

Abstract
Blast traumatic brain injury (bTBI) has been shown to contribute to progressive neurodegenerative disease. Recent evidence suggests that endoplasmic reticulum (ER) stress is a mechanistic link between acute neurotrauma and progressive tauopathy. We propose that ER stress contributes to extensive behavioral changes associated with a chronic traumatic encephalopathy (CTE)-like phenotype. Targeting ER stress is a promising option for the treatment of neurotrauma-related neurodegeneration, which warrants investigation. Utilizing our validated and clinically relevant Sprague–Dawley blast model, we investigated a time course of mechanistic changes that occur following bTBI (50 psi) including: ER stress activation, iron-mediated toxicity, and tauopathy via Western blot and immunohistochemistry. These changes were associated with behavioral alterations measured by the Elevated Plus Maze (EPM), Forced Swim Test (FST), and Morris Water Maze (MWM). Following characterization, salubrinal, an ER stress modulator, was given at a concentration of 1 mg/kg post-blast, and its mechanism of action was determined in vitro. bTBI significantly increased markers of injury in the cortex of the left hemisphere: p-PERK and p-eIF2α at 30 min, p-T205 tau at 6 h, and iron at 24 h. bTBI animals spent more time immobile on the FST at 72 h and more time in the open arm of the EPM at 7 days. Further, bTBI caused a significant learning disruption measured with MWM at 21 days post-blast, with persistent tau changes. Salubrinal successfully reduced ER stress markers in vivo and in vitro while significantly improving performance on the EPM. bTBI causes robust biochemical changes that contribute to neurodegeneration, but these changes may be targeted with ER stress modulators.
Keywords: : bTBI; ER stress; iron toxicity, salubrinal; tauopathy
Introduction
Blast exposure has been linked to progressive neurodegenerative changes. Several former war fighters have been diagnosed with neurodegenerative tauopathy and chronic traumatic encephalopathy (CTE) on postmortem examination.1 Shively and colleagues found that war fighters exposed to blast had extensive gliosis at the gray/white junction and vessel/brain interfaces.2 The injury associated with blast exposure is significantly different than that obtained from rotational brain injury alone.3 Blast exposure causes a unique pressure spike that can disrupt the blood–brain barrier (BBB) and contribute to the activation of secondary injury cascades.4 What is unknown, is how these secondary injury cascades contribute to the pathologic and behavioral deficits associated with CTE.
One of the most significant and characteristic pathologic changes is the accumulation of tau neurofibrillary tangles at the glia limitans and perivascular spaces.5 Puvenna and colleagues recently showed that glymphatic clearance of tau is impaired following repetitive traumatic brain injury in CTE specimens.6 We expanded upon these findings showing that TBI can contribute to tauopathy leading to CTE, Alzheimer's disease (AD), or, in rare cases, CTE/AD.7 Hyperphosphorylated tau aggregates around venules and begins to form oligomers and neurofibrillary tangles. We recently showed that this perivascular tau was associated with an increase in endoplasmic reticulum (ER) stress activation.8 Additionally, we found that nicotinamide adenine dinucleotide phosphate (NADPH) oxidative stress was increased in neuronal membranes surrounding areas of acute BBB disruption.9
In this article, we show evidence of how secondary injury produces a CTE phenotype with our clinically relevant and validated Sprague–Dawley blast model.10 Blast exposure caused early activation of ER stress in response to acute BBB disruption. The opening of the BBB that we have previously reported occurs early post-injury.11 This disruption allows further toxic particles, such as iron, to enter the brain and cause additional damage. Iron enters the brain from damaged red blood cells and heme degradation induced by blast exposure. Nisenbaum and colleagues have reported that iron-mediated toxicity can contribute to tau hyperphosphorylation and additional biochemical changes such as the formation of reactive oxygen species.12 ER stress weakens the cell, allowing iron to accumulate within the cytoplasm and damage the plasma membrane. We found that iron-mediated toxicity contributed to both tau hyperphosphorylation and conformational changes with our model. Salubrinal (Sal), an ER stress modulator, significantly reduced the iron-mediated toxicity.
