

Abstract
Cytochrome P450 27A1 (CYP27A1) is a ubiquitous enzyme that hydroxylates cholesterol and other sterols. Complete CYP27A1 deficiency owing to genetic mutations is detrimental to human health, whereas 50% of activity retention is not and does not affect the whole body cholesterol levels. CYP27A1 is considered a potential therapeutic target in breast cancer and age-related neurodegenerative diseases; however, CYP27A1 inhibition should be ≤50%. Herein, 131 pharmaceuticals were tested for their effect on CYP27A1-mediated cholesterol 27-hydroxylation by in vitro enzyme assay. Of them, 14 drugs inhibited CYP27A1 by ≥75% and were evaluated for in vitro binding to the enzyme active site and for inhibition constants. All drugs except one (dasatinib) elicited a spectral response in CYP27A1 and had Ki values for cholesterol 27-hydroxylation either in the submicromolar (clevidipine, delavirdine, etravirine, felodipine, nicardipine, nilotinib, and sorafenib) or low micromolar range (abiratone, candesartan, celecoxib, dasatinib, nilvadipine, nimodipine, and regorafenib). Clevidipine, felodipine, nicardipine, nilvadipine, and nimodipine have the same 1,4-dihydropyridine scaffold and are indicated for hypertension. We used two of these antihypertensives (felodipine and nilvadipine) for administration to mice at a 1-mg/kg of body weight dose, daily, for 7 days. Mouse 27-hydroxycholesterol levels in the plasma, brain, and liver were reduced, whereas tissue levels of total cholesterol were unchanged. Structure-activity relationships within the 1,4-dihydropyridine scaffold were investigated, and features important for CY27A1 inhibition were identified. We confirmed our previous finding that CYP27A1 is a druggable enzyme and found additional drugs as well as the scaffold with potential for partial CYP27A1 inhibition in humans.
Introduction
Cytochrome P450 27A1 (CYP27A1) or sterol 27-hydroxylase is a ubiquitous mitochondrial enzyme whose substrate preferences are tissue-specific and include bile-acid intermediates (in the liver), cholesterol (in many extrahepatic tissues), and vitamin D3 (in the kidneys) (Wikvall, 1984; Masumoto et al., 1988; Okuda et al., 1988). Broad sterol specificity determines the multiple physiologic roles of CYP27A1, which are reflected in part in clinical and biochemical manifestations of cerebrotendinous xanthomatosis (CTX), an autosomal recessive disease resulting from mutations in CYP27A1, which disrupt or abolish enzyme activity (Björkhem, 2013). A clinical hallmark of CTX is deposits of cholesterol and its metabolite cholestanol in the brain and tendons. In addition, CTX frequently leads to progressive dementia, juvenile bilateral cataracts, retinal abnormalities, chronic diarrhea, osteoporosis, and premature atherosclerosis (Björkhem, 2013). Biochemically, CTX is characterized by specific changes in the plasma sterol profile: the concentration of 27-hydroxycholesterol (27HC, the CYP27A1 product) becomes very low, whereas that of cholesterol (a CYP27A1 substrate) remains normal or below normal; there is also a significant increase in the levels of cholestanol, some bile alcohols, and several other sterols (Björkhem, 2013). People heterozygous for CYP27A1 mutations are usually free of CTX symptoms and have a 2-fold reduction in plasma 27HC with normal or slightly increased concentrations of plasma cholestanol and bile alcohols (Björkhem, 2013).
CYP27A1 and its product 27HC have been linked to hormonal cancers and neurodegenerative diseases. In several breast cancer studies, high CYP27A1 expression was significantly associated with high tumor grade and was shown to affect estrogen receptor (ER) signaling, a process that facilitates the growth of most luminal breast cancers (Nelson et al., 2013; Kimbung et al., 2017). 27HC is a selective ER modulator and a partial ER agonist in the breast (Umetani et al., 2007; Wu et al., 2013). Accordingly, in women of a reproductive age and postmenopausal women, high levels of 27HC in breast tumors seem to play an opposite role: they attenuate ER action in the former and facilitate ER signaling in the latter (Umetani et al., 2007; DuSell et al., 2008; Kimbung et al., 2017). Conversely, in prostate cancer, a bioinformatics approach revealed a significantly lower level of CYP27A1 transcripts in tumor samples than benign prostate tissue and correlated this change with shorter disease-free survival and higher tumor grade (Alfaqih et al., 2017). Cellular cholesterol is thought to be critical for production of intratumoral androgens and proliferation of prostate cancer cells, whose growth was found to be affected by CYP27A1 expression (Montero et al., 2008; Mostaghel, 2013). Restoration of CYP27A1 expression in this cell type increased the production of 27HC and decreased cellular cholesterol by decreasing cellular cholesterol biosynthesis and uptake (Alfaqih et al., 2017). Finally, a recent study showed that in patients with varying degrees of cognitive impairment, elevated levels of 27HC in the cerebrospinal fluid negatively correlate with the brain glucose uptake (Ismail et al., 2017). Evidence was presented that 27HC activates the transcription factors liver X receptors and affects the brain glucose uptake via the brain renin-angiotensin system, which plays a role in learning, memory, emotional responses, and other brain functions (Mateos et al., 2009, 2011a,b; Ismail et al., 2017). Reduction of 27HC levels was suggested as an approach to prevent age-related changes in the brain glucose uptake and mitigate cognitive problems in some of the progressive neurodegenerative diseases (Ismail et al., 2017).
