

Abstract
Germline mutations in the gene encoding tumor suppressor kinase LKB1 lead to gastrointestinal tumorigenesis in Peutz-Jeghers syndrome (PJS) patients and mouse models; however, the cell types and signaling pathways underlying tumor formation are unknown. Here, we demonstrated that mesenchymal progenitor- or stromal fibroblast–specific deletion of Lkb1 results in fully penetrant polyposis in mice. Lineage tracing and immunohistochemical analyses revealed clonal expansion of Lkb1-deficient myofibroblast-like cell foci in the tumor stroma. Loss of Lkb1 in stromal cells was associated with induction of an inflammatory program including IL-11 production and activation of the JAK/STAT3 pathway in tumor epithelia concomitant with proliferation. Importantly, treatment of LKB1-defcient mice with the JAK1/2 inhibitor ruxolitinib dramatically decreased polyposis. These data indicate that IL-11–mediated induction of JAK/STAT3 is critical in gastrointestinal tumorigenesis following Lkb1 mutations and suggest that targeting this pathway has therapeutic potential in Peutz-Jeghers syndrome.
Keywords: Gastroenterology, Oncology
Keywords: Gastric cancer, Mouse models, Tumor suppressors
Introduction
Germline mutations in LKB1 underlie the gastrointestinal (GI) hamartomatous polyposis and increased cancer risk in Peutz-Jeghers syndrome (PJS) (1). PJS and other hamartomatous polyps manifest as abnormal increase in number and length of epithelial glands together with increased stroma. By contrast to 2-hit tumor suppressor genes, the wild-type copy of LKB1 is typically retained in the epithelium of nondysplastic PJS polyps (2, 3), and accordingly Lkb1 acts as a haploinsufficient tumor suppressor in PJS mouse models (4). This together with the complex architecture of hamartomas has hampered studies investigating which cell type(s) initiate polyp formation. Indeed, it has even been proposed that PJS polyps represent prolapses of the mucosa and not tumors at all (5). A role for stromal smooth muscle cells was suggested by appearance of polyps at a low frequency in mice where Lkb1 deletion was limited to smooth muscle cells (6). The incomplete penetrance and small polyp size, however, suggested that other cell types may be involved. Also, Glu-Cre–mediated Lkb1 deletion — aimed to target epithelial enteroendocrine L cells — was tumorigenic (7), but interpretation of the result was complicated by targeting also to the mesenchyme. Of note, Lkb1 loss in both epithelial and stromal cells has been shown to induce tumorigenesis in different tissue contexts, e.g., lung epithelial deletion promoting KRAS-mutant lung cancer (8) and stromal deletion in the female reproductive tract driving ovarian and endometrial tumors (9).
The LKB1 kinase phosphorylates and activates a number of intracellular kinases including AMP-activated protein kinase α 1 (AMPKα1) and AMPKα2, NUAK1/2, SIK1–3, SNRK, BRSK1/2, and MARK1–4 and thereby regulates, for example, cell metabolism and polarity in a context-dependent fashion (10). Despite the multitude of substrates and pathways Lkb1 has been found to regulate, it is not clear which of these are critical for suppressing GI polyposis, although AMPK has been proposed as a likely candidate (11).
Recently, several reports have identified increased inflammatory signaling upon Lkb1 loss in different cell and tissue contexts, such as in skeletal muscle (12), macrophages (13), T cells (14), and lung cancer (15), implicating Lkb1 as a suppressor of inflammatory pathways. In PJS patients and in Lkb1+/– mice, inhibition of cyclooxygenase-2 with the selective inhibitor celecoxib demonstrated partial efficiency in reducing PJS polyposis by reducing the size but not the number of tumors (16). The potential role of inflammatory pathways in PJS pathogenesis is currently unknown.
In this study, we address the mechanism by which Lkb1 mutations lead to PJS tumorigenesis. Our results indicate clonal expansion of stromal cells and activation of JAK/STAT3 signaling in polyps, and indicate therapeutic efficiency with a JAK inhibitor.
Results
To investigate the role of stromal Lkb1 loss in PJS tumorigenesis we used 2 alternative strategies to delete Lkb1: the Twist2-Cre (also known as Dermo1-Cre) allele, aimed to target mesenchymal progenitor cells, and the fibroblast-specific protein-Cre (Fsp1-Cre, also known as S1004A-Cre) allele, aimed to target fibroblasts. As expected, reporter analysis in gastric mucosa, the predominant site of polyp development in PJS models (4), revealed that the Twist2-Cre allele induced Cre expression in multiple stromal lineages including smooth muscle cells and fibroblasts (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI93597DS1). Fsp1-Cre, on the other hand, induced Cre expression in a restricted subset of gastric stromal cells (Supplemental Figure 1B). Of note, unlike the exclusively stromal activity of the Twist2-Cre–knockin allele, the recombination pattern of the transgenic Fsp1-Cre allele also revealed rare activity in epithelial stem or progenitor cells, identified by about 50 (range 35–65) LacZ-positive gastric glands appearing across the entire glandular stomach (representing at most 0.05% of all glands) (Supplemental Figure 1C), emphasizing the importance of reporter alleles when analyzing the results using transgenic Cre lines.
We crossed the Twist2-Cre and Fsp1-Cre mice with a floxed Lkb1 allele and observed both Twist2-Cre;Lkb1fl/+ (hereafter referred to as Lkb1TwKO/+) and Fsp1-Cre;Lkb1fl/+ (hereafter referred to as Lkb1FspKO/+) mice born at expected frequency without noticeable abnormalities. To investigate the survival and possible tumor formation, we followed the well-being of the mice over time and euthanized them when signs of discomfort were visible. All Lkb1TwKO/+ mice (n = 7) were euthanized latest at 16 months of age due to poor health resulting from large gastric polyps, similarly to reports from Lkb1+/– mice (4), with a mean survival time of 13.3 months (Figure 1, A and B). In contrast, Lkb1FspKO/+ mice (n = 27) survived without signs of discomfort until the last point of observation at 17 months (Figure 1B). Next, we analyzed the gastric tumor burden from Lkb1TwKO/+ (n = 6) and Lkb1FspKO/+ mice (n = 8) at 11 months of age for comparison with earlier studies (4, 6). At this age, all Lkb1TwKO/+ mice had developed multiple polyps (12 to 28 per mouse, average 16), demonstrating full penetrance similar to Lkb1+/– mice (Figure 1, A, C, and D). Gastric polyps were also noted in Lkb1FspKO/+ mice but only in 50% (4 of 8) of mice and with only 1.3 polyps on average per mouse (Figure 1, C and D), indicating substantially lower tumorigenic potential, consistent with the survival analysis and low recombination frequency. As previously reported (4, 6, 7), intestinal polyps were rare in both mouse models. Remarkably, both the full penetrance and tumor burden in Lkb1TwKO/+ mice were comparable to Lkb1+/– mice (4, 6), demonstrating that heterozygous Lkb1 loss in stroma is sufficient for the full manifestation of PJS polyposis. Importantly, reporter analysis confirmed the exclusively stromal recombination also in the polyps of the Lkb1TwKO/+ mice (Figure 1E). The tumors in Lkb1TwKO/+ mice contained limited amounts of tumor-infiltrating immune cells, of which the vast majority were not recombined (Supplemental Figure 2A).
Figure 1. Mesenchymal loss of Lkb1 is sufficient to drive fully penetrant PJS polyposis in mice.

(A) Representative macroscopic images of wild-type, Lkb1+/–, Lkb1TwKO/+, and Lkb1FspKO/+ mouse stomachs at 11 months of age. Scale bars: 5 mm. (B) Survival curve of Lkb1+/– (n = 15), Lkb1TwKO/+ (TwKO/+, n = 7), and Lkb1FspKO/+ mice (FspKO/+, n = 27). Lkb1FspKO/+ mice were followed until 17 months, with no mortality observed. (C and D) Comparison of polyp number (nr) (C) and diameter (D) in Lkb1+/– (n = 15), Lkb1TwKO/+ (TwKO/+, n = 6), and Lkb1FspKO/+ mice (FspKO/+, n = 8) at 11 months of age. Lines depict mean and standard deviation. (E) Cre activity representing Lkb1 heterozygous cells as depicted by GFP signal in Lkb1TwKO/+;R26R-mTmG mouse antral polyp. Representative image is shown. Scale bars: 500 μm and 100 μm (zoom-ins).
Clonally expanding Lkb1-deficient stromal cells underlie polyp development.
