

Abstract
Genetic investigations of fibrotic diseases, including those of late onset, often yield unanticipated insights into disease pathogenesis. This Review focuses on pathways underlying lung fibrosis that are generalizable to other organs. Herein, we discuss genetic variants subdivided into those that shorten telomeres, activate the DNA damage response, change resident protein expression or function, or affect organelle activity. Genetic studies provide a window into the downstream cascade of maladaptive responses and pathways that lead to tissue fibrosis. In addition, these studies reveal interactions between genetic variants, environmental factors, and age that influence the phenotypic spectrum of disease. The discovery of forces counterbalancing inherited risk alleles identifies potential therapeutic targets, thus providing hope for future prevention or reversal of fibrosis.
End-stage organ fibrosis generates formidable social, economic and personal costs. In the modern era, organ fibrosis is an important cause of patient morbidity and mortality. A better understanding of disease pathogenesis may lead to more effective therapies aimed at preventing or reversing the scarring process.
Diseases characterized by progressive fibrosis, especially those of late onset, are complex and difficult to study. Many represent a heterogeneous collection of disorders with imprecise classification criteria and overlapping clinical presentations. Environmental triggers of disease may be obscure or difficult to quantify. In the absence of gene-environmental studies (such as family-based monozygotic and dizygotic twin studies), estimates of genetic and environmental contributions to disease phenotypes and the interactions between the two are difficult to partition and quantify. Most patients with adult-onset organ fibrosis do not have an affected family member. For these sporadic patients, weaker genetic risk alleles contribute to the pathogenesis of fibrosis. Less commonly, patients have similarly affected family members and share more highly penetrant genetic risk alleles. However, even in family studies, locus heterogeneity, reduced penetrance, and lack of parental samples present obstacles to gene discovery.
Advances in genetics have increased the ease and decreased the cost of identifying genetic differences that contribute to disease susceptibility. High-throughput, “next-generation” technologies make it possible to sequence entire human genomes, allowing for the identification of many new genetic variants that contribute to organ fibrosis. Genetic variants are subclassified by their frequency in the general population. Variants that are found with an allele frequency greater than 1% are classified as “common.” Most common variants linked to fibrotic diseases by genome-wide association studies (GWAS) are located in noncoding regions of the genome. Genetic variants that occur at lower frequencies are termed “rare.” Although rare variants are individually uncommon, they represent the larger class of genetic variants. Over 99% of variants have a frequency of less than 1%, and over half have been found only once in a large population database (1). Tools that compare evolutionarily conserved sequences and predict how changes in protein coding sequences affect folding or function are used to predict pathogenicity. While an understanding of the functional effect of a genetic variant may be initially elusive, these studies yield rich information regarding the inherited pathophysiology of fibrosis.
This Review focuses on biologic pathways implicated by human genetic studies of pulmonary fibrosis. The prototypic lung disease characterized by progressive fibrosis is idiopathic pulmonary fibrosis (IPF). This disorder affects older adults and results in the replacement of lung parenchyma with a dense extracellular matrix (2). The mechanisms responsible for the development of fibrosis in the lung can be generalized to diseases that lead to fibrosis of other organs (Figure 1). Fibrosis involves many different cell types and leads to a cascade of maladaptive responses that ultimately results in activated myofibroblasts, deposition of extracellular matrix proteins, aberrant remodeling, and organ failure (Figure 2). Thus, genetic studies provide an entry point for investigating complex downstream mechanisms that lead to tissue fibrosis.
Figure 1. Genetic variants define an inherited susceptibility to pulmonary fibrosis and to different manifestations of organ fibrosis.

Genes linked to an inherited risk of pulmonary fibrosis can be broadly classified into those that lead to a DNA damage response, those that are expressed in lung epithelium, and those that are expressed in lamellar bodies — or organelles crucial for type II alveolar epithelial cell function. Examples of other manifestations of organ fibrosis using this same broad classification scheme of variants affecting DNA, protein, and organelle function are indicated to the right. Genetic disorders characterized by the involvement of multiple different organs are listed with each panel.
Figure 2. Cascade of maladaptive responses originating from inherited genetic variants and leading to tissue fibrosis.

The process of fibrosis involves many different cell types, ultimately resulting in activation of myofibroblasts, deposition of extracellular matrix proteins, aberrant remodeling, and organ failure. Regardless of underlying pathogenic variant, fibrosis results from activation of a number of different downstream maladaptive responses. These involve both cell-autonomous changes within cells directly affected by the mutation and nonautonomous changes in cells affected by perturbations in cell signaling or the extracellular milieu. Even if only a restricted population of cells is affected by the pathogenic variant(s), its effects may spill over into overlapping and interacting mechanisms of cell injury, inflammation, and fibrosis.
Genetic variants associated with lung fibrosis
Telomere-shortening or DNA damage response–activating genetic variants.
Genetic alterations that lead to shortened telomere lengths or the activation of the DNA damage response can result in fibrosis. Family-based studies first demonstrated the unanticipated link between pulmonary fibrosis and telomere shortening. Subsequent studies have affirmed the central role of this mechanism in a wide spectrum of lung diseases. Overall, rare pathogenic DNA variants in telomere-related genes are found in about one-fourth of familial (3) and one-tenth of sporadic IPF patients (4). Fibrosis of other organs, especially the liver (cirrhosis) and bone marrow (myelofibrosis), may be seen in isolation or in conjunction with pulmonary fibrosis in patients with telomere-related gene mutations (5–9).
