

Abstract
Foods high in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) exacerbate symptoms of irritable bowel syndrome (IBS); however, their mechanism of action is unknown. We hypothesized that a high-FODMAP (HFM) diet increases visceral nociception by inducing dysbiosis and that the FODMAP-altered gut microbial community leads to intestinal pathology. We fed rats an HFM and showed that HFM increases rat fecal Gram-negative bacteria, elevates lipopolysaccharides (LPS), and induces intestinal pathology, as indicated by inflammation, barrier dysfunction, and visceral hypersensitivity (VH). These manifestations were prevented by antibiotics and reversed by low-FODMAP (LFM) diet. Additionally, intracolonic administration of LPS or fecal supernatant (FS) from HFM-fed rats caused intestinal barrier dysfunction and VH, which were blocked by the LPS antagonist LPS-RS or by TLR4 knockdown. Fecal LPS was higher in IBS patients than in healthy subjects (HS), and IBS patients on a 4-week LFM diet had improved IBS symptoms and reduced fecal LPS levels. Intracolonic administration of FS from IBS patients, but not FS from HS or LFM-treated IBS patients, induced VH in rats, which was ameliorated by LPS-RS. Our findings indicate that HFM-associated gut dysbiosis and elevated fecal LPS levels induce intestinal pathology, thereby modulating visceral nociception and IBS symptomatology, and might provide an explanation for the success of LFM diet in IBS patients.
Keywords: Gastroenterology
Keywords: Cytokines, Pain, Translation
Introduction
It is established that food may serve as an important trigger of symptoms in patients with irritable bowel syndrome (IBS) (1–3). Ong et al. (2) reported the association of a diet rich in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) with the generation of characteristic IBS symptoms. Research has shown that fructose or fructans induce IBS symptoms in a dose-dependent manner (3). In contrast, increasing evidence suggests a low-FODMAP (LFM) diet reduces IBS symptoms. A randomized controlled trial of a 4-week LFM diet improved symptoms in 68% of IBS patients compared with 23% on a habitual diet (4). Patients on the restricted diet experienced a reduction in bloating, borborygmi, urgency, and overall symptoms. Researchers in the United Kingdom reported the LFM diet to be superior to national guidelines for IBS management (5). These observations were confirmed by a controlled crossover study of 30 IBS patients and 18 healthy controls, in which an LFM diet significantly reduced bloating, pain, passage of gas, and diarrhea (6). Symptoms were minimal and unaffected by an LFM diet in healthy individuals (6). These observations demonstrate the impressive efficacy of LFM diet for many IBS patients and may justify the recommendation of an LFM diet as first-line therapy for IBS patients by some investigators (6). The mechanisms by which an LFM diet benefits IBS patients are not well understood.
Lipopolysaccharides (LPS) are large molecules consisting of a hydrophobic lipid A and a hydrophilic carbohydrate core with polysaccharide O-antigen. LPS are found in the outer membrane of most Gram-negative bacteria (7) and are released from the bacterial cell wall during lysis. LPS plays an important role in the inflammatory process of inflammatory bowel disease (IBD). Endotoxemia was present in 48% of patients with Crohn’s disease and 28% of patients with ulcerative colitis (8). Administration of Gram-negative bacterial LPS to human volunteers induced TNF and IL-6 secretion (9). An increased serum LPS level in patients with diarrhea-predominant IBS (IBS-D) has been reported (10). Indeed, phylogenetic microarray analysis revealed a significant change in the fecal microbiota of IBS-D patients compared with healthy subjects (HS). The one major difference in the IBS-type microbiota was the 12-fold increase in several Gram-negative Bacteroidetes species (11).
The effects of LPS on intestinal inflammation remain unclear. In enterocytes, LPS binds to TLR4, which is known to be an important pattern recognition receptor for LPS (12). Stimulation of TLR4 results in NF-κB activation via multiple downstream intracellular signals, which then stimulates synthesis and secretion of inflammatory cytokines (13, 14). The intestinal epithelium is a physical barrier that separates the commensal bacteria in the intestinal lumen from the underlying lamina propria and deeper intestinal layers. Evidence shows that increased LPS levels compromise intestinal tight junction permeability (15), thus allowing toxic substances such as endotoxins and microorganisms to cross the intestinal wall.
We hypothesize that FODMAPs cause endotoxemia and evoke intestinal inflammation, which in turn modulates visceral nociception. To test this hypothesis, we conducted experiments in rats to determine if a diet with high levels of FODMAPs can cause dysbiosis and lead to the production of endotoxins that cause gut inflammation and induce visceral hypersensitivity. In separate studies, we examined the effects of fecal supernatant obtained from rats fed a high-FODMAP (HFM) diet on visceral sensitivity in naive rats and the permeability of cultured human colonoids. We performed intervention studies in 2 chronic stress rat models to determine if FODMAP restriction prevents low-grade mucosal inflammation and improves gut barrier function.
To further test our hypothesis, we examined the effects of fecal supernatant obtained from IBS-D patients before and after 4 weeks of LFM diet treatment on visceral sensitivity in naive rats. The results were compared to HS. We conclude that increases in fecal LPS after ingestion of an HFM diet and in IBS-D patients mediate intestinal barrier dysfunction and visceral hypersensitivity due to dysbiosis. These abnormalities can be prevented by an LFM diet.
Results
HFM increases mucosal inflammation, gut permeability, and visceral hyperalgesia in the colon.
To study the impact of HFM on mucosal inflammatory tone, gene expression of inflammatory cytokines was measured in colonic tissues. IL-1β, IL-6, IL-17, TNF-α, and IFN-γ mRNA levels increased significantly in HFM rats, compared with regular chow–fed (RC-fed) rats (n = 6, P < 0.05, Student’s t test) (Figure 1A).
Figure 1. Effects of HFM on cytokine expression and gut permeability of the rat colon.

(A) RT-PCR measurement of cytokines in rats fed for 2 weeks with RC, HFM, or HFM with rifaximin (HFM+Rif). mRNA levels represented as fold change in each target mRNA after normalization to GAPDH. HFM induced increases in IL-1β, IL-6, IL-17, TNF-α, and IFN-γ gene expression, indicating low-grade mucosal inflammation. (B) HFM increased gut permeability, as measured by TEER. (C) HFM caused decreased ZO-1 and (D) OCLN expression. (E) HFM increased the appearance of FITC–dextran in serum, accompanied by increased serum LPS (F), indicating endotoxemia. Rifaximin prevented HFM-induced changes in cytokine, gut permeability, and endotoxemia (A–E) (n = 6 per group). *P < 0.05 versus RC; #P < 0.05 versus HFM. HFM, high-FODMAP diet; OCLN, occludin; LPS, lipopolysaccharide; RC, regular chow; TEER, transepithelial electrical resistance. P values determined by 2-tailed Student’s t test.
Intestinal barrier function was evaluated by measuring the transepithelial electrical resistance (TEER) of ex vivo colonic tissues. TEER reflects paracellular resistance imparted by tight junctions and the lateral paracellular space and is a sensitive measure of barrier integrity. Intestinal resistance was reduced in HFM rats compared with RC rats (n = 6, P < 0.05) (Figure 1B). A reduced TEER was accompanied by reduced expression of the epithelial tight junction proteins zonula occludens–1 (ZO-1) and occludin (OCLN) by 54.0% ± 13.4% and 35.3% ± 11.8%, respectively (n = 6) (Figure 1, C and D). In vivo assessment of gut permeability revealed a significant increase in serum FITC–dextran in HFM rats (n = 6, P < 0.05), indicating impairment of intestinal barrier function (Figure 1E).
Serum LPS in rats fed RC or HFM was measured. The LPS concentration was significantly increased in HFM rats, compared with RC rats (0.84 ± 0.10 vs. 0.40 ± 0.08 endotoxin units [EU]/ml, n = 6, P < 0.05) (Figure 1F). These data indicate that impaired mucosal permeability was accompanied by increased serum LPS. HFM evoked mucosal inflammation and impaired intestinal permeability, resulting in metabolic endotoxemia.
Histological analysis revealed increased numbers of mononuclear cells, neutrophils, eosinophils, and mast cells in the lamina propria of the colon from HFM compared with RC rats, suggesting low-grade mucosal inflammation after HFM exposure (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI92390DS1).
Lastly, the effect of HFM on visceromotor response (VMR) to graded colorectal distension (CRD) was examined. Rats showed pressure-dependent increases in VMR to CRD (Figure 2, A and B). The VMR to 20, 40, 60, and 80 mmHg CRD significantly increased 2.5-, 2.8-, 2.5-, and 2.4-fold after 2-week HFM (P < 0.05, n = 6 per group), suggesting the development of visceral hypersensitivity.