Next, we studied the viability and tumorigenic potential upon homozygous loss of stromal Lkb1 expression. Twist2-Cre;Lkb1fl/fl offspring were not observed, indicating embryonic lethality similarly to Lkb1 full knockout mice (17), whereas Fsp1-Cre;Lkb1fl/fl (Lkb1FspKO/FspKO) mice were born at expected frequencies. Remarkably, in contrast to Lkb1FspKO/+ mice with low tumorigenic potential (Figure 1), we observed full penetrance of polyp development in the Lkb1FspKO/FspKO mice already at 4 months of age (n = 19; Figure 2, A and B). Thus, tumorigenesis in Lkb1FspKO/FspKO mice occurs earlier than in any previously described PJS model (4, 6, 7). This result demonstrates that loss of heterozygosity (LOH) of Lkb1 increases tumorigenic potential in targeted fibroblasts, consistent with observations in smooth muscle cells (6). Remarkably, analysis of tumors in Lkb1FspKO/FspKO;R26R-LacZ reporter mice revealed a striking expansion of stromal Lkb1-deficient cells filling the entire polyp lamina propria, a pattern that was repeated in all studied polyps (n > 20) (Figure 2C). On the other hand, the rare Cre activity in the epithelial glands driven by the Fsp1-Cre allele (Supplemental Figure 1C) was observed only in a subset of polyps that contained a low number of recombined glands appearing at a frequency comparable to that in the normal gastric tissue in Fsp1-Cre;R26R-LacZ reporter mice (Supplemental Figure 1D). Tumor-infiltrating immune cells and smooth muscle beneath the polyps were negative for X-gal (Supplemental Figure 2B). These results strongly argue that stromal, not epithelial, loss of Lkb1 is the main driver of PJS polyps.
Figure 2. Biallelic Fsp1-Cre driven loss of Lkb1 results in polyp development and robust expansion of Lkb1-deficient stroma.

(A) Representative macroscopic image of Lkb1FspKO/FspKO mouse stomach at 4 months (mo) of age. Scale bar: 5 mm. (B) Polyp number (nr) and diameter in Lkb1FspKO/FspKO mice (n = 18) at 4 months of age. Lines depict mean and standard deviation. The polyp number was significantly increased (P < 0.05) in comparison with Lkb1FspKO/+ mice at 11 months (Figure 1C). Polyp size was not significantly different between these cohorts (unpaired 2-tailed t test). (C) Histological section of an X-gal–stained polyp in an Lkb1FspKO/FspKO;R26R-LacZ mouse. Representative image is shown. Scale bars: 100 μm and 20 μm (zoom-in).
The rapid induction of polyposis in Lkb1FspKO/FspKO mice provided an opportunity to follow the fate of Lkb1-deficient stromal cells during tumorigenesis in Lkb1FspKO/FspKO;R26R-LacZ reporter mice. Interestingly, X-gal staining of sections from macroscopically normal stomach of Lkb1FspKO/FspKO;R26R-LacZ mice demonstrated occasional patchy accumulation of recombined cells in lamina propria between normal-looking antral glands (Figure 3A, left panel) as well as areas with mucosal alterations containing larger numbers of recombined cells (Figure 3A, middle panel). In tumors, the entire stroma was filled with Lkb1-deficient cells (Figures 2C and Figure 3A, right panel). This observation led us to hypothesize that stroma of the polyps is composed of 1 or multiple clones of Lkb1-deficient stromal cells.
Figure 3. Lkb1-deficient stromal cells expand clonally during polyp development.

(A) Representative X-gal–stained antral sections from Lkb1FspKO/FspKO;R26R-LacZ mice with increasing severity of polyposis at 3–4 months of age. Left: Macroscopically normal-looking gastric mucosa with local accumulation of Lkb1-deficient stroma. Middle: Larger expansion of Lkb1-deficient stroma with disorganized glands. Right: Antral polyp demonstrating stroma filled by Lkb1-deficient cells while epithelium remained entirely wild type. Scale bars: 100 μm. (B) Schematic presentation of R26R-Confetti allele. Cre-mediated recombination leads to excision of one of the 2-color cassettes and allows reorientation of the remaining cassette, resulting in the expression of 4 alternative fluorescent colors, nGFP, YFP, RFP, or mCFP (25). Cells expressing similar color are derived from a clonal origin. (C) Left: Low-magnification image of an Lkb1FspKO/FspKO;R26R-Confetti polyp. Note large foci expressing similar fluorescent colors. Right: Zoom-in images of areas with RFP and nGFP/YFP stroma. Representative image is shown. Scale bar: 50 μm.
To test whether the increase of the Lkb1-deficient stroma resulted from clonal expansion, we generated Lkb1FspKO/FspKO mice carrying the R26R-Confetti reporter. With this reporter, Cre-mediated recombination leads to expression of 1 of 4 possible fluorochromes (nuclear GFP, yellow fluorescent protein [YFP], red fluorescent protein [RFP], or cyan fluorescent protein [CFP]; Figure 3B) and areas of single color indicate clonal origin. Upon analysis of the Lkb1FspKO/FspKO;R26R-Confetti tumors, we noted large single-fluorochrome-expressing foci of stromal cells representing stromal clonal expansion events (Figure 3C), indicating oligoclonal origin of the tumor stroma. By contrast, the epithelial compartment in polyps did not display clonal growth, as demonstrated in polyps of Lkb1+/– mice carrying the Lgr5-EGFP-IRES-ERT2 allele with R26R-tdTomato reporter, where lineage tracing demonstrated a similar pattern in normal and tumor epithelium (Supplemental Figure 3A).
Confirming the tumors arising in Lkb1FspKO/FspKO and Lkb1TwKO/+ mice as PJS polyps, histological analysis demonstrated lobular structures (18) and branching stroma indistinguishable from Lkb1+/– and PJS polyps (4) (Supplemental Figure 3B). The stroma of all PJS models also expressed vimentin and α smooth muscle actin (αSMA) (Supplemental Figure 4A), indicating that the stromal cells have characteristics of activated and contractile myofibroblasts (19) consistent with PJS polyp stroma (6). Notably, in all models only a subset of mesenchymal cells expressed Fsp1 (Supplemental Figure 4A). The Fsp1-expressing cells were almost exclusively distinct from αSMA-expressing populations, with only 2.4% of cells expressing both markers, consistent with previous studies addressing the appearance of these markers in fibroblast populations (20) (Supplemental Figure 4B). Analysis of proliferation in polyps by Ki67 staining indicated an expanded epithelial proliferative zone as previously noted (21), as well as active stromal proliferation (Supplemental Figure 5). These results indicate that clonally expanding stromal myofibroblasts, together with reactively hyperproliferating epithelium, form polyps in PJS.
Loss of AMPK activity does not induce or augment PJS polyposis.
As an initial approach to decipher molecular mechanisms underlying stromal expansion and subsequent epithelial hyperproliferation following Lkb1 loss, we investigated the possible role of AMPK. Activation of AMPK depends on phosphorylation by Lkb1 (22) and AMPK has been hypothesized as a mediator of Lkb1 tumor suppressor function (11), mainly because of its capability to regulate mTORC1 signaling (23). To examine the role of AMPK, we reasoned that if Lkb1-loss-mediated reduction of AMPK activity drove PJS polyp development, mice lacking AMPK catalytic subunits AMPKα1 or AMPKα2 would develop polyps and/or augment polyposis when combined with Lkb1 heterozygosity. When analyzing the experimental cohorts, however, we noted that deletion of AMPKα1 did not result in polyp formation, nor did the Lkb1+/–;AMPKa1-/- double mutant show an increase in tumor number or size as compared with Lkb1+/– mice (Figure 4, A and B). Interestingly, the tumor number in AMPKa1-/-;Lkb1+/– mice was even lower than in Lkb1+/–mice. Deletion of AMPKα2 did not result in tumors or modify the tumor development in Lkb1+/– mice (Figure 4, A and C). These results show that deletion of one the catalytic subunits of AMPK is not sufficient for polyp development, nor does it synergize with Lkb1 heterozygosity to drive tumorigenesis.
Figure 4. Inactivation of AMPK does not lead to polyposis.

(A) Representative images of stomachs at 8 months of age with genotypes indicated. (B) Polyp number (left) and diameter (right) of mice of indicated genotypes at 8 months of age. Lkb1+/+;AMPKa1–/– (n = 4), Lkb1+/–; AMPKa1+/+ (n = 9), Lkb1+/-; AMPKa1–/– (n = 8). Lines depict mean and standard deviation. (C) Average polyp number (left) and diameter (right) of mice of indicated genotypes at 8 months of age. Lkb1+/+; AMPKa2–/– (n = 6), Lkb1+/–; AMPKa2+/+ (n = 10), Lkb1+/–; AMPKa2–/– (n = 10). Lines depict mean and standard deviation. (D) Representative images of stomachs at 17 months of age with genotypes indicated. (E) Average polyp number (left) and diameter (right) of mice of indicated genotypes at 17 months of age. Lkb1FspKO/+ (n = 27), AMPKa1–/–; AMPKa2FspKO/FspKO (n = 12). *P < 0.05 as assessed by unpaired t test. n.s., not significant. Lines in graphs depict mean and standard deviation. Two-tailed unpaired t test was used as a statistical test. Scale bars: 5 mm. nr, number; n.s., not significant.
Combined loss of AMPKα1 or AMPKα2 leads to embryonic lethality (24), prohibiting the study of full AMPK inactivity using whole-body-KO mice. Next, we took advantage of our finding that Lkb1FspKO/FspKO mice develop tumors fast with full penetrance (Figure 2) and created AMPKa1–/–;AMPKa2FspKO/FspKO mice, displaying null AMPK activity in Fsp1-Cre–expressing cells. The mice were born at expected ratios and did not display signs of declining health up to 17 months of age. Autopsy at 17 months did not reveal any tumors, in contrast to Lkb1FspKO/+ mice investigated at the same age (Figure 4, D and E). Thus, partial or complete loss of AMPK activity does not recapitulate Lkb1 loss in terms of polyposis phenotype, suggesting other pathways as critical mediators in PJS tumorigenesis.