Telomeres are the specialized ends of chromosomes composed of stretches of repetitive DNA sequences (5′-TTAGGG-3′ in vertebrates). Standard DNA polymerases cannot completely replicate chromosomes to their distal ends, a phenomenon known as the end replication problem. Telomerase, composed of a catalytic protein component (encoded in the human by the TERT gene) and an RNA template (TERC), solves the end replication problem by directly adding telomeric DNA repeats to the ends of the chromosomes (10, 11). Most cells do not express telomerase, or they express it at levels so low as to be undetectable. In contrast, cancer cells and germ cells express TERT at high levels, leading to telomere elongation and cell immortality (12, 13). Cells lose up to several hundred base pairs of telomeric DNA with each cycle of replication (14). If telomerase expression is limited, after multiple rounds of cell division one or more of the telomere ends becomes sufficiently short or dysfunctional to trigger a DNA damage response (15–17).
The gene defects in telomerase (TERT, TERC) were first described in kindreds in whom pulmonary fibrosis segregated in an autosomal dominant pattern with incomplete penetrance (18, 19). More recently, rare variants in RTEL1, which encodes a helicase that can unwind the G-quadruplex and T-loop secondary structures at the telomere end, and PARN, which encodes for an enzyme that removes oligo(A) tails from precursor RNAs including TERC (20), have been discovered in familial pulmonary fibrosis kindreds (3, 21, 22). Pathogenic rare variants in these genes are highly represented in familial and sporadic cohorts, as they are found above genome-wide significance thresholds (3, 4). Rare, likely damaging variants in TERT, RTEL1, and PARN occur with large effect sizes (odds ratio 23–97) (4). Case reports have implicated other genes in the telomere-related pathway. These include genes that affect the biogenesis of telomerase (DKC1, NAF1) or the integrity of the telomere end (TINF2) (23–26). All pathogenic rare variants lead to alterations in protein function, as measured by decreased telomerase catalytic activity (18, 19, 27), decreased telomerase processivity (28), increased aberrant T-circle formations (21), and short telomere lengths (3, 18, 19, 21, 22, 23–27).
Whereas patients with adult-onset pulmonary fibrosis are heterozygous for pathogenic variants, some pediatric patients with dyskeratosis congenita or Hoyeraal-Hreidarsson syndrome have been described with biallelic mutations in TERT, PARN, and RTEL1 (20, 29–32). This 2-fold genetic burden leads to presentations of a characteristic triad of oral leukoplakia, skin hyperpigmentation, and nail dystrophy. These patients also have bone marrow failure, short stature, intrauterine growth retardation, developmental delay, cerebellar hypoplasia, dental caries, osteoporosis, esophageal strictures, enterocolitis, liver dysfunction or cirrhosis, immunodeficiency, and premature hair graying (33). Individuals with these syndromes reflect a phenotypic extreme, with more severe phenotypes affecting multiple organ systems and manifesting at a young age (Figure 3).
Figure 3. Distribution of phenotypic variation implicates multiple different alleles and roles of environment and aging.
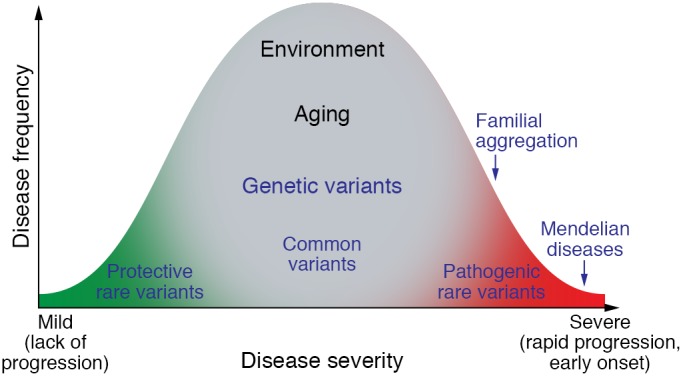
Extreme phenotypes, such as those that are seen in patients with Mendelian disorders, occur with an earlier onset and demonstrate more rapid progression. Individuals with familial aggregation of fibrosis phenotypes show not only enrichment of pathogenic rare variants, but also effects from common variants. Environmental factors influence the spectrum of phenotypes and include direct physical or cellular injury, infection, inflammation, and nutritional effects. Aging likely influences fibrosis through genomic-independent and -dependent mechanisms. Protective genetic variants are uncommonly identified but provide information regarding forces that balance disease susceptibility factors.
In contrast, familial pulmonary fibrosis typically presents with lung fibrosis between 40 and 60 years of age (34). In these families, incomplete penetrance of pulmonary fibrosis is the rule, although a personal or family history of the less severe short telomere phenotypes (macrocytosis, thrombocytopenia, premature graying of hair, liver disease) is frequently observed (34–36). In some families, older generations demonstrate adult-onset symptoms and younger generations display dyskeratosis congenita phenotypes. The onset of more severe phenotypes occurring at an earlier age in affected individuals in subsequent generations (genetic anticipation) is due to progressive telomere shortening (37).