Figure 2. HFM-induced visceral hyperalgesia is reversed by rifaximin.

(A) Representative electromyogram recordings of responses to colorectal distension in rats fed RC, HFM, or HFM with rifaximin (HFM+Rif). (B) Mean amplitudes of abdominal muscle contractions are expressed as area under the curve (AUC) after baseline subtraction (n = 6 per group). *P < 0.05 compared with RC rats; #P < 0.05 compared with HFM. AUC, area under the curve; EMG, electromyographic activity; HFM, high-FODMAP diet; RC, regular chow. P < 0.05, by 2-tailed Student’s t test or 2-way repeated-measures ANOVA.
Rifaximin prevents HFM-induced mucosal inflammation, impaired gut permeability, and visceral hyperalgesia.
To determine if gut mucosal changes and development of visceral hypersensitivity are caused by HFM acting directly on the mucosa or indirectly due to bacterial community changes in the gut, we investigated the effects of rifaximin, which has been shown to alter bacterial communities and prevent stress-induced gut inflammation and visceral hyperalgesia in rats (16).
Oral gavage of rifaximin prevented an increase in the numbers of colonic mucosal inflammatory cells (Supplemental Table 1) and elevated IL-1β, IL-6, IL-17, TNF-α, and IFN-γ mRNA levels in HFM rats (P < 0.05) (Figure 1A). Rifaximin also prevented a reduction of ZO-1 and OCLN (Figure 1, C and D) and a decrease in TEER in the colon (P < 0.05) (Figure 1B) and impairment of intestinal barrier functions measured by serum FITC–dextran (Figure 1E). Rifaximin also blocked the increase in serum LPS (Figure 1F).
We next assessed the preventive effect of rifaximin on the development of visceral hyperalgesia in HFM rats. Repeated oral gavage of rifaximin significantly attenuated the increased VMR to CRD induced by HFM at 20, 40, 60, and 80 mmHg (Figure 2, A and B). These data suggest that HFM causes mucosal inflammation and visceral hypersensitivity by inducing dysbiosis, which rifaximin prevents.
HFM diet alters the bacterial community.
The Illumina MiSeq sequencing platform was used to determine if HFM altered the luminal microbial communities of the ascending colon. The major phyla identified in the ascending colon of rats fed RC were Actinobacteria, Bacteroidetes, Firmicutes, and Verrucomicrobia (Figure 3A). Firmicutes significantly decreased after HFM (Figure 3B). In contrast, after HFM, the relative abundance of Actinobacteria (Gram-positive) and Verrucomicrobia (Gram-negative) significantly increased (Figure 3B). There was an enrichment in the abundance of Gram-negative bacteria after HFM and reduction of the Gram-positive (Figure 3, C and D). Among Gram-negative bacteria, Verrucomicrobia (Akkermancia muciniphila) was increased by 4.7-fold from 8.2% ± 6.5% to 38.3% ± 5.3% (percentage of total Gram-negative bacteria) (Figure 3E).
Figure 3. HFM-induced dysbiosis in the colon.

(A) Relative abundance of OTUs classified at the level of phylum. (B) Relative abundance of Firmicutes, Actinobacteria, and Verrucomicrobia. (C) Relative abundance of Gram-positive communities. (D) Relative abundance of Gram-negative communities. (E) Relative abundance of Verrucomicrobia in total Gram-negative bacteria. (n = 6 per group). *P < 0.05 versus RC; #P < 0.05 versus HFM. P values determined by 2-tailed Student’s t test.
Effects of rifaximin on HFM-induced microbiota changes and mucosal inflammation.
Rifaximin was shown to alter bacterial communities in the ascending colon. Compared with HFM, the rats given rifaximin had a significantly higher relative abundance of the Firmicutes (Figure 3B). There was also a significantly lower abundance of the Actinobacteria and Verrucomicrobia in the rifaximin group compared with HFM (Figure 3, B and E).
Elevated LPS in fecal samples from HFM rats.
To determine whether gut dysbiosis results in an increase in fecal LPS levels, the fecal samples from rats fed RC or HFM were obtained and endotoxin (LPS) levels were measured. Fecal LPS measurements showed an endotoxin level approximately 2-fold higher in HFM compared with RC rats (8.6 vs. 5.1 EU/mg fecal content, n = 6, P < 0.05) (Figure 4A).
Figure 4. Effects of fecal supernatant from HFM rats and LPS on gut permeability and visceral sensitivity.

(A) Endotoxin level (LPS) in fecal contents was elevated in HFM compared with RC rats (n = 6) *P < 0.05. (B–D) Intracolonic infusion of fecal supernatant from HFM rats caused changes in cytokines and junction proteins, ZO-1 and OCLN. These changes were prevented by LPS antagonist, LPS-RS. (E) HFM fecal supernatant decreased the transepithelial electrical resistance (TEER), indicating an increase in epithelium permeability of colon mucosa in naive rats (n = 6, P < 0.05). This increase was prevented by LPS-RS. (F) Intracolonic infusion of fecal supernatant from HFM rats induced visceral hyperalgesia in naive rats; this was prevented by LPS-RS or siRNA targeting TLR4 (n = 6, P < 0.05). (G) Intracolonic LPS infusion induced increased epithelium permeability of colonic mucosa (i.e., reduced TEER) in naive rats; this was prevented by LPS-RS. (H) Intracolonic infusion of LPS (10 μg/kg) induced visceral hyperalgesia in naive rats; this was prevented by LPS-RS (100 μg/kg). *P < 0.05 versus RC or PBS treated; #P < 0.05 versus HFM-supernatant or LPS treated. AUC, area under the curve; EMG, electromyographic activity; HFM, high-FODMAP diet; LPS, lipopolysaccharide; RC, regular chow; siRNA, small (or short) interfering RNA; TLR4, Toll-like receptor 4. P < 0.05, by 2-tailed Student’s t test or 2-way repeated-measures ANOVA.
Effects of fecal supernatant from HFM rats on intestinal barrier function and visceral sensitivity.
To evaluate the effects of fecal material on intestinal barrier function and visceral sensitivity, fecal supernatant (0.3 ml) from HFM or RC rats was administered intracolonically to naive rats. Colonic mucosa inflammatory tone and permeability were measured, and the VMR to CRD was performed 4–6 hours after supernatant administration.
Intracolonic administration of supernatant from HFM rats increased gene expression of IL-1β, IL-6, and TNF-α in the ascending colon compared with administration of fecal supernatant from RC rats (Student’s t test, n = 6, P < 0.05) (Figure 4B). It also reduced gene expression of the epithelial tight junction proteins ZO-1 and OCLN (Figure 4, C and D). Intracolonic HFM supernatant caused intestinal barrier dysfunction by decreasing TEER in the colonic mucosa, compared with RC rats (29.9 ± 0.85 vs. 36.2 ± 0.9 Ω/cm2, n = 6, P < 0.05) (Figure 4E) and 2- to 4-fold increases in VMR to CRD at 20, 40, 60, and 80 mmHg (Figure 4F). A potent LPS antagonist, LPS-RS (100 μg/kg), prevented an increase in mucosal cytokine expression, intestinal barrier dysfunction, and visceral hypersensitivity caused by HFM (Figure 4, C–F). The effects of LPS-RS are specific, since intracolonic administration of LPS-RS (1.0 mg/kg, 0.3 ml) failed to block visceral hypersensitivity induced by intracolonic infusion of protease-activated receptor 2 (PAR2) agonist, SLIGKV-NH2 (0.5 mg/kg, intracolonically) (Supplemental Figure 1). Separate studies showed that intracolonic administration of LPS (10 μg/kg, 0.3 ml) impaired colonic permeability (Figure 4G) and induced an increase in VMR to CRD, which was prevented by LPS-RS (100 μg/kg, 0.3 ml) (Figure 4H).
To examine whether TLR4 is involved, rat colon was pretreated with the intracolonic administration of siRNA targeting TLR4 (10 nmol per rat in 0.3 ml) for 3 days, which resulted in a 40% reduction in TLR4 gene expression in the ascending colonic mucosa (Supplemental Figure 2). Pretreatment of TLR4 siRNA prevented visceral hypersensitivity caused by fecal supernatant from HFM rats, indicating the involvement of TLR4 (Figure 4F).
Effects of fecal supernatant from HFM rats on epithelial barrier function of cultured human colonic organoids.