These biochemical changes were linked to depressive and impulsive behavior measured with Forced Swim Test (FST) and Elevated Plus Maze (EPM), respectively. The tau changes were also associated with worsened cognitive performance measured with the Morris Water Maze (MWM). Importantly, we present data showing that ER stress modulation with Sal reduces acute injury cascades and improves behavior on the EPM. Using an in vitro assay, we found that Sal effectively reduced the neuroinflammatory marker nuclear factor kappa B (NFκB). This is in agreement with our recent article showing important “cross talk” between ER stress and neuroinflammatory cascades.8 Further work will mechanistically verify how ER stress modulation functions to improve behavior.
Methods
Model and drug
The Sprague–Dawley tabletop blast model produces a peak impulse of 50 psi. Finite element modeling reveals that the wave produced is 1 ms in duration (Fig. 1). The blast was delivered to the right side of the rat's skull with the head freely mobile and body protected by PVC tubing. The West Virginia University Animal Care and Use Committee approved this study, and all experiments were conducted in accordance with the national guidelines for the use and care of laboratory animals. A total of 120 3–6-month-old male Sprague– Dawley rats were used for this study. Sal (Tocris) was dissolved in 0.9% saline and administered at a concentration of 1mg/kg at a single time 5 minutes post-blast. 0.9% saline was used for the vehicle in rats not receiving Sal.
FIG. 1.

Blast model schematic and finite element modeling representations showing that the tabletop model produces short duration waves, a rapid spike in pressure, and consistency in pressure readings at the tube exit. The shockwave combines with the contact wave and expansion wave to form a blast wave over time.
Study design
Rats for the biochemical characterization studies were divided into control or blast groups based on time of euthanasia (30 min, 24 h, or 21 days). The time points were chosen to look at 1) acute ER stress activation, 2) subsequent iron toxicity, and 3) tauopathy. Sham rats were anesthetized but not exposed to blast. The n numbers for each assay are outlined in detail subsequently. The rats for behavior were divided according to assay and time point (72 h, 7 days, and 1 month).
Cell culture
NG-108 cells were cultured in a humidified incubator (5% CO2 at 37°C) in T75 culture flasks. High-glucose (4.5 g/L) Dulbecco's Modified Eagle Medium (DMEM) was supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and hypoxanthine-aminopterin-thymidine (HAT) medium. When cells were 70% confluent, the media was replaced with differentiation media (DMEM with 5% FBS, penicillin, streptomycin, HAT, and 0.5% dimethyl sulfoxide [DMSO]) and allowed to differentiate for an additional 3 days.
Differentiated cells were seeded (1 × 105) onto 12 well plates for treatment (n = 5). On each plate, wells were divided into four groups: 1) control, 2) Sal, 3) tunicamycin (TUN) + DMSO, and (4) TUN + SAL. TUN was administered (1 μg/mL) to cells for 6 h prior to collection to activate the ER stress response, and Sal was administered (100 μM) to cells 30 min prior to collection. Control and TUN cells were administered DMSO (0.5%) 30 min prior to collection. Cells were collected in 1% sodium dodecyl sulfate (SDS) for measurement of protein expression.
Western blot analysis
For Western blot, frontal cortex was collected from blast and control rats, which has been shown previously to correlate with behavioral findings.13 In vitro cell collection was also conducted as stated. Protein samples were prepared in 1% SDS, and the assay was performed as previously described.14 Primary antibodies were rabbit anti-phosphoPERK mAB (1:1000), anti-PERK mAB (1:1000), and anti-phosphoeiF2α (1:1000) (Cell Signaling); mouse anti-NFκB p65 (1:200) (Santa Cruz); mouse anti-AT8 (Thermo), and mouse anti-CP13 (kindly gifted by Peter Davies). A rabbit anti-β-actin mAB (1:10,000) (Cell Signaling) was used as an endogenous control to normalize protein loading. Secondary antibodies were IRDye® 800CW (goat anti-rabbit) and IRDye® 680RD (goat anti-mouse) (LI-COR Biosciences). Images were collected and analyzed with an Odyssey fluorescent scanner. Images were converted to gray scale and the values calculated after background subtraction and then normalized to β-actin to measure relative intensity.