Emerging roles of CYP27A1 and 27HC in different diseases justified our previous studies of CYP27A1 as a therapeutic target that requires a partial inhibition of cholesterol 27-hydroxylation (Mast et al., 2015). We tested 26, mostly anticancer, medications and found that several of them have potential to partially inhibit CYP27A1 in humans. In the present work, we expanded our search for CYP27A1 inhibitors to noncancer medications and found a class of antihypertensive drugs that inhibits CYP27A1 in vitro and in vivo in mice.
Materials and Methods
Materials.
Cymal-7 was purchased from Anatrace (Maumee, OH). Cholesterol and [3H]cholesterol were from Steraloids (Newport, RI) and Perkin Elmer (Waltham, MA), respectively. [25,26,26,26,27,27,27-2H7]Cholesterol and [25,26,26,26,27,27-2H6]27HC were from Avanti Polar Lipids, Inc. (Alabaster, Al). All of the drugs for screening were ordered from either Toronto Research Chemicals (Toronto, Canada) or Sigma-Aldrich (St. Louis, MO) (Supplemental Table 1). Felodipine and nilvadipine for animal treatment were purchased from Abcam (Cambridge, MA). Felodipine analogs were purchased from ChemDiv (San Diego, CA). Human recombinant CYP27A1, bovine adrenodoxin reductase, and bovine adrenodoxin were expressed and purified as described (Sagara et al., 1992, 1993; Mast et al., 2006).
Animals.
All animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health and were approved by Case Western Reserve University’s Animal Care and Use Committee. C57BL/6J female mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in the Animal Resource Center at Case Western Reserve University until they reached the age of 4–5 months. Animals were maintained in a standard 12-hour light (∼10 lux)/12-hour dark cycle environment with water and food provided ad libitum. The female gender was chosen in consideration of CYP27A1 as a potential therapeutic target in breast cancer and possible future experiments in a mouse model of this disease. Also, we wanted to make sure that in our in vivo experiments, the drug treatment effect would be significant even in female mice, which typically have a higher data variability owing to monthly hormonal fluctuations.
Enzyme Assay for Drug Screening.
This assay was carried out as described (Mast et al., 2015) in 1 ml of 50 mM potassium phosphate buffer (KPi; pH 7.2) containing 1 mM EDTA and 0.01% CYMAL-7. Other reagents were as follows: 0.07 μM CYP27A1, 3.5 μM adrenodoxin, 0.35 μM adrenodoxin reductase, 46 μM drug under study, 2.3 μM cholesterol (a concentration equal to 0.5 Km CYP27A1 for this substrate), 13 nM (or ∼250,000 cpm) [3H]cholesterol, and 1 mM NADPH. Cholesterol was added from 1 mM stock in 4.5% aqueous 2-hydroxypropyl-β-cyclodextrin, and drugs were added from 5 mM stocks in either water, methanol, or dimethyl sulfoxide (Fig. 1). The volumes of the added cholesterol and drug solutions were 2.3 and 9.2 μl, respectively. Enzymatic reaction proceeded for 4 minutes at 37°C. Product formation was linear with time and CYP27A1 concentration.
Fig. 1.

Effect of drugs on CYP27A1 activity in the in vitro screening assay. Assay conditions are described in Materials and Methods. Control incubations contained only drug vehicle (1%, v/v). The results represent the mean ± S.D. of the three independent measurements. Drugs inhibiting CYP27A1 by ≥75% (the horizontal dashed line) are in bold. ND, not detectable; the limit of detection is 1% of cholesterol 27-hydroxylation. Superscripts indicate drug vehicles: D, dimethyl sulfoxide; M, methanol; W, water.
Spectral Binding Assay.
Drug binding to CYP27A1 (Mast et al., 2015) or any other P450 (Schenkman et al., 1967) can in many cases be monitored spectrally because of drug-induced changes in the coordination chemistry of the P450 heme iron, typically six-coordinated in the P450 ground state (Dawson et al., 1982; Poulos et al., 1986). Displacement of the water molecule serving as the sixth heme ligand and leaving the heme iron five-coordinated produces a so-called type I spectral response (a peak at 380–393 nm and a trough at 415–420 nm in the P450 difference spectrum), whereas coordinating the heme iron with a nitrogen atom from an added drug elicits a type II spectral response (a trough at 390–410 nm and a peak at 421–435 nm in the P450 difference spectrum) (Schenkman et al., 1967). A lack of drug-induced P450 spectral response does not necessarily mean a lack of drug binding; this could be the result of drug positioning at a distance from the heme iron (Isin and Guengerich, 2008). Binding of drugs in the CYP27A1 spectral assay was monitored as described (Mast et al., 2015), except only one drug concentration (5 μM) was used. The buffer was 50 mM KPi (pH 7.2) and contained 0.4 μM CYP27A1, 1 mM EDTA, 0.01% CYMAL-7, 10% glycerol, and 0.1 M NaCl; the solution temperature was 30°C.
Enzyme Assay for the Ki Determination.