We used inside-out human colonoids to test the permeability of the colonic epithelium. Human colonoids were injected with fecal supernatant containing FITC–dextran, and fluorescence images were obtained at different time points. When exposed to fecal supernatant from RC rats, the colonoids retained 85% of FITC–dextran at 12 hours. LPS reduced FITC–dextran retention to 40% (n = 8, P < 0.05) (Figure 5). Similarly, fecal supernatant from HFM rats reduced FITC–dextran retention to 43% (n = 8, P < 0.05). The increase in permeability evoked by fecal supernatant from HFM rats was prevented by coadministration of LPS-RS with fecal supernatant from HFM rats (Figure 5).
Figure 5. Fecal supernatant from rats fed HFM or LPS disrupted the paracellular barrier function in human colonoids.

(A) Representative images of FITC–dextran leakage from the lumen of human colonoid spheres. Colonoid spheres were injected with a mixture of FITC–dextran and fecal supernatant obtained from rats subjected to RC, HFM, LPS, or HFM with LPS-RS. (B) Data summary shows that HFM supernatant or LPS caused significant loss of paracellular barrier function compared with RC supernatant (n = 8) or HFM supernatant with LPS-RS (n = 8). *P < 0.05 versus RC supernatant; #P < 0.05 versus HFM supernatant. HFM, high-FODMAP diet; LPS, lipopolysaccharide; LPS-RS, LPS antagonist; RC, regular chow. P < 0.05, by 2-tailed Student’s t test or 2-way repeated-measures ANOVA.
LFM prevents restraint stress–induced mucosal inflammation, intestinal barrier impairment, and visceral hyperalgesia.
A diet with restricted FODMAP content has been reported to reduce symptoms in IBS patients (3, 5). We used a rodent restraint stress (RS) model to determine if LFM prevents intestinal inflammation and visceral hypersensitivity in RS rats.
RS showed evidence of mucosal inflammation characterized by increased numbers of neutrophils and mononuclear cells (Supplemental Table 2) accompanied by increased gene expression of IL-6 (1.3 ± 0.1–fold), IL-17 (1.4 ± 0.1–fold), TNF-α (1.5 ± 0.2–fold), and IFN-γ (1.5 ± 0.4–fold) (Figure 6A). This was accompanied by a 17% decrease in TEER (n = 6, P < 0.05) (Figure 6B). In vivo assessment of gut permeability revealed a significant increase in serum FITC–dextran in RS rats (0.19 ± 0.03 vs. 0.04 ± 0.01 μg/ml, n = 6, P < 0.05, Student’s t test) (Figure 6C). The serum LPS level was significantly increased in RS compared with control rats (0.58 ± 0.04 vs. 0.38 ± 0.02 EU/ml, n = 6, P < 0.05), indicating metabolic endotoxemia (Figure 6D). Western blot analysis of the tight junction protein ZO-1 in proximal colon tissues showed a significant decrease in protein expression after chronic RS (n = 6, P < 0.05) (Figure 6E), indicating impairment of intestinal barrier function after chronic stress.
Figure 6. Effects of LFM on RS-induced mucosal inflammation, permeability alteration, and visceral hypersensitivity.

(A) RS caused mucosal inflammation characterized by increased cytokine expression. LFM prevented these changes. RS increased colon permeability, as measured by TEER (B) and FITC–dextran in serum (C), accompanied by increased serum LPS (D), indicating endotoxemia. (E) RS caused a decrease in ZO-1 expression. LFM prevented RS-induced changes in cytokine levels, gut permeability, and endotoxemia (A–E). (F) RS caused an increase in the visceromotor response to colorectal distension, compared with sham RS controls. LFM prevented RS-induced visceral hypersensitivity. n = 6 per group. *P < 0.05 compared with sham RS as control and RS+LFM; #P < 0.05 compared with RS. AUC, area under the curve; EMG, electromyographic activity; LFM, low-FODMAP diet; LPS, lipopolysaccharide; RS, restraint stress; TEER, transepithelial electrical resistance. P < 0.05, by 2-tailed Student’s t test or 2-way repeated-measures ANOVA.
Two-week LFM feeding prevented RS-provoked cytokine gene expression (Figure 6A). LFM normalized the RS-induced increase in TEER (35.4 ± 1.5 vs. 41.0 ± 1.2 Ω/cm2, n = 6, P < 0.05) (Figure 6B) and the in vivo increased gut permeability to FITC–dextran (n = 6, P < 0.05) (Figure 6C). In addition, LFM prevented an increase in serum LPS (Figure 6D) and a reduction in ZO-1 expression in the proximal colon (Figure 6E).
Lastly, pain behavior studies showed VMR to 60 and 80 mmHg CRD significantly increased in RS rats compared with controls (sham RS, n = 6, P < 0.05). Two-week LFM normalized the VMR to CRD in RS rats (Figure 6F).
LFM prevents RS-induced alteration of the bacterial community.
Again, Illumina MiSeq was used to determine if repeated RS altered luminal microbial communities of the ascending colon. The major phyla identified in the ascending colon of the control rats were Firmicutes, Bacteroidetes, Proteobacteria, and Verrucomicrobia (Figure 7A). Of these major groups, the Bacteroidetes significantly increased in the RS group compared with the controls and RS rats with LFM (Figure 7B). Conversely, the Firmicutes significantly decreased in the RS compared with the other groups (Figure 7B). Overall, in the RS group, the Gram-positive bacteria were depleted (Figure 7C) while the Gram-negative bacteria were significantly enriched (Figure 7D). After RS+LFM the relative abundance of the Gram-positive and Gram-negative communities returned to a level similar to that of the controls (Figure 7, C and D). Rats subjected to RS showed evidence of increased fecal LPS, which was prevented by LFM (Figure 7E).
Figure 7. Restraint stress–induced dysbiosis that is prevented by LFM.

(A) Relative abundance of OTUs classified at the level of phylum. (B) Relative abundance of Firmicutes and Bacteroidetes. (C) Relative abundance of Gram-positive communities. (D) Relative abundance Gram-negative communities. (E) Restraint stress increased fecal LPS, which was prevented by LFM. n = 3 per group. *P < 0.05 compared with control; #P < 0.05 compared with restraint stress. LFM, low-FODMAP diet; RC, regular chow; RS, restraint stress. P values determined by 2-tailed Student’s t test.
LFM prevents water avoidance stress–induced mucosal inflammation, intestinal barrier impairment, and visceral hyperalgesia.
We used a second rodent stress model to determine if LFM prevents intestinal inflammation and visceral hypersensitivity. Rats subjected to water avoidance stress (WAS) showed evidence of mucosal inflammation in the colon (Supplemental Table 3 and Figure 8A), impaired barrier function (Figure 8, B and C), increased serum LPS (Figure 8D), reduction of ZO-1 protein (Figure 8E), and development of visceral hypersensitivity (Figure 8F). These changes were prevented by LFM (Figure 8, A–F).
Figure 8. Effects of LFM on WAS-induced mucosal inflammation, permeability alteration, and visceral hypersensitivity.

(A) Real-time quantitative RT–PCR measurement of colonic cytokine levels in rats subjected to sham WAS, WAS, or WAS+LFM. WAS increased IL-1β, IL-6, IL-17, TNF-α, and IFN-γ gene expression, indicating low-grade mucosal inflammation. WAS increased colon permeability, as measured by TEER (B) and the appearance of FITC–dextran in serum (C), and was accompanied by increased serum LPS, indicating endotoxemia (D). WAS caused a decrease in ZO-1 expression (E) and visceral hypersensitivity (F). LFM prevented WAS-induced changes in cytokine levels, gut permeability, and visceral hypersensitivity. n = 6 per group. *P < 0.05 versus sham WAS; #P < 0.05 versus WAS. AUC, area under the curve; EMG, electromyographic activity; LFM, low-FODMAP diet; LPS, lipopolysaccharide; TEER, transepithelial electrical resistance; WAS, water avoidance stress. P < 0.05, by 2-tailed Student’s t test or 2-way repeated-measures ANOVA.
Patient demographics and IBS symptoms before and after LFM.