Immunohistochemistry
Brains were rapidly removed and placed into an ice-cold protease/phosphatase inhibitor cocktail mix (Halt™; Thermo Scientific; Pittsburgh, PA). Tissues were flash frozen in liquid nitrogen for storage at −80°C. Fixed brain tissue for the TBI and control rats were prepared as previously described.14 Briefly, rats were anesthetized with 4% isoflurane and cardiac perfused with ice-cold 0.9% saline, followed by 4% paraformaldehyde for 15 min. Following perfusion, brains were removed and placed in 4% paraformaldehyde for 24 h. Following fixation, the frontal cortex was sectioned into 4 mm slabs, which were then processed and embedded in paraffin, as previously described.15 Brain slabs were sectioned (10 μm), mounted onto slides, and prepared for staining. Standard protocols for Perls DAB and cresyl violet were used for the iron staining (Sigma Aldrich).
FST
Depressive-like behavior was assessed with the FST assay. Two sets of rats were assessed: control and 72 h post-blast (n = 6 per group). The apparatus was filled with water so that the rat could not touch the floor. On habituation day, the rat was placed in the apparatus for 15 min and monitored continuously. It was dried and warmed on a heating blanket following the trial. On test day, the rat was placed in the apparatus for 5 min and tracked with Any-Maze Video Tracking software (Stoelting Co.). Time spent immobile and time to first immobility episode was analyzed.
EPM
Impulsive-like behavior was investigated with the EPM assay. Four sets of rats were assessed: control, Sal, 7 day blast, and 7 day Sal + blast (n = 9 per group). The two arms of the EPM were 50 × 10 cm and raised 60 cm from the floor. The closed arms had black siding 30 cm tall. The rat was placed in the middle of the EPM and tracking was performed with Any-Maze software for 5 min. The percentage time spent in the open arms and distance traveled was recorded and quantified.
MWM
Cognitive performance was evaluated with the MWM assay. Three sets of rats were assessed: control, 7 days post-blast, and 21 days post-blast (n = 10 per group). The circular pool was 180 cm in diameter. In the pool, a 10 × 10 cm platform was submerged 2.5 cm below the surface of the water (20°C). The training paradigm consisted of 6 days of acquisition with a hidden platform followed by a probe trial on day 7. During spatial acquisition, rats were placed into the maze apparatus four times from four different locations. Each rat had a 2 min trial to locate the platform. Timing stopped if the platform was found, but if the rat did not find it during the 2 min session, it was placed on the platform for 15 sec. On probe day, the rat was placed in the apparatus from a novel entrance point for 1 min. An area encompassing 300% of where the platform had been was outlined to indicate time spent in the appropriate region. Any-Maze was used to measure the distance traveled and the latency to reach the platform on acquisition days. On the probe trial day, it was used to measure how long the rat spent in the area 300% of where the platform had been.
Statistical analysis
Graphpad Prism software 5.0 (Graphpad Software, Inc.) was used for statistical analysis. A t test, one way ANOVA, or two way ANOVA was used depending on the assay. χ2 analysis was used to quantify the iron staining. Tukey's post-hoc comparison was used for the one way and two way ANOVA. A p value of <0.05 was considered statistically significant.
Results
Phosphorylation states of PERK and eIF2α are elevated after bTBI
To confirm ER stress activation following bTBI, two upstream components of the PERK-mediated ER stress pathway, PERK and eIF2α, were investigated for their respective phosphorylation levels. Western blot analyses showed a significant increase in PERK phosphorylation at 0.5 h following blast in the left prefrontal cortex (PFC) (t = 8.59; p < 0.01) (Fig. 2A). At 0.5 h following blast exposure, a significant increase in eIF2α phosphorylation was also found in the left PFC (t = 5.45; p < 0.05) (Fig. 2B). No differences were seen in total PERK or total eIF2α expression from the same samples (Fig. 2C,D), indicating a difference in phosphorylation state, but not in total protein expression (Fig. 2E,F). PERK and eIF2α phosphorylation levels returned to baseline after 6 h post-blast (data not shown). Results showed a single moderate blast exposure to activate upstream constituents of the PERK-mediated ER stress pathway.
FIG. 2.

Western blot analysis shows an increase in PERK and eIF2α phosphorylation following blast exposure at acute time points. (A) Shows an increase in PERK phosphorylation at 30 min following blast exposure when compared with control animals (t = 8.591; p < 0.01). (B) Shows an increase in eIF2α phosphorylation at 30 min following blast exposure when compared with control animals (t = 5.447; p < 0.05). (C) Shows no increase in the expression of total PERK following blast exposure. (D) Shows no increase in the expression of total eIF2α following blast exposure. **p < 0.01, *p < 0.05. (E, F) Representative graphs showing relative differences.