The assay conditions conformed to the guidelines of pharmaceutical industry for in vitro assessment of P450 inhibition during drug discovery and development process (Nettleton and Einolf, 2011) and were identical to those of the screening assay. The two exceptions were the cholesterol concentration, which was changed to 4.6 μM, and the drug concentration, which varied from 0.0005 to 160 μM. Typically, 15 drug concentrations (0.0005, 0.005, 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10, 20, 40, 80, 120, and 160 µM) were used to generate the IC50 curve. Cholesterol was added from the same stock as in the screening assay; drugs were added from the 25, 250 μM, and 5 mM stocks. The total volume of the added drug and the vehicle solution was equal to 32 μl per assay. The data were fitted to the following equation using the GraphPad Prism software (La Jolla, CA):
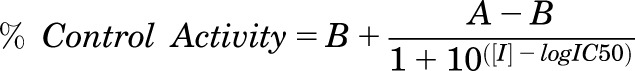 |
(1) |
in which [I] is the drug concentration, IC50 is the inflection point, A is the maximal activity of CYP27A1 (100%) in the absence of a drug, and B is the residual CYP27A1 activity (%) at the maximum drug concentration. Since the measurement of the IC50 was done at a cholesterol concentration equal to the Km, Ki = IC50/2, assuming a model of competitive inhibition.
Mouse Treatment with Felodipine and Nilvadipine.
Felodipine and nilvadipine stocks (0.2 mg/ml) were in 20% (v/v) ethanol, 10% (v/v) polyethylene glycol 400, 1.3 mM KPi (pH 7.4), and 96 mM NaCl. Each drug was given orally by gavage at a 1-mg/kg of mouse body weight dose, once a day for 7 days; the control group received only the drug vehicle. At the end of treatment, animals were fasted overnight and sacrificed the next morning. The brain and liver were isolated as described (Zheng et al., 2015); the blood was collected in EDTA-coated tubes and subjected to 1000g centrifugation for 10 minutes at 4°C to obtain plasma. The brain, liver, and plasma were then processed for the measurements of cholesterol and 27HC by gas chromatography-mass spectrometry as described (Mast et al., 2011).
Statistical Analysis.
All in vitro assays were carried out in triplicates. All quantifications in mice represent the measurements in at least six animals. Statistical analyses were experiment-specific and are indicated in figure legends along with the details of data presentation. Statistical significance was defined as *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Results
Drug Selection.
Drug selection was iterative and made on the basis of intuitive predictions used successfully in our previous investigation (Mast et al., 2015). The analyzed drugs were only those approved for use in the US by the Food and Drug Administration (FDA); neither of these compounds was a controlled substance or peptide/protein-based pharmaceutical. Chemical structures of a total of ∼1600 FDA-approved drugs were visually inspected to identify flexible linear structures with multiple rings that can fit the curvature of the presumably banana-shaped CYP27A1 active site, which connects the protein surface to the active site (Charvet et al., 2013). We also paid attention to compounds with oxygen- and nitrogen-containing functionalities that can interact with the protein side chains and/or coordinate the P450 heme iron. This is usually possible when there are no bulky substitutions in the vicinity of these functionalities that obstruct interactions with a cholesterol-metabolizing P450 (Mast et al., 2012). In addition, we decided to evaluate some of steroid-based structures and systems with conjugated rings. Accordingly, in the first round of screening, a total of 40 drugs was selected and tested for CYP27A1 inhibition in the screening assay. Of them, five inhibited CYP27A1 activity by ≥50% (we called them hits). In the second round of screening, we evaluated drugs with chemical structures similar to those of the hits from the first round of screening and from the hits discovered in our previous study (Mast et al., 2015). A total of 20 drugs were assessed, and four hits were identified. In the third round of screening, drugs with the same clinical indications as the hits identified in the first and second round were chosen, as opposed to the structural similarity search used in the previous rounds of screening. Additionally, preference was given for drugs that require a long-term use. The hit-to-tested drug ratio in this round was 3–22. In the fourth to sixth round of screenings, we continued to test drugs on the basis of the same clinical indication and had a hit-to-tested drug ratio of 7:25, 7:17, and 0:7 respectively.
Drug Evaluation by Screening Enzyme Assay.
A total of 131 drugs were screened for CYP27A1 inhibition. Of them, 14 inhibited enzyme activity by ≥75% (Fig. 1), the extent of inhibition indicative of a strong CYP27A1 inhibitor (Mast et al., 2015). These compounds were six antihypertensive drugs (candesartan, clevidipine, felodipine, nicardipine, nilvadipine, and nimodipine), five anticancer pharmaceuticals (abiratone, dasatinib, nilotinib, regorafenib, and sorafenib), two anti-HIV medications (delavirdine and etravirine), and one nonsteroidal anti-inflammatory drug (celecoxib) (Table 1).
TABLE 1 .