RNA sequencing reveals upregulation of cytokine signaling in the polyps.
As a second approach to identify molecular mechanisms leading to stromal expansion and epithelial hyperproliferation, we analyzed the transcriptome from Lkb1FspKO/FspKO polyps where Lkb1 loss is biallelic and restricted to the stroma. RNA sequencing (RNA-seq) was performed from Lkb1FspKO/FspKO polyps (n = 6), adjacent mucosa (n = 4), and mucosa from wild-type littermates (n = 5). Principal component analysis indicated marked differences between polyps and nonaffected mucosa (Supplemental Figure 6, A and B), whereas the predisposed mucosa did not separate from control. The latter results were not unexpected considering the low number of recombined cells in the predisposed gastric mucosa (Supplemental Figure 1B).
Analysis of gene expression differences in polyps compared with adjacent normal mucosa indicated 2,045 significantly upregulated and 2,153 significantly downregulated genes. Of these, 1,104 (Supplemental Table 1) were upregulated and 926 (Supplemental Table 2) downregulated more than 2-fold. Our results correlated well with previous microarray analyses of intestinal polyps of PJS patients (21, 25) and gastric polyps of Lkb1+/– mice (21, 25), further validating the Lkb1FspKO/FspKO mouse as a PJS model (Supplemental Figure 6C). Importantly, we identified substantially larger significantly altered gene sets (Figure 5E), probably due to limited patient material used previously (25), highlighting the usefulness of disease models where tissue-specific genetic targeting may reduce the variability and help to distinguish disease-driving events from secondary changes. The RNA-seq results and the validity of the 3 mouse models used in this study were further confirmed by quantitative PCR (qPCR) of Wnt5a and Lrg1 (upregulated in PJS polyps), as well as Lgr5 and Grem2 (downregulated in PJS polyps) (25), demonstrating that stromally induced polyps recapitulate the findings from PJS patients also on the molecular level (Supplemental Figure 6D).
Figure 5. Activation of JAK/STAT3 in polyps.

(A and B) Heatmap of significantly overexpressed KEGG (A) and GO molecular function (B) signatures in our RNA-seq data set, shared with previously published PJS polyp Affymetrix data set and 2 Lkb1+/– mouse polyp Affymetrix data sets. (C) Western blot analysis of STAT3 (p-STAT3-Y705) and MAPK pathway (p-ERK1/2) activation in Lkb1FspKO/FspKO and Lkb1+/– polyps. (D) Representative immunohistochemical analysis of Ki67 and p-STAT3-Y705 in consecutive sections of an Lkb1FspKO/FspKO mouse gastric polyp. Yellow arrows, examples of stromal staining; black arrows, examples of epithelial staining. Scale bars: 100 μm. (E) Venn diagram of overexpressed genes identified in indicated experiments. Genes represented in the RNA-seq data set and at least one of the other data sets (21, 25) (136 genes) are listed in Supplemental Table 3.
Unbiased analysis of the deregulated gene sets indicated highest enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) cytokine-cytokine receptor interaction genes (upregulated) and oxidative phosphorylation genes (downregulated) in Lkb1FspKO/FspKO polyps (Supplemental Figure 7A). Similar analysis with Gene Ontology (GO) gene sets indicated highest enrichment of receptor-binding genes (upregulated) and oxidoreductase activity (downregulated) (Supplemental Figure 7B). In addition to increased glycolysis and reduced oxidative phosphorylation, previously reported in PJS tumors (23), both analyses pointed towards robust upregulation of cytokine signaling in the polyps. Importantly, combined analysis of our data set and the previously published data sets revealed that both KEGG Cytokine-Cytokine Receptor Interaction and GO Receptor Binding sets were top hits in gene set analysis (Figure 5, A and B). Interestingly, the JAK/STAT signaling pathway was significantly upregulated both in our RNA-seq data set and in the PJS polyp data set (Figure 5A), representing the only significantly activated molecular signaling pathway identified from both signatures in this analysis. Western blotting analysis confirmed that levels of activated (phosphorylated at Y705) STAT3 were clearly elevated in polyps from both Lkb1+/– and Lkb1FspKO/FspKO mice (Figure 5C) as compared with adjacent mucosa and/or the antral mucosa from wild-type littermates. We also observed the previously noted increase in ERK1/2 phosphorylation, indicating activated MAPK pathways (Figure 5C) (4). Interestingly, immunohistochemical analysis revealed activated STAT3 signaling not only in the Lkb1-deleted stroma, but also in the adjacent proliferative epithelium (Figure 5D), suggesting that stromal paracrine signaling leads also to epithelial STAT3 activation and proliferation in polyps.
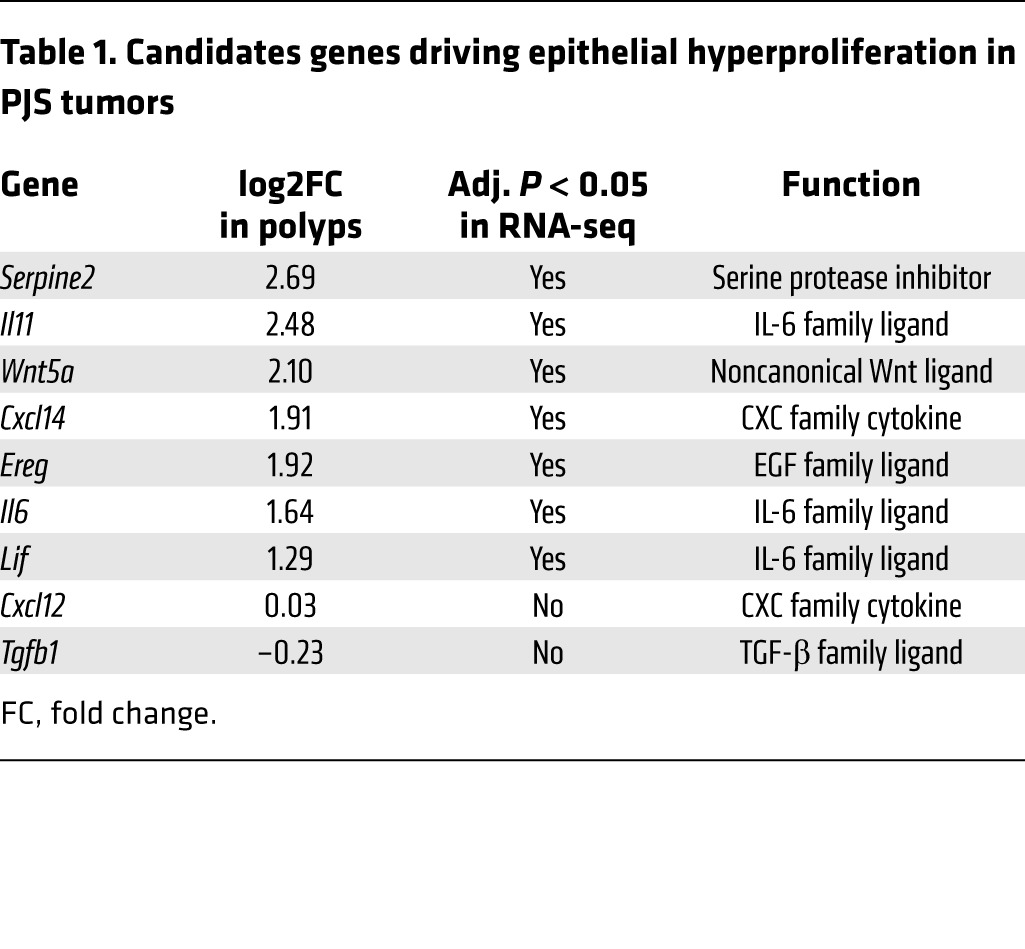
Next, we sought to identify potential paracrine factors responsible for epithelial proliferation and STAT3 activation in polyps. We constructed a list of all genes significantly overexpressed in our RNA-seq data set and at least one other available microarray data set derived from PJS (25) or Lkb1+/– mouse polyps (21, 25), to enrich for core changes taking place in more than one analysis (Figure 5E). From this list of 136 genes (Supplemental Table 3), we selected those whose products are known to be secreted, and which are expressed in the tumorigenic cell type, (myo)fibroblasts. These criteria resulted in identification of Serpine2, Il11, Wnt5a, Cxcl14, and Ereg as top candidates. In addition, we complemented the candidate list with Il6 and Lif, 2 members of IL-6 family that were upregulated in our RNA-seq analysis and are known activators of the JAK/STAT pathway. Further, we included Tgfb1, downregulated in PJS tumors and Lkb1-deficient fibroblasts (6, 26) and Cxcl12 (Sdf1), described to promote tumorigenesis through secretion from cancer-associated fibroblasts (CAFs) (27). The list of candidate paracrine factors included in further studies is given in Table 1.
