

Abstract
The ability to repair tissues is essential for the survival of organisms. In chronic settings, the failure of the repair process to terminate results in overproduction of collagen, a pathology known as fibrosis, which compromises organ recovery and impairs function. The origin of the collagen-overproducing cell has been debated for years. Here we review recent insights gained from the use of lineage tracing approaches in several organs. The resulting evidence points toward specific subsets of tissue-resident mesenchymal cells, mainly localized in a perivascular position, as the major source for collagen-producing cells after injury. We discuss these findings in view of the functional heterogeneity of mesenchymal cells of the perivascular niche, which have essential vascular, immune, and regenerative functions that need to be preserved for efficient repair.
Introduction
Tissue damage induces a repair process aimed at restoring tissue architecture and function. This process involves an inflammation phase to remove dead cells and control potential pathogens, a remodeling/scarring phase to generate a transient collagenous matrix, and a regeneration phase to replace damaged parenchymal cells. Failure to terminate such a repair process induces pathological scarring, termed fibrosis, leading to dysregulated inflammation and excess collagen deposition. Fibrosis can affect most organs and become a life-threatening condition. However, therapeutic options remain limited. Controlling the level of scarring is therefore a priority in a wide array of chronic inflammatory and fibrotic diseases, such as cardiovascular diseases, pulmonary fibrosis, kidney diseases, liver diseases, systemic sclerosis/scleroderma, and muscular dystrophies.
A major cause hindering therapeutic progress is the lack of understanding of the biological process involved in fibrosis. Tissue damage can result from insults of different natures, such as mechanical injury, infection, ischemia/reperfusion, toxins, or autoimmunity. Irrespective of the initiating insult and targeted organ, injury induces local activation and proliferation of specialized subsets of mesenchymal cells, which produces extracellular matrix (ECM) comprising fibrillar collagens and nonstructural proteins with regulatory roles in ECM, proinflammatory cytokines, chemokines, and growth/angiogenic factors, all of which are essential for repair (1, 2). These injury-induced mesenchymal cells have been historically referred to as “activated fibroblasts” or myofibroblasts, as they were initially identified in tissues by expression of α-smooth muscle actin (αSMA), an actin isoform also expressed in smooth muscle cells (3, 4). Increasing evidence indicates that αSMA+ myofibroblasts are only a subset of activated fibroblasts, which varies spatiotemporally after injury, and that other subsets of activated fibroblasts contribute to collagen deposition and repair as well (3, 5, 6). Nevertheless, in the absence of more specific markers, expression of αSMA is commonly used to identify activated mesenchymal cells at sites of injury, as mesenchymal cells at steady state do not express it.
In addition to secreting collagen and other ECM proteins, myofibroblasts contribute to repair by generating contractile forces that are transmitted to the surrounding ECM and activate integrin-bound latent TGF-β, a key cytokine in wound healing and fibrosis (7–10). Besides active TGF-β1, other factors released by damaged epithelial and endothelial cells, platelets, innate immune cells, and lymphocytes (such as IL-25, IL-33, PDGFs, IL-4, and IL-13), as well as pathogen-associated molecular patterns, directly or indirectly contribute to myofibroblast activation (11, 12). Initially beneficial, persistence or dysregulation of this process leads to fibrosis. The cellular origin of the matrix-producing cells is therefore a central issue. Reported potential progenitors for myofibroblasts include epithelial cells and endothelial cells, through processes termed epithelial-mesenchymal or endothelial-mesenchymal transition; circulating bone marrow–derived (BM-derived) fibrocytes; tissue-resident fibroblasts; and other mesenchymal cells related to blood vessels, such as pericytes, adventitial cells, and mesenchymal stem cells (MSCs) (13–16).
The development of genetic mouse models expressing Cre recombinase in putative progenitor cells has allowed researchers to map the fate of cells in vivo without removing them from their normal microenvironment. Genetic fate mapping strategies rely on site-specific recombinase-mediated DNA excision to activate a silenced reporter transgene, thereby labeling selectively and permanently the Cre-expressing cell population and their progeny (17). In this Review, we discuss insights gained from genetically engineered mouse models that allow more precise identification of the cell lineages activated toward a myofibroblastic phenotype in repair/fibrosis. We also discuss common issues of genetic fate mapping that have caused confusion in the field, such as Cre-expressing systems that lack specificity or show expression in unexpected cell types. The emerging picture suggests that a majority of injury-activated, matrix-producing cells in different organs, including in the skeletal muscle, skin, liver, kidney, heart, lung, and spinal cord, originate from specific subsets of tissue-resident mesenchymal cells mainly localized close to blood vessels (18–21). While these findings open new opportunities for therapeutic treatment, they also raise a number of challenging questions related to the functional heterogeneity of mesenchymal cells of the perivascular niche, such as pericytes, adventitial cells, and MSCs, which have vascular, immune, and regenerative roles that are essential for repair.
Identification of tissue-resident mesenchymal cells at homeostasis
Although the role of the mesenchymal compartment in tissue homeostasis or disease is increasingly recognized, the relative contribution of distinct mesenchymal subsets to these processes remains poorly understood. The main limitation has been the lack of specific markers available to discriminate between different mesenchymal cell types. Mesenchymal cells have been initially isolated based on adherence and capability to expand in vitro. Another common feature of mesenchymal cells is the lack of endothelial (CD31) or hematopoietic (CD45) markers. However, the resulting adherent CD31–CD45– population is neither homogeneous nor cell type–specific. Positive selection using a number of markers, as well as localization relative to the vasculature, further allows discrimination between distinct subsets of mesenchymal cells (described in the following sections).
Fibroblasts
Fibroblasts are tissue-resident mesenchymal cells found in the interstitial space of all organs. They contribute to their structural framework by producing ECM. Fibroblasts are morphologically and functionally distinct from myofibroblasts, as they do not express αSMA and lack the contractile microfilamentous apparatus (i.e., stress fibers) observed in myofibroblasts. Markers first used to identify fibroblasts included thymocyte differentiation antigen 1 (Thy-1, also known as CD90) and fibroblast-specific protein 1 (FSP1, also known as S100A4). However, these markers are not specific, as CD90 and FSP1 are also expressed by immune and endothelial cells (22–24), leading to some confusion in lineage tracing experiments (25). Other mesenchymal markers include vimentin, PDGFRα, and type I collagen, which are more specific for mesenchymal populations but are expressed by several subsets (discussed below). Some mesenchymal markers are tissue-specific, such as Tcf21 and periostin in cardiac fibroblasts and myofibroblasts, respectively (26).