Telomere shortening is a mechanism that has relevance to many different lung diseases. Interstitial lung diseases (ILD) define a very large and heterogeneous collection of over 100 separate lung diseases presenting with similar clinical, physiologic, and radiographic characteristics. The relevance of a telomere-centric mechanism underlying a wide spectrum of ILDs is illustrated by the finding that common variants near genes with telomere-related function (TERT, TERC, OBFC1) occur above genome-wide significance in patients with various fibrotic idiopathic interstitial pneumonias (38). Approximately one-half of all patients with a heterozygous rare variant in one of four genes (TERT, TERC, PARN, RTEL1) have a non-IPF diagnosis, but regardless of ILD diagnosis, rates of progression, lung function decline, and survival characteristics are similar (39). An excess of rare variants in the telomere genes has been reported in patients with rheumatoid arthritis–associated ILD, suggesting that telomere-related genetic susceptibility may promote this extra-articular fibrotic manifestation (40). Notably, deleterious mutations in TERT have been found in 1% of smokers with severe emphysema (41).
Telomere length, measured from genomic DNA isolated from blood leukocytes, is a biomarker that predicts survival characteristics of sporadic IPF patients (42). Only approximately 10% of sporadic IPF patients carry a rare qualifying variant in a telomere-related gene (4), yet a much higher percentage (25%–50%) have age-adjusted telomere lengths below the 10th percentile (43, 44). Shorter age-adjusted telomere lengths correlate with reduced overall and lung transplant–free survival. These associations are independent of usual clinical predictors. As has been seen in IPF, shorter telomere lengths predict worse survival for patients with chronic hypersensitivity pneumonitis (another specific type of ILD) after adjustment for age, sex, and baseline pulmonary function (45).
Proper telomere capping requires a group of proteins, collectively termed the shelterin complex, that prevent the telomere end from activating the DNA damage response (46). Uncapped telomeres activate signaling cascades that lead to cell cycle arrest and cell senescence (17, 47–49). While the initial studies of TERT-KO mice did not show an increase in susceptibility to bleomycin-induced lung fibrosis (50, 51), a more recent study demonstrates that short telomere lengths synergize with bleomycin to induce lung fibrosis in telomerase-deficient mice (52). Deletion of the shelterin protein TRF1 (telomeric repeat–binding factor 1) leads to an immediate and persistent DNA damage response at chromosome ends, cellular senescence, and proliferative defects (53, 54). Acute conditional deletion of TRF1 from epithelial type II alveolar cells leads to an influx of inflammatory cells (macrophages, polymorphonuclear leukocytes, lymphocytes) and collagen deposition in the absence of telomere shortening (52), suggesting that DNA damage, and not telomere shortening, is the key driver of lung fibrosis in this model. After 3 to 9 months, deletion of TRF1 from type II alveolar epithelial cells (AECs) results in epithelial cell turnover, shorter telomere lengths, and lung remodeling characterized by collagen deposition and accumulation of senescence-associated β-galactosidase–positive cells (55). The development of pulmonary fibrosis in older TRF1-deficient mice apparently occurs spontaneously and without a precipitating injury, such as exposure to cigarette smoke or bleomycin, suggesting that the genetic alteration is sufficient to drive lung fibrosis in older animals.
Other rare monogenic diseases that activate the DNA damage response also result in organ fibrosis. RIDDLE syndrome is an autosomal recessive disorder due to mutations in RNF168, which encodes an E3 ubiquitin ligase that mediates double-stranded DNA break repair. RNF168 promotes ubiquitination of proteins that mediates accumulation of repair factors at the site of DNA double-stranded breaks (56). This disorder is characterized by radiation sensitivity and adult-onset pulmonary fibrosis accompanied by immunodeficiency, learning difficulties, dysmorphic features, and short stature (57). More recently, RNF168 has been implicated in shelterin TRF2–mediated protection of telomere ends (58). Fanconi anemia protein 1 (FAN1) is recruited to DNA intrastrand cross-links and interacts with other proteins to repair DNA breaks. Mutations in this gene cause karyomegalic interstitial nephritis, a rare form of progressive tubulointerstitial fibrosis that culminates in end-stage renal disease (59).
Genetic variants affecting resident protein expression or function.
Certain alterations of intrinsic proteins lead to cellular damage and organ fibrosis. Cystic fibrosis, the autosomal recessive disease caused by mutations in the gene encoding the cystic fibrosis transmembrane regulator (CFTR), is the archetypal example. Alterations of airway mucus due to CFTR defects trigger a cascade of pathogenic mechanisms that ultimately lead to severe bronchiectasis, lung fibrosis, and respiratory failure (discussed below). Other organs affected by this disease include the male genital tract, sweat gland, pancreas, intestine, and hepatobiliary system.
The most strongly associated and the most widely replicated common variant linked to IPF is a single-nucleotide polymorphism (SNP; rs35705950) in the promoter of the gene encoding MUC5B, a large, highly glycosylated component of mucus. Individuals with this variant have an increased predisposition for developing sporadic or familial IPF. Individuals homozygous for the MUC5B risk allele have a greater risk for developing pulmonary fibrosis (odds ratio 21–22) than those who are heterozygous (odds ratio 7–9) (60). The association between the MUC5B rs35705950 SNP and IPF has been demonstrated in independent cohorts from different geographic locations and composed of patients of different ethnicities (38, 61–66). It is more strongly associated with IPF than ILD secondary to other causes (63, 64, 66). The variant is also associated with subclinical radiographically apparent pulmonary fibrosis, which is hypothesized to be an early form of IPF, in large population cohorts (67). IPF patients with the MUC5B risk allele have improved survival in independent cohorts after adjustment for age, sex, baseline lung function, and treatment (68).