Table 1. Candidates genes driving epithelial hyperproliferation in PJS tumors.
IL-11 as a potential activator of JAK/STAT3 signaling in polyps.
We wanted to investigate which of the identified candidate genes represent primary changes caused by loss of Lkb1 in the tumorigenic cell type (fibroblasts) rather than secondary changes reflecting the complex alterations of the polyp tissue. To this end, we utilized primary mouse embryonic fibroblasts (MEFs) isolated from Lkb1fl/fl embryos and deleted Lkb1 using AdCre (Figure 6, A and B). qPCR analysis indicated significant upregulation of Serpine2 (1.5-fold), Il11 (7.7-fold), Cxcl14 (7.7-fold), and Il6 (3.8-fold) in Lkb1-deficient cells (Figure 6C). Tgfb1 expression was slightly but significantly downregulated, consistent with previous reports (6, 26), while no significant differences were noted for Wnt5a, Ereg, Lif, and Cxcl12 expression. Of genes identified to be regulated by Lkb1, Il11 was particularly interesting as it is the critical cytokine promoting GI tumorigenesis in several mouse models (28). ELISA assay confirmed that in addition to mRNA expression, IL-11 secretion was dramatically (9-fold) increased in response to Lkb1 loss in primary MEFs (Figure 6F). Verifying the RNA-seq results, qPCR analysis of polyps indicated a prominent upregulation of Il11 mRNA in Lkb1+/–, Lkb1TwKO/+, and Lkb1FspKO/FspKO polyps compared with adjacent mucosa (Figure 6E).
Figure 6. Increased expression of Il11 in polyps and Lkb1-deficient fibroblasts.

(A) Western blot and (B) mRNA expression of Lkb1 in primary Lkb1fl/fl MEFs 2 passages after AdCre transduction. Control cells were transduced with AdGFP. (C) Relative mRNA expression of indicated genes after Lkb1 deletion. Data are shown relative to β-actin mRNA levels for triplicate samples and normalized relative to control (AdGFP) cells. MEFs derived from 3 independent embryos of the same genotype were used and data shown are the average of 3 experiments. (D) Relative mRNA expression of indicated genes after Lkb1 deletion. Average of triplicate samples is shown relative to control (AdGFP) cells. Primary gastric fibroblasts derived from 3 independent mice of the same genotype were used and data shown are the average of 3 experiments. (E) Relative expression of Il11 mRNA in PJS mouse model polyps compared with adjacent normal mucosa (genotypes indicated). n = 3–5 mice per data point. (F) ELISA measurement of secreted IL-11 from primary Lkb1fl/fl MEFs after Lkb1 deletion. MEFs derived from 3 independent embryos of the same genotype were used and data shown are the average of 3 experiments. (G) Western blot analysis of p-STAT3-Y705 in wild-type intestinal epithelial crypts incubated for 45 minutes with IL-6 or IL-11 (20 ng/ml). Representative blot of 2 independent experiments is shown. (H) qPCR analysis of Reg3b, Reg3b, and Lrg1 mRNA expression in intestinal organoids cultured with IL-6 and IL-11 (20 ng/ml). Average of 3 experiments is shown. Error bars denote SEM. *P < 0.05 as assessed by paired (C, D, F, and H) or unpaired (E) 2-tailed t test. In qPCR experiments, β-actin was used as the normalizing control. n.d., not detected
Despite being originally identified as secreted from fibroblasts, IL-11 can also be expressed by other cell types such as immune cells (29). Our transcriptional profiling experiments (Supplemental Figure 7) as well as histological analysis (Supplemental Figure 2) suggested immune cell infiltration in polyps as previously described in PJS and other hamartomatous polyps (30). To determine whether the tumor-initiating cells (fibroblasts) in the tumorigenic site (stomach) express IL-11, and whether IL-11 expression is induced by Lkb1 loss in these cells, we isolated primary gastric fibroblasts from adult (4- to 5-month-old) Lkb1fl/fl mice and deleted Lkb1 using AdCre. Expression levels of Il11, Cxcl14, and Il6 — genes with the strongest overexpression upon Lkb1 loss in primary MEFs — were analyzed. Of these genes, Il11 expression was significantly increased, demonstrating that Lkb1 inhibits Il11 expression also in gastric fibroblasts (Figure 6D). In addition, to verify that Lkb1 loss induces Il11 expression in gastric fibroblasts also in vivo, we isolated gastric fibroblasts from 2 Lkb1FspKO/FspKO mice and 1 control Fsp1-Cre mouse carrying the R26R-mTmG reporter, where mEGFP expression indicates Cre activity and mTomato expression in nonrecombined cells. These cells were immortalized by continuous passaging, allowing sufficient numbers for FACS. We sorted the fibroblasts based on membrane fluorescence expression and measured the expression of Lkb1 and Il11 by qPCR. We observed an approximately 50% reduction of Lkb1 mRNA and 7- and 24-fold increases in Il11 expression in the population of Lkb1FspKO/FspKO fibroblasts expressing GFP (mGFP) but not in the control, supporting our finding that Lkb1 deficiency leads to increased levels of IL-11 in gastric fibroblasts where Lkb1 is deleted in vivo (Supplemental Figure 8).
Next, we sought to study the potential downstream pathways leading to overexpression of inflammatory cytokines upon Lkb1 deletion. We targeted phosphorylation substrates of Lkb1 using conditional targeting (AMPKα1, AMPKα2) or small hairpin (sh)RNA approaches (Nuak1, Nuak2, Mark1, Mark2, Mark3, Mark4, Sik1, Sik2, and Sik3) and compared the transcriptional changes to Lkb1-loss-induced changes in primary MEFs (Supplemental Figure 9A). Simultaneous deletion of both catalytic AMPK subunits did not result in changes in Il11, nor of Il6 or Cxcl14 expression (Supplemental Figure 9B), consistent with our in vivo data suggesting that AMPK deletion is not sufficient for polyposis (Figure 4). Instead, we observed that silencing of Mark1 resulted in a significant increase in Il11 expression, and silencing of Mark4 and Sik1 led to similar trends (Supplemental Figure 9C). Cxcl14 was significantly upregulated by downregulation of Nuak2, Mark4, and Sik1 (Supplemental Figure 9C). These results suggest that multiple downstream pathways may contribute to the increased expression of Il11 and other proinflammatory factors in response to Lkb1 loss in fibroblasts, potentially including Mark1-, Mark4-, and Sik1-mediated effects.
IL-11 signaling is mediated via its binding to the transmembrane receptor Gp130, resulting in activation of the JAK/STAT and MAPK pathways (31). Of these, the induction of JAK/STAT3 activation appears most critical in GI tumorigenesis (32). To investigate whether IL-11 is sufficient to induce STAT3 activation in isolated wild-type epithelium, we incubated primary epithelial crypts isolated from mouse small intestine with recombinant IL-11. As expected, IL-11 resulted in activation of STAT3, comparably to IL-6 (Figure 6G). Next, we addressed whether STAT3-dependent transcriptional changes observed in polyps can be induced by IL-11 in primary epithelial organoid cultures. We focused our analysis on Reg3b, Reg3g, and Lrg1 genes, owing to their reported expression in inflammatory epithelium in a STAT3-dependent manner (33) and overexpression in PJS model polyps (Supplemental Figure 6D and Supplemental Table 3). Interestingly, IL-11 stimulation (but not IL-6) led to robust induction of Reg3b, Reg3g, and Lrg1 expression (Figure 6H), consistent with previous reports showing induction of Reg3g and Reg3b in vivo by ectopic IL-11 (34). These results demonstrate that IL-11 is sufficient to induce STAT3 activation and a regenerative program in adjacent epithelial cells.
Pharmacological inhibition of JAKs reduces PJS polyp development in mice.
STAT3 is phosphorylated and activated by Janus kinases (JAKs), which are potent drug targets for inflammatory and neoplastic diseases (35). The JAK1/2 inhibitor ruxolitinib (INCB018424) (36) has been approved for treatment of myeloproliferative diseases (37) and is currently being studied in clinical trials for treatment of several other diseases, including GI neoplasias. To address whether the observed STAT3 activation in PJS model polyps could be targeted therapeutically by inhibiting JAK, we fed Lkb1FspKO/FspKO mice with ruxolitinib-containing chow and analyzed tumor burden after treatment (Figure 7A). Intriguingly, after 6 weeks of treatment, 55% (6 of 11) of the ruxolitinib-treated mice were tumor free, while all (n = 11) untreated mice developed polyps. The average tumor number was reduced to 1.27 in treated compared with 6.54 in untreated animals (Figure 7, B and D). Also the average tumor area was dramatically reduced from 47.4 to 3.58 mm2 in the ruxolitinib-treated cohort (Figure 7, C and D). The average polyp diameter was also smaller (1.68 mm in treated vs. 2.51 mm in untreated mice), although this did not reach statistical significance (P = 0.082, Supplemental Figure 10A). The drug treatment did not alter the weight of the animals (Supplemental Figure 10, B and D), but did reduce spleen size in both Lkb1FspKO/FspKO and control mice (Supplemental Figure 10, C and E), consistent with the efficiency of ruxolitinib in treating splenomegaly in myeloproliferative disease (38). These experiments uncover a central and targetable role for JAK/STAT pathway activation in PJS polyp development.