Increasing evidence suggests that the historically defined fibroblast is actually not a cell type, but a general name for heterogeneous populations of mesenchymal cells. In the skin, initial experiments addressing the molecular basis of fibroblast diversity showed that fibroblast transcriptional profiles cluster into groups defined by anatomic site (27, 28), suggesting that fibroblasts have a transcriptionally imprinted memory that define cell position. Single-cell transcriptomics will most likely reveal additional heterogeneity, and potentially diverse function, in fibroblastic populations. In lymphoid organs, specialized subsets of fibroblastic mesenchymal cells produce chemokines and growth factors that organize the localization, survival, and interactions of immune cells (29). During ontogeny, the development of lymphoid organs requires a specialized fibroblastic mesenchyme expressing lymphotoxin β receptor (LTβR), ICAM1, VCAM1, and Gp38 (also known as podoplanin) (30, 31). Mesenchymal cells with similar phenotypes are induced in inflamed/fibrotic tissues as well as in tertiary lymphoid organs developing during chronic inflammation (32–37), suggesting an active role in the inflammatory process. The intestinal lamina propria, which harbors a major reservoir of immune cells, contains an abundant population of Gp38+ mesenchymal cells that contribute to intestinal homeostasis and gut immunity (32, 38, 39).
Perivascular mesenchymal cells
The blood vasculature is organized into networks of arteries, veins, and interconnected capillaries. Arterioles, capillaries, and venules constitute the microvasculature, where most intercellular communication occurs, and are covered by vascular basement membrane (vBM) and pericytes to various extents (described below). Larger arteries and veins have three structural layers: the tunica intima (endothelial cells), the tunica media (smooth muscle cells, which express high levels of αSMA), and the tunica externa (called the adventitia) (40)(Figure 1).
Figure 1. The perivascular niche.

At steady state, the perivascular niche contains different subsets of mesenchymal cells, depending on the vessel type and size. In the microvasculature, pericytes are embedded within the vascular basement membrane and are essential for vascular development and stability. The outer covering of larger vessels (the adventitia) contains adventitial mesenchymal cells in a collagenous matrix. Other mesenchymal subsets such as FAPs and MSCs are localized in close proximity to blood vessels. At steady state, perivascular mesenchymal cells have essential roles in maintenance of tissue homeostasis. SMC: smooth muscle cells.
Pericytes.
In the microvasculature, endothelial cells are surrounded by a discrete subset of contractile mesenchymal cells termed pericytes. Pericytes are embedded within the vBM and establish close contacts with endothelial cells through specialized membrane invaginations called peg-socket contacts, which contain adherens junctions. Pericytes were first described by Charles Rouget in 1873 (41). Electron microscopy enabled the currently accepted definition of a mature pericyte as a mesenchymal cell embedded within the vBM of capillaries and venules (42). Identification of pericytes remains challenging but can be addressed using a combination of criteria, including location relative to endothelial cells and the vBM, morphology, exclusion of the lineage markers CD45 and CD31, and expression of markers such as PDGFRβ, the cell surface glycoprotein MUC18 (CD146), GTPase signaling 5 (RGS5), chondroitin sulfate proteoglycan 4 (NG2), or αSMAlo. However, none of these markers are unique to pericytes, and expression levels vary with pericyte activation state and vessel type (43, 44).
As integral constituents of blood vessels, pericytes are essential regulators of vascular development, stabilization, maturation, and remodeling (reviewed in ref. 45). The density of pericytes on vessels varies between organs, with the CNS vasculature displaying the highest ratio of pericyte/endothelial cell coverage (46). In the CNS, capillary pericytes are required for the formation and regulation of the blood-brain barrier, maintenance of vascular permeability, and regulation of cerebral blood flow (47–49). Pericyte loss or dysfunction is commonly observed in diverse fibrotic diseases, CNS disorders such as diabetic retinopathy and neurodegenerative diseases, and solid tumors (50–53). Consistent with a major role in vascular stability, pathological microvessels that lack pericytes or have pericytes loosely attached to endothelial cells are leaky and poorly functional (50–53).
In addition to their vascular functions, pericytes regulate different aspects of immune responses. Stark et al. showed that NG2-expressing capillary and arteriolar pericytes support the immunosurveillance and effector function of extravasating neutrophils and macrophages (54). These NG2+ pericytes also express TNFR and various pattern recognition receptors, such as TLR2, TLR4, and NLRP3, as well as ICAM1 and chemoattractants such as CXCL1, CXCL8, MIF, and CCL2, allowing them to sense inflammatory stimuli and promote monocyte and neutrophil migration and survival. Confocal intravital microscopy studies of direct pericyte-neutrophil interaction demonstrated that neutrophils transmigrate through gaps regulated by pericyte shape and activation (55). In the kidney, pericytes mediate a TLR2/4- and MyD88-dependent proinflammatory program, thereby regulating profibrotic responses to acute renal injury (56). Conditional knockin mice with activating mutations at the PDGFRβ locus, which increases PDGFRβ signaling, have enhanced proinflammatory genes in pericytes and mesenchymal cells (57), further suggesting a role for pericytes in immune response.
Adventitial cells.
The outer covering of arteries and veins is composed of a connective tissue termed the adventitia. The adventitia is the most complex and heterogeneous compartment of blood vessels, containing a collagen-rich matrix and different cell types including mesenchymal cells (expressing CD34, Sca1, and PDGFRα), small blood vessels (also called vasa vasorum), lymphatic vessels, nerve fibers, and immune cells. The adventitia has essential roles in vascular structure and function (58). Adventitial mesenchymal cells are the primary sensors of vascular stress/injury and contribute to vascular remodeling and low-grade chronic inflammation by stimulating expansion of microvessels and producing chemokines such as CCL2, which recruit BM-derived monocytes and other immune cells to the vessel wall (58–60). In chronic inflammatory settings such as atherosclerosis, vascular adventitial inflammation is involved in the local generation of tertiary lymphoid organs in the vessel wall, which perpetuate inflammation (61, 62). Adventitial-like cells, termed veil cells, have been described around dermal microvessels (63).
MSCs.