Some of the properties of the strong CYP27A1 inhibitors
The alphabetical list of submicromolar CYP27A1 inhibitors is followed by the alphabetical list of low micromolar CYP27A1 inhibitors. The black line separates the two groups of inhibitors. Assay conditions are described under Material and Methods. Ki values represent the geometric mean of measurements from three independent experiments; the geometric S.D. factor is shown in parenthesis. Residual CYP27A1 activity in the screening assay represents the means ± S.D. of measurements from three independent experiments.
| Drug | Indication (Drug Target) | Ki | CYP27A1 Spectral Response | Cholesterol 27-Hydroxylation |
|---|---|---|---|---|
| μM | λmax /λmin in Difference Spectrum | %a | ||
| No drug | 100 ± 3 | |||
| Clevidipine | Hypertension (calcium channel) | 0.10 (1.04) | 433/412 | NDb |
| Delavirdine | HIV (reverse transcriptase) | 0.46 (1.09) | 426/403 | 2 ± 1 |
| Etravirine | HIV (reverse transcriptase) | 0.10 (1.14) | 385/418 | 2 ± 1 |
| Felodipine | Hypertension (calcium channel) | 0.47 (1.07) | 433/412 | ND |
| Nicardipine | Hypertension (calcium channel) | 0.76 (1.20) | 433/412 | ND |
| Nilotinib | Leukemia (tyrosine kinase) | 0.19 (1.09) | 435/412 | 2 ± 1 |
| Sorafenib |
Thyroid cancer (tyrosine kinase) |
0.18 (1.02) |
393/427 |
2 ± 1 |
| Abiratone | Prostate cancer (CYP17A1) | 1.57 (1.06) | 417/407 | 5 ± 1 |
| Candesartan | Hypertension (angiotensin II receptor) | 3.71 (1.13) | 436/414 | 11 ± 5 |
| Celecoxib | NSAID (cyclo-oxygenase 2)c | 7.72 (1.04) | 385/419 | 14 ± 5 |
| Dasatinib | Leukemia (tyrosine kinase) | 10.1 (1.06) | ND | 27 ± 2 |
| Nilvadipine | Hypertension (calcium channel) | 3.35 (1.03) | 433/412 | 7 ± 3 |
| Nimodipine | Hypertension (calcium channel) | 4.64 (1.07) | 433/413 | 10 ± 2 |
| Regorafenib | Colorectal cancer (tyrosine kinase) | 1.77 (1.07) | 406/426 | NDb |
Activity in the screening enzyme assay.
ND, not detectable; the limit of detection was 1% of cholesterol 27-hydroxylation in the screening enzyme assay and 0.002 absorbance units in the spectral assay.
NSAID, nonsteroidal anti-inflammatory drug.
Drug Evaluation by Spectral Binding Assay.
Only potentially strong CYP27A1 inhibitors were assessed (Fig. 2; Table 1). Of them, one (dasatinib) did not induce in CYP27A1 any spectral response under the experimental conditions used, whereas the remaining 13 elicited a perturbation around the P450 heme iron. Nine drugs induced a type II spectral response (abiratone, candesartan, clevidipine, delavirdine, felodipine, nicardipine, nilotinib, nilvadipine, and nimodipine) and four elicited a type I (celecoxib, etravirine, and sorafenib) or type I-like (regorafenib) spectral response. Thus, at least 13 tested drugs seem to enter the CYP27A1 active site and act as competitive inhibitors.
Fig. 2.

Effect of strong inhibitors on CYP27A1 difference spectra. Assay conditions are described in Materials and Methods; drug concentration was 5 μM. Chemical structure of drugs are shown above difference spectra.
Drug Evaluation by In Vitro Ki Measurements.
All of the 14 potentially strong CYP27A1 inhibitors were characterized by the inhibition constants. All completely inhibited CYP27A1 in the IC50 measurements and yielded Ki values ranging from submicromolar (a total of seven drugs) to low micromolar (a total of seven drugs) (Table 1). The submicromolar CYP27A1 inhibitors were clevidipine, delavirdine, etravirine, felodipine, nicardipine, nilotinib, and sorafenib, and the low micromolar inhibitors were abiratone, candesartan, celecoxib, dasatinib, nilvadipine, nimodipine, and regorafenib.
Drug Evaluation by In Vivo CYP27A1 Inhibition.
Felodipine and nilvadipine were selected for administration to mice. Both felodipine and nilvadipine are antihypertensive drugs, belonging to the same class of 1,4-dihydropyridine (DHP) calcium channel blockers, and have the Ki values for cholesterol 27-hydroxylation (0.5 and 3.4 μM respectively, Table 1) representative of the Ki range (0.1–4.7 μM) for this class of drugs (clevidipine, felodipine, nicardipine, nilvadipine, and nimodipine, Table 1). The drug dose was 1 mg/kg of mouse body weight, the same for felodipine and nilvadipine, which was shown previously for nilvadipine to decrease arterial blood pressure in mice (Paris et al., 2004). For comparison, drug doses that are typically given to humans are 2.5–10 mg for felodipine and 4–16 mg for nilvadipine (Brogden and McTavish, 1995; Kirsten et al., 1998). Total cholesterol and 27HC were measured to assess drug effects on cholesterol homeostasis and CYP27A1 inhibition, respectively, in the whole body (plasma) and two organs (liver and brain). After a 7-day treatment with either felodipine or nilvadipine, the levels of cholesterol in the plasma, liver, and brain were not altered relative to the control group, whereas those of 27HC were decreased (Fig. 3). For felodipine and nilvadipine, these decreases were by 2.1- and 1.4-fold in the plasma, 2.7- and 2.2-fold in the liver, and 1.4- and 1.8-fold in the brain, respectively. No statistically significant differences were found between felodipine and nilvadipine for 27HC lowering in the liver and brain, yet in the plasma, felodipine lowered 27HC to a greater extent than nilvadipine. Overall, we confirmed our previous findings that it is possible to partially inhibit CYP27A1 in vivo without increasing cholesterol levels in the plasma and liver (the major site of cholesterol elimination) (Mast et al., 2015). The data obtained represent preclinical indication for off label use of felodipine and nilvadipine in humans.