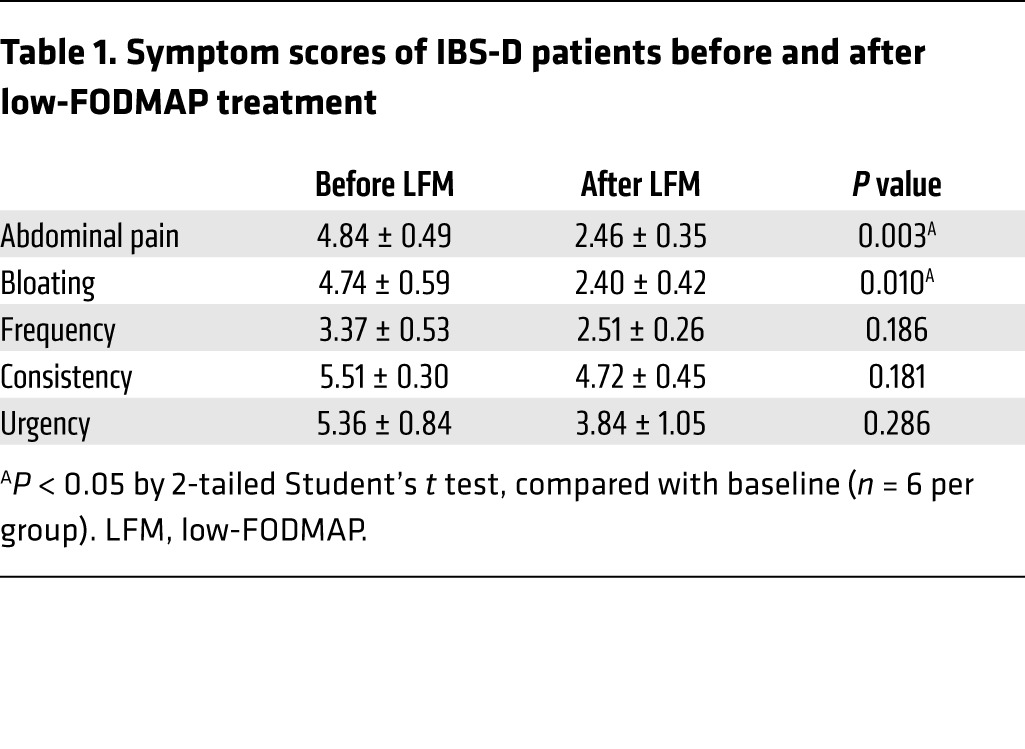
Six IBS-D patients (4 females and 2 males), ages ranging from 43 to 59 years old, were recruited from the University of Michigan GI outpatient clinic and primary care clinics. Six sex- and age-matched HS were recruited to serve as controls. All 6 IBS-D subjects were symptomatic, with symptom scores presented in Table 1. Following 4 weeks of LFM, all 6 patients experienced significant improvement of abdominal pain and bloating (Table 1). There was a trend towards symptomatic improvement with stool frequency, consistency, and urgency, but the differences did not reach statistical significance (Table 1).
Table 1. Symptom scores of IBS-D patients before and after low-FODMAP treatment.
Effects of fecal supernatant from IBS-D patients on visceral sensitivity of naive rats.
Endotoxin assays were performed on fecal samples from 6 HS and 6 IBS-D patients. These assays revealed a higher level of fecal LPS among IBS-D patients compared with HS (16.2 EU/μg vs. 7.9 EU/μg, P < 0.05). Four-week treatment with LFM diet lowered fecal LPS to a level similar to that of HS (Figure 9A).
Figure 9. Effects of fecal supernatant from IBS-D patients on visceral sensitivity.

(A) Endotoxin level in fecal contents was elevated in IBS-D compared with HS (n = 6). (B) Representative electromyogram recordings of responses to colorectal distension showing that an intracolonic infusion of fecal supernatant from IBS-D patients induced visceral hyperalgesia in naive rats. This action was abolished by LPS-RS or treatment with low-FODMAP diet. (C) Bar chart showing that an intracolonic infusion of fecal supernatant from IBS-D patients induced visceral hyperalgesia in naive rats; this was prevented by LPS-RS or treatment with 4 weeks of low-FODMAP diet. Mean amplitudes of abdominal muscle contractions are expressed as area under the curve (AUC) after baseline subtraction (n = 5 per group). *P < 0.05 compared with HS; #P < 0.05 compared with IBS (IBS-D); ‡P < 0.05 and †P < 0.05 compared with IBS. HC, healthy controls; HS, healthy subjects; LFM, low-FODMAP diet; LPS, lipopolysaccharide; LPS-RS, LPS antagonist. P < 0.05, by 2-tailed Student’s t test or 2-way repeated-measures ANOVA.
To evaluate the effects of fecal material on visceral sensitivity, fecal supernatant (0.3 ml) from each of the 6 IBS-D patients and each of the 6 HS was intracolonically administered to the colon of naive rats and then the VMR to CRD was evaluated 4–6 hours after supernatant administration. Intracolonic injection of IBS fecal supernatant evoked visceral hypersensitivity and caused an increase in VMR to CRD at 20, 40, 60, and 80 mmHg compared with fecal supernatant from HS (Figure 9, B and C). In contrast, the VMR to intracolonic administration of fecal supernatant from IBS-D patients following 4-week LFM diet caused an increase in VMR to CRD, which was similar to that observed in HS (Figure 9, B and C). Furthermore, coadministration of the LPS antagonist LPS-RS (100 μg/kg, intracolonically) prevented the development of visceral hypersensitivity caused by fecal supernatant from IBS-D, which indicates that elevated fecal LPS is responsible for the induction of visceral hypersensitivity. Thus, similar to our rat data, our human studies suggest that IBS-D patients have elevated fecal LPS levels resulting from gut dysbiosis and this appears to be responsible for the development of visceral hypersensitivity in IBS-D patients.
Discussion
Food is associated with symptom onset or exacerbation in many IBS patients. Ingestion of sugar (lactose or fructose), sorbitol, and oligosaccharides (fructans) alone or in combination may trigger abdominal symptoms (1, 17, 18). However, the mechanisms responsible for symptom generation in IBS remain poorly understood. In general, luminal water retention by osmotically active short-chain carbohydrates and increased gas production due to bacterial fermentation are believed to induce gastrointestinal symptoms in patients with visceral hypersensitivity (1).
Our study shows for the first time to our knowledge that HFM induces mucosal inflammation and impaired gut permeability. These changes are similar to subclinical mucosal inflammation and increased permeability in a subset of IBS patients (19, 20). Furthermore, we also observed a reduction of ZO-1 and OCLN gene expression in the colon of rats receiving an HFM. A similar reduction of ZO-1 and OCLN has been reported in IBS patients (21). It is conceivable that other tight junction proteins such as claudin-1, -3, -4, -5, and -8 that decrease paracellular permeability and claudin-2 that forms charge-selective paracellular pores could also be affected by HFM and this should be evaluated in future studies. In addition, the recognized correlation between impaired gut permeability and visceral hypersensitivity (22) was also observed in HFM rats. A cause-and-effect relationship between mucosal barrier alteration and visceral hypersensitivity has been described in a rodent model in which chemical blockade of enhanced stress-induced paracellular permeability was accompanied by reduced sensitivity to colonic distension (23). It is conceivable that HFM-induced endotoxemia contributes to hyperexcitability of dorsal root ganglia innervating the distal colon.
An increased serum LPS concentration has been reported in patients with IBS-D compared with HS (24). Endotoxins, which are a component of the outer membrane of Gram-negative bacteria, stimulate various inflammatory mediators. It is conceivable that LPS from Gram-negative bacteria gain access to the gut lamina propria to induce local inflammation (25). Inflammation leads to further elevation of blood endotoxin levels as a result of increased intestinal permeability due to mucosal damage (26).
We observed an overgrowth of Gram-negative bacteria and increased LPS concentration in fecal samples collected from the colon of HFM rats. We also noted that Akkermansia muciniphila, a genus in the phylum Verrucomicrobia, was significantly increased from 9% up to 38% in HFM-fed rats compared with the rats fed RC (Figure 3E). It is interesting to note that Halmos et al. observed that a typical Australian diet containing FODMAPs increased the relative abundance of Akkermansia muciniphila in IBS patients (27). Akkermansia muciniphila, a Gram-negative anaerobe and a mucin degrader that uses mucin as nutrients (28), is found in the gastrointestinal tract of more than 90% of humans. Usually, it only constitutes 1% to 4% of the fecal microbiota (29). The dramatic increase in Akkermansia muciniphila may have clinical significance. Although previous studies showed that Akkermansia muciniphila prevents colonic mucosal barrier dysfunction in obese mice and normalized metabolic endotoxemia (30), more recent studies reported that Akkermansia muciniphila has a deleterious effect on dextran sulfate sodium–induced colitis (31). Furthermore, LPS purified from Akkermansia muciniphila produces a strong inflammatory response characterized by the production of the NF-κB–dependent cytokines IL-10, TNF-α, IL-1β, and IL-6 similar to that elicited by Escherichia coli (32). In human studies, it has been reported that an increased number of Akkermansia muciniphila in IBS patients is closely correlated with episodes of abdominal pain (33, 34).