Figure 7. Pharmacological JAK inhibition reduces polyposis.

(A) Outline of the experiment. Lkb1FspKO/FspKO mice were treated with ruxolitinib or control for 6 weeks and sacrificed at about 20 weeks of age. (B) Quantification of polyp number (nr) in control and ruxolitinib-treated mice. Lines depict mean and standard deviation. (C) Quantification of total polyp area in control and ruxolitinib-treated mice. Two-tailed unpaired t test was used as a statistical test. (D) Macroscopic images of representative untreated and ruxolitinib-treated mouse stomachs. Scale bars: 50 mm.
Discussion
In this study using previously uncharacterized PJS models we demonstrate that loss of Lkb1 in stroma leads to (a) clonal expansion of stromal cells and (b) activation of an inflammatory program involving the IL-11–JAK/STAT3 pathway critical for tumorigenesis. The clonal expansion of stroma driving hyperproliferation of epithelial glands can be seen as a reversal from traditional epithelial tumors where activated but nonclonal stromal fibroblasts support the growth of clonally expanding epithelial cells, and is interesting in light of previous suggestions of genetic coevolution (39) and even initiating roles proposed for stroma (40). Our findings are supported by the lack of clonal growth of polyp epithelium addressed in this study, and in PJS polyps previously analyzed with the HUMARA assay (3). Also, the observation that deletion of Lkb1 in GI epithelial cells does not result in a significant decrease in mouse lifespan (41) and that Lkb1 loss from smooth muscle cells using Tagln-CreERT2 led to development of PJS-type polyps, strongly support our view that stroma is the critical site of tumor suppression by Lkb1 (6). Interestingly, the observations here with Fsp1-Cre and earlier with Tagln-CreERT2 (6) suggest that homozygous deletion of Lkb1 in stroma enhances tumorigenicity while heterozygous deletion is sufficient for polyposis. In this light it will be important to focus future LOH studies in PJS on tumor stroma.
Our observations expand the findings deciphering the complex interplay of epithelium and stroma in hamartomatous tumors. In juvenile polyposis syndrome caused by mutations of SMAD4, second hits are not required for polyposis or progression (42) and either the epithelial component alone (43) or together with the mesenchyme (44) drives tumorigenesis based on clonality analysis. In PTEN hamartoma tumor syndrome (PHTS/Cowden syndrome), polyposis is initiated by PTEN-deficient epithelial stem cells (45) and epithelial loss of PTEN is sufficient for polyposis and to recapitulate PHTS features (46), suggesting a secondary role for the stroma. By contrast, in tuberous sclerosis hamartomas, biallelic stromal deletion of TSC1 or TSC2 drives hamartoma formation (47) through induction of paracrine factors activating the adjacent epidermis. Together, these results indicate gene- and context-specific etiologies for hamartomas and suggest that studies on hamartomas will provide insights into tissue morphogenesis, regeneration, and mesenchymal-epithelial interactions.
Regarding the stromal deletion approaches resulting in PJS polyps, Twist2-Cre is widely expressed in mesenchymal lineages, while Fsp1-Cre and Tagln-CreERT2 mainly target different cell types (fibroblasts vs. mature smooth muscle, respectively), raising a question about the stromal cell type(s) able to initiate PJS polyposis. On one hand, a differentiation program towards the tumorigenic myofibroblast could be initiated in different contexts following Lkb1 deletion; alternatively, the deletors may overlap in expression. Consistent with the latter model, Tagln can be expressed in a gastric myofibroblasts (48); thus, it is possible that myofibroblasts represent the tumorigenic cells in all PJS models.
The induction of an inflammatory program and JAK/STAT signaling was a major alteration in PJS and mouse model polyps noted in unbiased analyses in this study. This finding was interesting considering that secretion of inflammatory cytokines, many with capacity to trigger JAK/STAT pathway activation, represents an important tumor-promoting mechanism of activated fibroblasts (49), suggesting that similar activities may be important in the fibroblast-epithelium interplay in PJS polyps. Interestingly, a recent study distinguished 2 populations of CAFs in pancreatic cancer: CAFs with high expression of ɑSMA (termed myCAFs) and CAFs with lower levels of ɑSMA (termed iCAFs) (50). The iCAF cells expressed high levels of inflammatory cytokines and induced JAK/STAT signaling in pancreatic epithelial cells. Considering that loss of Lkb1 in mesenchymal cells leads both to a reduced expression of ɑSMA (26) and induces an inflammatory signature (this study), it would be interesting to study whether the iCAF population in pancreatic cancer shares other similarities with Lkb1-deficient fibroblasts in PJS polyps.
Emphasizing the importance of activated STAT3 signaling in GI tumorigenesis, mice carrying a STAT3-activating mutation in the Gp130 gene develop tumors in the gastric antrum (51), also representing the most frequent site of PJS-type polyp development. Gp130 is the receptor for IL-6–family cytokines, including IL-11. Regarding the mechanisms of STAT3 activation in PJS models, we showed here that Lkb1-deficient fibroblasts express and secrete increased amounts of IL-11, which was also strongly upregulated in polyps and sufficient to induce epithelial STAT3 activation. Further, recombinant IL-11 but not IL-6 induced a set of STAT3-dependent epithelial genes in cultured epithelial organoids; this set was also upregulated in polyps. Notably, previous studies have demonstrated the importance of IL-11 in GI tumorigenesis; antral tumorigenesis in gp130-mutant mice (51) can be blocked by deletion of the gp130 coreceptor IL-11Ra critical for IL-11 signaling, but not by deleting IL-6 (52, 53). IL-11 signaling appears to play an important role also in colitis-associated colon cancer as well as in intestinal tumorigenesis in Apc-min mice (54). In the future, it would be interesting to address the requirement of IL-11 in PJS tumorigenesis by deleting Lkb1 together with IL-11 from stromal fibroblasts.
Our finding that JAK inhibition using ruxolitinib dramatically reduced polyp development in Lkb1FspKO/FspKO mice confirmed that activated JAK/STAT signaling is a central pathway driving polyposis. Previous preclinical studies using the COX-2 inhibitor celecoxib (16) and the mTOR inhibitor rapamycin (23, 55) have not resulted in a comparable reduction, and could partly represent unspecific inhibition of growth. Our mechanistic studies indicating that the AMPK/mTOR axis is not sufficient for polyposis and rather implicating cooperation of other Lkb1 substrate kinases may partly explain these results. The identification of a clinically approved drug that efficiently inhibits tumorigenesis in PJS models suggests therapeutic opportunities also for PJS patients.
In summary, our study demonstrates that PJS polyposis is a stromal disease mediated by abnormal stroma-epithelium crosstalk including an activated IL-11–JAK/STAT3 axis. These findings significantly advance our knowledge of PJS, provide therapeutic opportunities, and may have broader implications for other tumors.
Methods
Animals.
The following mouse alleles were used in this study: Fsp1-Cre Twist2-Cre (Dermo1-Cre) (57), Lgr5-EGFP-IRES-CreERT2 (58), Lkb1fl/fl (4), Lkb1 KO (59), Rosa26R-LacZ reporter (60) Rosa26R-mTmG reporter (61), Rosa26R-Confetti reporter (62), Rosa26R-tdTomato reporter (63), AMPKɑ1 KO (64), AMPKɑ2 KO (65), and AMPKɑ2fl/fl (65). All experimental cohorts were maintained at mixed genetic background, and littermate controls were used in all analyses unless indicated otherwise. Both male and female mice were used. The mice had free access to food (Harlan Teklad, 2916) and water throughout the studies and were group housed when possible. Tamoxifen induction was performed by intraperitoneally injecting 2- to 3-month-old mice with 0.1 mg tamoxifen (25 mg/ml) per 1 g body weight every other day for 5 days. All animals were housed in a specific pathogen–free facility at the University of Helsinki. Animals were housed in individually ventilated cages containing 2–4 mice per cage, except in rare cases where single housing was needed.
Ruxolitinib treatment.
The ruxolitinib (Incyte, INCB18424) treatment was done in littermate male and female Lkb1FspKO/FspKO mice. The drug was given orally as a chow mixed in the rodent chow (LabDiet, 5002) in the ratio of 2 g of ruxolitinib to 1 kg of chow as previously described (66) except that the treatment period was 6 weeks (n = 11, 6 females and 5 males). The control group (n = 11, 5 females and 6 males) received the same rodent chow without ruxolitinib for 6 weeks. Both groups were fed ad libitum. Wild-type (Lkb1fl/fl without the Cre allele) littermate mice were also treated with ruxolitinib and control diet (n = 5 each, 3 females and 2 males) for 6 weeks to control for adverse effects. At the endpoint, body weight and spleen weight were recorded and tumor number and size counted.
Analysis of tumor size and multiplicity.