Fig. 3.

Felodipine and nilvadipine treatment (Tx) of mice. Drugs effect on the levels of cholesterol and 27-hydroxycholesterol in the plasma, liver, and brain. Mice were C57BL/6J females of 4–5 months of age. Drugs were given orally by gavage as described in Materials and Methods. The drug dose was 1 mg/kg body weight given once a day for 7 days. The results represent the mean ± S.D. of the measurements in individual animals. The control group received only the vehicle. **P ≤ 0.01; ***P ≤ 0.001 by one-way analysis of variance followed by a Tukey posthoc multiple comparison test.
Studies of Felodipine Analogs.
To gain insight into the structural features of felodipine, one of the strongest CYP27A1 inhibitors, three commercially available felodipine analogs were investigated (Fig. 4A). Analogs A and B have the modifications at the carboxyl groups of the pyridine ring (positions 3 and 5), and analog C lacks the chlorine atoms at positions 2′ and 3′ of the 4-phenyl ring. Eliminating the ester functionality at position 3 of the pyridine ring and yielding free carboxylic acid abolished CYP27A1 inhibition in the screening assay (Fig. 4B) and resulted in essentially no P450 spectral response (Fig. 4C). Either the negative charge prevents analog A from entering the CYP27A1 active site or this analog is not retained in the enzyme active site because of a smaller size compared with felodipine. Conversely, increasing the chain length at position 5 of the pyridine ring by replacing the methyl ester with a larger ethyl ester still enabled CYP27A1 inhibition and preserved the spectral response. Yet, the extent of CYP27A1 inhibition by analog B in the screening enzyme assay was lower than that by felodipine (90 vs. 100%) and the Ki of analog B was increased about 5-fold (Fig. 4D). Finally, the size-reducing removal of the two chlorine atoms at positions 2′ and 3′ in the 4-phenyl ring (analog C) abolished CYP27A1 inhibition in the screening and elicited only very weak P450 spectral response (Fig. 4, B and C). Thus, the length and possibly nature of the functionalities at positions 3 and 5 of the pyridine ring and 2′ and 3′ of the 4-phenyl ring are important for CYP27A1 inhibition by felodipine.
Fig. 4.

Studies of felodipine analogs. (A) Chemical structures of the studied compounds; structural differences relative to felodipine are highlighted in orange (analog A), blue (analog B), and magenta (analog C). (B) Effect of felodipine analogs on CYP27A1 activity in the screening enzyme assay. Assay conditions are described in Materials and Methods. The results represent the mean ± S.D. of three independent measurements. (C) Difference spectra of CYP27A1 in the presence of felodipine and its analogs. (D) Representative IC50 plots for the inhibition of cholesterol 27-hydroxylation. All assays conditions are described in Materials and Methods. Ki values represent geometric mean of triplicate measurements; the geometric S.D. factor is shown in parenthesis. ND, not detectable; the limit of detection is 1% of cholesterol 27-hydroxylation.
Discussion
The major outcome of the present work is the identification of the scaffold that inhibits CYP27A1 both in vitro and in vivo in mice. We also confirmed our previous finding that CYP27A1 is a druggable enzyme (Mast et al., 2015) and found additional pharmaceuticals with potential for CYP27A1 inhibition in humans.
The identified CYP27A1 inhibitory scaffold represents the 1,4-DHP core from a class of antihypertensive drugs that block the L-type voltage-dependent Ca2+ channels. In vitro, all five tested drugs from this class can bind to the CYP27A1 active site and ligate the heme iron as indicated by the spectral assay (Fig. 2). Moreover, these drugs have in vitro Ki values from 0.1 to 4.7 μM (Table 1), a range in the pharmaceutical industry for compounds of a high risk (Ki < 0.5 μM; clevidipine and felodipine) and potential risk (0.5 μM < Ki < 5 μM; nicardipine, nilvadipine, and nimodipine) for in vivo P450 inhibition (Obach et al., 2005; Nettleton and Einolf, 2011). The capability of the 1,4-DHP scaffold to inhibit CYP27A1 in humans was further confirmed by studies in mice revealing that at a therapeutically relevant dose of 1 mg/kg of body weight, both felodipine and nilvadipine partially inhibited CYP27A1 in vivo (Fig. 3). The extent of the CYP27A1 inhibition in mouse whole body (the plasma 27HC levels) was higher for felodipine than nilvadipine and consistent with the Ki values of these compounds (0.47 and 3.38 μM, respectively, Table 1). Remarkably, a greater decrease in the plasma 27HC in the case of felodipine did not translate into the lower sterol levels in the brain (Fig. 3), which uptakes 27HC from the systemic circulation in addition to local 27HC synthesis (Heverin et al., 2005). Since both felodipine and nilvadipine cross the blood-brain barrier (Takakura et al., 1992; Lindgren et al., 2000), perhaps in the brain, nilvadipine is a stronger inhibitor of CYP27A1 than felodipine.
