

Abstract
Duchenne muscular dystrophy (DMD) is a severe, progressive muscle disease caused by mutations in the dystrophin gene. The majority of DMD mutations are deletions that prematurely terminate the dystrophin protein. Deletions of exon 50 of the dystrophin gene are among the most common single exon deletions causing DMD. Such mutations can be corrected by skipping exon 51, thereby restoring the dystrophin reading frame. Using clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9), we generated a DMD mouse model by deleting exon 50. These ΔEx50 mice displayed severe muscle dysfunction, which was corrected by systemic delivery of adeno-associated virus encoding CRISPR/Cas9 genome editing components. We optimized the method for dystrophin reading frame correction using a single guide RNA that created reframing mutations and allowed skipping of exon 51. In conjunction with muscle-specific expression of Cas9, this approach restored up to 90% of dystrophin protein expression throughout skeletal muscles and the heart of ΔEx50 mice. This method of permanently bypassing DMD mutations using a single cut in genomic DNA represents a step toward clinical correction of DMD mutations and potentially those of other neuromuscular disorders.
INTRODUCTION
Muscular dystrophies comprise a group of more than 30 genetic diseases characterized by progressive weakness and degeneration of skeletal muscles (1–3). Duchenne muscular dystrophy (DMD) is one of the most severe muscle diseases, affecting ~1 in 5000 boys and causing premature death from respiratory failure caused by diaphragm insufficiency, cardiomyopathy, and heart failure. The disease is caused by mutations in the DMD gene encoding dystrophin, a large intracellular protein that links the dystroglycan complex at the cell surface with the underlying cytoskeleton, thereby maintaining integrity of muscle cell membranes during contraction (4, 5). The absence of dystrophin results in membrane fragility and muscle degeneration.
Despite numerous therapeutic strategies to restore muscle function in DMD, including myoblast transfer, viral delivery of missing proteins, and oligonucleotide-mediated exon skipping, there remains no cure for the disease (6–9). A variety of gene therapy approaches for DMD are in development, including delivery of microdystrophin by an adeno-associated virus (AAV9) vector (10–12). However, these truncated forms of dystrophin are not fully functional, and episomal AAV9 vectors are gradually lost over time (13). We and others have recently used clustered regularly interspaced short palindromic repeat/CRISPR-associated 9 (CRISPR/Cas9)–mediated genome editing to correct the dystrophin gene (Dmd) mutation in cells (14, 15) and postnatal mdx mice, a model of DMD that harbors a nonsense mutation in exon 23 (16–18). Others have applied similar strategies in mdx4cv mice, which contain a nonsense mutation in exon 53 (19). In addition, we have shown that the Cas9-related endonuclease Cpf1 can correct the Dmd mutation in mdx mice (20).
More than 4000 DMD mutations have been identified in humans. The majority of mutations are deletions that cluster in hotspots, such that skipping of out-of-frame exons can potentially restore the reading frame of the dystrophin protein (21). The rationale for exon skipping is based on the genetic difference between patients with DMD and patients with Becker muscular dystrophy (BMD). In DMD patients, the reading frame of dystrophin mRNA is disrupted, resulting in prematurely truncated and unstable proteins that are degraded in muscle cells. In contrast, BMD patients harbor mutations in the DMD gene that maintain the reading frame, allowing the production of internally deleted but partially functional dystrophin proteins, often leading to milder disease symptoms than in DMD patients (22–24). The mouse Dmd and human DMD genes each contain 79 exons, which are conserved with respect to intron-exon junctions and splicing patterns. The most common hotspot region for DMD mutations lies between exons 45 and 55, where skipping or restoring the reading frame of exon 51 can restore the open reading frame in 13 to 14% of DMD patients.
Exon deletions account for ~60 to 70% of human DMD mutations, with deletion of exon 50 representing one of the most common single exon deletions (25, 26). All previous studies of CRISPR/Cas9-mediated exon skipping in mice focused on mdx or mdx4cv mice, which harbor nonsense mutations, so we used CRISPR/Cas9 to generate a new mouse model of DMD that lacks exon 50 (referred to as ΔEx50 mice). These mice exhibited severely dystrophic myofibers, providing a model for this prevalent human DMD mutation. We found that systemic delivery of Cas9 and an expression cassette encoding a single guide RNA (sgRNA) targeting genomic sequences adjacent to the splice acceptor of exon 51 created reframing mutations and allowed skipping of exon 51 and reframing of the gene. This resulted in the restoration of up to 90% of normal wild-type dystrophin expression in ΔEx50 mouse skeletal and heart muscles.
RESULTS
Engineering a mouse model with a common human DMD exon deletion
To optimize gene editing in vivo, we first generated mice lacking exon 50 of the Dmd gene, using the CRISPR/Cas9 system directed by two sgRNAs targeting sequences in the surrounding introns (Fig. 1A and fig. S1A). F0 generation pups were screened for indels, and a founder with a 245–base pair (bp) deletion that completely eliminated exon 50 was chosen for further analysis (fig. S1). These mice, referred to as ΔEx50 mice, genomically mimic one of the most common single exon deletions in a mutational hotspot region for DMD in humans. Deletion of exon 50 was confirmed by DNA sequencing of reverse transcription polymerase chain reaction (RT-PCR) products using primers for sequences in exons 48 and 55 (Fig. 1B). Deletion of exon 50 placed the Dmd gene out of frame, preventing production of dystrophin protein in the skeletal muscle and the heart (Fig.1, C to E). As in the mdx mouse model, muscles from ΔEx50 mice displayed necrosis by 3 weeks of age (fig. S1B). ΔEx50 mice showed severely dystrophic muscle with necrotic fibers and inflammatory infiltration by 2 months of age (Fig. 1D), and elevation of serum creatine kinase (CK) (Fig. 1F), indicative of muscle damage. Similarly, as seen in mdx mice, muscles from ΔEx50 mice displayed variability in fiber size with aggregated necrotic, regenerative fibers recognizable by centralized nuclei, and the presence of few revertant fibers (figs. S1 to S3). Grip strength analysis compared to wild-type mice revealed a similar decrease in muscle strength in both mdx mice and ΔEx50 mice by 2 and 5 months of age, respectively (fig. S3, B and C). Thus, by all evaluated parameters, the severity and progression of disease were comparable in ΔEx50 mice and mdx mice (Fig. 1F and figs. S1 to S3).
Fig. 1. Generation of the ΔEx50 mouse model.
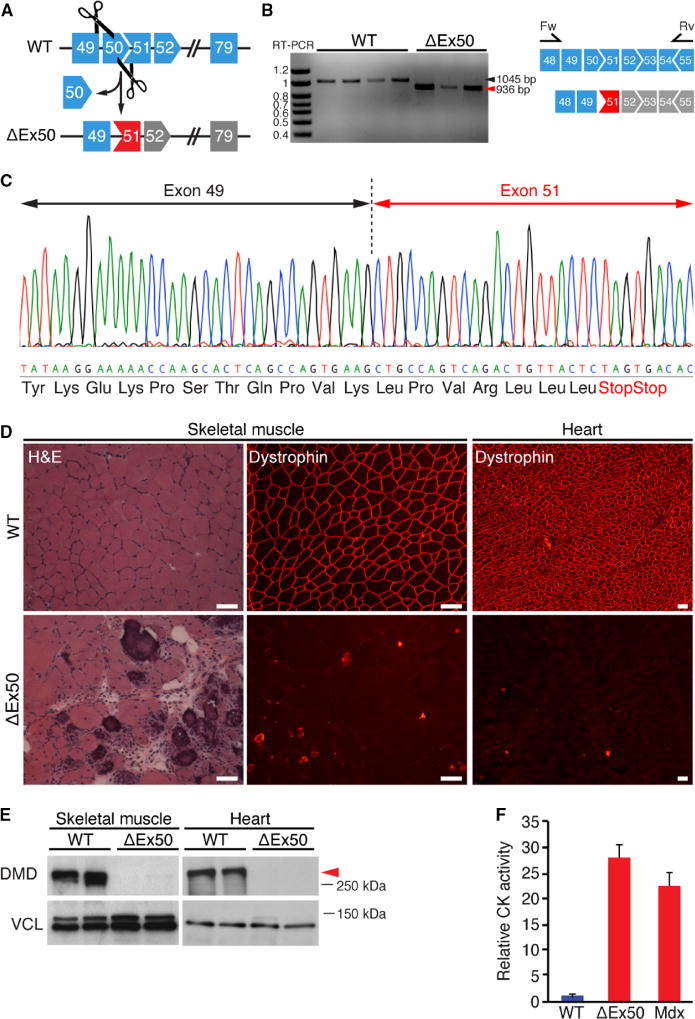
(A) Strategy showing clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9)–mediated genome editing approach to generate ΔEx50 mice. (B) Reverse transcription polymerase chain reaction (RT-PCR) analysis of muscle RNA using primers in exons 48 and 55 to validate deletion of exon 50 (ΔEx50). Bands for wild-type (WT) and ΔEx50 mouse dystrophin are 1045 and 936 base pair (bp), respectively. (C) RT-PCR products from ΔEx50 mouse muscle were sequenced to validate exon 50 deletion and generation of an out-of-frame sequence. (Δ) Hematoxylin and eosin (H&E) staining and immunostaining of dystrophin in the cardiac and skeletal muscles from WT (upper) and ΔEx50 (lower) mice at 2 months of age. (E) Western blot analysis of dystrophin (DMD) and vinculin (VCL) expression in the tibialis anterior and heart muscle from 2-month-old ΔEx50 mice. VCL expression in the skeletal muscle displays two isoforms compared to the cardiac muscle. (F) Serum creatine kinase (CK), a marker of muscle damage and membrane leakage, in WT, ΔEx50, and mdx mice at 2 months of age. n = 5. Data are represented as means ± SEM. Red arrow indicates dystrophin protein. Scale bars, 50 mm.