MSCs are multipotent, self-renewing cells that are capable of generating, in single-cell assays, a complete heterotopic BM organ, including bone, cartilage, adipocyte, fibroblasts, and a hematopoiesis-supporting stroma. First identified in the BM over 40 years ago as cells able to form fibroblastic colonies (colony-forming unit fibroblasts, CFU-F) (64), MSCs were subsequently identified as CD146+CD45– cells localized close to the BM blood vessels (65). In mice, PDGFRα+Sca1+CD45–Ter119– or nestin+CD31–CD45– perivascular stromal cells isolated from BM were shown to be the major source for osteoblasts, adipocytes, and reticular cells in vivo upon transplantation into irradiated mice (66), and to contain the CFU-F activity and capacity to form clonal spheres (67). In addition to their progenitor functions, MSCs are essential regulators of the hematopoietic stem cell niche (reviewed in ref. 68). Currently, several surface markers allow for the prospective isolation of MSCs from the BM, including CD146, CD105, alkaline phosphatase, Stro-1, and VCAM1 in humans, and CD105, PDGFRα, Sca1, CD44, CD29, and VCAM1 in mouse. At the transcriptomic level, MSCs express genes characteristic of committed early osteogenic cells, such as RUNX2, and genes characteristic of perivascular mesenchymal cells, such as angiopoietin-1 (65). However, most of these markers are not stable in culture, which increases the difficulty of studying these cells in vitro. In the past few years, evidence emerged that mesenchymal cells similar to MSCs are found in most organs, localized in the perivascular niche of small and larger blood vessels in a position compatible with pericytes and adventitial cells (13, 69, 70). Whether MSCs from the BM and perivascular mesenchymal cells from other organs that express the same markers have a similar progenitor potential remains controversial (71). Also, it remains unclear whether MSCs and pericytes have a lineage relationship, such as that one generates the other one during development/injury, or whether they represent functionally distinct mesenchymal populations that share a perivascular location. Identification of more specific markers and development of novel fate mapping strategies might help in answering this question. Nevertheless, putative MSCs isolated and expanded in vitro from diverse organs, particularly from the adipose tissue, where they are abundant, can efficiently promote repair and tissue regeneration in several organs, including the heart, bone, and lung (44, 72, 73). Although the underlying mechanism(s) remain unclear, MSCs have paracrine immunomodulatory and angiogenic functions that are likely essential for their beneficial role in repair (reviewed in refs. 74, 75). Some tissue-specific perivascular mesenchymal progenitors have a restricted potential, such as the fibro-adipogenic progenitors (FAPs, not to be confused with fibroblast-activating protein) initially described in the skeletal muscle (76, 77). FAPs express PDGFRα, Sca1, and CD34, and have an essential role in muscle repair (described in the following section).
Stellate cells.
The liver contains a specific subset of perivascular mesenchymal cells called hepatic stellate cells (HSCs) (78). HSCs are located in the perisinusoidal space, between the basolateral surface of hepatocytes and the antiluminal side of sinusoidal endothelial cells. They have subendothelial processes that wrap around sinusoids between endothelial cells and hepatocytes, allowing them to sense and rapidly adapt to the microenvironment. HSCs are abundant, constituting one-third of the nonparenchymal CD31–CD45– population in the liver. They do not express αSMA at homeostasis and can be identified by expression of glial fibrillary acidic protein (GFAP), nerve growth factor receptor (NGFR; p75), desmin, and PDGFRβ. Similar cells are found in the pancreas (79). The close proximity of this perivascular cell type to collagen fibers in injured liver was described over 40 years ago (80, 81), suggesting an important role in liver fibrosis. These early discoveries leading to the identification of HSCs as major contributors to liver fibrosis were subsequently confirmed by many other studies (82).
Origin of collagen-producing mesenchymal cells in repair/fibrosis
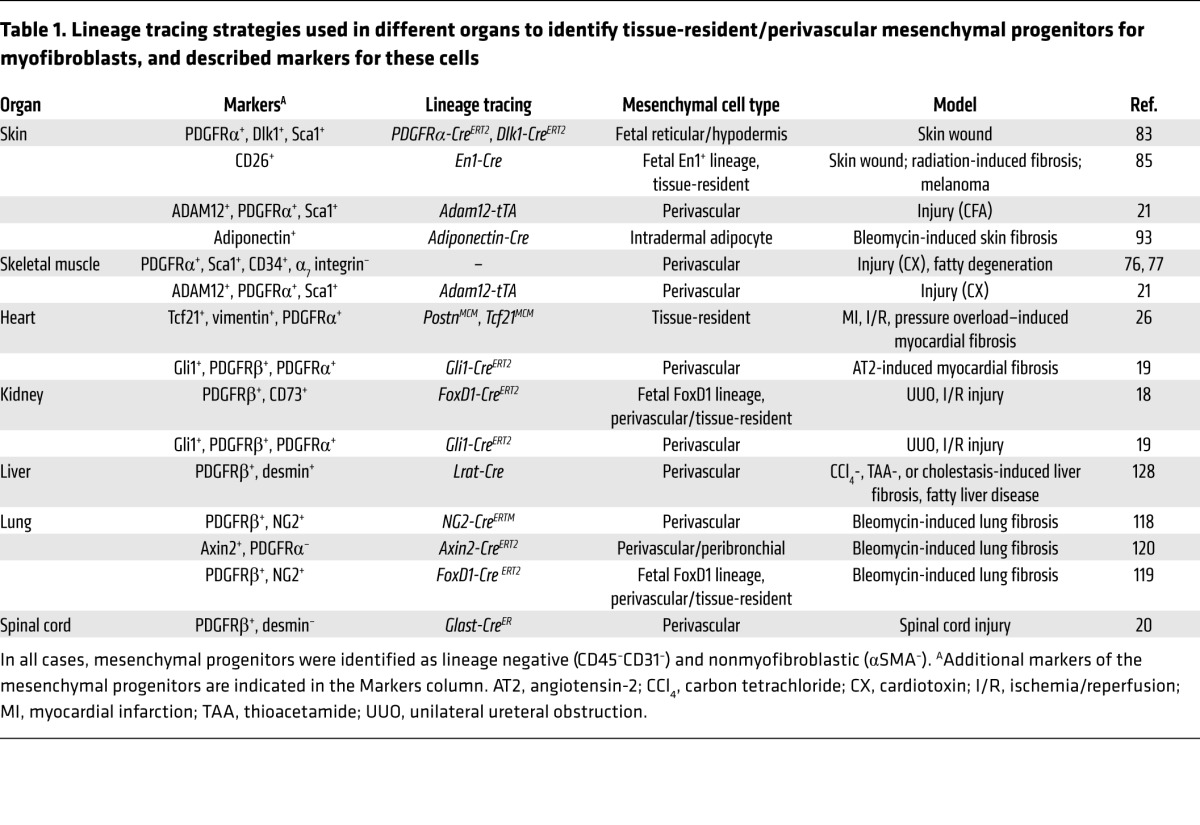
Activated mesenchymal cells such as myofibroblasts are not usually present in normal tissues, except in a few organs that require localized contractile force, such as the uterine submucosa, the intestinal lamina propria, and the lung septa. When present in normal organs, localization and numbers of myofibroblasts are strictly controlled, suggesting that proper regulation of these cells is required to maintain tissue homeostasis. In contrast, tissue damage induces massive development of αSMAmed myofibroblasts and other collagen-producing mesenchymal cells at the site of injury (Figure 2). Below, we discuss a number of reports investigating the cellular origin of injury-induced mesenchymal cells in different organs as well as the ontogeny of these lineages (summarized in Table 1). Most studies relied on expression of αSMA to identify myofibroblasts or upon collagen to identify collagen-producing cells. Strikingly, the two populations do not always overlap, suggesting that nonmyofibroblastic mesenchymal populations produce a substantial amount of collagen, or that production of collagen and expression of αSMA in the myofibroblastic lineage are variable over time (5, 21). A few functional studies further quantified the amount of matrix in the absence of specific mesenchymal types, providing compelling evidence of the role of these specific mesenchymal subsets in repair or fibrosis. Details are provided in the sections below.