Intracolonic administration of fecal supernatant from HFM rats impaired colonic permeability, as reflected by reduced TEER, and increased leakage of fluorescent dye in human colonoids. The VMR to CRD was also increased. These abnormalities were prevented by coadministration of the LPS antagonist LPS-RS. Hence, HFM likely induces gut dysbiosis, causing an increase in fecal LPS, which may impair gut barrier function and induce visceral hypersensitivity.
LPS from Gram-negative bacteria bind to the LPS-binding protein coupled to CD14 on the cell surface. LPS–CD14 interacts with TLR4 to form a complex with another accessory protein, MD-2. The TLR4 signaling pathway is activated by the adaptor protein MyD88, which triggers the activation and translocation of NF-κB, and results in the transcription of cytokines, such as TNF-α (35). The molecular mechanisms by which LPS modulates the intestinal epithelium barrier function are unknown. Research suggests that LPS causes an increase in intestinal permeability via an intracellular mechanism involving a TLR4-dependent increase in membrane-associated protein CD14 expression (15). We show that silencing mucosal expression of TLR4 in the colon prevented the development of visceral hypersensitivity in rats receiving intracolonic administration of fecal supernatant from HFM rats. Thus, it appears that increased permeability of the intestinal mucosal barrier allows toxic substances and microorganisms to cross the intestinal epithelium, which results in elevation of the serum LPS and causes an increase in mucosal inflammatory tone and development of visceral hypersensitivity.
To determine if HFM and its metabolites induce mucosal changes and visceral hypersensitivity through direct actions on the gut epithelium and its immune system or through gut dysbiosis, we examined the effects of rifaximin, a nonabsorbable, broad-spectrum antibiotic. We have previously shown amelioration of mucosal inflammation and normalization of visceral hypersensitivity following rifaximin treatment in rats subjected to WAS by modulating host gut bacterial communities (16). In the current study, we show that rifaximin prevents HFM-induced mucosal inflammation, impaired gut permeability, and visceral hypersensitivity. It should be noted that under certain experimental conditions, rifaximin may exert its effects independently of its ability to alter the microbial composition. It was reported that rifaximin therapy ameliorated gut inflammation and endotoxemia in mice treated with humanized microbiota (36). This occurred without alteration in the relative microbial composition. However, it is well known that rifaximin causes a reduction in the total fecal bacteria count (16). Our studies show that in rats fed an HFM, rifaximin therapy reduced endotoxemia accompanied by a decrease in Gram-negative bacteria such as Verrucomicrobia. Hence, our data suggest that HFM causes these changes by inducing dysbiosis, which rifaximin prevents.
Although LFM appears to be effective in the treatment of IBS, little is known about the mechanism. Recognizing the lack of suitable IBS animal models, we chose 2 rat models of visceral hyperalgesia, and mucosal inflammation induced by RS or repeated WAS, to determine if LFM feeding alters gut microbiota, prevents subclinical intestinal inflammation, improves gut barrier function, and reduces visceral hyperalgesia. Psychological stressors have been shown to alter the bacterial community. Previous research using 454 pyrosequencing has shown that stress caused by prolonged restraint significantly changes the bacterial community structure, reduces species richness and diversity, and facilitates host susceptibility to a bacterial pathogen (37). Social stress also significantly alters bacterial community composition and decreases species richness and diversity (38). We have shown that chronic stress caused a significant loss of several bacterial family groups and decreased community diversity and evenness (16). These changes in bacterial community composition can contribute to mucosal barrier impairment and inflammation. In the current study, rats subjected to RS showed an increased relative abundance of Gram-negative bacteria, as well as an increase in LPS levels in the colon, accompanied by mucosal inflammation and impaired gut permeability. These abnormalities are similar to those observed in IBS patients (17, 19, 20). In the current study, we show for the first time to our knowledge that 2-week LFM prevents the development of mucosal inflammation, impaired permeability, and visceral hypersensitivity evoked by stress. Interestingly, LFM also prevents the increase in colonic Gram-negative bacteria and LPS. This suggests that LFM acts by modulating the gut microbiota, which seems to be affected by stress. This is not surprising, as HFM is poorly absorbed and likely serves as nutrients for proinflammatory commensal bacteria in the colon. In contrast, LFM is well absorbed in the proximal gut and is not available to colonic bacteria. Thus, it is conceivable that LFM modulates visceral nociception by altering gut microbiota and preventing intestinal inflammation.
It should be noted that rodent and human microbiota may differ significantly. Hence, caution should be exercised in extrapolating our data to explain human disease. However, it appears that foods differing in FODMAP composition induce some similar changes in colonic microbial compositions in rats and humans (27). In a human clinical study, we observed elevated fecal LPS levels among IBS-D patients compared with HS (Figure 9A). Four weeks of treatment with LFM diet may not only reduce abdominal pain and bloating but also normalize the fecal LPS to a level similar to that of HS. Similar to our findings in rats, we observed that intracolonic administration of fecal supernatant from IBS-D patients caused visceral hypersensitivity (Figure 9, B and C). Administration of LPS inhibitor, LPS-RS, prevented the development of visceral hypersensitivity caused by fecal supernatant of IBS-D patients (Figure 9, B and C). Following 4 weeks of LFM treatment, the fecal supernatant from the same 6 IBS-D patients did not evoke an abnormal increase in visceral pain in response to CRD (Figure 9, B and C). These findings confirm our observation in rats that FODMAP diet modulates visceral nociception by LPS-mediated intestinal barrier dysfunction and gut inflammation.
In conclusion, we have shown that an HFM diet causes an increase in fecal LPS, likely from gut dysbiosis. This induces mucosal inflammation, impairs permeability, and contributes to the development of visceral hypersensitivity. In contrast, an LFM diet reduces fecal LPS by modulating gut microbial composition. This decreases mucosal inflammation, improves gut barrier function, and prevents stress-induced visceral hyperalgesia. Similar observations were made in IBS-D patients who show an elevated fecal LPS level that was normalized following 4 weeks of treatment with LFM. Fecal supernatant from IBS-D patients receiving LFM diet also failed to evoke visceral hypersensitivity in naive rats. These observations may explain how an LFM diet benefits IBS patients.
Methods
Study approval.
All experimental procedures were performed in accordance with NIH guidelines and approved by the University Committee on Use and Care of Animals at the University of Michigan. The protocol for human colonoid culture was approved by the Institutional Review Board of the University of Michigan Hospital with written informed consent.
Animals, diets, and experimental procedures.
Adult male Wistar rats (200–220 g) were housed 3 per cage in a controlled environment (12-hour daylight cycle, lights off at 18:00) with free access to food and water. The composition of the HFM diet was based on a human clinical study (2): 10% w/w FODMAPs, comprising 3.5% w/w fructose, 3.5% w/w lactose, and 3% w/w oligofructose (D12020103, Research Diets). Rats were randomized into groups, and for 14 days were fed HFM or RC (2.1% FODMAPs). Rats were treated with oral gavage of rifaximin (150 mg/kg) (16) or water twice daily, 8 hours apart, for 14 days. In a separate study, rats were fed RC or an LFM diet (0% FODMAPs) (D12020102, Research Diets) for 14 days, and then exposed to daily WAS for 10 days or daily RS for 10 days. Each gram of HFM and LFM contains 3.89 and 3.96 kcal, respectively, with 6.7% and 6.9% of calories, respectively, provided by fat, 61.4% and 63.8% of calories provided by carbohydrate, and 19.5% and 19.9% of calories provided by protein.
Repeated RS.
Repeated RS was conducted as described previously (16).Each rat was submitted to a 120-minute restraint period in a plastic rat restrainer (diameter 6.5 cm and length 15 cm), daily, for 10 consecutive days. This restraint procedure minimized the space around the animal, prevented turning, and provided a rather strong stressful stimulus without causing lasting harm. After restraint, the rats were returned to their cages. The control group remained in their cages, undisturbed, throughout the experiment.
Chronic WAS.
Chronic WAS was conducted as described previously (16). Rats were placed on a block in the middle of a Plexiglas tank filled with water (25°C) to 1 cm below platform height. Animals were maintained on the block for 1 hour daily for 10 consecutive days. Sham WAS rats were placed similarly in a tank, but without water, for 1 hour daily for 10 days.
Microbial DNA isolation.