Studies of felodipine analogs (Fig. 4) along with data on five 1,4-DHP antihypertensives (Table 1) provide insight into structure-activity relationships of the 1,4-DHP scaffold for CYP27A1 inhibition. All 1,4-DHP compounds with submicromolar Ki for cholesterol 27-hydroxylation (clevidipine, felodipine, and nicardine) have a long, at least a four atom-, flexible ester-containing chain at position 3 of the pyridine ring and a short methyl ester chain at position 5. Shortening the chain length at position 3 to carboxylic acid and introducing the negative charge eliminated CYP27A1 inhibition (analog A), whereas increasing the chain length at position 5 by one methylene group increased the Ki ∼5-fold (analog B). The chlorine atom at position 2′ of the 4-phenyl ring could be lacking (nicardipine) but position 3′ must be occupied (analog C) and can have the nitro group (nicardipine). The two 1,4-DHP antihypertensives with low micromolar Kis (nilvadipine and nimodipine) had the same substitutions in the 4-phenyl ring as nicardine and identical structure at position 3 of the pyridine ring. The structural differences between the two drugs were that nilvadipine had a short methyl ester chain at position 5 and the cyano group at position 6 of the of the pyridine ring. Nimodipine had a long, six-atom chain at position 5 and the methyl group at position 6 of the of the pyridine ring. It is possible that the cyano group in nilvadipine and the long side chain at position 5 of the nimodipine pyridine ring lessen the inhibition of CYP27A1 by these drugs compared with clevidipine, felodipine, and nicardine. Of importance for understanding the structures that could inhibit CYP27A1 is the finding that the nitrogen atom in the pyridine ring can have on each side the adjacent methyl group, which apparently does not hinder this atom from the coordination of the CYP27A1 heme iron.
While a novel finding, in retrospect, CYP27A1 inhibition by the 1,4-DHP compounds should not be surprising; in addition to lowering blood pressure, the 1,4-DHP scaffold possesses a wide variety of other biologic and pharmacological effects including antibacterial, anticancer, anticoagulant, anticonvulsant, antitubercular, antiulcer, antimalarial, and anti-HIV activities (Khedkar and Auti, 2014). Moreover, in a small clinical study nilvadipine but not amlodipine stabilized cognitive decline and reduced the incidence of Alzheimer’s disease (Hanyu et al., 2007); possibly beneficial effects of nilvadipine were not related to its antihypertensive activity. Preclinical work in mice suggests that the potential anti-Alzheimer’s disease mechanisms of some of the 1,4-DHP antihypertensives could be realized through lowering of amyloid β accumulation and enhancing amyloid β clearance from the brain (Bachmeier et al., 2011; Paris et al., 2011). A large phase III clinical trial of nilvadipine to treat Alzheimer’s disease has recently been completed (ClinicalTrials.gov identifier: NCT02017340) with no study results yet posted. If available, plasma analysis of the trial participants for 27HC levels would provide insight into nilvadipine effect on CYP27A1 and suggest, if plasma 27HC is lowered, studies of additional mechanisms of drug effects.
Not every class of antihypertensive drugs inhibited CYP27A1 in vitro as indicated by our evaluation of angiotensin II receptor antagonists (eprosartan, irbesartan, losartan, and telmisartan, Fig. 1). These antihypertensives have a scaffold different from that of calcium channel blockers with only candesartan showing a significant (>25%) CYP27A1 inhibition in the screening assay (Fig. 1) and having a low Ki value of 3. 7 μM (Table 1). Angiotensin-converting-enzyme inhibitors, the third class of antihypertensive drugs, were not evaluated in the present work because their scaffold (small pyrrolidine moiety with a hindered tertiary amine) was not believed to inhibit CYP27A1.
Of the remaining 6 out of the 12 drugs with potential of a partial CYP27A1 inhibition in humans (Ki < 5 μM, Table 1), four were against different types of cancers (abiratone, dasatinib, nilotinib, and regorafenib) and two (delavirdine and etravirine) were the anti-HIV medications. With the exception of abiratone, which has a steroid-based structure similar to that of the CYP27A1 substrate cholesterol, all these potential inhibitors have a long, ring-containing flexible chain with nitrogen atoms, either unhindered or partially hindered. Two of these potential inhibitors (etravirine and sorafenib) induced a classic type I P450 spectral response (Fig. 2) and both had very low Ki values of 0.11 and 0.19 μM, respectively; these compounds are certainly interesting candidates for cocrystallization with CYP27A1 to evaluate how they bind to the enzyme active site. Also, etravirine and delavirdine represent the only class of the anti-HIV agents (the non-nucleoside reverse transcriptase inhibitors) that significantly inhibited CYP27A1 in vitro. The representatives from other classes of the anti-HIV agents—nucleoside/nucleotide reverse transcriptase inhibitors (abacavir, lamivudine, and tenofovir) and integrase inhibitors (dolutegravir and raltegravir)—did not substantially inhibit CYP27A1 in the screening assay.