Figure 2. The perivascular niche after injury.

Injury induces activation and differentiation of specific subsets of perivascular mesenchymal cells toward a myofibroblastic phenotype, which regulate scar tissue formation and immune cells recruitment/activity through production of ECM, chemokines and growth factors. Whether all perivascular mesenchymal cell types are similarly involved in this process remain unclear. Signals produced by damaged epithelial cells, endothelial cells, and inflammatory cells further contribute to this transition. Failure to terminate this repair program leads to fibrosis.
Table 1. Lineage tracing strategies used in different organs to identify tissue-resident/perivascular mesenchymal progenitors for myofibroblasts, and described markers for these cells.
Skin fibrosis
Mammalian skin is composed of the epidermis, which consists of a multilayered epithelium and associated hair follicles; the dermis, a connective tissue rich in collagen fibers; and a thin intradermal adipose layer. The dermal ECM is heterogeneous, harboring thin collagen fibers in the region closest to the epidermis, known as the papillary dermis, and dense collagen fibers in the lower layer close to the intradermal adipose tissue, known as the reticular dermis. The embryonic origin of dermal fibroblasts depends on their body location. The neural crest gives rise to the mesenchyme of the head, and the lateral plate mesoderm and dermomyotome generate ventral and dorsal body skin mesenchyme, respectively.
Using lineage tracing experiments in murine dorsal skin, Driskell et al. reported that further lineage restriction occurs a few days before birth, so that mesenchymal cells expressing BLIMP1 and IRIG give rise to the upper papillary dermis and CD26+ papillary fibroblasts after birth, while DLK1+Sca1+ mesenchymal cells generate the lower reticular dermis and dermal adipose tissue (83). In a full-thickness wound model, the DLK1+Sca1+ reticular lineage, which also expresses PDGFRα, generated the ECM-producing myofibroblasts that mediated the initial phase of wound repair (83) in a TGF-β–dependent process (84). More recently, lineage tracing of Engrailed-1–positive (EN1-positive) embryonic fibroblasts, which migrate during development from the somites to the dorsal dermis, indicated that these cells are major contributors to ECM production during wounding, radiation fibrosis, or skin cancer (85). These data are consistent with studies performed about 50 years ago showing that myofibroblasts in skin wounds derive not from circulating cells, but rather from tissue-resident mesenchymal cells (86, 87). Using a K14-CreERT2 mouse line, our group confirmed that keratinocytes are not involved in the generation of mesenchymal cells expanding in the ear skin following CFA-induced injury (32). We further showed that a subset of PDGFRα+ perivascular mesenchymal cells expressing ADAM12, a membrane-bound metalloprotease normally expressed during organ morphogenesis, generate a major fraction of myofibroblasts after CFA-induced injury (21). ADAM12+ cells in the ear originate from the cranial neural crest and uniformly express PDGFRα and Sca1, suggesting a developmental relationship with the lower reticular skin lineage. Interestingly, immunohistochemistry studies performed two decades ago suggested that subsets of perivascular mesenchymal cells migrate from their perivascular location and become myofibroblasts, synthesizing collagen in patients with excessive dermal scarring (88). Consistent with this hypothesis, increased numbers of PDGFRβ+ perivascular cells are found in patients with early systemic sclerosis, and expression of ADAM12 in the dermis, as well as serum levels of soluble ADAM12, is increased in scleroderma patients (89–92).
During skin fibrosis, a loss of intradermal adipocytes precedes expansion of fibrous tissue. In the bleomycin model for skin fibrosis, cell fate mapping studies using adiponectin-Cre transgenic mice showed that the adiponectin+ adipocyte lineage is capable of generating myofibroblasts (93). As expression of adiponectin is restricted to mature adipocytes, it will be interesting to determine whether perivascular adipocyte mesenchymal progenitors, which express PDGFRα, Sca, and CD34, are involved in the generation of myofibroblasts after injury. Consistent with a close interaction between adipose tissue and fibrous tissue, a loss of adipose tissue is observed in a variety of human fibrotic diseases, including systemic sclerosis, laminopathies, and lipodystrophies (94–96). Accordingly, injection of the adipose stromal vascular fraction is being tried out as antifibrotic therapy in scleroderma (97). In mice, stimulation of LTβR improves engraftment of adipose-derived stromal cells and partially reverses fibrosis, further reflecting the heterogeneity of the adipose vascular fraction and an essential crosstalk with lymphotoxin-expressing immune cells (98).