The mice were euthanized by CO2 followed by cervical dislocation at indicated time points or when showing poor health. The GI tract was removed and flushed with ice-cold PBS and fixed with 4% paraformaldehyde (PFA) overnight. The stomach and proximal part of the duodenum were inflated with 4% PFA. The stomach was opened under a stereomicroscope (Leica MZFIII), imaged (Leica ES3 camera and LAS software), and tumor number and size recorded before embedding the tissues in paraffin. Graphs were drawn and statistical analysis performed using Prism 7 software (GraphPad).
Histological analysis and immunohistochemistry.
PFA-fixed samples were embedded in paraffin using standard procedures. Tissue sections were cut (5 μm) and stained with hematoxylin and eosin using routine protocols. For immunohistochemistry, sections were deparaffinized in xylene and ethanol series. Antigen retrieval was done in 1× antigen retrieval solution pH 6.5 (Dako) in a 95°C water bath for 20 minutes followed by incubation at room temperature for 25 minutes. Endogenous peroxidase activity was quenched with 3% H2O2 for 10 minutes. The following antibodies were used: anti-Fsp1 (S1004A) (DAKO, A5114, 1:500 dilution), anti-αSMA (Sigma, A2547, 1:1,000 dilution), anti-vimentin (Abcam, ab92547, 1:1,000 dilution), anti-Ki67 (Abcam, ab15580, 1:500 dilution), anti-GFP (Abcam, ab5450, 1:500 dilution), and anti-RFP (Rockland, 600-401-379, 1:400 dilution). Antibodies were incubated overnight at 4°C and Mouse-on-Mouse or Rabbit-on-Rodent polymers (Biocare) and DAB reagent (Dako) were used for signal detection. For Fsp1 and αSMA costaining, a confocal microscope (Leica TCS SP8 C-ARS) was used. For immunofluorescent detection of GFP and TdTomato, antigen retrieval was performed by using 1× antigen retrieval solution pH 9.0 (Dako) in a 95°C water bath for 20 minutes followed by incubation at room temperature for 30 minutes. Alexa Fluor 488–conjugated anti-goat and Alexa Fluor 546–conjugated anti-rabbit secondary antibodies were used for visualization. The slides were counterstained with hematoxylin and scanned with a digital slide scanner (3D Histech Pannoramic 250 FLASH II). Pannoramic Viewer software (3DHistech) was used to export images and set scale bars. Sections from at least 3 different polyps per genotype were stained to ensure reproducibility, and representative images are shown in the figures.
Frozen sections.
Frozen sections were used to image the fluorescent reporters (R26R-mTmG and R26R-Confetti). Tissues were fixed in ice-cold 4% PFA for 4 hours, incubated in 30% sucrose in PBS for 1 hour, and embedded in OCT compound (Algol). The OCT blocks were stored at –80°C. Sections (5 μm) were cut using a cryotome (Leica) and stored at –80°C. After Hoechst staining, direct fluorescence was imaged using a Zeiss Axioplan epifluorescence microscope and Zeiss Axio Imager software, or slides were scanned with a digital slide scanner (3D Histech Pannoramic 250 FLASH II). ImageJ (NIH) and Pannoramic Viewer were used to process the images. Sections from at least 3 different polyps were analyzed and representative images are shown in the figures. For immunofluorescent staining, sections were incubated overnight with anti-CD45 antibody (Abcam, ab10588, dilution 1:150). Alexa Fluor 647–conjugated anti-rabbit was used as secondary antibody.
X-gal staining.
For whole-mount X-gal staining, tissues were incubated in 4% PFA/0.2% glutaraldehyde for 1 hour, followed by 3 washes in wash buffer (2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40 in PBS pH 7.4). Staining was done overnight at room temperature using X-gal staining solution (5 mM potassium ferricyanide [K3Fe(CN)6], 5 mM potassium ferrocyanide [K4Fe(CN)6-3H2O], and 1 mg/ml X-gal in wash buffer). Tissues were rinsed with wash buffer and imaged with a stereomicroscope (Leica MZFIII) followed by postfixation in 4% PFA and paraffin embedding. Tissue sections were cut (5 μm) and counterstained with Nuclear Fast Red (Sigma). Slides were imaged using a light microscope (Olympus BX41) and an attached camera (Leica DFC480) or scanned with a digital slide scanner (3D Histech Pannoramic 250 FLASH II). Pannoramic Viewer software was used to export images and set scale bars.
RNA extraction from tissues and cultured cells.
Gastric polyps and adjacent normal tissue were dissected and immersed in 10 volumes of RNAlater tissue preserving reagent (Ambion). After a minimum of 24 hours at 4°C, gastric mucosa was separated from external muscle layers under a stereomicroscope. Total RNA from mucosa and polyps was extracted using an RNAeasy Plus Kit (Qiagen). Total RNA from cultured cells was extracted using TRI Reagent (MilliporeSigma) according to the manufacturer’s recommendations. RNA concentrations were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific).
cDNA synthesis and qPCR.
A total of 1,000 ng RNA was used as template in cDNA synthesis in a total volume of 50 μl using a Taqman reverse transcription kit according to the manufacturer’s recommendations (Applied Biosciences). qPCR was performed in 18 μl using Kapa SybrFAST reagents (Kapa Biosystems) and StepOnePlus quantitative PCR platform (Thermo Fisher Scientific). The cycle threshold was set to 0.1 and samples were run in duplicate. β-Actin was used as the normalization control. All primers were validated to be at expected length using agarose gel electrophoresis, and a single peak was observed in melt curve analysis. All primers were intron spanning and controls without reverse transcriptase were used to ensure efficient depletion of genomic DNA. Primer sequences are listed in Supplemental Table 4.
RNA-seq and data acquisition.
For RNA-seq, RNA from 6 polyps and 4 adjacent mucosa samples was isolated from 4 tumor-bearing mice (Lkb1FspKO/FspKO) and normal mucosa samples from 5 wild-type littermates. From each mRNA sample, a library was constructed using a TruSeq Stranded mRNA Library Prep Kit according to the manufacturer’s instructions (Illumina, Inc.). The libraries were single-end sequenced using a NextSeq 500 sequencer (Illumina, Inc.). RNA-seq single-end reads were aligned to the ENSEMBL mouse reference genome (GRCm38 release 81) using the ultrafast universal RNA-seq aligner (STAR_2.4.2) (67). Genewise counts were assessed with HTSeq-count_v.0.6.1 in union mode. Raw count reads were imported to R/Bioconductor and analyzed for differential gene expression using the DESeq2 package (68). Principal component analysis and hierarchical clustering of the Euclidean distances between samples were performed upon regularized-logarithm-transformed data, as suggested in the DESeq2 package documentation. The data files are available in ArrayExpress with accession number E-MTAB-5549.
GSEA analyses and overrepresentation analyses of GO and KEGG pathways.
Gene set enrichment analysis (GSEA) software (69) was used to compare previously published data sets to the fold-change ranked list of our RNA-seq results. Our lists of significantly up- and downregulated genes were compared to GO Molecular Function and KEGG gene lists using Molecular Signatures Database (70). Significance of overlaps was assessed by hypergeometric test.
Protein extraction and Western blotting.
Tissues were dissected on ice, snap frozen, and stored at –80°C. Tissue samples were homogenized by sonication in RIPA buffer (25 mM Tris-HCl pH 7, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) containing 1× protease inhibitor (Pierce) and 1× phosphatase inhibitor cocktail (Roche) and incubated 20 minutes on ice before centrifugation at 18,000 g for 15 minutes. Supernatants were collected and snap frozen. For cultured cells, protein extracts were prepared by suspending cell pellets in boiling SDS lysis buffer (2% SDS, 60 mM Tris-HCl pH 6.8, 10% glycerol). Samples were incubated 5 minutes on a 95°C heat block and homogenized by passing them through a 27-gauge needle 5 times, and cleared by centrifugation at 18,000 g for 15 minutes. The supernatant was collected and snap frozen. Protein concentrations were measured using a Bio-Rad DC protein assay. Lysates (20 μg) were run in precast 10% acrylamide gels (Bio-Rad) and wet blotted overnight onto nitrocellulose membranes (PerkinElmer). After Ponceau staining, the membranes were blocked with 5% milk in TBS-Tween (0.1%) and incubated with primary antibodies overnight at 4°C. HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (MilliporeSigma) and Supersignal Femto chemiluminescence reagents (Thermo Fisher Scientific) were used before exposing the membranes on films (Figure 5) and scanning the membranes with a chemiluminescence scanner (Bio-Rad) (Figure 6). In primary crypt isolations, crypts were pelleted and lysed in 100 μl 2× Laemmli buffer, boiled for 5 minutes, and homogenized by passing through a 20-gauge needle 5 times. Ten microliters of each lysate was used for Wester blotting. The primary antibodies and dilutions were as follows: anti-GAPDH (Cell Signaling Technology, 2118S, 1:10,000), anti–STAT3-phosphoY705 (Cell Signaling Technology, 9145, 1:1,000), anti-STAT3 (Cell Signaling Technology, 9132, 1:1,000), anti-ERK1/2-phosphoT202/204 (Cell Signaling Technology, 4695, 1:1,000), anti-ERK1 (Santa Cruz Biotechnology, sc-94, 1:1,000), and anti-Lkb1 (Abcam, ab15095, 1:1,000).
