: a role for melatonin and trimetazidine cocktail. Journal of Pineal Research. 2013;55(1):65–78. doi: 10.1111/jpi.12051. [DOI] [PubMed] [Google Scholar]
- 67.Ma S., Wang Y., Chen Y., Cao F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2015;1852(2):271–276. doi: 10.1016/j.bbadis.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 68.Ma X., Liu H., Foyil S. R., et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125(25):3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim J.-S., Wang J.-H., Biel T. G., et al. Carbamazepine suppresses calpain-mediated autophagy impairment after ischemia/reperfusion in mouse livers. Toxicology and Applied Pharmacology. 2013;273(3):600–610. doi: 10.1016/j.taap.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu S., Hartleben B., Kretz O., et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8(5):826–837. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 71.Sciarretta S., Hariharan N., Monden Y., Zablocki D., Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatric Cardiology. 2011;32(3):275–281. doi: 10.1007/s00246-010-9855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scherz-Shouval R., Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends in Cell Biology. 2007;17(9):422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 73.Filomeni G., De Zio D., Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death and Differentiation. 2015;22(3):377–388. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiffin R., Bandyopadhyay U., Cuervo A. M. Oxidative stress and autophagy. Antioxidants & Redox Signaling. 2006;8(1-2):152–162. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 75.Nacarelli T., Torres C., Sell C. Mitochondrial reactive oxygen species in cellular senescence. Switzerland: Springer, Cham; 2016. pp. 169–185. (Healthy Ageing and Longevity). [DOI] [Google Scholar]
- 76.Hoshino A., Mita Y., Okawa Y., et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nature Communications. 2013;4:p. 2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 77.Youle R. J., Narendra D. P. Mechanisms of mitophagy. Nature Reviews Molecular Cell Biology. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ichimura Y., Waguri S., Sou Y.-S., et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Molecular Cell. 2013;51(5):618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 79.Cui J., Shi S., Sun X., et al. Mitochondrial autophagy involving renal injury and aging is modulated by caloric intake in aged rat kidneys. PLoS One. 2013;8(7, article e69720) doi: 10.1371/journal.pone.0069720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akhtar M. Z., Huang H., Kaisar M., et al. Using an integrated -omics approach to identify key cellular processes that are disturbed in the kidney after brain death. American Journal of Transplantation. 2016;16(5):1421–1440. doi: 10.1111/ajt.13626. [DOI] [PubMed] [Google Scholar]
- 81.Rebolledo R. A., Van Erp A. C., Ottens P. J., Wiersema-Buist J., Leuvenink H. G. D., Romanque P. Anti-apoptotic effects of 3,3′,5-triiodo-L-thyronine in the liver of brain-dead rats. PLoS One. 2015;10(10, article e0138749) doi: 10.1371/journal.pone.0138749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sinha R. A., You S.-H., Zhou J., et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. The Journal Clinical Investigation. 2012;122(7):2428–2438. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sinha R. A., Singh B. K., Zhou J., et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy. 2015;11(8):1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Southard J. H., Belzer F. O. Organ preservation. Annual Review of Medicine. 1995;46:235–247. doi: 10.1146/annurev.med.46.1.235. [DOI] [PubMed] [Google Scholar]
- 85.Semmelmann A., Neeff H., Sommer O., Thomusch O., Hopt U. T., Dobschuetz Von E. Evaluation of preservation solutions by ESR-spectroscopy: superior effects of University of Wisconsin over histidine-tryptophan-ketoglutarate in reducing renal reactive oxygen species. Kidney International. 2007;71(9):875–881. doi: 10.1038/sj.ki.5002129. [DOI] [PubMed] [Google Scholar]
- 86.Belzer UW® cold storage solution (University of Wisconsin), bridge to life. 2017. http://www.bridgetolife.eu/wp-content/uploads/2012/12/english-cold-storage-belzer-uw-solution-usage-instructions-ifu.pdf.