Skeletal muscle
Skeletal muscle has a remarkable ability to repair after injury. Regeneration of skeletal muscle is a highly orchestrated process involving muscle stem cells and other cell types (99). In muscular pathologies such as dystrophies and chronic injuries, continuous cycles of degeneration and regeneration result in uncontrolled expansion of fibrous/adipogenic tissue, which impairs muscle repair and function. Such fibrous tissue was suggested to originate from muscle stem cells exposed to environmental modifications associated with injury or aging (100, 101). In 2010, two groups reported that PDGFRα+Sca1+ mesenchymal cells of the skeletal muscle, localized in proximity to blood vessels, are major progenitors for fibrogenic and adipogenic cells (therefore termed fibro-adipogenic progenitors, or FAPs) when transplanted into mice with injured muscle, as well as in clonal assays in vitro (76, 77, 102). FAPs are abundant in the normal muscle and further expand after injury to facilitate myogenesis (76, 77), in a process dependent on type 2 innate signals such as IL-4 and IL-33 (103, 104). Consistent with an essential role for PDGFRα in FAPs, chronic activation of PDGFRα is sufficient to generate widespread organ fibrosis in mice (105), and conditional expression of a constitutively active mutant of PDGFRα in PDGFRα+ mesenchymal cells hinders the repair process and promotes muscle fibrosis (106). By generating reporter mice for ADAM12, our group showed that expression of ADAM12 identifies a distinct subset (<5%) of perivascular PDGFRα+Sca1+ mesenchymal cells after cardiotoxin-induced (CX-induced) muscle injury (21). Inducible, tetracycline transactivator–based cell fate mapping demonstrated that injury-induced ADAM12+ cells were progenitors to a major fraction of myofibroblasts accumulating following CX muscle injury, which were eliminated after healing (21). Consistent with a functional role in scarring, diphtheria toxin–mediated ablation of ADAM12+ cells was sufficient to decrease injury-induced collagen accumulation. Interestingly, ADAM12+ cells downregulated expression of type I collagen while acquiring expression of αSMA, suggesting that collagen production precede expression of αSMA during differentiation toward a myofibroblastic phenotype. Injury-induced ADAM12+ cells, which are perivascular, expressed markers of MSCs such as PDGFRα, CD44, and CD29. However, the progeny of ADAM12+ cells was restricted to fibrogenic cells after muscle injury or fatty degeneration, suggesting that either ADAM12+ cells are a subset of resident MSC-like cells with a restricted differentiation potential, or unidentified factors influence their fate. Consistent with a role in pathological scarring, transgenic overexpression of ADAM12 under control of the muscle creatine kinase promoter aggravates fibrosis, a phenotype further amplified in the dystrophic background of mdx mice (107).
The heart
Excessive deposition of ECM in interstitial and perivascular cardiac regions is common in diverse heart pathologies involving adverse remodeling of the myocardium, as observed after myocardial infarction (MI) or pressure overload stress. During development, heart fibroblasts are derived from epicardial progenitors expressing the transcription factor Tcf21 (108). A number of cell types, including BM-derived cells and endothelial cells, were suggested to contribute to heart disease by converting to a myofibroblastic fate. However, several experimental approaches using BM chimeras, parabiosis, and lineage tracing of BM-derived cells with LysM-Cre (109), Kit-Cre (110), or Vav-Cre showed no significant contribution of BM-derived cells to the injury-responsive fibroblast population after MI or pressure overload stress (25, 111). Similarly, fate mapping of endothelial cells using different endothelial Cre–expressing mouse lines, such as Cdh5-Cre, VE-cadherin-CreERT2, or Tie2-Cre mice, indicated that endothelial conversion to the mesenchymal type after heart injury is a rare event (25, 26, 111). Periostin (POSTN) was described as a marker for myofibroblasts that is expressed in adult tissues only after injury (112). By generating a mouse model where tamoxifen-regulated mutated estrogen receptor–Cre (MCM) was inserted into the Postn locus, as well as other Cre lines, Kanisicack and colleagues showed that nearly all of the periostin-labeled myofibroblasts developing in the heart subjected to pressure overload, MI injury, or neuroendocrine stimulation arise from tissue-resident mesenchymal cells of the Tcf21 lineage. Using an inducible Cre-dependent strategy for ablating cells in vivo (PostnMCM;Rosa26fl-DTA mice), these cells were shown to be required for healing and scar formation after MI injury (26). These data support the hypothesis that resident mesenchymal cells derived from the epicardium during embryonic development are the major source of disease-associated myofibroblasts after cardiac injury.
Kidney
While the above study did not address the location of these progenitors with respect to the vasculature, Kramann and colleagues used expression of Gli1, a transcription factor involved in the Hedgehog signaling pathway, to specifically mark perivascular mesenchymal cells and adventitial cells in several organs, including the heart and kidney. Fate mapping of Gli1+ cells, which had characteristics of MSCs, indicated that these cells generate a majority of αSMA+ myofibroblasts in models for heart, renal, lung, and liver fibrosis (19). Genetic depletion of the Gli1+ lineage using a heritable cell-specific expression of the human diphtheria toxin receptor (Gli1-CreER;iDTR) resulted in reduced fibrosis following unilateral ureteral obstruction (UUO) in the kidney and ascending aortic constriction in the heart. The Gli1+ population does not seem to be restricted to myofibroblasts, as depletion of Gli1+ cells leads to capillary rarefaction and increased tubular injury in the kidney, an effect attributed to loss of normal pericytes (113). BM transplantation and parabiosis experiments confirmed that Gli1+ cells are tissue-resident, not circulating cells. These results are consistent with initial observations that a majority of injury-induced myofibroblasts developing after UUO are derived from resident mesenchymal cells (114). However, whether all tissue-resident mesenchymal cells are equal in this process remains unclear, because of the lack of truly specific markers in the kidney mesenchyme (115). Fate mapping of embryonic mesenchymal cells expressing FoxD1, a transcription factor expressed during nephrogenesis, indicated that FoxD1+ cells give rise to PDGFRβ+ pericytes, fibroblasts, and mesangial cells of the kidney, which in turn generate αSMA+ myofibroblasts in renal fibrosis (18). These results suggest that embryonic seeded kidney mesenchyme plays an essential role in the generation of collagen-producing fibroblasts in kidney disease. In addition to mesenchymal cells, tubular epithelial cells were suggested to generate myofibroblasts through epithelial-mesenchymal transition (EMT) in kidney disease (116). However, the use of various Cre lines to perform lineage tracing of different subsets of kidney epithelial cells showed only marginal progeny of epithelial cells in the pool of αSMA+ myofibroblasts after kidney injury (18, 117). Rather, EMT occurring in tubular epithelial cells following injury contributes to fibrosis mainly by altering the intracellular metabolism of epithelial cells and inducing cell cycle arrest (117).