The MUC5B risk variant is also associated with increased expression of mucin in the lung (60). MUC5B is highly expressed in mucus-secreting cells of the distal airways and epithelial cells lining the honeycomb cysts in patients with IPF (69).
Another common variant associated with IPF is located within an intron of the gene encoding desmoplakin (DSP) (38). Like MUC5B, desmoplakin is highly expressed in airway epithelial cells. But in contrast with the MUC5B risk allele, the disease-associated DSP SNP is associated with decreased protein expression, implicating a mechanism involving altered cell-cell adhesion and epithelial barrier function (70).
Pathogenic variants in SFTPB, SFTPC, and SFTPA1/2, which respectively encode the surfactant proteins SP-B, SP-C, and SP-A1/2, were among the first variants to be associated with various lung diseases (71). Surfactant proteins are almost exclusively expressed by type II AECs. In rare case reports, heterozygous variants in SP-A1 or SP-A2 cosegregate with pulmonary fibrosis and adenocarcinoma and are implicated in defective posttranslational modification and secretion of SP-A from type II AECs (72–74). Over 40 different pathogenic SP-C variants have been described, although a single variant, Ile73Thr, predominates and represents approximately 25%–35% of abnormal alleles (75–77). These variants lead to protein misfolding, increased ER stress, activation of the unfolded protein response, and cell toxicity (78). Transgenic expression of the pathogenic SP-C variant Leu188Gln in mouse type II AECs leads to increased sensitivity to bleomycin-induced lung fibrosis (79).
There are many other examples of organ fibrosis due to genetic defects in intrinsic proteins. Variants in over 100 different genes cause various subtypes of cardiomyopathies (dilated, hypertrophic, restrictive, arrhythmogenic right ventricular, and others) (80, 81). Defective liver proteins lead to autosomal recessive forms of cirrhosis, such as α1-antitrypsin disease, Wilson’s disease, hereditary forms of hemochromatosis, progressive familial intrahepatic cholestasis, and inborn errors of metabolism (82). A common missense allele (Ile148Met) of a lipid droplet–associated triglyceride lipase (PNPLA3) confers susceptibility to fatty liver disease and risk of liver cirrhosis (83). Medullary cystic kidney disease associated with heterozygous rare variants in MUC1 and UMOD leads to chronic tubulointerstitial kidney disease and end-stage renal disease (84, 85).
Genetic variants affecting organelle function.
Some genetic variants that cause fibrosis directly affect organelle function. The lamellar body is a key organelle within type II AECs that mediates the packaging and secretion of pulmonary surfactant. The A3 member of the ATP-binding cassette family of transporters (ABCA3) is localized to the membranes of lamellar bodies and facilitates transport of lipids, especially phosphotidylcholine, across the lamellar membrane. Mutations in ABCA3 can present in the neonatal period with respiratory distress or later in life as interstitial lung disease (76). ABCA3-deficient children and mice lack normal lamellar bodies, implicating a role for this protein in lamellar body biogenesis (86–89).
Pulmonary fibrosis is a feature of Hermansky-Pudlak syndrome, an autosomal recessive disease caused by defective lysosome-related organelles and disrupted trafficking of vesicular cargo proteins. This disease is also characterized by ocular albinism and platelet dysfunction (90). Patients with pulmonary fibrosis have evidence of giant lamellar body degeneration, thus linking lysosome function with lamellar body integrity. Lung fibrosis is seen most frequently in patients with HPS1 mutations, and less frequently in those with HPS2 or HPS4 mutations. Mice homozygous for both Hps1/2 mutations have abnormal type II AEC lamellar bodies and demonstrate spontaneous fibrosis at 9 months (91). More recently, mouse models have highlighted the central role of epithelial-macrophage interactions in leading to lung fibrosis (92, 93).
Congenital fibrosis of the extraocular muscles is a disorder that prevents the normal development and function of muscles that control eye movement. Patients may have difficulty looking upward or to the side or have misalignments of the eyes (strabismus) or ptosis of the upper eyelid. Mutations in genes important for cranial nerve development (PHOX2A), and those important for microtubule function (KIF21A, TUBB3), lead to fibrosis of the extraocular muscles (94–96). Mutations in these genes highlight the importance of the microtubule organelle in the pathogenesis of eye muscle fibrosis.
Nephronophthisis is the most common monogenic form of end-stage renal disease affecting children. It represents a type of ciliopathy characterized by degeneration of the tubular basement membrane, interstitial fibrosis, tubular atrophy, and cysts at the corticomedullary junction. The ciliopathies, which include a wide range of malformation and degeneration phenotypes affecting the kidney, eye, cerebellum, and liver, are all caused by disruption of the cilium-centrosome complex, which serves as a detection system for external signals (97). Defects in this organelle lead to activation of multiple different downstream signaling mechanisms, including planar cell polarity, Wnt, sonic hedgehog (SHH), focal adhesion signaling pathways, and cell cycle regulation. Mutations in three genes (MRE11, ZNF423, and CEP164) encoding proteins that colocalize to sites of DNA damage link certain ciliopathies with activation of the DNA damage response (98).