Tailoring a single DNA cut genome editing strategy
Proper exon splicing requires 3′ and 5′ splice acceptor and donor sites of exons. Exons also contain splicing regulatory sequences, such as an exonic splicing enhancer (ESE) sequence that is responsible for proper recognition by the splicing machinery and inclusion of the exon into the mRNA. A Web-based ESE-Finder helps predict putative ESE sequences within exons, based on the target sequence motifs of the most abundant serine-arginine proteins that bind to ESE motifs (fig. S4) (27, 28). The ESE sequences of exon 51 are conserved between the mouse (fig. S4, A and B) and human (fig. S4, C and D) Dmd genes.
To correct the dystrophin reading frame in ΔEx50 mice (Fig. 2A), we designed a sgRNA that targets a region adjacent to the exon 51 splice acceptor site (referred to as sgRNA-51) and cuts 9 bp away from the intron-exon junction (Fig. 2B). We used Streptococcus pyogenes Cas9, which requires NAG/NGG as a protospacer adjacent motif (PAM) sequence, to generate a double-strand DNA break (29–31).
Fig. 2. Strategy for CRISPR/Cas9-mediated genome editing in ΔEx50 mice.
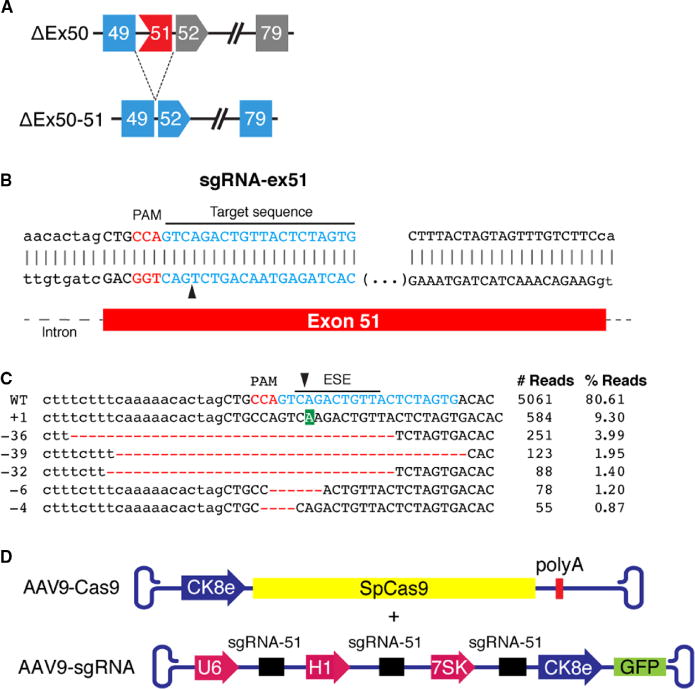
(A) Scheme showing the CRISPR/Cas9-mediated genome editing approach to correct the reading frame in ΔEx50 mice by skipping exon 51. Gray exons are out of frame. (B) Illustration of single guide RNA (sgRNA) binding position and sequence for sgRNA-ex51. Protospacer adjacent motif (PAM) sequence for sgRNA is indicated in red. Black arrow indicates the cleavage site. Fw, foward primer; Rv, reverse primer. (C) Genomic deep-sequencing analysis of PCR amplicons generated across the exon 51 target site in 10T1/2 cells. Sequence of representative indels aligned with sgRNA sequence (indicated in blue) revealing insertions (highlighted in green) and deletions (highlighted in red). The line indicates the predicted exonic splicing enhancer (ESE) sequences located at the site of sgRNA. Black arrowhead indicates the cleavage site. (D) The muscle creatine kinase 8 (CK8e) promoter was used to express SpCas9. The U6, H1, and 7SK promoters for RNA polymerase III were used to express sgRNAs. GFP, green fluorescent protein.
First, we evaluated the DNA cutting activity of Cas9 coupled with sgRNA-51 in 10T1/2 mouse fibroblasts using the mismatch-specific T7 endonuclease I (T7E1) assay (fig. S5A). To investigate the type of mutations generated by Cas9 coupled with sgRNA-51, we performed genomic deep-sequencing analysis. The sequencing analysis revealed that 9.3% of mutations contained a single adenosine (A) insertion three nucleotides 3′ of the PAM sequence. In addition, 7.3% of mutations contained deletions covering the splice acceptor site and a highly predicted ESE site for exon 51 (Fig. 2C). The sgRNA-51 corresponds to a highly conserved region of the Dmd gene (fig. S5C). Therefore, we tested the ability of Cas9 and human sgRNA-51 to cut the human Dmd locus in 293T cells. The T7E1 assay revealed clear cleavage at the predicted site (fig. S5B). Similarly, sequence analysis revealed that Cas9 coupled with human sgRNA-51 generated the same adenosine insertion and a different range of deletions around the cleavage site (fig. S5C).
For the in vivo delivery of Cas9 and sgRNA-51 to the skeletal muscle and heart tissue, we used AAV9, which displays preferential tropism for these tissues (32). To further enhance muscle-specific expression, we used an AAV9-Cas9 vector (CK8e-Cas9-shortPolyA), which contains a muscle-specific CK regulatory cassette, referred to as the CK8e promoter, which is highly specific for expression in the muscle and the heart (Fig. 2D) (33). Together, this 436-bp muscle-specific cassette and the 4101-bp Cas9 complementary DNA (cDNA) are within the packaging limit of AAV9 (32, 34). Expression of each sgRNA was driven by three RNA polymerase III promoters (U6, H1, and 7SK) (Fig. 2D).
Correction of the dystrophin reading frame in ΔEx50 mice by a single DNA cut
After intramuscular injection of mice at postnatal day 12 (P12) with 5 × 1010 AAV9 viral genomes (vg), we analyzed the tibialis anterior muscles. Western blot analysis of the tibialis anterior muscles confirmed Cas9 expression in the injected muscles (fig. S6). In vivo targeting efficiency was estimated by RT-PCR with primers for sequences in exons 48 and 53 and the T7E1 assay for the targeted genomic regions. To investigate whether we achieved efficient target cleavage, we amplified a 771-bp region spanning the target site and analyzed it using the T7EI assay (fig. S7A). The activity of SpCas9 with the corresponding sgRNA was analyzed on the target site. T7EI assays revealed mutagenesis of the Dmd locus after delivery of AAV-Cas9 and AAV9-sgRNA-51 (fig. S7A). Control mice were injected with AAV9-Cas9 without AAV9-sgRNA-51. To investigate the type of mutations generated in ΔEx50 mice injected with Cas9 and sgRNA-expressing AAV9s, we analyzed genomic PCR amplification products spanning the target site by amplicon deep-sequencing analysis. Deep sequencing of the targeted region indicated that 27.9% of total reads contained changes at the targeted genomic site (fig. S7B). On average, 15% of the identified mutations contained the same adenosine insertion seen in mouse 10T1/2 and human 293 cells in vitro. The deletions identified using this method encompassed a highly predicted ESE site for exon 51 (figs. S5B and S7B). However, this method did not identify larger deletions that might occur beyond the annealing sites of the primers used for PCR. Given that these tissue biopsies contained a mixture of muscle and nonmuscle cells, the method probably underrepresented the actual efficiency of gene editing within muscle cells.