MEF isolation and adenoviral transduction.
Primary MEFs were prepared and genotyped from E13.5 pregnant Lkb1fl/fl females mated with Lkb1fl/+ males or from AMPKɑ1fl/fl;AMPKɑ2fl/fl females mated with AMPKɑ1fl/+;AMPKɑ2fl/+ males using routine protocols and frozen at passage 2. MEFs were maintained in DMEM with 10% FBS, 1× penicillin-streptomycin, and L-glutamine. Cells were transduced with CMV5-AdCre virus (Viral Vector Core Facility, University of Iowa, Iowa City, Iowa, USA) at MOI 1,000. Control cells were transduced with CMV5-AdGFP viruses. For adenoviral transduction, 1,000,000 cells from passage 2 or 3 were seeded on 10-cm2 dishes 1 day before transduction. Gene and protein expression was assessed 2 passages after transduction. For each single transduction, MEFs were derived from a single embryo. Independent isolations were used for triplicate experiments.
Crypt isolation and organoid culture.
Small intestinal crypts were isolated and cultured as described previously (71) with minor modifications. Intestinal pieces were incubated in 10 mM EDTA in PBS for 105–115 minutes with 3 changes of buffer. Crypts were dissociated by gentle shaking and filtered through 70-μm filters, pelleted, and suspended in crypt basal media (72) containing 10 μM Y-27632 (MilliporeSigma). The suspension was mixed with 2 volumes of growth factor–reduced Matrigel (Corning), and 25–30 μl drops were plated on 48-well plates. Drops were overlaid with 300 μl ENR media as described previously (71) except that R-spondin was used at 250 ng/ml. When applicable, 20 ng/ml of IL-6 and IL-11 (both R&D Systems) were added to the growth media. Media were replaced every 2 or 3 days. For p-STAT3 induction, freshly isolated crypts (approximately 10,000 crypts per condition) were incubated for 45 minutes at 37°C with 100 μl basal crypt media containing 10 μM Y-27632 and, when applicable, 20 ng/ml of IL-6 or IL-11.
shRNA constructs and lentiviral transduction.
The following shRNA sequences subcloned into the pLKO.1 vectors were obtained from The RNAi Consortium (TRC) library (https://www.broadinstitute.org/rnai-consortium/rnai-consortium-shrna-library): Nuak1, TRCN0000024112; Nuak2, TRCN0000024271; Mark1, TRCN0000024173; Mark2, TRCN0000023988; Mark3, TRCN0000024107; Mark4, TRCN0000024281; Sik1, TRCN0000024098; Sik2, TRCN0000024288; and Sik3, TRCN0000079132. For production of lentiviral particles, 293FT cells were seeded at a density of 6 × 106 cells per 10-cm plate in antibiotic-free DMEM with 10% FBS. The next day, cells were cotransfected with the shRNA plasmids and lentiviral packaging plasmids pCMV-dR8.91 and VSV-G according to recommendations by the Broad Institute (http://www.broadinstitute.org/rnai/public/resources/protocols). Lentiviral particles were collected 48 hours after transfection. For transduction, 180,000 MEFs were seeded in a 6-well dish in DMEM containing 10% FBS and 1× penicillin-streptomycin. The next day, cells were washed with PBS and transduced with 1 ml of lentiviral stock mixed with 1 ml of complete growth media and 8 μg/ml polybrene. Media were replaced after 24 hours and 1 μg/ml puromycin selection was started after 48 hours. Two rounds of selection (48 hours each) were done before collection of RNA.
IL-11 ELISA.
Primary MEFs (200,000 cells) were seeded in duplicate on 6-well plates in 2 ml complete media 2 passages after AdCre/AdGFP transduction. Media were harvested after 24 hours, centrifuged, aliquoted, and stored at –80°C. A Mouse IL-11 ELISA Kit (MilliporeSigma) was used to quantify the IL-11 in the culture media.
Isolation and FACS of gastric fibroblasts.
Gastric fibroblasts were isolated as described previously (73) with minor modifications. Mouse glandular stomach was isolated approximately at 4 months of age and washed thoroughly with PBS. The muscular layer was removed under a dissection microscope, and the mucosa chopped to pieces and incubated with 1 mg/ml collagenase I (Life Technologies) at 37°C for 30–60 minutes. Tissue pieces were washed with DMEM containing 10% FBS and plated on collagen-coated plates in DMEM with 20% FBS. Fibroblasts grew to confluence within 3 weeks. Primary cultures were used at passage 2 or 3. Continuous passaging led to spontaneous immortalization allowing FACS and subsequent culture. For FACS isolation, cells were suspended in DMEM without supplements and filtered through a 40-μm mesh. EGFP- and Tomato-expressing cells were isolated using a BD FACSAria II cell sorter (BD Biosciences).
Statistics.
A paired or unpaired 2-tailed t test was used to determine statistical significance as detailed in the figure legends. P less than 0.05 was considered statistically significant. In RNA-seq analysis, P values were adjusted for multiple testing using the Benjamini-Hochberg method implemented within the DESeq2 package and log2 fold changes were considered significant when the adjusted P value was less than 0.05.
Study approval.
All described animal experiments were approved by the National Animal Experiment Board of Finland.
Author contributions
SO and TPM conceived and designed the study. SO, EDM, KL, IPLW, ST, NP, YG, YY, EHN, and KV acquired the data. SO, EDM, KV, and TPM analyzed and interpreted the data. SO and TPM wrote the manuscript. EDM, TCW, PK, and KV critically revised the manuscript for important intellectual content. SO and EDM performed the statistical analyses. BV and GL provided material support. TPM supervised the study.
Supplementary Material
Acknowledgments
This study was supported by Academy of Finland, Awards number 259278 and 271845 (to TM), Award Number 252494 (to SO); Biocentrum Helsinki, Award Number: 730401905 (to TM); and Sigrid Juselius Foundation, Award Number: 4704116 (to TM). The authors would like to thank Andrew Silver and James Robinson for kind assistance in retrieving the PJS patient polyp expression data set. We thank Incyte Corporation for providing ruxolitinib and Hans Clevers for providing the Lgr5-EGFP-IRES-CreERT2 mice. Saana Ruusulampi and Susanna Räsänen are thanked for excellent technical assistance, and Jaan-Olle Andressoo for helpful comments on the manuscript.
Version 1. 12/04/2017
Electronic publication
Version 2. 01/02/2018
Print issue publication
Footnotes
GL’s present address is: The Hollings Cancer Center, Department of Biochemistry & Molecular Biology, Medical University of South Carolina, Charleston, South Carolina, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(1):402–414.https://doi.org/10.1172/JCI93597.
Contributor Information
Saara Ollila, Email: saara.ollila@helsinki.fi.
Kaisa Laajanen, Email: kaisa.laajanen@helsinki.fi.
Sushil Tripathi, Email: sushil.tripathi@helsinki.fi.
Nalle Pentinmikko, Email: nalle.pentinmikko@helsinki.fi.
Yajing Gao, Email: yajing.gao@helsinki.fi.
Yan Yan, Email: yan.yan@helsinki.fi.
Benoit Viollet, Email: benoit.viollet@inserm.fr.
Gustavo Leone, Email: leoneg@musc.edu.
Pekka Katajisto, Email: pekka.katajisto@helsinki.fi.
Kari Vaahtomeri, Email: kari.vaahtomeri@helsinki.fi.