Pathogenic signaling pathways
Identification of linked genetic variants provides the first step in the long process of investigating the underlying mechanisms leading to fibrosis. Sometimes, these mechanisms are obscure and require animal models developed decades after the initial gene discovery. Regardless of the underlying genetic mutation, fibrosis results from activation of a number of different downstream maladaptive responses (Figure 2). These involve both cell-autonomous changes within the cells directly affected by the genetic mutation, and nonautonomous changes in cells affected by perturbations in cell signaling or the extracellular milieu. Even if only a restricted population of cells is affected by the pathogenic variant or variants, its effects may spill over into overlapping and interacting pathways of cell injury, inflammation, and fibrosis. Uncovering the diverse molecular mechanisms downstream of a particular pathogenic variant provides new insights into fibrosis. Examples of genetic variants linked to inherited pulmonary fibrosis that lead to telomere shortening, activation of the DNA damage response, cellular senescence, ER stress, and the unfolded protein response are discussed above. Other genetic defects illustrate additional dysregulated signaling pathways that lead to tissue fibrosis. For example, a missense mutation in platelet-derived growth factor receptor B (PDGFRB) leads to ligand-independent constitutive signaling of the PDGFR through STAT3 and PLCγ and causes Penttinen syndrome, an autosomal dominant disease with premature aging, digital contractures, and hyperkeratotic scars (99). Missense mutations in the gene encoding fibrillin (FBN1), clustered in the integrin-binding domain, cause stiff skin syndrome, an autosomal-dominantly inherited, congenital form of scleroderma (100). These mutations lead to excess extracellular deposition (fibrillin, elastin) and increased TGF-β signaling in the dermis.
Fibrosis is also a dreaded consequence of autoimmune disease. Tissue fibrosis results from a complex interplay between genetic predisposition, activation of the innate and adaptive immune systems, unbridled inflammation, metamorphosis of fibroblasts into myofibroblasts, and secretion of excessive extracellular matrix proteins (101). Genetic variants within the HLA genomic region predispose to a variety of autoimmune diseases (102). Recent discoveries have uncovered two new unanticipated links between non-HLA loci, autoimmunity, and dysregulated intercellular signaling. Autoimmune interstitial lung, joint, and kidney disease is a rare syndrome affecting children and young adults that is characterized by high-titer autoantibodies, lymphocytic lung infiltrates, inflammatory arthritis, and renal disease. Patients with heterozygous mutations in the coatomer subunit α (COPA) gene demonstrate impaired retrograde Golgi-to-ER transport, upregulation of Th17 cytokines, and increased lung infiltration of CD4+ T cells and CD20+ B cells (103). Another recently described disorder, STING-associated vasculopathy, presents in infancy and leads to severe ulcerative skin lesions, lung involvement ranging from radiographic abnormalities to fibrosis, arthritis, autoantibodies, and immune complex deposition (104, 105). All cases are due to heterozygous gain-of-function variants in the TMEM173 gene that lead to stimulation of interferon genes and activation of STAT1.
Two major drivers of fibrosis in the lung are chronic respiratory infections and persistent inflammation. Both can lead to tissue destruction and pulmonary fibrosis. When both infection and inflammation are the sequelae of a genetic mutation, the pathophysiologic pathways leading to fibrosis can be difficult to dissect and untangle. The recent development of a new piglet model of cystic fibrosis finally solved a more-than-20-year-old puzzle (106–108). Using the piglet model, Welsh and his colleagues showed that bacterial infection of the airway precedes lung inflammation, tissue remodeling, and lung fibrosis. The reduced chloride and bicarbonate secretion in this model leads to an acidic airway-surface liquid that impairs bacterial killing and reduces mucociliary transport. These findings directly link CFTR function to mucosal host defense. The diseased airway epithelium demonstrates constitutive activation of NF-κB, dysregulated TLR4 signaling, altered regulation of type I interferon gene products, increased oxidative stress, and exaggerated inflammation, which all contribute to fibrosis (109).
Common variable immunodeficiency syndromes lead to recurrent sinopulmonary infections and sequelae of bronchiectasis, pneumatoceles, lung fibrosis, and chronic respiratory failure. The immunodeficiency syndromes can lead to dysregulation of the immunoregulatory environment with paradoxical increases in inflammatory cell infiltrates in the lung. This is a very genetically heterogeneous group of disorders. A few illustrative examples of rare Mendelian disorders illustrate the coexistence of immunodeficiency and exaggerated inflammation that result from a single germline mutation. Autosomal dominant hyper-IgE syndrome is due to heterozygous variants in the STAT3 gene, leading to failure of proper differentiation of native T cells into Th17 cells, elevated IgE, eosinophilia, recurrent staphylococcal infections, and a number of nonimmune features, including retained primary teeth, scoliosis, joint hyperextensibility, bone fragility, and vascular abnormalities (110, 111). Cytotoxic T lymphocyte antigen 4 (CTLA4) is a key negative regulator of the immune system. Heterozygous loss-of-function mutations in CTLA4 cause immunodeficiency, recurrent respiratory infections, lymphocytic infiltrates, including granulomatous-lymphocytic ILD in the lung, and autoantibodies (112, 113). X-linked reticulate pigmentary disorder presents in childhood with immunodeficiency, recurrent pneumonias, bronchiectasis, diffuse skin hyperpigmentation, and a variety of other fibrotic phenotypes (corneal scarring, urethral strictures). The disease is caused by an intronic mutation that disrupts expression of the catalytic subunit of DNA polymerase-α (POLA1), which leads to decreased RNA:DNA hybrid synthesis and persistent activation of the type I interferon pathway (114). In all of these cases of an inherited immunodeficiency syndrome, the pathophysiology of fibrosis is due to both recurrent pulmonary bacterial infections and an exaggerated inflammatory response.