RT-PCR products of RNA from the muscle of ΔEx50 mice injected intramuscularly with AAV9-Cas9 and AAV9-sgRNA-51 showed that deletion of exon 51 (ΔEx50–51) allowed splicing from exon 49 to 52 (Fig. 3A, lower band). By sequencing RT-PCR products of the ΔEx50–51 band, we confirmed that exon 49 was spliced to exon 52 (Fig. 3C). To further define the mutations introduced by our gene editing strategy, RT-PCR amplification products from four samples were directly subjected to topoisomerase-based thymidine to adenosine (TOPO-TA) cloning without gel purification, and then sequenced. Sequence analysis of 40 clones from each sample showed that in addition to exon 51–skipped cDNA products (ΔEx50–51) identified in 15% of sequenced clones, ΔEx50 mice injected with AAV9-Cas9 and AAV9-sgRNA-51 showed a high frequency of reframing events. Of the sequenced clones, 63% contained a single nucleotide insertion in the sequence of exon 51 (Fig. 3, B and C). The most dominant insertion mutation seen was an adenosine insertion.
Fig. 3. RT-PCR analysis of correction of reading frame.
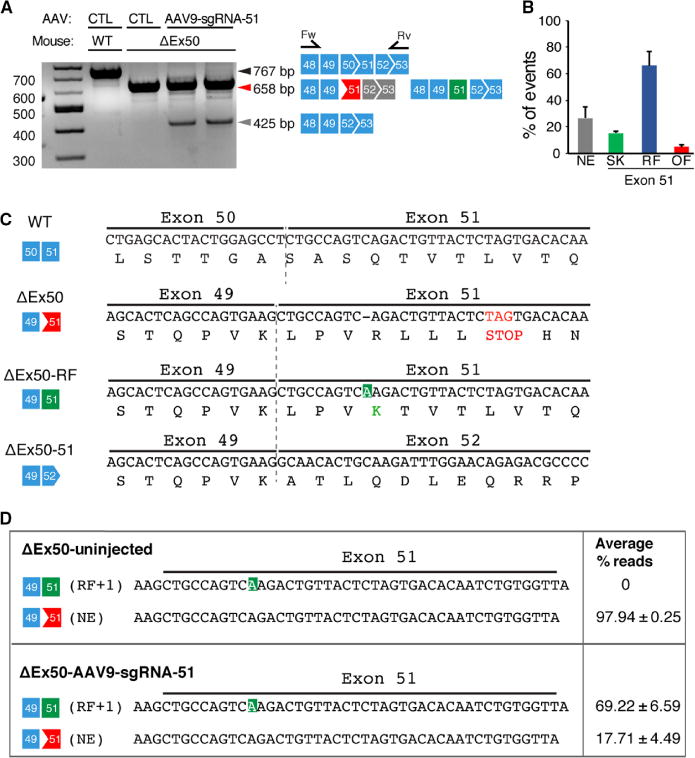
(A) RT-PCR of RNA from the tibialis anterior muscles of WT and ΔEx50 mice 3 weeks after intramuscular injection of the AAV9-sgRNA-51 and AAV9-Cas9 expression vectors. Lower dystrophin bands indicate deletion of exon 51. Primer positions in exons 48 and 53 are indicated. (B) Percentage of events detected at exon 51 after AAV9-Cas9/sgRNA-51 treatment using RT-PCR sequence analysis of TOPO-TA (topoisomerase-based thymidine to adenosine) generated clones. For each of four different samples, we generated and sequenced 40 clones. RT-PCR products were divided into four groups: Not edited (NE), exon 51–skipped (SK), reframed (RF), and out of frame (OF). (C) Sequence of the RT-PCR products of the ΔEx50–51 mouse dystrophin lower band confirmed that exon 49 spliced directly to exon 52, excluding exon 51. Sequence of RT-PCR products of ΔEx50 reframed (ΔEx50-RF) is also shown. (D) Deep-sequencing analysis of RT-PCR products from the upper band containing ΔEx50-NE and ΔEx50-RF was shown. Sequence of RT-PCR products revealing insertions (highlighted in green) is also depicted. n = 4. Data are represented as means ± SEM.
On gels, the PCR products with the adenosine insertion were indistinguishable in size from nonedited cDNA products, so we performed deep-sequencing analysis to determine their abundance compared to other small insertions. Deep sequencing of the upper band containing the nonedited cDNA product and reframed cDNA products indicated that 69.22% of total reads contained reframed cDNA products with an adenosine insertion, 17.71% contained nonedited cDNA product, and the rest contained small deletions and insertions (Fig. 3D). The deep-sequencing analysis of uninjected ΔEx50 mice confirmed that the adenosine insertion was a result of Cas9-generated editing. These amplicon deep-sequencing results confirmed our results from TOPO-TA cloning and sequencing. Together, our RT-PCR analysis revealed that ΔEx50 mice injected with AAV9-Cas9 and AAV9-sgRNA-51 showed a high requency of reframing events with cDNA products containing an adenosine insertion in the sequence of exon 51 in addition to exon 51 skipping events resulting from deletion in a highly conserved ESE region.
Defining the off-target activity of the Cas9 single-cut DNA strategy in vivo To
evaluate the specificity of our gene editing approach, we analyzed predicted off-target genomic sites for possible promiscuous editing. A total of 6 potential genome-wide off-target sites (OT1 to OT6) (table S1) were predicted in coding exons and 47 in noncoding regions (table S2) by the CRISPR design tool (http://crispr.mit.edu/). Using PCR followed by the T7E1 assay, only the target site for Dmd exon 51 in ΔEx50 mice treated with AAV9-Cas9 and AAV9-sgRNA-51 showed cleavage bands, and no off-target effects were detected in the six potential off-target sites (fig. S8). Genomic PCR amplification products spanning target sites were also isolated and cloned. DNA sequence analysis of the genomic PCR amplicons confirmed the absence of sgRNA/Cas9-mediated mutations at the predicted sites (fig. S9). In addition, we performed deep sequencing at the top predicted off-target sites within protein-coding exons. None of these sites revealed significantly more sequence alterations than the background analysis performed with other regions of the amplicons (fig. S10).
Restoration of dystrophin expression after intramuscular AAV9 delivery of Cas9 and sgRNA-51
We performed histological analysis of the tibialis anterior muscle of ΔEx50 mice injected with AAV9-Cas9 and AAV9-sgRNA-51 to evaluate the number of muscle fibers that expressed dystrophin. Immunohistochemical staining of dystrophin in the muscle from ΔEx50 mice injected with AAV9-Cas9 and AAV9-sgRNA-51 revealed restoration of dystrophin in an average of 99.5± 0.7% of fibers (Fig. 4, A and B and fig. S11). It should be noted that although 99.5% of the total fibers exhibited dystrophin immunostaining, the close packing of fibers introduced the possibility that some negative fibers surrounded by positive fibers might appear falsely positive. Thus, the actual number of positive fibers could be slightly lower than 99.5%. Hematoxylin and eosin (H&E) staining showed that histopathologic hallmarks of muscular dystrophy, such as necrotic myofibers and regenerated fibers with central nuclei, were diminished in the tibialis anterior muscle at 3 weeks after AAV9-Cas9/sgRNA-51 delivery (Fig. 4 and fig. S12). Quantitative analysis of the distribution of myofiber area and centrally nucleated fibers showed an improvement in AAV9-Cas9/sgRNA-51–treated muscles compared to ΔEx50 mouse control muscle injected with AAV9-Cas9 alone without sgRNAs (fig. S13). Western blot analysis confirmed the restoration of dystrophin expression in skeletal muscle (Fig. 4, C and D).
Fig. 4. Intramuscular injection of AAV9-Cas9/sgRNA-51 corrects dystrophin expression.