Lung
Using inducible lineage tracing of alveolar epithelial cells of the lung with an Sftpc-CreERT2 knockin allele, Rock and colleagues found no evidence for EMT in the bleomycin model for pulmonary fibrosis. Rather, multiple mesenchymal populations were activated and proliferated after injury, including NG2+ cells (presumably pericytes) and PDGFRα+ mesenchymal cells (118). Similar results were obtained by lineage tracing of fetal FoxD1+ progenitor-derived mesenchymal cells in the adult lung (119). Recently, lineage tracing analysis of different mesenchymal subsets of the lung using Axin2-CreERT2, Pdgfra-CreERT2, and Wnt2-CreERT2 lines showed that Axin2+PDGFRα– mesenchymal cells, primarily found surrounding blood vessels or airways, are major contributors to the pool of myofibroblasts developing after bleomycin-induced pulmonary fibrosis (120). In pulmonary, renal, and liver fibrosis, depletion of αv integrin from PDGFRβ lineage–derived mesenchymal cells (using Pdgfrb-Cre × Itgavfl/fl mice) that were identified as myofibroblasts after injury improved fibrosis (121).
Liver
In the liver, lineage tracing studies have excluded the contribution of hepatocytes and cholangiocytes to the pool of myofibroblasts developing during fibrosis (122–124). The use of collagen-driven Cre or Wt1-Cre allowed the identification of the mesenchymal compartment as the major source for myofibroblasts after injury (125). Initial attempts to specifically trace hepatic stellate cells (HSCs), a subset of perivascular mesenchymal cells suspected to become major producers of collagen after liver injury (126), were based on Cre expression under the GFAP promoter, as GFAP is expressed by HSCs at steady state. However, lineage tracing studies using the human or murine GFAP promoter (hGFAP-Cre or mGfap-Cre mice) showed that bile ducts and cholangiocytes are marked much more efficiently than HSCs in this model (127, 128), thereby limiting interpretation. A novel lineage tracing strategy targeting Cre expression to Lrat (lecithin-retinol acyltransferase), also expressed by HSCs, showed that Lrat+ cells gave rise to up to 95% of myofibroblasts in multiple models of liver injury (toxic, cholestatic, and fatty liver disease), confirming that Lrat+ HSCs are progenitors for myofibroblasts after injury.
Spinal cord
In a model of spinal cord injury, fate mapping studies using Glast-CreER mice confirmed that mesenchymal cells surrounding the spinal cord blood vessels are a major source for injury-induced myofibroblasts after injury (20).
Summary
Recent research has provided compelling evidence that specific subsets of tissue-resident mesenchymal cells, rather than epithelial cells, endothelial cells, or BM-derived cells, are the major source for injury-induced matrix-producing fibroblasts and myofibroblasts in multiple organs. Interestingly, the majority of these mesenchymal progenitors are localized in proximity to blood vessels, and share several characteristics with pericytes and adventitial mesenchymal cells. These findings highlight a predominant role for the perivascular niche in the scarring process. They also raise a number of challenging questions. It is still unclear whether all pericyte/adventitial mesenchymal cells have a similar ability to react to injury and acquire myofibroblastic features, or whether specific subsets are endowed with a profibrotic fate and function. As pericytes and adventitial cells are essential for vascular stability and function, and overall tissue homeostasis, the second hypothesis seems more likely. It is also possible that myofibroblast progenitors belong to a distinct subset of mesenchymal cells also localized in the perivascular niche, related to MSCs. Even though MSCs can be generated in vitro from pericytes or adventitial cells, their precise identity and function in vivo remain unclear and will require further investigation.
While these findings hold great promise for potential novel therapeutic avenues in fibrotic diseases, a number of pitfalls must be considered. Notably, pericytes, adventitial cells, and other perivascular mesenchymal subsets such as MSCs are present in all organs and have essential vascular, immunomodulatory, and regenerative roles. Therefore, a major goal is to specifically target profibrotic subsets while preserving those essential for homeostasis and repair. Also, a similar subset has most likely both beneficial and pathological roles in repair, depending on the time after injury and, potentially, the type of injury. Clearly, the discovery of more specific markers, both in murine models and in human pathology, will be instrumental in answering these questions. Targeting profibrotic mesenchymal subsets might also be beneficial in regenerative medicine, as decreasing a fibrotic perivascular microenvironment most likely alters the way stem cells react to injury. To reach these objectives, the functional heterogeneity of perivascular mesenchymal cells, fibroblasts, and myofibroblasts, together with their lineage relationship and crosstalk with immune cells, needs to be better understood.
Acknowledgments
The laboratory of L. Peduto has received funding from Institut Pasteur, Institut National de la Santé et de la Recherche Médicale (INSERM), and the European Research Council (Consolidator Grant 648428-PERIF).
Version 1. 01/02/2018
Print issue publication
Footnotes
Conflict of interest: L. Peduto is co-inventor of the patent (no. 9777276) “The use of ADAM12 inhibitors to control inflammation-induced fibrosis” at Institut Pasteur.
Reference information: J Clin Invest. 2018;128(1):54–63.https://doi.org/10.1172/JCI93558.