- 87.Du H., Sheng M., Wu L., et al. Hydrogen-rich saline attenuates acute kidney injury after liver transplantation via activating p53-mediated autophagy. Transplantation. 2016;100(3):563–570. doi: 10.1097/TP.0000000000001052. [DOI] [PubMed] [Google Scholar]
- 88.Guixé-Muntet S., de Mesquita F. C., Vila S., et al. Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. Journal of Hepatology. 2017;66(1):86–94. doi: 10.1016/j.jhep.2016.07.051. [DOI] [PubMed] [Google Scholar]
- 89.Gracia-Sancho J., García-Calderó H., Hide D., et al. Simvastatin maintains function and viability of steatotic rat livers procured for transplantation. Journal of Hepatology. 2013;58(6):1140–1146. doi: 10.1016/j.jhep.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 90.Pantazi E., Zaouali M. A., Bejaoui M., et al. Sirtuin 1 in rat orthotopic liver transplantation: an IGL-1 preservation solution approach. World Journal of Gastroenterology. 2015;21(6):1765–1774. doi: 10.3748/wjg.v21.i6.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schlegel A., Kron P., Dutkowski P. Hypothermic oxygenated liver perfusion: basic mechanisms and clinical application. Current Transplantation Reports. 2015;2(1):52–62. doi: 10.1007/s40472-014-0046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dutkowski P., Schönfeld S., Odermatt B., Heinrich T., Junginger T. Rat liver preservation by hypothermic oscillating liver perfusion compared to simple cold storage. Cryobiology. 1998;36(1):61–70. doi: 10.1006/cryo.1997.2066. [DOI] [PubMed] [Google Scholar]
- 93.Lüer B., Koetting M., Efferz P., Minor T. Role of oxygen during hypothermic machine perfusion preservation of the liver. Transplant International. 2010;23(9):944–950. doi: 10.1111/j.1432-2277.2010.01067.x. [DOI] [PubMed] [Google Scholar]
- 94.Shi S., Xue F. Current antioxidant treatments in organ transplantation. Oxidative Medicine and Cellular Longevity. 2016;2016:9. doi: 10.1155/2016/8678510.8678510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vairetti M., Ferrigno A., Carlucci F., et al. Subnormothermic machine perfusion protects steatotic livers against preservation injury: a potential for donor pool increase? Liver Transplantation. 2009;15(1):20–29. doi: 10.1002/lt.21581. [DOI] [PubMed] [Google Scholar]
- 96.Lenaz G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Advances in Experimental Medicine and Biology. 2012;942:93–136. doi: 10.1007/978-94-007-2869-1_5. [DOI] [PubMed] [Google Scholar]
- 97.Petrosillo G., Di Venosa N., Ruggiero F. M., et al. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2005;1710(2-3):78–86. doi: 10.1016/j.bbabio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 98.Brown D. A., Sabbah H. N., Shaikh S. R. Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacology & Therapeutics. 2013;140(3):258–266. doi: 10.1016/j.pharmthera.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 99.Chu C. T., Ji J., Dagda R. K., et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology. 2013;15(10):1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Y., Shoji-Kawata S., Sumpter R. M., et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J., Wang F., Xia Y., et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Marine Drugs. 2015;13(6):3368–3387. doi: 10.3390/md13063368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mitchell T., Saba H., Laakman J., Parajuli N., MacMillan-Crow L. A. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radical Biology & Medicine. 2010;49(8):1273–1282. doi: 10.1016/j.freeradbiomed.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hariharan N., Zhai P., Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxidants & Redox Signaling. 2011;14(11):2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van den Akker E. K., van den Akker E. K., Hesselink D. A., et al. Ischemic postconditioning in human DCD kidney transplantation is feasible and appears safe. Transplant International. 2014;27(2):226–234. doi: 10.1111/tri.12242. [DOI] [PubMed] [Google Scholar]