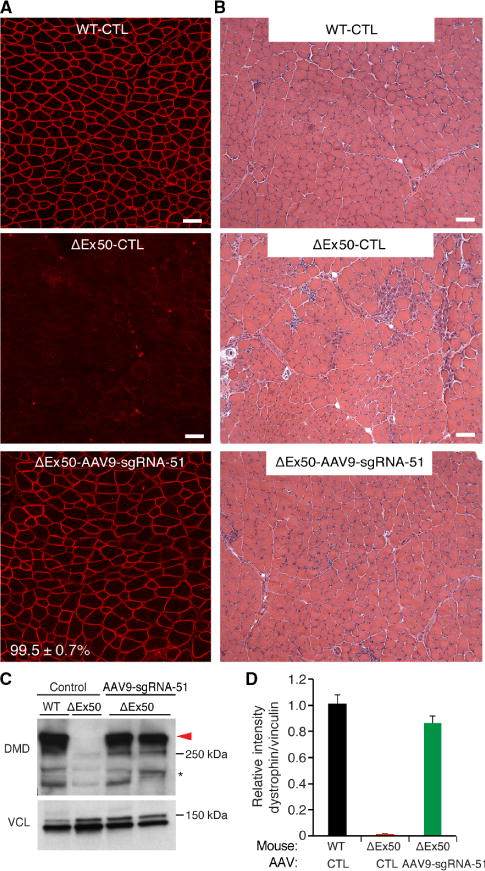
(A) Tibialis anterior muscles of ΔEx50 mice were injected with AAV9 vector encoding sgRNA-51 and Cas9 (see Fig. 2) and were analyzed 3 weeks later by immunostaining for dystrophin. WT control (WT-CTL) mice and ΔEx50 control mice (ΔEx50-CTL) were injected with AAV9-Cas9 alone without sgRNAs. Indicated are percentages of dystrophin-positive myofibers in ΔEx50 mice receiving intramuscular injections of AAV9-Cas9/sgRNA-51 (ΔEx50-AAV9-sgRNA-51) compared to WT-CTL. (B) H&E staining of tibialis anterior muscles. (C) Western blot analysis of dystrophin (DMD) and VCL expression in tibialis anterior muscles 3 weeks after intramuscular injection of AAV9-Cas9 control or AAV9-Cas9/sgRNA-51. (D) Quantification of dystrophin expression from Western blots after normalization to VCL. Asterisk indicates nonspecific immunoreactive bands. n = 5 for AAV9-sgRNA-51. Scale bars, 50 μm.
AAV9-Cas9/sgRNA-51 delivered by intramuscular injection leaked into the bloodstream, allowing efficient gene correction in the heart tissue. Dystrophin immunohistochemistry of the heart tissue from ΔEx50 mice injected intramuscularly with AAV9-Cas9/sgRNA-51 revealed extensive dystrophin-positive cardiomyocytes (fig. S14A). Western blot analysis confirmed the restoration of dystrophin expression in the cardiac muscle (fig. S14B).
Dystrophin gene editing in ΔEx50 mice by systemic AAV delivery of Cas9 and sgRNA-51
On the basis of the high dystrophin correction efficiency observed for ΔEx50 mice after intramuscular injection of AAV9-Cas9 and AAV9-sgRNA-51, we next tested for rescue of the disease phenotype by intra-peritoneal injection of AAV9-Cas9 and AAV9-sgRNA-51 into ΔEx50 mice, which allowed for systemic distribution of the AAV9 vectors. Systemic delivery of 6.3 × 1010 vg of AAV to ΔEx50 mice at P4 of age (2.63 × 1013 vg/kg) yielded widespread expression of the virus in the heart, triceps, gastrocnemius-plantaris, quadriceps, and diaphragm, as shown by Western blot for Cas9 (fig. S15). T7EI assays performed in the tibialis anterior muscle, diaphragm, and heart muscles revealed mutagenesis of the Dmd-targeted locus after systemic delivery of AAV9-Cas9/sgRNA-51 (fig. S16A).
To characterize the mutations generated after systemic delivery of Cas9 and sgRNA-expressing AAV9 vectors to ΔEx50 mice, we performed DNA amplicon deep-sequencing analysis of heart muscle DNA samples. Deep sequencing of the targeted region showed that, on average, 21% of total reads contained changes at the targeted genomic site (fig. S16B). Moreover, 7.9% of identified mutations contained an adenosine insertion, as seen in the other analyses. In addition, the deletions identified covered the ESE site and splice acceptor region of exon 51 (fig. S16B). RT-PCR products of RNA from the cardiac muscle of ΔEx50 mice injected with Cas9 and sgRNA-expressing AAV9 vectors revealed splicing events from exons 49 to 52 (fig. S16C, lower band). To investigate whether the cardiac muscle contained reframing events similar to those in the skeletal muscle, we performed deep sequencing of the upper band containing nonedited cDNA product and potential reframed cDNA products from the cardiac muscle. Sequencing results indicated that 66.8% of total reads contained reframed cDNA products with an adenosine insertion, 24.89% contained nonedited cDNA product, and the rest contained small deletions and insertions (fig. S16D).
Rescue of muscle structure and function after systemic AAV delivery of Cas9 and sgRNA-51
Systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 to P4 ΔEx50 mice yielded widespread dystrophin expression in the heart, triceps, tibialis anterior muscle, and diaphragm at 4 and 8 weeks after injection (Fig. 5 and fig. S17). Western blot analysis confirmed the restoration of dystrophin expression in the skeletal and heart muscles (Fig. 6A and fig. S17). H&E staining of multiple skeletal muscles showed that histopathologic hallmarks of muscular dystrophy, such as necrotic myofibers, were also largely corrected 4 and 8 weeks after delivery of AAV9-Cas9 and AAV9-sgRNA-51 (Fig. 6B and figs. S17 to S19). Grip strength testing also showed a significant increase in muscle strength of ΔEx50 mice at 4 weeks after intraperitoneal injection of AAV9-Cas9/sgRNA-51 compared with ΔEx50 control mice receiving AAV9-Cas9 alone (wild-type control, 92.6 ± 1.63; ΔEx50 control, 50.5 ± 1.85; ΔEx50-AAV9-Cas9/sgRNA-51, 79.7 ± 2.63; P < 0.05) (Fig. 6C). Consistently, AAV9-Cas9/sgRNA-51 gene-edited ΔEx50 mice also showed significant reductions in serum CK concentrations compared with ΔEx50 control mice (P < 0.05; Fig. 6D).
Fig. 5. Systemic delivery of AAV9-Cas9/sgRNA-51 to ΔEx50 mice rescues dystrophin expression.
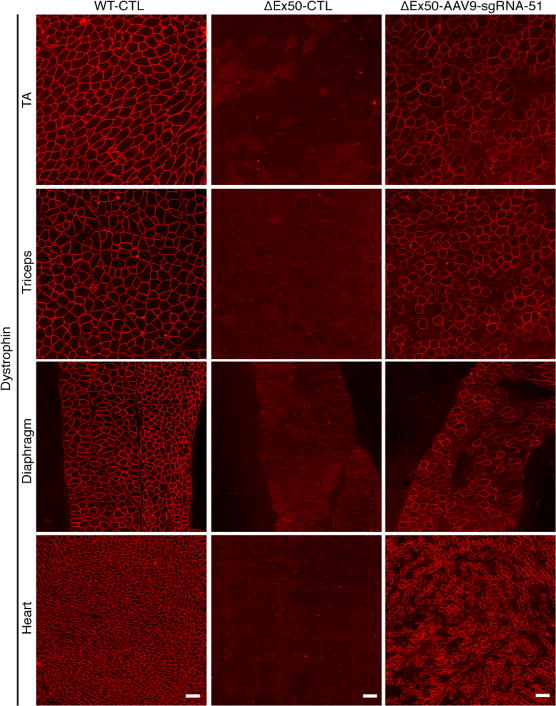
Immunostaining for dystrophin in tibialis anterior, triceps, diaphragm, and cardiac muscles of ΔEx50 mice is shown. Immunostaining was performed 4 weeks after systemic injection of AAV9-Cas9 only (WT-CTL and ΔEx50-CTL) or AAV9-Cas9/sgRNA-51 (ΔEx50-AAV9-sgRNA-51). n = 5 for each group. Scale bars, 50 μm.
Fig. 6. Histological and functional analysis of dystrophin correction after systemic delivery of AAV9-Cas9/sgRNA-51 to ΔEx50 mice.