Iron-mediated toxicity is increased following bTBI but is ameliorated by Sal administration
It has previously been reported that non-heme iron accumulates in the brain after injury and contributes to robust cell loss.16 The mechanisms by which this occurs are not fully known. In Figure 3 we show robust iron accumulation within neurons of the frontal cortex at 24 h post-TBI. A χ2 test revealed that severe TBI (sTBI) significantly increased the ratio of iron-positive stained cells (Control = 4/104, Blast = 63/103, and Blast + Sal = 24/108; χ2 = 59.46; p < 0.001). Not surprisingly, ER stress modulation with Sal significantly prevented an increase in iron-positive cells, showing enhanced neuroprotective properties.
FIG. 3.

Iron staining in the frontal cortex shows a significant increase following blast traumatic brain injury (bTBI). Scale bar = 50μm. (A) Iron accumulation in vehicle control is minimal (4/104 cells). (B) Iron accumulation following bTBI is significant (63/103 cells). (C) Iron accumulation following bTBI is prevented by the administration of salubrinal (Sal) (24/108 cells). χ2 = 59.46; p < 0.001. Arrows indicate representative cells where iron accumulation is pronounced in the cytoplasm.
Sal attenuated NFκB administration after in vitro TUN administration
TUN is a potent inducer of ER stress. We recently reported that ER stress activation contributes to robust neuroinflammation in vivo mediated by NFκB.16 What was unknown, was how ER stress modification altered the neuroinflammatory cascade. The activation of the neuroinflammatory cascade may be the contributing factor leading to iron toxicity. Figure 4 shows a significant difference in NFκB activity between groups of NG108 cells (F(3,16) = 4.987; p < 0.05). TUN + DMSO produced a significant increase in NFκB activity compared with control cells treated with DMSO (q = 4.766; p < 0.05) or SAL (q = 4.432; p < 0.05). TUN + Sal significantly attenuated NFκB activity compared with TUN + DMSO cells (q = 4.101; p < 0.05). No difference between DMSO + SAL and TUN + SAL was observed. Future studies will examine this potential mechanism in relation to iron-mediated toxicity in vivo.
FIG. 4.
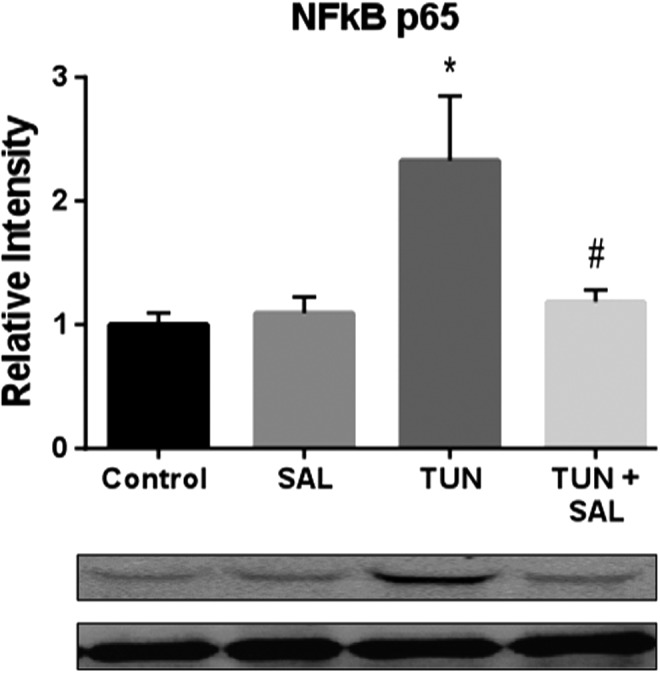
Salubrinal (Sal) significantly reduced the activation of nuclear factor kappa B (NFκB) in vitro following endoplasmic reticulum (ER) stress induction with tunicamycin (TUN). A significant difference between TUN and control was seen for NFκB activation in NG-108 cells following treatment (q = 4.432; p < 0.05). Sal significantly reduced the increase by TUN (t = 4.101; p < 0.05). *p < 0.05 for Control versus TUN groups. #p < 0.05 for TUN versus TUN + Sal groups.