- 105.Soendergaard P., Krogstrup N. V., Secher N. G., et al. Improved GFR and renal plasma perfusion following remote ischaemic conditioning in a porcine kidney transplantation model. Transplant International. 2012;25(9):1002–1012. doi: 10.1111/j.1432-2277.2012.01522.x. [DOI] [PubMed] [Google Scholar]
- 106.Krogstrup N. V., Oltean M., Nieuwenhuijs-Moeke G. J., et al. Remote ischemic conditioning on recipients of deceased renal transplants does not improve early graft function: a multicentre randomised, controlled clinical trial. American Journal of Transplantation. 2016;17(4):1042–1049. doi: 10.1111/ajt.14075. [DOI] [PubMed] [Google Scholar]
- 107.Wei C., Li H., Han L., Zhang L., Yang X. Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. Journal of Cardiovascular Pharmacology. 2013;61(5):416–422. doi: 10.1097/FJC.0b013e318287d501. [DOI] [PubMed] [Google Scholar]
- 108.Wei C., Gao J., Li M., et al. Dopamine D2 receptors contribute to cardioprotection of ischemic post-conditioning via activating autophagy in isolated rat hearts. International Journal of Cardiology. 2016;203:837–839. doi: 10.1016/j.ijcard.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 109.Guo L., Xu J.-M., Mo X.-Y. Ischemic postconditioning regulates cardiomyocyte autophagic activity following ischemia/reperfusion injury. Molecular Medicine Reports. 2015;12(1):1169–1176. doi: 10.3892/mmr.2015.3533. [DOI] [PubMed] [Google Scholar]
- 110.Land W., Schneeberger H., Schleibner S., et al. The beneficial effect of human recombinant superoxide dismutase on acute and chronic rejection events in recipients of cadaveric renal transplants. Transplantation. 1994;57(2):211–217. doi: 10.1097/00007890-199401001-00010. [DOI] [PubMed] [Google Scholar]
- 111.Chen Y., Azad M. B., Gibson S. B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death and Differentiation. 2009;16(7):1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 112.van Gelder T., van Schaik R. H., Hesselink D. A. Pharmacogenetics and immunosuppressive drugs in solid organ transplantation. Nature Reviews Nephrology. 2014;10(12):725–731. doi: 10.1038/nrneph.2014.172. [DOI] [PubMed] [Google Scholar]
- 113.Lim S. W., Hyoung B. J., Piao S. G., Doh K. C., Chung B. H., Yang C. W. Chronic cyclosporine nephropathy is characterized by excessive autophagosome formation and decreased autophagic clearance. Transplantation. 2012;94(3):218–225. doi: 10.1097/TP.0b013e31825ace5c. [DOI] [PubMed] [Google Scholar]
- 114.Settembre C., Di Malta C., Polito V. A., et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim D., Hwang H.-Y., Kim J. Y., et al. FK506, an immunosuppressive drug, induces autophagy by binding to the V-ATPase catalytic subunit a in neuronal cells. Journal of Proteome Research. 2017;16(1):55–64. doi: 10.1021/acs.jproteome.6b00638. [DOI] [PubMed] [Google Scholar]
- 116.Długosz A., Srednicka D., Boratyński J. The influence of tacrolimus on oxidative stress and free-radical processes. Postȩpy Higieny i Medycyny Doświadczalnej. 2007;61:466–471. [PubMed] [Google Scholar]
- 117.McTaggart R. A., Gottlieb D., Brooks J., et al. Sirolimus prolongs recovery from delayed graft function after cadaveric renal transplantation. American Journal of Transplantation. 2003;3(4):416–423. doi: 10.1034/j.1600-6143.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 118.Jolicoeur E. M., Qi S., Xu D., Dumont L., Daloze P., Chen H. Combination therapy of mycophenolate mofetil and rapamycin in prevention of chronic renal allograft rejection in the rat. Transplantation. 2003;75(1):54–59. doi: 10.1097/00007890-200301150-00010. [DOI] [PubMed] [Google Scholar]
- 119.Steiner R. W., Awdishu L. Steroids in kidney transplant patients. Seminars in Immunopathology. 2011;33(2):157–167. doi: 10.1007/s00281-011-0259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Torres R. L., Torres I. L. D. S., Laste G., Ferreira M. B. C., Cardoso P. F. G., Belló-Klein A. Effects of acute and chronic administration of methylprednisolone on oxidative stress in rat lungs. Jornal Brasileiro de Pneumologia. 2014;40(3):238–243. doi: 10.1590/s1806-37132014000300006. [DOI] [PMC free article] [PubMed] [Google Scholar]