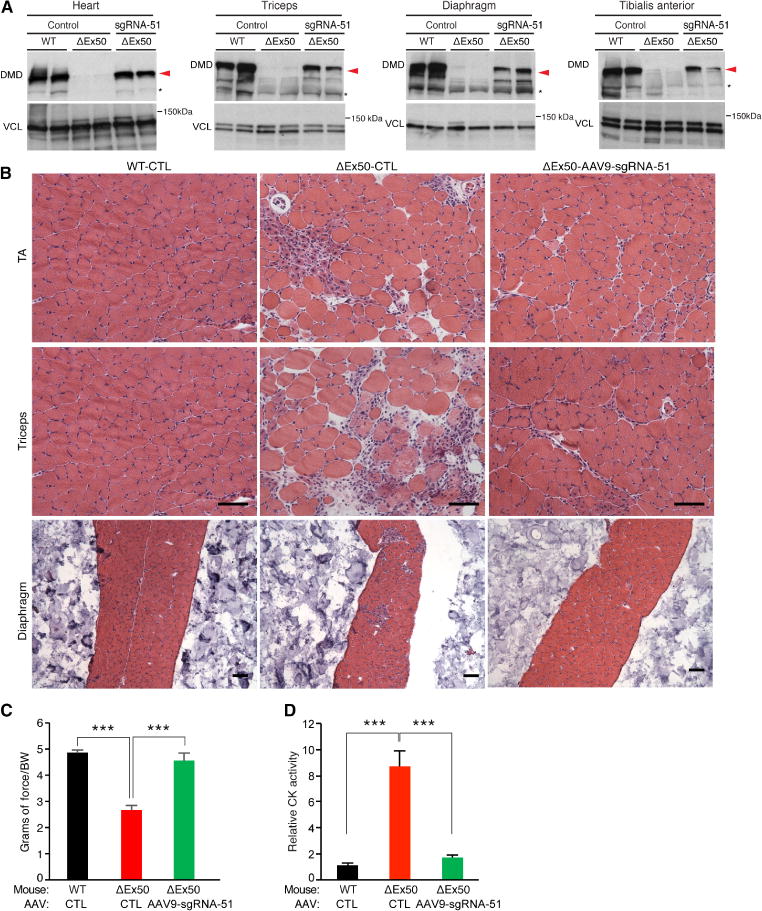
(A) Western blot analysis of dystrophin (DMD) and VCL expression in tibialis anterior (TA) muscle, triceps, diaphragm, and cardiac muscle of ΔEx50 mice 4 weeks after systemic delivery of AAV9-Cas9 or AAV9-Cas9/sgRNA-51. (B) H&E staining of the TA muscle, triceps, and diaphragm muscle of ΔEx50 mice 4 weeks after systemic delivery of AAV9-Cas9 or AAV9-Cas9-sgRNA-51. (C) WT-CTL mice, ΔEx50 control mice, and ΔEx50 mice treated with AAV9-Cas9/sgRNA-51 (ΔEx50-AAV9-sgRNA-51) were subjected to grip strength testing to measure muscle performance (grams of force) that was normalized by body weight (BW). (D) Serum CK was measured in WT-CTL, ΔEx50-CTL, and ΔEx50-AAV9-sgRNA-51 mice. n = 5. Asterisk indicates nonspecific immunoreactive bands. Data are represented as means ± SEM. Significant differences between conditions are indicated by asterisk. ***P < 0.0005 using unpaired two-tailed Student’s t tests. Scale bars, 50 μm.
Dystrophin assembles a series of proteins into the dystrophin-associated glycoprotein complex, which links the cytoskeleton and extracellular matrix. In ΔEx50 mice, mdx mice, and DMD patients, these proteins are destabilized and fail to appropriately localize to the subsarcolemmal region (4, 5). In contrast, immunostained muscle sections of gene-edited ΔEx50 mice showed recovery of the dystrophin-associated glycoprotein complex proteins α-sarcoglycan and β-dystroglycan in AAV9-Cas9/sgRNA-51–treated mice, but not in ΔEx50 control mice treated with AAV9-Cas9 alone without sgRNA-51(Fig. 7). We concluded that single-cut genomic editing using AAV9-Cas9 and AAV9-sgRNA-51 was highly efficient at restoring dystrophin expression, dystrophin-associated glycoprotein complex proteins, and muscle function after systemic delivery to P4 ΔEx50 mice.
Fig. 7. Systemic delivery of AAV9-Cas9/sgRNA-51 to ΔEx50 mice rescues dystroglycan complex protein expression.
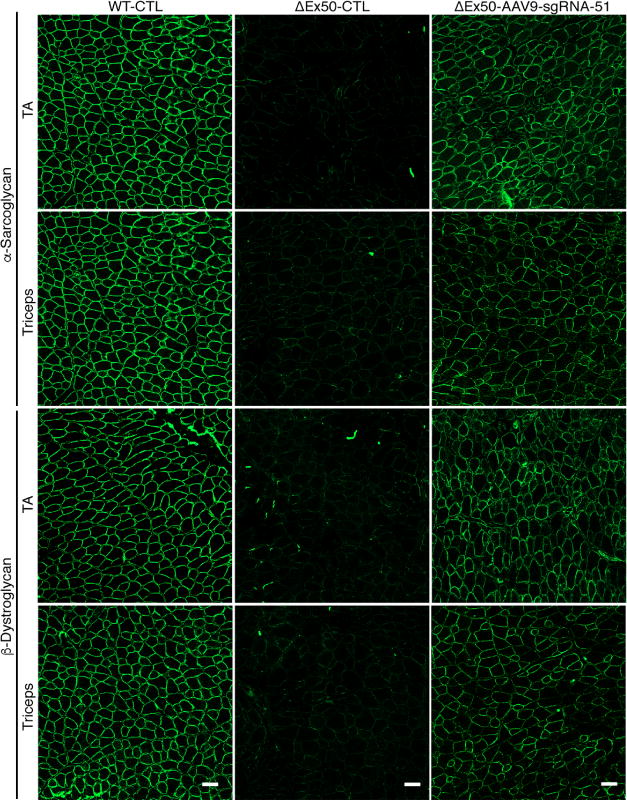
Immunohistochemical staining for α-sarcoglycan and β-dystroglycan in tibialis anterior (TA) and triceps muscles 4 weeks after systemic injection of ΔEx50 mice with AAV9-Cas9 only (WT-CTL and ΔEx50-CTL) or AAV9-Cas9/sgRNA-51 (ΔEx50-AAV9-sgRNA-51). n = 5. Scale bars, 50 μm.
DISCUSSION
The results of this study establish ΔEx50 mice as a new mouse model representing a prominent mutational hotspot associated with DMD in humans. The ΔEx50 mice will serve as a valuable toolfor testing many different strategies for amelioration of DMD pathogenesis. Our results revealed that the systemic delivery of AAV9 encoding a sgRNA directed against a region adjacent to the splice acceptor sequence of exon 51 in young mice efficiently restored dystrophin expression and prevented the onset of DMD histopathology and loss of muscle function. This optimized approach allowed for the restoration of dystrophin expression in up to 90% of skeletal myofibers, as well as efficient expression in cardiomyocytes.
Previously, we used AAV9 with a minicytomegalovirus (miniCMV) promoter to individually direct the expression of SpCas9 and two sgRNAs directed against exon 23 in mdx mice (15). The successful excision of exon 23 required that muscle nuclei received all three viruses simultaneously (AAV9-Cas9 and two AAV9-sgRNAs), as well as two concurrent cuts in the genomic DNA surrounding the mutant exon. Here, we optimized our approach by using the CK8e promoter to drive the expression of SpCas9 specifically in muscle nuclei. We also changed our sgRNA strategy of correction such that introduction of indels in the splice acceptor site region allowed reframing of exon 51 with a single DNA cut. We also used three different promoters to drive the expression of the single exon 51 sgRNA. Combining these modifications significantly enhanced the efficiency of gene correction.