References
- 1.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darby I, Skalli O, Gabbiani G. α-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990;63(1):21–29. [PubMed] [Google Scholar]
- 4.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 5.Sun KH, Chang Y, Reed NI, Sheppard D. α-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGF-β activation or collagen production across multiple models of organ fibrosis. Am J Physiol Lung Cell Mol Physiol. 2016;310(9):L824–L836. doi: 10.1152/ajplung.00350.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skalli O, et al. Myofibroblasts from diverse pathologic settings are heterogeneous in their content of actin isoforms and intermediate filament proteins. Lab Invest. 1989;60(2):275–285. [PubMed] [Google Scholar]
- 7.Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister JJ. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol Biol Cell. 2004;15(9):4310–4320. doi: 10.1091/mbc.E04-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol. 2006;85(3–4):175–181. doi: 10.1016/j.ejcb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. 2014;32:51–82. doi: 10.1146/annurev-immunol-032713-120257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8(5):a021873. doi: 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta. 2013;1832(7):1049–1060. doi: 10.1016/j.bbadis.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gieseck RL, 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair fibrosis. Nat Rev Immunol. doi: 10.1038/nri.2017. [published online ahead of print August 30, 2017]. https://doi.org/10.1038/nri.2017.90. [DOI] [PubMed] [Google Scholar]
- 13.Crisan M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. 2010;38(7):548–556. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Jensen P, Dymecki SM. Essentials of recombinase-based genetic fate mapping in mice. Methods Mol Biol. 2014;1092:437–454. doi: 10.1007/978-1-60327-292-6_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphreys BD, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramann R, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16(1):51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science. 2011;333(6039):238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 21.Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18(8):1262–1270. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- 22.Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Heart Circ Physiol. 2013;305(9):H1363–H1372. doi: 10.1152/ajpheart.00395.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrera-Molina R, et al. Thy-1-interacting molecules and cellular signaling in cis and trans. Int Rev Cell Mol Biol. 2013;305:163–216. doi: 10.1016/B978-0-12-407695-2.00004-4. [DOI] [PubMed] [Google Scholar]
- 24.Österreicher CH, et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A. 2011;108(1):308–313. doi: 10.1073/pnas.1017547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore-Morris T, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124(7):2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanisicak O, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2(7):e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsonage G, et al. Global gene expression profiles in fibroblasts from synovial, skin and lymphoid tissue reveals distinct cytokine and chemokine expression patterns. Thromb Haemost. 2003;90(4):688–697. doi: 10.1160/TH03-04-0208. [DOI] [PubMed] [Google Scholar]
- 29.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9(9):618–629. doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bénézech C, et al. Ontogeny of stromal organizer cells during lymph node development. J Immunol. 2010;184(8):4521–4530. doi: 10.4049/jimmunol.0903113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3(4):292–303. doi: 10.1038/nri1054. [DOI] [PubMed] [Google Scholar]
- 32.Peduto L, et al. Inflammation recapitulates the ontogeny of lymphoid stromal cells. J Immunol. 2009;182(9):5789–5799. doi: 10.4049/jimmunol.0803974. [DOI] [PubMed] [Google Scholar]
- 33.Buckley CD, Barone F, Nayar S, Bénézech C, Caamaño J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol. 2015;33:715–745. doi: 10.1146/annurev-immunol-032713-120252. [DOI] [PubMed] [Google Scholar]
- 34.Nazari B, et al. Altered dermal fibroblasts in systemic sclerosis display podoplanin and CD90. Am J Pathol. 2016;186(10):2650–2664. doi: 10.1016/j.ajpath.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6(3):205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 36.Takemura S, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167(2):1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 37.Ruddle NH. Lymphoid neo-organogenesis: lymphotoxin’s role in inflammation and development. Immunol Res. 1999;19(2-3):119–125. doi: 10.1007/BF02786481. [DOI] [PubMed] [Google Scholar]
- 38.Stzepourginski I, et al. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc Natl Acad Sci U S A. 2017;114(4):E506–E513. doi: 10.1073/pnas.1620059114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vicente-Suarez I, et al. Unique lamina propria stromal cells imprint the functional phenotype of mucosal dendritic cells. Mucosal Immunol. 2015;8(1):141–151. doi: 10.1038/mi.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potente M, Mäkinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. 2017;18(8):477–494. doi: 10.1038/nrm.2017.36. [DOI] [PubMed] [Google Scholar]
- 41.Rouget C. Mémoire sur le développement, la structure et les propriétés physiologiques des capillaires sanguins et lymphatiques. Arch Physiol Norm et Path. 1873;5:603–663. [Google Scholar]
- 42.Sims DE. The pericyte — a review. Tissue Cell. 1986;18(2):153–174. doi: 10.1016/0040-8166(86)90026-1. [DOI] [PubMed] [Google Scholar]
- 43.Murfee WL, Skalak TC, Peirce SM. Differential arterial/venous expression of NG2 proteoglycan in perivascular cells along microvessels: identifying a venule-specific phenotype. Microcirculation. 2005;12(2):151–160. doi: 10.1080/10739680590904955. [DOI] [PubMed] [Google Scholar]
- 44.Crisan M, Corselli M, Chen WC, Peault B. Perivascular cells for regenerative medicine. J Cell Mol Med. 2012;16(12):2851–2860. doi: 10.1111/j.1582-4934.2012.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58(9):1094–1103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- 47.Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 48.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hall CN, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508(7494):55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lange S, et al. Brain pericyte plasticity as a potential drug target in CNS repair. Drug Discov Today. 2013;18(9-10):456–463. doi: 10.1016/j.drudis.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 53.Schrimpf C, Teebken OE, Wilhelmi M, Duffield JS. The role of pericyte detachment in vascular rarefaction. J Vasc Res. 2014;51(4):247–258. doi: 10.1159/000365149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stark K, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol. 2013;14(1):41–51. doi: 10.1038/ni.2477. [DOI] [PubMed] [Google Scholar]
- 55.Proebstl D, et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209(6):1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leaf IA, et al. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J Clin Invest. 2017;127(1):321–334. doi: 10.1172/JCI87532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olson LE, Soriano P. PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev Cell. 2011;20(6):815–826. doi: 10.1016/j.devcel.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stenmark KR, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47. doi: 10.1146/annurev-physiol-030212-183802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203(9):2073–2083. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tieu BC, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119(12):3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gräbner R, et al. Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE–/– mice. J Exp Med. 2009;206(1):233–248. doi: 10.1084/jem.20080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 63.Braverman IM. The cutaneous microcirculation. J Investig Dermatol Symp Proc. 2000;5(1):3–9. doi: 10.1046/j.1087-0024.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 64.Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–247. doi: 10.1097/00007890-196803000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Sacchetti B, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131(2):324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 66.Morikawa S, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. 2009;206(11):2483–2496. doi: 10.1084/jem.20091046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Méndez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125(17):2621–2629. doi: 10.1182/blood-2014-09-570192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crisan M, Chen CW, Corselli M, Andriolo G, Lazzari L, Péault B. Perivascular multipotent progenitor cells in human organs. Ann N Y Acad Sci. 2009;1176:118–123. doi: 10.1111/j.1749-6632.2009.04967.x. [DOI] [PubMed] [Google Scholar]