bTBI increases depressive-like behavior
A prominent symptom after blast TBI is the onset of depressive behavior. Recently, Tucker and colleagues reported depressive-like behavior in mice following bTBI.18 We show a similar phenotype of depressive-like behavior in our rat blast model: 72 h after blast, a significant difference between blast and control groups was seen for time spent immobile (t = 2.49; p < 0.05). Blast rats spent 136.2 ± 35.84 sec immobile, whereas control rats spent 39.90 ± 14.45 sec immobile (Fig. 5A). No significant difference was seen for total immobility episodes (t = 1.2; p = 0.26) (Fig. 5B). A significant difference was seen for time to first immobility session (t = 2.24; p < 0.05). Blast-exposed rats had an average time of 9.35 ± 6.16 sec to first immobility episode, whereas control rats had an average time of 73.52 ± 28.03 sec to first immobility episode.
FIG. 5.

Blast exposure significantly increased depressive-like behavior (n = 6 per group). (A) Rats exposed to blast spent more time immobile compared with control rats (t = 2.49, p < 0.05). (B) No significant difference was seen on total number of immobility episodes. (C) The latency to first immobility episode was significantly shortened for the blast-exposed rats (t = 2.24, p < 0.05). *p < 0.05.
Impulsive-like behavior is prevented by Sal administration
A prominent symptom following blast exposure is the development of impulsivity.19 We have previously shown that the EPM is useful in determining impulsive-like behavior in rats.17 In this article, we found a significant difference between groups for time spent in the open arm F(2,24) = 4.715; p < 0.05 (Fig. 6). Multiple comparisons revealed a significant difference between blast and control groups (q = 4.09; p < 0.05). Blast-exposed rats spent on average 24.52 sec in the open arm, whereas control rats spent 3.244 sec in the open arm. Sal ameliorated the increase seen with blast (q = 3.31; p < 0.05). Sal-treated rats spent on average only 7.3 sec in the open arm.
FIG. 6.
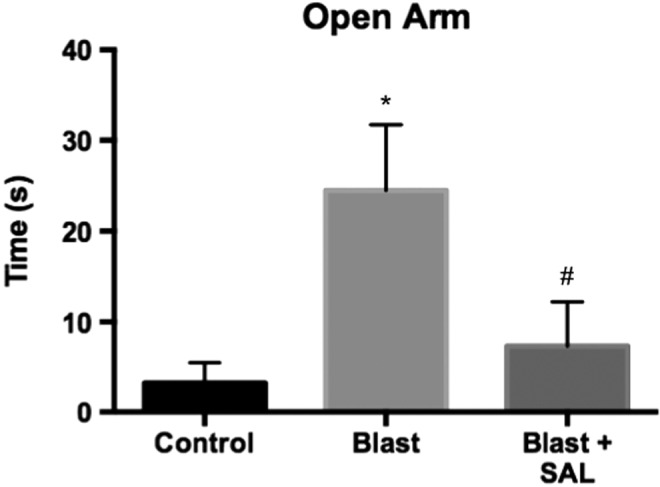
Salubrinal (Sal) administration prevented the development of impulsive-like behavior following blast exposure (n = 9 per group). A significant difference was seen in time spent in the open arm of the Elevated Plus Maze (EPM) between the control and blast groups (q = 4.09; p < 0.05). Sal administration successfully prevented this increase in exploratory behavior in the open arm (q = 3.31; p < 0.05). *p < 0.05 for Control versus Blast groups. #p < 0.05 for Blast versus Blast + Sal groups.
Blast exposure increases tau hyperphosphorylation and conformational change
Tau pathology is a characteristic change showing evidence of neurodegeneration following neurotrauma.20 Tau can undergo biochemical modifications such as nitration and hyperphosphorylation. These modifications can lead to conformational changes in tau that predispose to pathologic aggregation.21 We observed a significant difference in tau changes between control and blast-exposed rats at 3 weeks post-injury. AT8, a marker of tau hyperphosphorylation, was significantly increased following blast (t = 3.59; p < 0.05). CP13, a marker of tau comformational change, was also significantly increased following blast (t = 3.25; p < 0.05) (Fig. 7).
FIG. 7.