Recently, the excision of large genomic regions encompassing mutational hotspots in the Dmd locus using pairs of sgRNAs to direct CRISPR/Cas9 cutting has been reported (19, 35). Correction of DMD mutations by reframing and exon skipping using a single sgRNA against a genomic sequence in the close vicinity of the intron-exon region offers several advantages over the use of multiple sgRNAs to introduce two DNA cuts and excision of relatively large intervening genomic sequences. The single sgRNA gene editing approach requires minimal modification of the genome to induce reframing events and exon skipping that lead to restoration of the reading frame. Therefore, a single cut within the vicinity of the intron-exon junction and splice acceptor region is sufficient to induce exon skipping and reframing, thereby increasing the efficiency of correction of dystrophin. Thus, to bypass mutant exons, it is not necessary to simultaneously introduce two cuts separated by large intervening genomic regions. Simply targeting essential splice acceptor and enhancer regions within the exon mitigates possible genomic rearrangements or insertions that might result from deletion of large genomic regions. In addition, this method can potentially be adapted to the editing of any exon by generating a sgRNA that targets the exon sequence and allows potential reframing of the exon or skipping by deletion of essential splicing sequences, such as highly conserved ESE sequences. Finally, the use of a single sgRNA reduces the likelihood of OT effects that might be incurred with multiple sgRNAs. It should also be pointed out that DNA repair of indel mutations can often restore the correct DNA sequence, thereby rendering the initial DNA cut ineffective. However, proper DNA repair recreates the original PAM site, enabling Cas9 to cut again until the PAM site or target sequence is eliminated. This reiterative cutting allows for more efficient editing of a single site, compared with editing two separate genomic sites separated by a distance.
A large fraction of edited Dmd alleles contained a single adenosine insertion at the same position in mouse and human cells. This adenosine insertion restored the dystrophin open reading frame without exon skipping. Thus, whereas skipping of exon 51 removed 78 amino acids within the rod domain of the dystrophin protein, the adenosine insertion allowed the retention of exon 51–encoded sequence that would otherwise be deleted. This finding might be explained by a phenomenon known as the A rule, in which an adenosine is preferentially inserted at sites of double-strand DNA breaks (36).
Because the CK8e regulatory cassette is active only in differentiated muscle cells and not in myoblasts or satellite cells (37, 38), we conclude that the efficient reading frame correction in ΔEx50 mice with AAV9 delivery of gene editing components reflected transduction of myofibers, rather than of satellite cells. As a multinuclear tissue, skeletal muscle may allow for efficient restoration of missing gene products after gene editing, even when only a small subset of nuclear genomes are edited (39). Similarly, adult cardiomyocytes are commonly polyploid, providing multiple genomic targets for gene editing in each cell.
Previous studies indicated that AAV6, AAV8, or AAV9 vectors do not transduce satellite cells (40), but Wagers and colleagues found that AAV9 could target these stem cells with modest efficiency (18). Future comprehensive analysis should be performed to investigate the potential contribution of muscle satellite cells to gene-edited skeletal muscle. Nevertheless, the efficient correction of dystrophin in the heart, which lacks an endogenous stem cell population (41), indicates that gene editing occurs efficiently in postmitotic muscle cells. Efficient cardiac dystrophin correction is especially important, because dilated cardiomyopathy due to the lack of dystrophin in cardiomyocytes is the primary cause of death of DMD patients. Our findings demonstrate that CRISPR/Cas9-mediated gene editing in vivo in young mice before the onset of the acute necrotic phase of the disease preserves muscle structure and function and prevents disease progression. In addition, histological and Western blot analyses of muscles at 8 weeks after injection demonstrated that dystrophin expression persisted and progressively increased. From a potential therapeutic perspective, early intervention would be the most effective approach to slow progression of this disease.
Off-target effects have been raised as a potential concern for gene editing in vivo. The use of the CK8e promoter, which allowed for highly muscle-specific expression of gene editing components, may minimize possible off-target toxicities in nonmuscle tissues. In this regard, skeletal muscle and the heart represent potentially attractive tissues for in vivo gene editing, especially with respect to possible oncogenic off-target effects, because these tissues are permanently postmitotic.
Several therapeutic approaches have been taken to restore dystrophin expression through exon 51 skipping in boys with DMD (42). Most recently, systemic delivery of an oligonucleotide that masks the exon 51 splice acceptor, referred to as eteplirsen, was approved for clinical use despite less than a 1% increase in dystrophin protein expression (43–45). Whereas many safety and efficacy challenges remain with respect to translating AAV9-mediated delivery of gene editing components for the correction of DMD, it is noteworthy that this approach allowed nearly complete restoration of dystrophin-positive myofibers within 4 weeks in ΔEx50 mice.
Among the more than 4000 DMD mutations (25), gene editing of exon 51 could potentially restore the open reading frame in 13 to14% of DMD patients who harbor different types of mutations, including deletions of single or multiple exons, missense mutations, or duplications. Because skipping or reframing exon 51 results in small in-frame deletions of the dystrophin protein, this strategy represents a potential means of converting the lethal DMD to a milder BMD.
This study has several limitations that will need to be addressed in future research, including the lack of data regarding the consequences of long-term expression of Cas9 in vivo. In addition, the longevity of rescue and possible immunological responses to Cas9 and to dystrophin protein in DMD patients (42) will eventually need to be addressed. Finally, it remains to be determined whether AAV delivery of gene editing components can be sufficiently scaled up to larger animals and, ultimately, patients with DMD.
Correcting DMD mutations by CRISPR/Cas9-mediated exon reframing and skipping is distinct from other potential therapeutic strategies that have been applied to DMD. In principle, this approach eliminates the disease-causing mutation after a single treatment and expresses the edited DMD gene under the endogenous cis-regulatory elements, thus allowing for proper temporal and spatial regulation throughout life. Finally, whereas here we have used ΔEx50 mice as a platform for optimizing CRISPR/Cas9-mediated genomic editing, these mice should also be useful to the scientific community for testing other strategies for correcting the most predominant human DMD mutation.
MATERIALS AND METHODS
Study Design
The objectives of the present study were to generate a DMD mouse model that mimics the most common human mutational hotspot and to identify the most efficient way to correct the mutation by gene editing. Male mice were used in all experiments. Animals were allocated to experimental groups based on genotype; we did not use exclusion, randomization, or blinding approaches to assign the animals for the experiments. AAV injection and dissection experiments were conducted in a non-blinded fashion. The sample sizes were determined on the basis of previous experience. Blinding approaches were used during grip tests and CK analysis. For each experiment, sample size reflects the number of independent biological replicates and was provided in the figure legend. In general, sample size was chosen to use the least number of animals to achieve statistical significance, and no statistical methods were used to predetermine sample size.
Mice
Mice were housed in a barrier facility with a 12-hour light/dark cycle and maintained on standard chow (2916 Teklad Global). ΔEx50 mice were generated in the C57/BL6J background using the CRISPR/Cas9 system. Two sgRNAs specific to the intronic regions surrounding exon 50 of the mouse Dmd locus were cloned into vector pSpCas9(BB)– 2A–green fluorescent protein(GFP) (PX458) (Addgene plasmid #48138) using the primers from table S2. For in vitro transcription of sgRNAs, T7 promoter sequence was added to the sgRNA template by PCR using the primers from table S3. Gel-purified PCR products were used as templates for in vitro transcription using the MEGA shortscript T7 Kit (Life Technologies). sgRNAs were purified by MEGA clear Kit (Life Technologies) and eluted with nuclease-free water (Ambion). The concentration of sgRNA was measured by a Nano Drop instrument (Thermo Scientific). Injection procedures were performed as described previously (16). ΔEx50 mice were backcrossed with C57/BL6J mice for more than three generations. Littermates were screened by genotyping.
Study approval
All experimental procedures involving animals in this study were reviewed and approved by the University of Texas Southwestern Medical Center’s Institutional Animal Care and Use Committee.
sgRNA identification, evaluation, and cloning
Dmd exon 51 sgRNAs were selected using crispr.mit.edu; these are listed in table S3. sgRNA sequences were cloned into Addgene plasmid #48138 (a gift from F. Zhang). sgRNAs were tested in tissue culture using 10T1/2 cells as previously described (16) before cloning into the rAAV9 backbone.
Cas9 plasmid
The pSpCas9(BB)-2A-GFP (PX458) plasmid was a gift from F. Zhang (Addgene plasmid #48138) (46) and contains the human codon–optimized SpCas9 gene with 2A-EGFP and the backbone of sgRNA. pGL3-CK8e plasmid has been previously described (33). A full description of this small regulatory cassette will be reported elsewhere.