References
- 1.Ollila S, Mäkelä TP. The tumor suppressor kinase LKB1: lessons from mouse models. J Mol Cell Biol. 2011;3(6):330–340. doi: 10.1093/jmcb/mjr016. [DOI] [PubMed] [Google Scholar]
- 2.Korsse SE, et al. Identification of molecular alterations in gastrointestinal carcinomas and dysplastic hamartomas in Peutz-Jeghers syndrome. Carcinogenesis. 2013;34(7):1611–1619. doi: 10.1093/carcin/bgt068. [DOI] [PubMed] [Google Scholar]
- 3.de Leng WW, et al. Peutz-Jeghers syndrome polyps are polyclonal with expanded progenitor cell compartment. Gut. 2007;56(10):1475–1476. doi: 10.1136/gut.2007.128132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi DJ, et al. Induction of cyclooxygenase-2 in a mouse model of Peutz-Jeghers polyposis. Proc Natl Acad Sci U S A. 2002;99(19):12327–12332. doi: 10.1073/pnas.192301399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansen M, et al. Mucosal prolapse in the pathogenesis of Peutz-Jeghers polyposis. Gut. 2006;55(1):1–5. doi: 10.1136/gut.2005.069062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katajisto P, et al. LKB1 signaling in mesenchymal cells required for suppression of gastrointestinal polyposis. Nat Genet. 2008;40(4):455–459. doi: 10.1038/ng.98. [DOI] [PubMed] [Google Scholar]
- 7.Zac-Varghese S, et al. The Peutz-Jeghers kinase LKB1 suppresses polyp growth from intestinal cells of a proglucagon-expressing lineage in mice. Dis Model Mech. 2014;7(11):1275–1286. doi: 10.1242/dmm.014720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji H, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 9.Tanwar PS, Kaneko-Tarui T, Zhang L, Tanaka Y, Crum CP, Teixeira JM. Stromal liver kinase B1 [STK11] signaling loss induces oviductal adenomas and endometrial cancer by activating mammalian Target of Rapamycin Complex 1. PLoS Genet. 2012;8(8):e1002906. doi: 10.1371/journal.pgen.1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katajisto P, et al. The LKB1 tumor suppressor kinase in human disease. Biochim Biophys Acta. 2007;1775(1):63–75. doi: 10.1016/j.bbcan.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen T, et al. Liver kinase B1 inhibits the expression of inflammation-related genes postcontraction in skeletal muscle. J Appl Physiol. 2016;120(8):876–888. doi: 10.1152/japplphysiol.00727.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Zhang W, Zhang M, Zhu H, Moriasi C, Zou MH. Liver kinase B1 suppresses lipopolysaccharide-induced nuclear factor κB (NF-κB) activation in macrophages. J Biol Chem. 2015;290(4):2312–2320. doi: 10.1074/jbc.M114.616441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacIver NJ, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 2011;187(8):4187–4198. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koyama S, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. doi: 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Udd L, et al. Suppression of Peutz-Jeghers polyposis by inhibition of cyclooxygenase-2. Gastroenterology. 2004;127(4):1030–1037. doi: 10.1053/j.gastro.2004.07.059. [DOI] [PubMed] [Google Scholar]
- 17.Ylikorkala A, et al. Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science. 2001;293(5533):1323–1326. doi: 10.1126/science.1062074. [DOI] [PubMed] [Google Scholar]
- 18.Tse JY, et al. Peutz-Jeghers syndrome: a critical look at colonic Peutz-Jeghers polyps. Mod Pathol. 2013;26(9):1235–1240. doi: 10.1038/modpathol.2013.44. [DOI] [PubMed] [Google Scholar]
- 19.Adegboyega PA, Mifflin RC, DiMari JF, Saada JI, Powell DW. Immunohistochemical study of myofibroblasts in normal colonic mucosa, hyperplastic polyps, and adenomatous colorectal polyps. Arch Pathol Lab Med. 2002;126(7):829–836. doi: 10.5858/2002-126-0829-ISOMIN. [DOI] [PubMed] [Google Scholar]
- 20.Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5(12):1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 21.Udd L, Katajisto P, Kyyrönen M, Ristimäki AP, Mäkelä TP. Impaired gastric gland differentiation in Peutz-Jeghers syndrome. Am J Pathol. 2010;176(5):2467–2476. doi: 10.2353/ajpath.2010.090519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lizcano JM, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23(4):833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shackelford DB, et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci U S A. 2009;106(27):11137–11142. doi: 10.1073/pnas.0900465106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viollet B, et al. Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans. 2003;31(Pt 1):216–219. doi: 10.1042/bst0310216. [DOI] [PubMed] [Google Scholar]
- 25.Lai C, Robinson J, Clark S, Stamp G, Poulsom R, Silver A. Elevation of WNT5A expression in polyp formation in Lkb1+/- mice and Peutz-Jeghers syndrome. J Pathol. 2011;223(5):584–592. doi: 10.1002/path.2835. [DOI] [PubMed] [Google Scholar]
- 26.Vaahtomeri K, et al. Lkb1 is required for TGFbeta-mediated myofibroblast differentiation. J Cell Sci. 2008;121(Pt 21):3531–3540. doi: 10.1242/jcs.032706. [DOI] [PubMed] [Google Scholar]
- 27.Scherz-Shouval R, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158(3):564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Putoczki TL, Ernst M. IL-11 signaling as a therapeutic target for cancer. Immunotherapy. 2015;7(4):441–453. doi: 10.2217/imt.15.17. [DOI] [PubMed] [Google Scholar]
- 29.Ernst M, Putoczki TL. Molecular pathways: IL11 as a tumor-promoting cytokine-translational implications for cancers. Clin Cancer Res. 2014;20(22):5579–5588. doi: 10.1158/1078-0432.CCR-13-2492. [DOI] [PubMed] [Google Scholar]
- 30.Shaco-Levy R, et al. Morphologic characterization of hamartomatous gastrointestinal polyps in Cowden syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome. Hum Pathol. 2016;49:39–48. doi: 10.1016/j.humpath.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Garbers C, Scheller J. Interleukin-6 and interleukin-11: same same but different. Biol Chem. 2013;394(9):1145–1161. doi: 10.1515/hsz-2013-0166. [DOI] [PubMed] [Google Scholar]
- 32.Ernst M, Putoczki TL. Stat3: linking inflammation to (gastrointestinal) tumourigenesis. Clin Exp Pharmacol Physiol. 2012;39(8):711–718. doi: 10.1111/j.1440-1681.2011.05659.x. [DOI] [PubMed] [Google Scholar]
- 33.Pickert G, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206(7):1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howlett M, et al. The interleukin-6 family cytokine interleukin-11 regulates homeostatic epithelial cell turnover and promotes gastric tumor development. Gastroenterology. 2009;136(3):967–977. doi: 10.1053/j.gastro.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Roskoski R. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res. 2016;111:784–803. doi: 10.1016/j.phrs.2016.07.038. [DOI] [PubMed] [Google Scholar]
- 36.Quintás-Cardama A, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Sullivan JM, Harrison CN. JAK-STAT signaling in the therapeutic landscape of myeloproliferative neoplasms. Mol Cell Endocrinol. 2017;451:71–79. doi: 10.1016/j.mce.2017.01.050. [DOI] [PubMed] [Google Scholar]
- 38.Verstovsek S, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10(1):55. doi: 10.1186/s13045-017-0417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tripathi M, Billet S, Bhowmick NA. Understanding the role of stromal fibroblasts in cancer progression. Cell Adh Migr. 2012;6(3):231–235. doi: 10.4161/cam.20419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rønnov-Jessen L, Bissell MJ. Breast cancer by proxy: can the microenvironment be both the cause and consequence? Trends Mol Med. 2009;15(1):5–13. doi: 10.1016/j.molmed.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shorning BY, Griffiths D, Clarke AR. Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS One. 2011;6(1):e16209. doi: 10.1371/journal.pone.0016209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langeveld D, et al. SMAD4 immunohistochemistry reflects genetic status in juvenile polyposis syndrome. Clin Cancer Res. 2010;16(16):4126–4134. doi: 10.1158/1078-0432.CCR-10-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodford-Richens K, et al. Allelic loss at SMAD4 in polyps from juvenile polyposis patients and use of fluorescence in situ hybridization to demonstrate clonal origin of the epithelium. Cancer Res. 2000;60(9):2477–2482. [PubMed] [Google Scholar]
- 44.Alberici P, et al. Smad4 haploinsufficiency: a matter of dosage. Pathogenetics. 2008;1(1):2. doi: 10.1186/1755-8417-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He XC, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39(2):189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marsh Durban V, Jansen M, Davies EJ, Morsink FH, Offerhaus GJ, Clarke AR. Epithelial-specific loss of PTEN results in colorectal juvenile polyp formation and invasive cancer. Am J Pathol. 2014;184(1):86–91. doi: 10.1016/j.ajpath.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 47.Li S, et al. Mesenchymal-epithelial interactions involving epiregulin in tuberous sclerosis complex hamartomas. Proc Natl Acad Sci U S A. 2008;105(9):3539–3544. doi: 10.1073/pnas.0712397105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu B, et al. Stromal fibroblasts in the microenvironment of gastric carcinomas promote tumor metastasis via upregulating TAGLN expression. BMC Cell Biol. 2013;14:17. doi: 10.1186/1471-2121-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 50.Öhlund D, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tebbutt NC, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8(10):1089–1097. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- 52.Howlett M, et al. Differential regulation of gastric tumor growth by cytokines that signal exclusively through the coreceptor gp130. Gastroenterology. 2005;129(3):1005–1018. doi: 10.1053/j.gastro.2005.06.068. [DOI] [PubMed] [Google Scholar]
- 53.Ernst M, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118(5):1727–1738. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Putoczki TL, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell. 2013;24(2):257–271. doi: 10.1016/j.ccr.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 55.Robinson J, Lai C, Martin A, Nye E, Tomlinson I, Silver A. Oral rapamycin reduces tumour burden and vascularization in Lkb1(+/-) mice. J Pathol. 2009;219(1):35–40. doi: 10.1002/path.2562. [DOI] [PubMed] [Google Scholar]
- 56.Trimboli AJ, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68(3):937–945. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- 57.Yu K, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130(13):3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- 58.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 59.Bardeesy N, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419(6903):162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 60.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 61.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 62.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143(1):134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 63.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jørgensen SB, et al. Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279(2):1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 65.Viollet B, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111(1):91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iacobucci I, et al. Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell. 2016;29(2):186–200. doi: 10.1016/j.ccell.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 72.Mahe MM, et al. Establishment of Gastrointestinal Epithelial Organoids. Curr Protoc Mouse Biol. 2013;3(4):217–240. doi: 10.1002/9780470942390.mo130179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quante M, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.