In summary, we screened 131 marketed drugs and identified 12 strong CYP27A1 inhibitors with potential for a partial CYP27A1 inhibition in humans. We found an inhibitory scaffold for CYP27A1 and that nitrogen atoms in inhibitory drugs that coordinate the P450 heme iron can be partially hindered with small methyl groups. The data obtained expand our knowledge of chemical structures that inhibit CYP27A1 and provide preclinical data for testing of the identified potential CYP27A1 inhibitors in humans for conditions that may benefit from partial enzyme inhibition.
Abbreviations
- CYMAL-7
7-cyclohexyl-1-heptyl-β-d-maltoside
- CTX
cerebrotendinous xanthomatosis
- DHP
1,4-dihydropyridine
- ER
estrogen receptor
- FDA
US Food and Drug Administration
- 27HC
27-hydroxycholesterol
- KPi
potassium phosphate
- P450
cytochrome P450
Authorship Contributions
Participated in research design: Lam, Mast, Pikuleva.
Conducted experiments: Lam, Mast.
Performed data analysis: Lam, Mast, Pikuleva.
Wrote or contributed to the writing of the manuscript: Lam, Pikuleva.
Footnotes
This work was supported in part by National Institutes of Health National Institute of General Medical Sciences [Grant GM62882] to (I.A.P.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Alfaqih MA, Nelson ER, Liu W, Safi R, Jasper JS, Macias E, Geradts J, Thompson JW, Dubois LG, Freeman MR, et al. (2017) CYP27A1 loss dysregulates cholesterol homeostasis in prostate cancer. Cancer Res 77:1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmeier C, Beaulieu-Abdelahad D, Mullan M, Paris D. (2011) Selective dihydropyiridine compounds facilitate the clearance of β-amyloid across the blood-brain barrier. Eur J Pharmacol 659:124–129. [DOI] [PubMed] [Google Scholar]
- Björkhem I. (2013) Cerebrotendinous xanthomatosis. Curr Opin Lipidol 24:283–287. [DOI] [PubMed] [Google Scholar]
- Brogden RN, McTavish D. (1995) Nilvadipine. A review of its pharmacodynamic and pharmacokinetic properties, therapeutic use in hypertension and potential in cerebrovascular disease and angina. Drugs Aging 6:150–171. [DOI] [PubMed] [Google Scholar]
- Charvet CD, Laird J, Xu Y, Salomon RG, Pikuleva IA. (2013) Posttranslational modification by an isolevuglandin diminishes activity of the mitochondrial cytochrome P450 27A1. J Lipid Res 54:1421–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson JH, Andersson LA, Sono M. (1982) Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J Biol Chem 257:3606–3617. [PubMed] [Google Scholar]
- DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP. (2008) 27-hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol Endocrinol 22:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanyu H, Hirao K, Shimizu S, Sato T, Kiuchi A, Iwamoto T. (2007) Nilvadipine prevents cognitive decline of patients with mild cognitive impairment. Int J Geriatr Psychiatry 22:1264–1266. [DOI] [PubMed] [Google Scholar]
- Heverin M, Meaney S, Lütjohann D, Diczfalusy U, Wahren J, Björkhem I. (2005) Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. J Lipid Res 46:1047–1052. [DOI] [PubMed] [Google Scholar]
- Isin EM, Guengerich FP. (2008) Substrate binding to cytochromes P450. Anal Bioanal Chem 392:1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail MA, Mateos L, Maioli S, Merino-Serrais P, Ali Z, Lodeiro M, Westman E, Leitersdorf E, Gulyás B, Olof-Wahlund L, et al. (2017) 27-Hydroxycholesterol impairs neuronal glucose uptake through an IRAP/GLUT4 system dysregulation. J Exp Med 214:699–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khedkar SA, Auti PB. (2014) 1, 4-Dihydropyridines: a class of pharmacologically important molecules. Mini Rev Med Chem 14:282–290. [PubMed] [Google Scholar]
- Kimbung S, Chang C, Bendahl PO, Dubois L, Thompson WJ, McDonnell DP, Borgquist S. (2017) Impact of 27-hydroxylase (CYP27A1) and 27-hydroxycholesterol in breast cancer. Endocr Relat Cancer 24:339–349. [DOI] [PubMed] [Google Scholar]
- Kirsten R, Nelson K, Kirsten D, Heintz B. (1998) Clinical pharmacokinetics of vasodilators. Part I. Clin Pharmacokinet 34:457–482. [DOI] [PubMed] [Google Scholar]
- Lindgren M, Thostrup H, Svannäs M, Fagher B. (2000) Felodipine in patients with organic solvent-induced chronic toxic encephalopathy. Clin Drug Investig 19:267–276. [Google Scholar]
- Mast N, Lin JB, Pikuleva IA. (2015) Marketed drugs can inhibit cytochrome P450 27A1, a potential new target for breast cancer adjuvant therapy. Mol Pharmacol 88:428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Linger M, Clark M, Wiseman J, Stout CD, Pikuleva IA. (2012) In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol Pharmacol 82:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Murtazina D, Liu H, Graham SE, Bjorkhem I, Halpert JR, Peterson J, Pikuleva IA. (2006) Distinct binding of cholesterol and 5beta-cholestane-3alpha,7alpha,12alpha-triol to cytochrome P450 27A1: evidence from modeling and site-directed mutagenesis studies. Biochemistry 45:4396–4404. [DOI] [PubMed] [Google Scholar]