To generate the AAV9-CK8-Cas9 vector, the AAV9-miniCMV-Cas9-shortPolyA (16) plasmid was digested with Pac I and Nhe I enzymes to remove the miniCMV promoter. The CK8e regulatory cassette was amplified from pGL3-CK8e plasmid using primers containing the Pac I and Nhe I sites, sequenced, and cloned into linearized vectors to generate the AAV9-CK8e-Cas9-shortPolyA plasmid.
sgRNA assembly in AAV9 backbone
Cloning of sgRNA was done using a Bbs I site. Assembly of the AAV9 TRISPR backbone cloning system relies on two consecutive steps of the Golden Gate Assembly (New England Biolabs). In the first step of assembling the sgRNA into the donor plasmid, annealing of oligonucleotides was performed by heating a reaction containing 2.5 μl of each oligo (0.5 μM), 5 μl of NEB uffer 2 (NEB), and 40 μl of ddH2O to 95°C for 5 min using a heating block. For the assembly reaction into the donor plasmid, 40 fmol (~100 ng) of destination backbone was mixed with 1 μl of annealed, diluted oligos, 0.75 μl of Esp3I (Thermo Scientific), 1 μl of buffer tango (Thermo Scientific), 1 μl of T4 DNA ligase (400 U/μl) (NEB), and ATP (adenosine 5′-triphosphate) and DTT (dithiothreitol) at a final concentration of 1 mM in 10-μl total volume. Using a thermo-cycler, PCR was performed for 25 to 50 cycles at 37°C for 3 min followed by 20°C for 5 min. Restriction enzyme and ligase were then denatured by heating to 80°C for 20 min. Three microliters of this reaction was used for transformation of chemo-competent bacteria, which were recovered in super optimal broth with catabolite repression (SOC) (37°C, 800 rpm, 40 min) and spread on LB agar plates containing chloramphenicol (25 μg/ml). Annealed oligonucleotides encoding for the sgRNA were cloned into donor plasmids that carry the negative selection marker ccdB (to reduce background during cloning) and the chloramphenicol resistance gene.
To test for correct assembly, the plasmid was sequenced using the primer Dono-R-5′-GTATGTTGTGTGGAATTGTGAG-3′. In the second step, three of these donor plasmids driving the expression of one sgRNA under transcriptional control of the U6, H1, or 7SK promoter were pooled in a second Golden Gate Assembly along with a recipient plasmid that carries AAV9 inverted terminal repeats (ITRs). The assembly reaction contained all four plasmids: donor plasmid-#1-U6-sgRNA, donor plasmid-#2-H1-sgRNA, donor plasmid-#3-7SK-sgRNA, and recipient plasmid containing the ITR. Digestion with Bbs I generated unique overhangs for each fragment (U6, H1, 7SK, and recipient backbone). During the ligation procedure, these overhangs annealed; a circularized plasmid was only obtained when the three cassettes matched each other.
AAV9 delivery to ΔEx50 mice
Before the AAV9 injections, the ΔEx50 mice were anesthetized by intraperitoneal injection of ketamine and xylazine anesthetic cocktail. For intramuscular injection, the tibialis anterior muscle of P12 male ΔEx50 mice was injected with 50 μl of AAV9 (1 × 1012 vg/ml) preparations or with saline solution. For intraperitoneal injection, the P4 ΔEx50 mice were injected using an ultrafine needle (31 gauge) with 80 μl of AAV9 preparations corresponding to 2.67 × 1013 vg/kg or with saline solution.
Statistics
Values are presented as means ± SEM. Differences between the respective two groups (wild-type and ΔEx50 mice, wild-type and ΔEx50-AAV9 mice, and ΔEx50 control and ΔEx50-AAV9 mice) were assessed using unpaired two-tailed Student’s t tests. P < 0.05 was regarded as significant. Statistical analysis was performed in Excel (Microsoft).
Supplementary Material
Fig. S1. ΔEx50 mouse model analyses.
Fig. S2. Comparison of dystrophin staining in ΔEx50 and mdx mouse models at 1 month of age.
Fig. S3. Characterization of ΔEx50 mice at different ages.
Fig. S4. ESEs of exon 51.
Fig. S5. Validation of sgRNAs in mouse 10T1/2 and human 293T cells.
Fig. S6. Cas9 expression in injected muscles.
Fig. S7. In vivo Dmd gene editing.
Fig. S8. Off-target analyses for sgRNA-51.
Fig. S9. Off-target sequence analyses for sgRNA-51.
Fig. S10. Amplicon PCR deep-sequencing analyses for sgRNA-51.
Fig. S11. Rescue of dystrophin expression after intramuscular injection of AAV9-Cas9 and AAV9-sgRNA-51 in the ΔEx50 mouse model.
Fig. S12. AAV9-Cas9– and AAV9-sgRNA-51–injected muscle shows histological improvement after 3 weeks.
Fig. S13. Quantification of histological improvement of AAV9-Cas9– and AAV9-sgRNA-51–injected muscle from ΔEx50 mice after 3 weeks.
Fig. S14. Intramuscular injection of AAV9-Cas9 and AAV9-sgRNA-51 in ΔEx50 mice corrects dystrophin expression in the heart.
Fig. S15. AAV9-Cas9 expression after systemic delivery in mice.
Fig. S16. Dmd gene editing 4 weeks after systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 in mice.
Fig. S17. Rescue of dystrophin expression 8 weeks after systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 in ΔEx50 mice.
Fig. S18. Histological analysis of dystrophin correction 8 weeks after systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 in ΔEx50 mice.
Fig. S19. Histological improvement of ΔEx50 mice 4 weeks after systemic injection of AAV9-Cas9 and AAV9-sgRNA-51.
Table S1. Sequences of potential exonic off-target (OT) sites in the mouse genome.
Table S2. Sequences of top 45 off-target (OT) sites in the mouse genome.
Table S3. Primer sequences.
Acknowledgments
We thank S. Rovinsky and E. Plautz [University of Texas (UT) Southwestern Neuro-Models Core Facility] for grip strength testing, the UT Southwestern Mouse Metabolic Phenotyping Core Facility for CK measurements, B. Chen (UT Southwestern Department of Clinical Sciences) for bioinformatics analysis advice, and J. Cabrera for the graphics.
Funding: This work was supported by grants from the NIH (HL130253, HL-077439, DK-099653, and AR-067294), Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant (U54 HD 087351), the Robert A. Welch Foundation (grant 1-0025 to E.N.O.), by grants to D.G. from the German Research Foundation (SFB1129, TRR179, and EXC81), the German Center for Infection Research (TTU HIV 04.803) and Deutsche Duchenne-Stiftung, and by grants to S.D.H. from the Muscular Dystrophy Association, the NIH (AR-18860), and a Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center grant (U54 AR-065139).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/418/eaan8081/DC1
Materials and Methods
Author contributions: L.A., C.L., R.B.-D., and E.N.O. designed the project. L.A. conceptualized and designed the experiments and performed the animal studies, genomic DNA, RT-PCR, protein experiments, and analysis. H.L. performed the cell culture sgRNA validation, T7EI assays, and RT-PCR analysis. A.A.M. performed the mouse genotyping and assisted in the tissue processing experiments. J.M.S. and E.S.-O. performed the immunohistochemistry and H&E staining. J.R.M. performed the zygote injection to generate the mice. S.B., F.S., D.G., and S.D.H. provided the reagents. L.A., R.B.-D., and E.N.O. wrote and edited the manuscript.
Competing interests: L.A., R.B.-D., and E.N.O. are consultants for Exonics Therapeutics. L.A., C.L., R.B.-D., and E.N.O. are listed as co-inventors on two filed patents regarding the mouse model (provisional filing application number 62/431,699) and strategy (provisional filing application number 62/442,606) presented in this study. The other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Requests for ΔEx50 mice, available through a material transfer agreement (MTA), should be directed to E.N.O. Requests for the Addgene plasmid #48138, available through an MTA, should be sent to F. Zhang at the Massachusetts Institute of Technology.