Genomic DNA was extracted by using a modified protocol of the Qiagen DNeasy Blood & Tissue Kit. These modifications included (a) adding a bead-beating step using Ultra Clean fecal DNA bead tubes (MO BIO Laboratories) that were shaken using a Mini-Beadbeater-16 (BioSpec Products) for 1.5 minutes, (b) increasing the amount of Buffer ATL (Qiagen) used in the initial steps of the protocol (from 180 μl to 360 μl), (c) increasing the volume of proteinase K used (from 20 μl to 40 μl), and (d) decreasing the amount of Buffer AE (Qiagen) used to elute the DNA at the end of the protocol (from 200 μl to 85 μl). All samples were weighed prior to the start of DNA isolation.
Illumina MiSeq sequencing.
Samples were submitted to the University of Michigan Host Microbiome Initiative and processed using the Illumina MiSeq platform. 16S rRNA gene libraries were constructed using primers specific for the V4 region.
Operational taxonomic units.
The open-source, platform-independent, community-supported software program, mothur (http://www.mothur.org) (39) was used and we followed the steps outlined in the mothurMiSeq SOP (http://www.mothur.org/wiki/MiSeq_SOP) (40). Sequences were classified using the Ribosomal Database Project (RDP) 16S rRNA gene training set (version 9) using a naive Bayesian approach with an 80% confidence threshold. Sequences that did not classify or were identified as Eukaryota, Mitochondria, Chloroplasts, Archaea, or Unknown were removed. We used the cluster.split command in mothur to cluster sequences into operational taxonomic units (OTUs) at a cutoff equal to 0.03. OTU sequence data were converted to relative abundance ± SEM.
Quantitative or semiquantitative RT-PCR for inflammatory cytokines.
Total RNA was extracted from proximal colon tissue samples using TRIzol reagent (Life Technologies), according to the manufacturer’s instructions. cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad Laboratories). Quantitative PCR (qPCR) for inflammatory cytokines and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed with a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories) using SYBR Green detection.
Primers used for qPCR, GAPDH, IL-1β, IL-6, IL-10, TNF-α, and IFN-γ were obtained from Qiagen. Primer sequences for IL-17 are as follows: forward IL-17, 5′-ACAGTGAAGGCAGCGGTACT-3′; reverse IL-17, 5′-GCTCAGAGTCCAGGGTGAAG-3′.
The PCR conditions were as follows: one cycle at 95°C for 10 minutes, followed by 40 two-temperature cycles at 95°C for 15 seconds and 60°C for 60 seconds. PCR amplifications were performed in a total volume of 25 μl, containing iQSYBR Green supermix (Bio-Rad Laboratories). Cytokine transcript levels were normalized to that of GADPH, and relative gene expression was expressed as the fold change (2−ΔΔCt) relative to expression in the control samples.
Western blot analysis.
Proteins were extracted from the proximal colon tissues and analyzed on Ready Gel Tris-HCl (Bio-Rad Laboratories). The tissues were homogenized in RIPA buffer (1% IGEPAL, 0.5% sodium deoxycholate, and 0.1% SDS in Tris-buffered saline solution [pH 7.4]), supplemented with protease inhibitor cocktail (Sigma-Aldrich). The homogenate was centrifuged at 14,000 g for 10 minutes. Equal amounts of protein (30 μg) were separated by 4%–20% Ready Gel Tris-HCl gels (Bio-Rad Laboratories), transferred to polyvinylidene difluoride membranes, and blocked with StartingBlockT20 blocking buffer (Thermo Fisher Scientific) for 60 minutes at room temperature. Membranes were incubated with rabbit anti–ZO-1 antibody (61-7300, Life Technologies) and rabbit anti-OCLN antibody (71-1500, Life Technologies) at 1:400 dilution at 4°C overnight, and then washed in Tris-buffered saline for 1 hour. The membranes were then probed with peroxidase-conjugated goat anti–rabbit IgG at 1:8,000 dilution for 1 hour at room temperature, and the bands were visualized by electrochemiluminescence (ECL, Thermo Fisher Scientific). Signals were quantified using ImageJ (NIH) and normalized to controls.
Histology and immunohistochemistry.
Segments of the proximal colon were fixed in 10% formalin. For morphologic analysis, colon segments were embedded in paraffin, sectioned at 5-μm thickness, and subsequently stained with H&E. Neutrophils, mononuclear cells, and eosinophils were identified in H&E-stained sections; mast cells were identified in toluidine blue–stained sections. Cells were counted at a magnification of ×400 in 8 different areas above the muscularis mucosae of each section using a micrometer grid and expressed as the number of cells/mm2.
Serum and fecal LPS levels.
LPS levels were measured with a quantitative chromogenic limulus amoebocyte lysate (LAL) QCL-1000 test kit (LONZA), following the manufacturer’s protocols. Serum and fecal samples were prepared in pyrogen-free water provided with the kit. Dilute serum samples were deactivated at 75°C for 10 minutes in a water bath. Samples were further incubated for 10 minutes with 50 μl of LAL reagent at 37°C, followed by the addition of 100 μl of LAL chromogenic substrate for 6 minutes. The reaction was terminated by adding 100 mg/ml of SDS, and the yellow color that developed due to cleavage of the substrate was measured spectrophotometrically at 405 nm.
TEER.
The intestinal barrier function was assessed by measuring the TEER of ex vivo tissues as reported previously (41, 42). TEER reflects paracellular resistance imparted by tight junctions and the lateral paracellular space and is a sensitive measure of barrier integrity (43).
Intestinal tissue from the proximal colon was isolated. Intestinal segments were opened along the mesenteric border, washed in phosphate-buffered saline (PBS), and cut into 5 × 7 mm pieces. Tissues were washed twice in sterilized PBS and transferred to Petri dishes containing DMEM culture medium. After a 30-minute incubation at 37°C and pH stabilization, the TEER was measured using the micro-Snapwell system with an Endohm SNAP electrode attached to an EVOM2 epithelial volt-ohm meter (World Precision Instruments) and expressed in ohms per square centimeter (Ω/cm2).
Gut permeability in vivo.
In vivo permeability measurement was modified from previously described methods based on gut permeability to 4-kDa FITC–dextran (16). Rats were fasted for 6 hours and gavaged with 4-kDa FITC–dextran (0.5 ml, 100 mg/ml). After 1 hour, whole blood was collected from the tip of the tail vein. Plasma was diluted in an equal volume of PBS (pH 7.4) and the FITC–dextran concentration was determined with a Synergy 2 microplate reader (BioTek), with serial dilutions of FITC–dextran used as a standard curve.
VMR to CRD.
The VMR to CRD was measured as previously described (16, 44–46). For surgical preparations, rats were anesthetized with a mixture of xylazine and ketamine. A 32-gauge stainless steel wire was implanted in the external oblique pelvic muscle 4–6 days before beginning the experimental procedure. Animals were habituated in the testing room and placed in a Plexiglas cylinder for 30 minutes before the CRD procedure was initiated. A latex balloon (5–6 cm in length) was attached to a flexible plastic tubing. The balloon was lubricated with mineral oil and inserted into the descending colon. The balloon was positioned such that its end was 1 cm proximal to the anus. The tubing was taped to the tail to prevent dislocation (44, 46). Rats were fully awake in a plastic cylinder during testing. A series of CRDs were conducted at constant pressures of 20, 40, 60, and 80 mmHg. Each distension consisted of 3 segments: a 20-second predistension baseline period, a 20-second distension period, and a 20-second period after termination of CRD with a 4-minute interstimulus period. Electromyographic (EMG) activity was amplified and digitized using a SPIKE2/CED 1401 data acquisition interface (Cambridge Electronic Design). The responses were considered stable if there was less than 20% variability between 2 consecutive trials of each CRD. EMG activity was rectified, and the increase in the area under the curve (AUC) of EMG amplitude during CRD over the baseline period before CRD was recorded as the response.
Clinical IBS studies.
IBS-D patients meeting the Rome III criteria were recruited from the gastroenterology and primary care clinics at the University of Michigan. After completing a 2-week screening period during which symptom severity was assessed based on daily abdominal symptom score and Bristol stool-scale score, patients were given 4 weeks of LFM diet under the supervision of an experienced research dietitian. Abdominal pain (11-point numerical rating scale [NRS]), bloating (11-point NRS), stool consistency (Bristol stool-form scale [BSFS]), stool frequency, and urgency (NRS) were assessed. Human fecal samples were collected from 6 patients showing significant reductions in abdominal symptoms before and following the 4-week LFM diet (Table 1). Fecal samples from 6 age- and sex-matched HS were also collected and stored in a freezer at –80°C. To prepare fecal supernatant, fecal samples were diluted (1 g fecal sample/5 ml PBS), homogenized on ice, and centrifuged (10, 000 g, 10 minutes, 4°C). Pellets were discarded, and supernatants were recovered. Bacteria were removed by 0.22-μm syringe filters. Supernatants were kept at –80°C.