Other insights in human fibrosis
Organ fibrosis is related to a complex interplay between intrinsic susceptibility and exposure to injury (Figure 3). As many of the above examples have illustrated, rare pathogenic variants are substantially enriched in patients with earlier-onset, more rapidly progressive, and severe disease. Although environmental injuries may be difficult to quantitate over the entire patient lifespan, they influence the organ-specific manifestations of fibrosis, even for those with rare Mendelian disorders. Population-based and genetically defined at-risk cohorts demonstrate a higher penetrance of pulmonary fibrosis associated with smoking and various inhaled “fibrogenic” exposures (27, 115, 116). Bone marrow transplant conditioning regimens are precipitants for pulmonary fibrosis in younger patients with dyskeratosis congenita (117, 118). Environmental injuries may take a variety of forms. In the case of liver cirrhosis, interactions between the PNPLA3 variant and ethanol ingestion or obesity promote the variant’s link to the development of liver fibrosis (119, 120).
Aging also contributes to the phenotypic spectrum of organ fibrosis in a number of different ways. Some of its effects are dependent on genomic changes, such as telomere shortening, accumulation of somatic variants, and alterations in epigenetic marks. These result in cell senescence, loss of self-renewing stem cells, and diminished tissue repair. Aging also leads to dysfunctional mitochondria and increased oxidative stress. Epidemiologic studies of IPF consistently demonstrate a significant stepwise increase in the disease with advancing age, which is inversely associated with survival (121, 122).
Genetics has a role in revealing forces that provide selective advantages to normal and diseased populations. The high frequencies of the mutant CFTR alleles may have allowed heterozygotes to be more resistant to deleterious effects of cholera- or lactose-induced diarrhea (123). Similarly, the high frequency of the MUC5B risk variant in populations susceptible to developing IPF (allele frequency of approximately 9% in the non-Hispanic white population) may be due to positive natural selection related to its indispensable role in maintaining immune homeostasis through mucociliary clearance of particulates and pathogens out of airways (124).
Somatic genetic variants, which are acquired over time, may provide an explanation for variable expressivity of phenotypes. Skewed X-inactivation, revertant mosaicism, and clonal hematopoiesis can be seen in patients with dyskeratosis congenita (125–127). In these cases, the somatic change results in the correction or deletion of the pathogenic allele. Mutations in the promoter of the TERT gene are some of the most common genetic alterations found in human cancers and occur very frequently in bladder cancer, melanoma, and glioblastoma (128). Approximately 5% of older adults with germline heterozygous TERT coding mutations have acquired one of two different TERT promoter mutations linked in cis with the wild-type allele in somatic blood leukocytes (129). As the promoter mutations lead to increased TERT activity, increased cellular proliferation, and clonal survival, their presence in blood leukocytes may be protective for the bone marrow failure or immunodeficiency phenotypes associated with telomerase insufficiency. Thus, compensatory somatic variants reveal selective forces that counteract germline disease mutations.
In contrast with pigs and humans, mice with a Cftr deletion do not develop cystic fibrosis lung disease (130). This difference stems from decreased expression of the gene encoding the nongastric H+/K+ adenosine triphosphatase (ATP12A) in mouse airways, resulting in minimal acidification of the airway surface liquid and an intact mucosal host defense (131). Inhibition of this transporter leads to improved mucosal properties in human and pig epithelia, and, conversely, expression of the transporter in mouse airways leads to airway acidification and increased number of bacteria in the lung. Thus, the lack of ATP12A expression is a protective factor in the pathogenesis of cystic fibrosis and provides a molecular explanation for species-specific difference in the expressivity of lung disease. Importantly, ATP12A provides another potential target for the amelioration of lung disease associated with cystic fibrosis.
Merging quantification of genetic and environmental risks with age will refine estimates of patient susceptibility to fibrosis. Although age and genetic risks cannot be readily altered, modification of environmental exposures, especially in individuals with a high genetic risk, may potentially delay or prevent organ fibrosis. Interventions that therapeutically target the cascade of maladaptive responses triggered by genetic mutations will lead to improved clinical outcomes. Thus, genetics offers cogent insights into strategies for risk stratification, prevention, and treatment of organ fibrosis.
Acknowledgments
The author acknowledges helpful discussions with Helen Hobbs, Chad Newton, Joseph Garcia, and members of the Garcia laboratory; Chelsea Burroughs for help with artwork; as well as support from NIH grants HL093096 and HL121267.
Version 1. 01/02/2018
Print issue publication
Footnotes
Conflict of interest: The author acknowledges past advisory board service for Pliant Therapeutics.
Reference information: J Clin Invest. 2018;128(1):36–44.https://doi.org/10.1172/JCI93556.