- Mast N, Reem R, Bederman I, Huang S, DiPatre PL, Bjorkhem I, Pikuleva IA. (2011) Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest Ophthalmol Vis Sci 52:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto O, Ohyama Y, Okuda K. (1988) Purification and characterization of vitamin D 25-hydroxylase from rat liver mitochondria. J Biol Chem 263:14256–14260. [PubMed] [Google Scholar]
- Mateos L, Akterin S, Gil-Bea FJ, Spulber S, Rahman A, Björkhem I, Schultzberg M, Flores-Morales A, Cedazo-Mínguez A. (2009) Activity-regulated cytoskeleton-associated protein in rodent brain is down-regulated by high fat diet in vivo and by 27-hydroxycholesterol in vitro. Brain Pathol 19:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos L, Ismail MA, Gil-Bea FJ, Leoni V, Winblad B, Björkhem I, Cedazo-Mínguez A. (2011a) Upregulation of brain renin angiotensin system by 27-hydroxycholesterol in Alzheimer’s disease. J Alzheimers Dis 24:669–679. [DOI] [PubMed] [Google Scholar]
- Mateos L, Ismail MA, Gil-Bea FJ, Schüle R, Schöls L, Heverin M, Folkesson R, Björkhem I, Cedazo-Mínguez A. (2011b) Side chain-oxidized oxysterols regulate the brain renin-angiotensin system through a liver X receptor-dependent mechanism. J Biol Chem 286:25574–25585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, Antonsson B, Prieto J, García-Ruiz C, Colell A, et al. (2008) Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res 68:5246–5256. [DOI] [PubMed] [Google Scholar]
- Mostaghel EA. (2013) Steroid hormone synthetic pathways in prostate cancer. Transl Androl Urol 2:212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, et al. (2013) 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342:1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton DO, Einolf HJ. (2011) Assessment of cytochrome p450 enzyme inhibition and inactivation in drug discovery and development. Curr Top Med Chem 11:382–403. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K, Houston JB, Tremaine LM. (2005) In vitro cytochrome P450 inhibition data and the prediction of drug-drug interactions: qualitative relationships, quantitative predictions, and the rank-order approach. Clin Pharmacol Ther 78:582–592. [DOI] [PubMed] [Google Scholar]
- Okuda K, Masumoto O, Ohyama Y. (1988) Purification and characterization of 5 beta-cholestane-3 alpha,7 alpha,12 alpha-triol 27-hydroxylase from female rat liver mitochondria. J Biol Chem 263:18138–18142. [PubMed] [Google Scholar]
- Paris D, Bachmeier C, Patel N, Quadros A, Volmar CH, Laporte V, Ganey J, Beaulieu-Abdelahad D, Ait-Ghezala G, Crawford F, et al. (2011) Selective antihypertensive dihydropyridines lower Aβ accumulation by targeting both the production and the clearance of Aβ across the blood-brain barrier. Mol Med 17:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris D, Quadros A, Humphrey J, Patel N, Crescentini R, Crawford F, Mullan M. (2004) Nilvadipine antagonizes both Abeta vasoactivity in isolated arteries, and the reduced cerebral blood flow in APPsw transgenic mice. Brain Res 999:53–61. [DOI] [PubMed] [Google Scholar]
- Poulos TL, Finzel BC, Howard AJ. (1986) Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry 25:5314–5322. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Hara T, Ariyasu Y, Ando F, Tokunaga N, Horiuchi T. (1992) Direct expression in Escherichia coli and characterization of bovine adrenodoxins with modified amino-terminal regions. FEBS Lett 300:208–212. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Wada A, Takata Y, Waterman MR, Sekimizu K, Horiuchi T. (1993) Direct expression of adrenodoxin reductase in Escherichia coli and the functional characterization. Biol Pharm Bull 16:627–630. [DOI] [PubMed] [Google Scholar]
- Schenkman JB, Remmer H, Estabrook RW. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol Pharmacol 3:113–123. [PubMed] [Google Scholar]
- Takakura S, Sogabe K, Satoh H, Mori J, Fujiwara T, Totsuka Z, Tokuma Y, Kohsaka M. (1992) Nilvadipine as a neuroprotective calcium entry blocker in a rat model of global cerebral ischemia. A comparative study with nicardipine hydrochloride. Neurosci Lett 141:199–202. [DOI] [PubMed] [Google Scholar]
- Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ. (2007) 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med 13:1185–1192. [DOI] [PubMed] [Google Scholar]
- Wikvall K. (1984) Hydroxylations in biosynthesis of bile acids. Isolation of a cytochrome P-450 from rabbit liver mitochondria catalyzing 26-hydroxylation of C27-steroids. J Biol Chem 259:3800–3804. [PubMed] [Google Scholar]
- Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA, et al. (2013) 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Reports 5:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Mast N, Saadane A, Pikuleva IA. (2015) Pathways of cholesterol homeostasis in mouse retina responsive to dietary and pharmacologic treatments. J Lipid Res 56:81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]