Intracolonic infusion.
Following 2-week RC or HFM feeding, rat fecal samples were collected from the proximal colon. Fecal samples were diluted (1 g fecal sample/5 ml PBS), homogenized on ice, and centrifuged (10,000 g, 10 minutes, 4°C). Pellets were discarded, and supernatants were recovered. Bacteria were removed by 0.22-μm syringe filters. Supernatants were kept at –80°C.
Rats were fasted overnight and then placed in a plastic rat holder for 30 minutes to empty the bowel contents. Intracolonic infusion of fecal supernatant was performed using a flexible plastic tube (18-gauge, 3 inches, Instech Laboratories). The plastic tube was inserted into the distal colon 3 inches from the anus. Fecal supernatant (0.3 ml) or PBS was injected slowly for 1 minute (47). LPS from the photosynthetic bacterium Rhodobacter sphaeroides (LPS-RS) is a potent antagonist of LPS. In separate studies, intracolonic injection of LPS-RS (100 μg/kg, 0.3 ml, InvivoGen) was given 30 minutes prior to fecal supernatant administration. The VMR to graded CRD was performed 4–6 hours after infusion of the fecal supernatant.
Intracolonic siRNA administration.
Intracolonic application of siRNA was performed as described previously (48, 49). Rats were fasted overnight and then placed in a plastic holder for 30 minutes to empty the bowel contents. Lipofectamine 2000 (Invitrogen) was diluted with OptiMEM (Invitrogen) according to the manufacturer’s instructions and incubated for 5 minutes at room temperature. siRNA targeting TLR4 (Thermo Fisher Scientific) was dissolved in OptiMEM. These 2 solutions were then mixed at a 1:1 ratio and incubated at room temperature for 20 minutes. The combined Liposomal siRNA (300 μl) containing 10 nmol siRNA was then immediately injected into the rat colon at day 0 and day 1 using a flexible plastic tube (18-gauge, 3 inches, Instech Laboratories) inserted into the distal colon 3 inches from the anus. Experiments were perfumed 3 days after the first siRNA injection.
Human colonoid culture.
The protocol for colonic crypt isolation and culture was a modification of previously described protocols (50, 51). Colonic crypts were obtained from colonoscopy biopsy samples at the University of Michigan Hospitals. The study was approved by the Institutional Review Board of the University of Michigan Hospital with written informed consent. Patient biopsy samples were collected from 5 normal subjects. Colon biopsies were washed with PBS containing penicillin-streptomycin (Pen/Strep, 1× Invitrogen), gentamicin (50 μg/ml, Life Technologies), Normocin (100 g/ml, InvivoGem), and amphotericin (2.5 μg/ml, Sigma-Aldrich) to control contamination, with thiazovivin (2.5 μM, Stemgent), a potent inhibitor of Rho-associated coiled-coil–containing protein kinase (ROCK). After 3 washes over 30 minutes, tissue was incubated in nonenzymatic-digestion PBS containing 8 mM EDTA (Lonza) with DTT (Sigma) for 5 minutes to remove mucus and in 8 mM EDTA for 5–10 minutes for further digestion. All digestion-buffer PBS contains Pen/Strep (1×) and Normocin (100 μg/ml). Crypts were released by rhythmic shaking in PBS containing Pen/Strep (1×) for 1–2 minutes. Isolated crypts were centrifuged at 500 g for 1 minute. Colonic crypts were collected and plated in 24-well plates with growth factor–reduced Matrigel (Corning). The Matrigel was polymerized for 10 minutes at 37°C. R-spondin (500 μl) and Wnt3a-conditioned advanced DMEM/F12 culture medium were added to each well. The culture medium contained 1× GlutaMax, HEPES, N2 supplement, B27, insulin-transferrin-selenium (ITS), Pen/Strep, N-acetylcysteine (1 mM), nicotinamide (5 mM), as well as growth factor combinations, including murine EGF (100 ng/ml), noggin (100 ng/ml), A8301 (0.5 μM), SB202190 (30 μM), Chiron (10 μM), PGE2 (10 μM), thiazovivin (2.5 μM), and diluted anoikis inhibitor. After several passages, colonoids were cultured with fecal supernatant. After 24 hours, colonoids were harvested, and RNA was extracted for qPCR.
Colonoid microinjection.
Microinjections were performed on human colonoids with a mixture of fecal supernatant, LPS, or LPS-RS with FITC–dextran. Thin-wall glass pipettes (TW100F-4; World Precision Instruments) were pulled using a Narishige PN-30 micropipette puller. The glass pipettes tips were cut open and filled with a mixture containing 27 μl of fecal supernatant, 3 μl of 4-kDa FITC–dextran (10 mM, Sigma-Aldrich), and LPS (10 μg/ml) or LPS-RS (100 μg/ml) using Eppendorf Microloader pipette tips. Microinjections were performed with a XenoWorks BRI analog microinjector (Sutter Instrument). After injection, colonoid spheres were selected for 24-hour live-cell imaging. To determine the epithelial barrier function of the colonoid spheres after application of the supernatant, images of FITC–dextran in the colonoid spheres were captured every 30 minutes using the DeltaVision epifluorescence imaging system (GE Healthcare Bio-Sciences) at ×4 magnification and the pixel intensity measured. Colonoid spheres (n = 6–10) were selected for 24-hour live-cell imaging. Images were recorded by SoftWoRx imaging software (Applied Precision, GE Healthcare Bio-Sciences) for off-line analysis. The disruption of barrier integrity was visualized by the loss of FITC–dextran in the lumen of each colonoid sphere compared with the starting point (52).
Statistics.
EMR amplitude is represented by the area under the curve. The effects of stress and/or dietary treatment on VMR to CRD were analyzed by comparing poststress and baseline values at each distension pressure, using 2-way repeated-measures ANOVA. To examine data for other studies, differences between groups were compared by 2-tailed Student’s t test or ANOVA for comparisons between 2 groups or more than 2 groups, respectively. Results are expressed as the mean ± SEM. P less than 0.05 was considered statistically significant.
Author contributions
CO and SYZ conceived and designed the study. SYZ, MG, XW, GZ, HZ, YL, BY, and PL acquired the data. SYZ and MG analyzed and interpreted the data. SYZ, MG, and CO wrote the manuscript. CO obtained funding.
Supplementary Material
Acknowledgments
The studies were supported by NIH grants R01DK058913 and P30DK34933.
Version 1. 11/27/2017
Electronic publication
Version 2. 01/02/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(1):267–280.https://doi.org/10.1172/JCI92390.
Contributor Information
Shi-Yi Zhou, Email: zhousy@med.umich.edu.
Merritt Gillilland, III, Email: mgillill@med.umich.edu.
Xiaoyin Wu, Email: wxy@med.umich.edu.
Pornchai Leelasinjaroen, Email: tee_pornchai@yahoo.com.
Guanpo Zhang, Email: zhangguanpo@163.com.
Hui Zhou, Email: mdzhouhui@163.com.
Bo Ye, Email: bobmaster819@126.com.
Yuanxu Lu, Email: lu1234us@yahoo.com.
Chung Owyang, Email: Cowyang@umich.edu.