References
- 1.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 3.Stuart BD, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512–517. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrovski S, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(1):82–93. doi: 10.1164/rccm.201610-2088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110(4):768–779. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 6.Calado RT, et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS One. 2009;4(11):e7926. doi: 10.1371/journal.pone.0007926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361(24):2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartmann D, et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology. 2011;53(5):1608–1617. doi: 10.1002/hep.24217. [DOI] [PubMed] [Google Scholar]
- 9.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775–2783. doi: 10.1182/blood-2014-05-526285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 pt 1):405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 11.Feng J, et al. The RNA component of human telomerase. Science. 1995;269(5228):1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 12.Kim NW, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 13.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18(2):173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 15.d’Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 16.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107(1):67–77. doi: 10.1016/S0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 17.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13(17):1549–1556. doi: 10.1016/S0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 18.Armanios MY, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 19.Tsakiri KD, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moon DH, et al. Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nat Genet. 2015;47(12):1482–1488. doi: 10.1038/ng.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cogan JD, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015;191(6):646–655. doi: 10.1164/rccm.201408-1510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kannengiesser C, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. 2015;46(2):474–485. doi: 10.1183/09031936.00040115. [DOI] [PubMed] [Google Scholar]
- 23.Kropski JA, et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest. 2014;146(1):e1–e7. doi: 10.1378/chest.13-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley SE, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8(351):351ra107. doi: 10.1126/scitranslmed.aaf7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015;147(5):1361–1368. doi: 10.1378/chest.14-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffman TW, et al. TINF2 gene mutation in a patient with pulmonary fibrosis. Case Rep Pulmonol. 2016;2016:1310862. doi: 10.1155/2016/1310862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz de Leon A, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE. 2010;5(5):e10680. doi: 10.1371/journal.pone.0010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alder JK, et al. Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet. 2011;7(3):e1001352. doi: 10.1371/journal.pgen.1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du HY, et al. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111(3):1128–1130. doi: 10.1182/blood-2007-10-120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tummala H, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151–2160. doi: 10.1172/JCI78963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Savage SAC, Elizabeth F, eds. Dyskeratosis Congenita and Telomere Biology Disorders: Diagnosis and Management Guidelines. New York, New York, USA: Dyskeratosis Congenita Outreach Inc.; 2015. [Google Scholar]
- 33.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–486. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 34.Borie R, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016;48(6):1721–1731. doi: 10.1183/13993003.02115-2015. [DOI] [PubMed] [Google Scholar]
- 35.Diaz de Leon A, et al. Subclinical lung disease, macrocytosis, and premature graying in kindreds with telomerase (TERT) mutations. Chest. 2011;140(3):753–763. doi: 10.1378/chest.10-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George G, et al. Short telomeres, telomeropathy, and subclinical extrapulmonary organ damage in patients with interstitial lung disease. Chest. 2015;147(6):1549–1557. doi: 10.1378/chest.14-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13(10):693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fingerlin TE, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newton CA, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710–1720. doi: 10.1183/13993003.00308-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Juge PA, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J. 2017;49(5):1602314. doi: 10.1183/13993003.02314-2016. [DOI] [PubMed] [Google Scholar]
- 41.Stanley SE, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest. 2015;125(2):563–570. doi: 10.1172/JCI78554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stuart BD, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. 2014;2(7):557–565. doi: 10.1016/S2213-2600(14)70124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cronkhite JT, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(7):729–737. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alder JK, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ley B, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5(8):639–647. doi: 10.1016/S2213-2600(17)30216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 47.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 48.Hewitt G, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alder JK, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112(16):5099–5104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu T, et al. Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J Clin Invest. 2007;117(12):3800–3809. doi: 10.1172/JCI32369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Degryse AL, et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp Lung Res. 2012;38(3):124–134. doi: 10.3109/01902148.2012.658148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015;12(2):286–299. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 53.Martínez P, et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23(17):2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood. 2012;120(15):2990–3000. doi: 10.1182/blood-2012-03-418038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naikawadi RP, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1(14):e86704. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doil C, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136(3):435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 57.Stewart GS, et al. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci U S A. 2007;104(43):16910–16915. doi: 10.1073/pnas.0708408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okamoto K, Bartocci C, Ouzounov I, Diedrich JK, Yates JR, Denchi EL. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature. 2013;494(7438):502–505. doi: 10.1038/nature11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou W, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44(8):910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seibold MA, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364(16):1576–1577. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noth I, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stock CJ, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68(5):436–441. doi: 10.1136/thoraxjnl-2012-201786. [DOI] [PubMed] [Google Scholar]
- 64.Borie R, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8(8):e70621. doi: 10.1371/journal.pone.0070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horimasu Y, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology. 2015;20(3):439–444. doi: 10.1111/resp.12466. [DOI] [PubMed] [Google Scholar]