- 70.Corselli M, Chen CW, Sun B, Yap S, Rubin JP, Péault B. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev. 2012;21(8):1299–1308. doi: 10.1089/scd.2011.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bianco P, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19(1):35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen CW, et al. Human pericytes for ischemic heart repair. Stem Cells. 2013;31(2):305–316. doi: 10.1002/stem.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hayes M, Curley G, Laffey JG. Mesenchymal stem cells — a promising therapy for Acute Respiratory Distress Syndrome. F1000 Med Rep. 2012;4:2. doi: 10.3410/M4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110(10):3499–3506. doi: 10.1182/blood-2007-02-069716. [DOI] [PubMed] [Google Scholar]
- 75.Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med. 2013;45:e54. doi: 10.1038/emm.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Joe AW, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12(2):153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12(2):143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 78.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol. 2012;3:344. doi: 10.3389/fphys.2012.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kent G, Gay S, Inouye T, Bahu R, Minick OT, Popper H. Vitamin A-containing lipocytes and formation of type III collagen in liver injury. Proc Natl Acad Sci U S A. 1976;73(10):3719–3722. doi: 10.1073/pnas.73.10.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McGee JO, Patrick RS. The role of perisinusoidal cells in hepatic fibrogenesis. An electron microscopic study of acute carbon tetrachloride liver injury. Lab Invest. 1972;26(4):429–440. [PubMed] [Google Scholar]
- 82.Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. doi: 10.1016/j.addr.2017.05. [published online ahead of print May 12, 2017]. https://doi.org/10.1016/j.addr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Driskell RR, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504(7479):277–281. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lichtenberger BM, Mastrogiannaki M, Watt FM. Epidermal β-catenin activation remodels the dermis via paracrine signalling to distinct fibroblast lineages. Nat Commun. 2016;7:10537. doi: 10.1038/ncomms10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rinkevich Y, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348(6232):aaa2151. doi: 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grillo HC. Origin of fibroblasts in wound healing. An autoradiographic study of inhibition of cellular proliferation by local x-irradiation. Ann Surg. 1963;157:453–467. doi: 10.1097/00000658-196303000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ross R, Everett NB, Tyler R. Wound healing and collagen formation. VI. The origin of the wound fibroblast studied in parabiosis. J Cell Biol. 1970;44(3):645–654. doi: 10.1083/jcb.44.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sundberg C, Ivarsson M, Gerdin B, Rubin K. Pericytes as collagen-producing cells in excessive dermal scarring. Lab Invest. 1996;74(2):452–466. [PubMed] [Google Scholar]
- 89.Shi-Wen X, et al. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol. 2007;26(8):625–632. doi: 10.1016/j.matbio.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 90.Cipriani P, et al. Perivascular cells in diffuse cutaneous systemic sclerosis overexpress activated ADAM12 and are involved in myofibroblast transdifferentiation and development of fibrosis. J Rheumatol. 2016;43(7):1340–1349. doi: 10.3899/jrheum.150996. [DOI] [PubMed] [Google Scholar]
- 91.Soudais C, et al. Stable and functional lymphoid reconstitution of common cytokine receptor γ chain deficient mice by retroviral-mediated gene transfer. Blood. 2000;95(10):3071–3077. [PubMed] [Google Scholar]
- 92.Taniguchi T, et al. Serum levels of ADAM12-S: possible association with the initiation and progression of dermal fibrosis and interstitial lung disease in patients with systemic sclerosis. J Eur Acad Dermatol Venereol. 2013;27(6):747–753. doi: 10.1111/j.1468-3083.2012.04558.x. [DOI] [PubMed] [Google Scholar]
- 93.Marangoni RG, et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. 2015;67(4):1062–1073. doi: 10.1002/art.38990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Béréziat V, et al. LMNA mutations induce a non-inflammatory fibrosis and a brown fat-like dystrophy of enlarged cervical adipose tissue. Am J Pathol. 2011;179(5):2443–2453. doi: 10.1016/j.ajpath.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009;1791(6):507–513. doi: 10.1016/j.bbalip.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Allanore Y, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1:15002. doi: 10.1038/nrdp.2015.2. [DOI] [PubMed] [Google Scholar]
- 97.Scuderi N, et al. Human adipose-derived stromal cells for cell-based therapies in the treatment of systemic sclerosis. Cell Transplant. 2013;22(5):779–795. doi: 10.3727/096368912X639017. [DOI] [PubMed] [Google Scholar]
- 98.Chia JJ, et al. Dendritic cells maintain dermal adipose-derived stromal cells in skin fibrosis. J Clin Invest. 2016;126(11):4331–4345. doi: 10.1172/JCI85740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93(1):23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brack AS, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317(5839):807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 101.Li Y, et al. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am J Pathol. 2004;164(3):1007–1019. doi: 10.1016/S0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Uezumi A, et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci. 2011;124(pt 21):3654–3664. doi: 10.1242/jcs.086629. [DOI] [PubMed] [Google Scholar]
- 103.Heredia JE, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153(2):376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kuswanto W, et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity. 2016;44(2):355–367. doi: 10.1016/j.immuni.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Olson LE, Soriano P. Increased PDGFRα activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell. 2009;16(2):303–313. doi: 10.1016/j.devcel.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ieronimakis N, Hays A, Prasad A, Janebodin K, Duffield JS, Reyes M. PDGFRα signalling promotes fibrogenic responses in collagen-producing cells in Duchenne muscular dystrophy. J Pathol. 2016;240(4):410–424. doi: 10.1002/path.4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jørgensen LH, Jensen CH, Wewer UM, Schrøder HD. Transgenic overexpression of ADAM12 suppresses muscle regeneration and aggravates dystrophy in aged mdx mice. Am J Pathol. 2007;171(5):1599–1607. doi: 10.2353/ajpath.2007.070435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Acharya A, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139(12):2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi: 10.1023/A:1008942828960. [DOI] [PubMed] [Google Scholar]
- 110.van Berlo JH, et al. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509(7500):337–341. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ali SR, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115(7):625–635. doi: 10.1161/CIRCRESAHA.115.303794. [DOI] [PubMed] [Google Scholar]
- 112.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105(10):934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kramann R, Wongboonsin J, Chang-Panesso M, Machado FG, Humphreys BD. Gli1(+) pericyte loss induces capillary rarefaction and proximal tubular injury. J Am Soc Nephrol. 2017;28(3):776–784. doi: 10.1681/ASN.2016030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.LeBleu VS, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124(6):2299–2306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. 2008;13:6991–6998. doi: 10.2741/3204. [DOI] [PubMed] [Google Scholar]
- 117.Lovisa S, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21(9):998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rock JR, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hung C, et al. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(7):820–830. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zepp JA, et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell. 2017;170(6):1134–1148.e10. doi: 10.1016/j.cell.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Henderson NC, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19(12):1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Scholten D, et al. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology. 2010;139(3):987–998. doi: 10.1053/j.gastro.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Taura K, et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51(3):1027–1036. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chu AS, et al. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. 2011;53(5):1685–1695. doi: 10.1002/hep.24206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kisseleva T, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(24):9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 127.Yang L, et al. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26(8):2104–2113. doi: 10.1634/stemcells.2008-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mederacke I, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]