REFERENCES AND NOTES
- 1.O’Brien KF, Kunkel LM. Dystrophin and muscular dystrophy: Past, present, and future. Mol Gen Metab. 2001;74:75–88. doi: 10.1006/mgme.2001.3220. [DOI] [PubMed] [Google Scholar]
- 2.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 3.Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–860. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 4.Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 5.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 6.Guiraud S, Aartsma-Rus A, Vieira NM, Davies KE, van Ommen GJB, Kunkel LM. The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet. 2015;16:281–308. doi: 10.1146/annurev-genom-090314-025003. [DOI] [PubMed] [Google Scholar]
- 7.Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Benson S, McAndrew PE, Rice R, Nagaraja H, Stephens R, Lantry L, Morris GE, Burghes AHM. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med. 1995;333:832–838. doi: 10.1056/NEJM199509283331303. [DOI] [PubMed] [Google Scholar]
- 8.Arechavala-Gomeza V, Graham IR, Popplewell LJ, Adams AM, Aartsma-Rus A, Kinali M, Morgan JE, van Deutekom JC, Wilton SD, Dickson G, Muntoni F. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum Gene Ther. 2007;18:798–810. doi: 10.1089/hum.2006.061. [DOI] [PubMed] [Google Scholar]
- 9.Wu B, Lu P, Benrashid E, Malik S, Ashar J, Doran TJ, Lu QL. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010;17:132–140. doi: 10.1038/gt.2009.120. [DOI] [PubMed] [Google Scholar]
- 10.Shin JH, Pan X, Hakim CH, Yang HT, Yue Y, Zhang K, Terjung RL, Duan D. Microdystrophin ameliorates muscular dystrophy in the canine model of Duchenne muscular dystrophy. Mol Ther. 2013;21:750–757. doi: 10.1038/mt.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yue Y, Pan X, Hakim CH, Kodippili K, Zhang K, Shin JH, Yang HT, McDonald T, Duan D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet. 2015;24:5880–5890. doi: 10.1093/hmg/ddv310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramos J, Chamberlain JS. Gene therapy for Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2015;3:1255–1266. doi: 10.1517/21678707.2015.1088780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Hir M, Goyenvalle A, Peccate C, Précigout G, Davies KE, Voit T, Garcia L, Lorain S. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther. 2013;21:1551–1558. doi: 10.1038/mt.2013.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyombe-Engembe JP, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P, Lagüe P, Tremblay JP. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol Ther Nucleic Acids. 2016;5:e283. doi: 10.1038/mtna.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RM Castellanos, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR, Chamberlain JS. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Long C, Li H, McAnally JR, Baskin KK, Shelton JM, Bassel-Duby R, Olson EN. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci Adv. 2017;3:e1602814. doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A, Stanley R, Chandler K, Marks SL, Muntoni F, Shelton GD, Piercy RJ. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient Cavalier King Charles Spaniels is amenable to exon 51 skipping. PLOS ONE. 2010;5:e8647. doi: 10.1371/journal.pone.0008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helderman-van den Enden ATJM, Straathof CSM, Aartsma-Rus A, den Dunnen JT, Verbist BM, Bakker E, Verschuuren JJGM, Ginjaar HB. Becker muscular dystrophy patients with deletions around exon 51; a promising outlook for exon skipping therapy in Duchenne patients. Neuromuscul Disord. 2010;20:251–254. doi: 10.1016/j.nmd.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Romitti PA, Zhu Y, Puzhankara S, James KA, Nabukera SK, Zamba GKD, Ciafaloni E, Cunniff C, Druschel CM, Mathews KD, Matthews DJ, Meaney FJ, Andrews JG, Conway KMC, Fox DJ, Street N, Adams MM, Bolen J, MD STARnet Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. 2015;135:513–521. doi: 10.1542/peds.2014-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat Rev Genet. 2013;14:373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- 25.Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barišić N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, Bergen JC van den, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet P-Y, Joncourt F, Díaz-Manera J, Gallardo E, Karaduman AA, Topaloğlu H, Sherif R El, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Béroud C, Lochmüller H. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Liu Z, OuYang S, Zhu Y, Wang L, Wu J. Distribution of dystrophin gene deletions in a Chinese population. J Int Med Res. 2016;44:99–108. doi: 10.1177/0300060515613223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 28.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 33.Martari M, Sagazio A, Mohamadi A, Nguyen Q, Hauschka SD, Kim E, Salvatori R. Partial rescue of growth failure in growth hormone (GH)-deficient mice by a single injection of a double-stranded adeno-associated viral vector expressing the GH gene driven by a muscle-specific regulatory cassette. Hum Gene Ther. 2009;20:759–766. doi: 10.1089/hum.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Büning H, Perabo L, Coutelle O, Quadt-Humme S, Hallek M. Recent developments in adeno-associated virus vector technology. J Gene Med. 2008;10:717–733. doi: 10.1002/jgm.1205. [DOI] [PubMed] [Google Scholar]
- 35.Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, Karumbayaram S, Kumagai-Cresse C, Wang D, Zack JA, Kohn DB, Nakano A, Nelson SF, Miceli MC, Spencer MJ, Pyle AD. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strauss BS. The “A” rule revisited: Polymerases as determinants of mutational specificity. DNA Repair. 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 37.Chamberlain JS, Jaynes JB, Hauschka SD. Regulation of creatine kinase induction in differentiating mouse myoblasts. Mol Cell Biol. 1985;5:484–492. doi: 10.1128/mcb.5.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaynes JB, Chamberlain JS, Buskin JN, Johnson JE, Hauschka SD. Transcriptional regulation of the muscle creatine kinase gene and regulated expression in transfected mouse myoblasts. Mol Cell Biol. 1986;6:2855–2864. doi: 10.1128/mcb.6.8.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnett ALH, Konieczny P, Ramos JN, Hall J, Odom G, Yablonka-Reuveni Z, Chamberlain JR, Chamberlain JS. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev. 2014;1:14038. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Berlo JH, Molkentin JD. An emerging consensus on cardiac regeneration. Nat Med. 2014;20:1386–1393. doi: 10.1038/nm.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S, Aoki Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am J Transl Res. 2016;8:2471–2489. [PMC free article] [PubMed] [Google Scholar]
- 43.Stein CA. Eteplirsen approved for Duchenne muscular dystrophy: The FDA faces a difficult choice. Mol Ther. 2016;24:1884–1885. doi: 10.1038/mt.2016.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowling JJ. Eteplirsen therapy for Duchenne muscular dystrophy: Skipping to the front of the line. Nat Rev Neurol. 2016;12:675–676. doi: 10.1038/nrneurol.2016.180. [DOI] [PubMed] [Google Scholar]
- 45.Aartsma-Rus A, Krieg AM. FDA approves eteplirsen for Duchenne muscular dystrophy: The next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, Yuan GC. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol. 2016;34:695–697. doi: 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kodippili K, Vince L, Shin JH, Yue Y, Morris GE, McIntosh MA, Duan D, McNeil P. Characterization of 65 epitope-specific dystrophin monoclonal antibodies in canine and murine models of Duchenne muscular dystrophy by immunostaining and western blot. PLOS ONE. 2014;9:e88280. doi: 10.1371/journal.pone.0088280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ΔEx50 mouse model analyses.
Fig. S2. Comparison of dystrophin staining in ΔEx50 and mdx mouse models at 1 month of age.
Fig. S3. Characterization of ΔEx50 mice at different ages.
Fig. S4. ESEs of exon 51.
Fig. S5. Validation of sgRNAs in mouse 10T1/2 and human 293T cells.
Fig. S6. Cas9 expression in injected muscles.
Fig. S7. In vivo Dmd gene editing.
Fig. S8. Off-target analyses for sgRNA-51.
Fig. S9. Off-target sequence analyses for sgRNA-51.
Fig. S10. Amplicon PCR deep-sequencing analyses for sgRNA-51.
Fig. S11. Rescue of dystrophin expression after intramuscular injection of AAV9-Cas9 and AAV9-sgRNA-51 in the ΔEx50 mouse model.
Fig. S12. AAV9-Cas9– and AAV9-sgRNA-51–injected muscle shows histological improvement after 3 weeks.
Fig. S13. Quantification of histological improvement of AAV9-Cas9– and AAV9-sgRNA-51–injected muscle from ΔEx50 mice after 3 weeks.
Fig. S14. Intramuscular injection of AAV9-Cas9 and AAV9-sgRNA-51 in ΔEx50 mice corrects dystrophin expression in the heart.
Fig. S15. AAV9-Cas9 expression after systemic delivery in mice.
Fig. S16. Dmd gene editing 4 weeks after systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 in mice.
Fig. S17. Rescue of dystrophin expression 8 weeks after systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 in ΔEx50 mice.
Fig. S18. Histological analysis of dystrophin correction 8 weeks after systemic delivery of AAV9-Cas9 and AAV9-sgRNA-51 in ΔEx50 mice.
Fig. S19. Histological improvement of ΔEx50 mice 4 weeks after systemic injection of AAV9-Cas9 and AAV9-sgRNA-51.
Table S1. Sequences of potential exonic off-target (OT) sites in the mouse genome.
Table S2. Sequences of top 45 off-target (OT) sites in the mouse genome.
Table S3. Primer sequences.