- 66.Wei R, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. 2014;163(5):494–502. doi: 10.1016/j.trsl.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hunninghake GM, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. 2013;368(23):2192–2200. doi: 10.1056/NEJMoa1216076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peljto AL, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309(21):2232–2239. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seibold MA, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8(3):e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathai SK, et al. Desmoplakin variants are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193(10):1151–1160. doi: 10.1164/rccm.201509-1863OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 72.Wang Y, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Moorsel CH, et al. SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2015;192(10):1249–1252. doi: 10.1164/rccm.201504-0675LE. [DOI] [PubMed] [Google Scholar]
- 74.Nathan N, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet. 2016;25(8):1457–1467. doi: 10.1093/hmg/ddw014. [DOI] [PubMed] [Google Scholar]
- 75.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med. 2010;61:105–119. doi: 10.1146/annurev.med.60.041807.123500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nogee LM. Genetic basis of children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23(1):15–24. doi: 10.1089/ped.2009.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Moorsel CH, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182(11):1419–1425. doi: 10.1164/rccm.200906-0953OC. [DOI] [PubMed] [Google Scholar]
- 78.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302(8):L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lawson WE, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108(26):10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liew CC, Dzau VJ. Molecular genetics and genomics of heart failure. Nat Rev Genet. 2004;5(11):811–825. doi: 10.1038/nrg1470. [DOI] [PubMed] [Google Scholar]
- 81.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115(3):518–526. doi: 10.1172/JCI24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karlsen TH, Lammert F, Thompson RJ. Genetics of liver disease: from pathophysiology to clinical practice. J Hepatol. 2015;62(1 suppl):S6–S14. doi: 10.1016/j.jhep.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 83.Romeo S, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kirby A, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45(3):299–303. doi: 10.1038/ng.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hart TC, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39(12):882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350(13):1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 87.Ban N, et al. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. 2007;282(13):9628–9634. doi: 10.1074/jbc.M611767200. [DOI] [PubMed] [Google Scholar]
- 88.Cheong N, et al. ABCA3 is critical for lamellar body biogenesis in vivo. J Biol Chem. 2007;282(33):23811–23817. doi: 10.1074/jbc.M703927200. [DOI] [PubMed] [Google Scholar]
- 89.Fitzgerald ML, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48(3):621–632. doi: 10.1194/jlr.M600449-JLR200. [DOI] [PubMed] [Google Scholar]
- 90.Gahl WA, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338(18):1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 91.Mahavadi P, et al. Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am J Respir Crit Care Med. 2010;182(2):207–219. doi: 10.1164/rccm.200909-1414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lyerla TA, et al. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L643–L653. doi: 10.1152/ajplung.00024.2003. [DOI] [PubMed] [Google Scholar]
- 93.Young LR, et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight. 2016;1(17):e88947. doi: 10.1172/jci.insight.88947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamada K, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1) Nat Genet. 2003;35(4):318–321. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 95.Tischfield MA, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakano M, et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29(3):315–320. doi: 10.1038/ng744. [DOI] [PubMed] [Google Scholar]
- 97.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chaki M, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150(3):533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnston JJ, et al. A point mutation in PDGFRB causes autosomal-dominant Penttinen syndrome. Am J Hum Genet. 2015;97(3):465–474. doi: 10.1016/j.ajhg.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Loeys BL, et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med. 2010;2(23):23ra20. doi: 10.1126/scitranslmed.3000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis—a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10(7):390–402. doi: 10.1038/nrrheum.2014.53. [DOI] [PubMed] [Google Scholar]
- 102.Ramos PS, Shedlock AM, Langefeld CD. Genetics of autoimmune diseases: insights from population genetics. J Hum Genet. 2015;60(11):657–664. doi: 10.1038/jhg.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watkin LB, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47(6):654–660. doi: 10.1038/ng.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu Y, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jeremiah N, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124(12):5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 107.Stoltz DA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2(29):29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372(16):1574–1575. doi: 10.1056/NEJMc1502191. [DOI] [PubMed] [Google Scholar]
- 109.Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med. 2012;18(4):509–519. doi: 10.1038/nm.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Minegishi Y, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 111.Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452(7188):773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuehn HS, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–1627. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Starokadomskyy P, et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. 2016;17(5):495–504. doi: 10.1038/ni.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Marshall RP, Puddicombe A, Cookson WO, Laurent GJ. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax. 2000;55(2):143–146. doi: 10.1136/thorax.55.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc. 2006;3(4):293–298. doi: 10.1513/pats.200512-131TK. [DOI] [PubMed] [Google Scholar]
- 117.de la Fuente J, Dokal I. Dyskeratosis congenita: advances in the understanding of the telomerase defect and the role of stem cell transplantation. Pediatr Transplant. 2007;11(6):584–594. doi: 10.1111/j.1399-3046.2007.00721.x. [DOI] [PubMed] [Google Scholar]
- 118.Dietz AC, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46(1):98–104. doi: 10.1038/bmt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42(1):21–23. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]
- 120.Stender S, Kozlitina J, Nordestgaard BG, Tybjærg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842–847. doi: 10.1038/ng.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Raghu G, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med. 2014;2(7):566–572. doi: 10.1016/S2213-2600(14)70101-8. [DOI] [PubMed] [Google Scholar]
- 122.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 123.Modiano G, Ciminelli BM, Pignatti PF. Cystic fibrosis and lactase persistence: a possible correlation. Eur J Hum Genet. 2007;15(3):255–259. doi: 10.1038/sj.ejhg.5201749. [DOI] [PubMed] [Google Scholar]
- 124.Roy MG, et al. Muc5b is required for airway defence. Nature. 2014;505(7483):412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vulliamy TJ, Knight SW, Dokal I, Mason PJ. Skewed X-inactivation in carriers of X-linked dyskeratosis congenita. Blood. 1997;90(6):2213–2216. [PubMed] [Google Scholar]
- 126.Jongmans MC, et al. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet. 2012;90(3):426–433. doi: 10.1016/j.ajhg.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Perdigones N, et al. Clonal hematopoiesis in patients with dyskeratosis congenita. Am J Hematol. 2016;91(12):1227–1233. doi: 10.1002/ajh.24552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vinagre J, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. doi: 10.1038/ncomms3185. [DOI] [PubMed] [Google Scholar]
- 129.Maryoung L, et al. Somatic mutations in telomerase promoter counterbalance germline loss-of-function mutations. J Clin Invest. 2017;127(3):982–986. doi: 10.1172/JCI91161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79(1 suppl):S193–S214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- 131.Shah VS, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. 2016;351(6272):503–507. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]