References
- 1.Shepherd SJ, Lomer MC, Gibson PR. Short-chain carbohydrates and functional gastrointestinal disorders. Am J Gastroenterol. 2013;108(5):707–717. doi: 10.1038/ajg.2013.96. [DOI] [PubMed] [Google Scholar]
- 2.Ong DK, et al. Manipulation of dietary short chain carbohydrates alters the pattern of gas production and genesis of symptoms in irritable bowel syndrome. J Gastroenterol Hepatol. 2010;25(8):1366–1373. doi: 10.1111/j.1440-1746.2010.06370.x. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd SJ, Parker FC, Muir JG, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence. Clin Gastroenterol Hepatol. 2008;6(7):765–771. doi: 10.1016/j.cgh.2008.02.058. [DOI] [PubMed] [Google Scholar]
- 4.Staudacher HM, et al. Fermentable carbohydrate restriction reduces luminal bifidobacteria and gastrointestinal symptoms in patients with irritable bowel syndrome. J Nutr. 2012;142(8):1510–1518. doi: 10.3945/jn.112.159285. [DOI] [PubMed] [Google Scholar]
- 5.Staudacher HM, Whelan K, Irving PM, Lomer MC. Comparison of symptom response following advice for a diet low in fermentable carbohydrates (FODMAPs) versus standard dietary advice in patients with irritable bowel syndrome. J Hum Nutr Diet. 2011;24(5):487–495. doi: 10.1111/j.1365-277X.2011.01162.x. [DOI] [PubMed] [Google Scholar]
- 6.Halmos EP, Power VA, Shepherd SJ, Gibson PR, Muir JG. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology. 2014;146(1):67–75.e5. doi: 10.1053/j.gastro.2013.09.046. [DOI] [PubMed] [Google Scholar]
- 7.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pastor Rojo O, López San Román A, Albéniz Arbizu E, de la Hera Martínez A, Ripoll Sevillano E, Albillos Martínez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(3):269–277. doi: 10.1002/ibd.20019. [DOI] [PubMed] [Google Scholar]
- 9.van Deventer SJ, Büller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990;76(12):2520–2526. [PubMed] [Google Scholar]
- 10.Dlugosz A, et al. Increased serum levels of lipopolysaccharide and antiflagellin antibodies in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil. 2015;27(12):1747–1754. doi: 10.1111/nmo.12670. [DOI] [PubMed] [Google Scholar]
- 11.Jalanka-Tuovinen J, et al. Faecal microbiota composition and host-microbe cross-talk following gastroenteritis and in postinfectious irritable bowel syndrome. Gut. 2014;63(11):1737–1745. doi: 10.1136/gutjnl-2013-305994. [DOI] [PubMed] [Google Scholar]
- 12.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10(2):131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharyya S, Dudeja PK, Tobacman JK. Lipopolysaccharide activates NF-kappaB by TLR4-Bcl10-dependent and independent pathways in colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G784–G790. doi: 10.1152/ajpgi.90434.2008. [DOI] [PubMed] [Google Scholar]
- 14.Lenoir C, et al. MD-2 controls bacterial lipopolysaccharide hyporesponsiveness in human intestinal epithelial cells. Life Sci. 2008;82(9–10):519–528. doi: 10.1016/j.lfs.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182(2):375–387. doi: 10.1016/j.ajpath.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu D, et al. Rifaximin alters intestinal bacteria and prevents stress-induced gut inflammation and visceral hyperalgesia in rats. Gastroenterology. 2014;146(2):484–96.e4. doi: 10.1053/j.gastro.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Symons P, Jones MP, Kellow JE. Symptom provocation in irritable bowel syndrome. Effects of differing doses of fructose-sorbitol. Scand J Gastroenterol. 1992;27(11):940–944. doi: 10.3109/00365529209000167. [DOI] [PubMed] [Google Scholar]
- 18.Ravich WJ, Bayless TM. Carbohydrate absorption and malabsorption. Clin Gastroenterol. 1983;12(2):335–356. [PubMed] [Google Scholar]
- 19.Ford AC, Talley NJ. Mucosal inflammation as a potential etiological factor in irritable bowel syndrome: a systematic review. J Gastroenterol. 2011;46(4):421–431. doi: 10.1007/s00535-011-0379-9. [DOI] [PubMed] [Google Scholar]
- 20.Dunlop SP, et al. Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am J Gastroenterol. 2006;101(6):1288–1294. doi: 10.1111/j.1572-0241.2006.00672.x. [DOI] [PubMed] [Google Scholar]
- 21.Bertiaux-Vandaële N, et al. The expression and the cellular distribution of the tight junction proteins are altered in irritable bowel syndrome patients with differences according to the disease subtype. Am J Gastroenterol. 2011;106(12):2165–2173. doi: 10.1038/ajg.2011.257. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Q, Zhang B, Verne GN. Intestinal membrane permeability and hypersensitivity in the irritable bowel syndrome. Pain. 2009;146(1–2):41–46. doi: 10.1016/j.pain.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ait-Belgnaoui A, Bradesi S, Fioramonti J, Theodorou V, Bueno L. Acute stress-induced hypersensitivity to colonic distension depends upon increase in paracellular permeability: role of myosin light chain kinase. Pain. 2005;113(1–2):141–147. doi: 10.1016/j.pain.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Ludidi S, et al. The intestinal barrier in irritable bowel syndrome: subtype-specific effects of the systemic compartment in an in vitro model. PLoS ONE. 2015;10(5):e0123498. doi: 10.1371/journal.pone.0123498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res. 2007;149(4):173–186. doi: 10.1016/j.trsl.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 26.van Deventer SJ, ten Cate JW, Tytgat GN. Intestinal endotoxemia. Clinical significance. Gastroenterology. 1988;94(3):825–831. doi: 10.1016/0016-5085(88)90261-2. [DOI] [PubMed] [Google Scholar]
- 27.Halmos EP, Christophersen CT, Bird AR, Shepherd SJ, Gibson PR, Muir JG. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut. 2015;64(1):93–100. doi: 10.1136/gutjnl-2014-307264. [DOI] [PubMed] [Google Scholar]
- 28.Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54(Pt 5):1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- 29.Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol. 2007;73(23):7767–7770. doi: 10.1128/AEM.01477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everard A, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. 2011;60(11):2775–2786. doi: 10.2337/db11-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang CS, et al. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS ONE. 2013;8(10):e76520. doi: 10.1371/journal.pone.0076520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vatanen T, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165(6):1551. doi: 10.1016/j.cell.2016.05.056. [DOI] [PubMed] [Google Scholar]
- 33.Saulnier DM, et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1782–1791. doi: 10.1053/j.gastro.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajilić-Stojanović M, et al. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1792–1801. doi: 10.1053/j.gastro.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 35.Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol. 2011;8(5):388–403. doi: 10.1038/cmi.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang DJ, et al. Rifaximin exerts beneficial effects independent of its ability to alter microbiota composition. Clin Transl Gastroenterol. 2016;7(8):e187. doi: 10.1038/ctg.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect Immun. 2010;78(4):1509–1519. doi: 10.1128/IAI.00862-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25(3):397–407. doi: 10.1016/j.bbi.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Söderholm JD, et al. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology. 2002;123(4):1099–1108. doi: 10.1053/gast.2002.36019. [DOI] [PubMed] [Google Scholar]
- 40.Bhunia A, Mohanram H, Domadia PN, Torres J, Bhattacharjya S. Designed beta-boomerang antiendotoxic and antimicrobial peptides: structures and activities in lipopolysaccharide. J Biol Chem. 2009;284(33):21991–22004. doi: 10.1074/jbc.M109.013573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El Asmar R, et al. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology. 2002;123(5):1607–1615. doi: 10.1053/gast.2002.36578. [DOI] [PubMed] [Google Scholar]
- 42.Tripathi A, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci USA. 2009;106(39):16799–16804. doi: 10.1073/pnas.0906773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buzza MS, et al. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci U S A. 2010;107(9):4200–4205. doi: 10.1073/pnas.0903923107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coutinho SV, et al. Neonatal maternal separation alters stress-induced responses to viscerosomatic nociceptive stimuli in rat. Am J Physiol Gastrointest Liver Physiol. 2002;282(2):G307–G316. doi: 10.1152/ajpgi.00240.2001. [DOI] [PubMed] [Google Scholar]
- 45.Chen SL, et al. Subdiaphragmatic vagal afferent nerves modulate visceral pain. Am J Physiol Gastrointest Liver Physiol. 2008;294(6):G1441–G1449. doi: 10.1152/ajpgi.00588.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bradesi S, et al. Repeated exposure to water avoidance stress in rats: a new model for sustained visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol. 2005;289(1):G42–G53. doi: 10.1152/ajpgi.00500.2004. [DOI] [PubMed] [Google Scholar]
- 47.Dabek M, et al. Intracolonic infusion of fecal supernatants from ulcerative colitis patients triggers altered permeability and inflammation in mice: role of cathepsin G and protease-activated receptor-4. Inflamm Bowel Dis. 2011;17(6):1409–1414. doi: 10.1002/ibd.21454. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, et al. Engineering mucosal RNA interference in vivo. Mol Ther. 2006;14(3):336–342. doi: 10.1016/j.ymthe.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 49.Palliser D, et al. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature. 2006;439(7072):89–94. doi: 10.1038/nature04263. [DOI] [PubMed] [Google Scholar]
- 50.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 51.Jung P, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17(10):1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 52.Leslie JL, et al. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun. 2015;83(1):138–145. doi: 10.1128/IAI.02561-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.