Blast exposure increases tauopathy changes. (A) AT8, a marker of tau hyperphosphorylation, was increased in the contralateral cortex 3 weeks after blast exposure (t = 3.59; p < 0.05). (B) CP13, a marker of tau conformational change, was increased in the cortex as well 3 weeks after blast exposure (t = 3.25; p < 0.05). *p < 0.05.
Cognitive deficits are increased following blast exposure
bTBI can increase learning deficits and accelerate degenerative changes.22 MWM is a sensitive assay to detect these changes. We found a significant difference between groups in escape latency (F(2,19) = 18.58; p < 0.001). At 3 weeks, blast-exposed rats had diminished performance on acquisition trials compared with control rats. Interestingly, a separate cohort of blast-exposed rats had no significant differences from controls when measured at 1 week post-injury. On the probe trial, a significant difference between groups was observed (F(2,7) = 13.61; p < 0.001). Rats exposed to blast and aged for 3 weeks spent less time in the area where the platform had been compared with controls (q = 6.51; P < 0.01) and with blast-exposed rats aged for 1 week (q = 6.44; p < 0.01). No significant difference in swim speed was seen between groups (F(2,7) = 0.209; p > 0.05) (Fig. 8).
FIG. 8.

Blast exposure causes learning impairment at 3 weeks post-injury but not as early as 7 days (n = 10 per group). A significant difference was seen between groups on (A) acquisition trials (F(2,19) = 18.58; p < 0.001) and (B) the probe trial (F(2,7) = 13.61; p < 0.001). No significant difference was seen in (C) swim speed. ***p < 0.001, **p < 0.01.
Discussion
Blast TBI can cause extensive changes to the brain that can lead to behavioral deficits such as executive dysfunction, memory deficits, and cognitive impairments in war fighters.23 The underlying pathophysiology behind these behavior changes is poorly understood. In this article, we found that rats exposed to blast had increased depressive-like and impulsive-like behavior and cognitive deficits. Interestingly, the timing of these changes was sequential. Rats displayed depressive-like symptoms at 72 h post-injury and impulsive-like behavior at 7 days. A separate cohort of rats with the same blast conditions was used for the MWM testing. Surprisingly, the 7 day blast group showed no changes in cognitive performance whereas the 3 week blast group did. These changes are in agreement with the progressive symptom development often reported in patients with CTE.24 The more rapid progression seen in the rodent model is likely the result of species differences and the shortened life span of the animals compared with humans.
To investigate the underlying pathophysiology linking neurotrauma to neurodegenerative disease, we looked at the activation of the ER stress response as well as iron-mediated toxicity. Chen and colleagues proposed that iron accumulation released from degrading red blood cells contributes to robust inflammatory and secondary injury responses in the brain.25 We found that ER stress contributed to the activation of iron toxicity, but that Sal prevented this activation. In our previous article, we found an association among ER stress, oxidative stress, and neuroinflammation.26 Using in vitro investigation, we verified that reducing TUN mediated ER stress significantly, mitigated the activation of NFκB. The reduction in neuroinflammation mediated by ER stress modulation may have beneficial effects on behavior. Faden and colleagues have shown that persistent neuroinflammation is the key player in the development of chronic neurodegeneration.27
Preliminary evidence showed that when we administered Sal post-blast, impulsive-like behavior was prevented. As the injury progressed with time, tau changes began to occur, and induced impairment in learning as measured with the MWM. Gerson and colleagues likewise showed that tau oligomer formation following blast exposure could contribute to cognitive impairment.21 Going forward, we want to investigate the progression of tauopathy at later time points. This will include an assessment of both oligomer formation and cis/trans tau configuration similar to that being performed by Kanaan and colleagues in human CTE tissue.28 We propose that as the rats age over a series of months, they will develop progressive pathologic tau oligomers.
Most importantly, we will investigate whether ER stress modulation can prevent long-term behavior. We have shown in our recent Journal of Neurosurgery and Frontiers in Neuroscience articles that reducing ER stress is protective at subacute time points.8,13 A multi-drug regimen targeting different secondary injury cascades at different sequential time points will likely have the most efficacy. We have previously shown benefit in targeting the NADPH oxidative stress response, which can preserve plasma membranes and prevent iron damage.9 Iron damage is further increased post-blast disruption. Targeting BBB disruption and oxidative stress may, therefore, be additional strategies to be used in conjunction with ER stress modulation.
Conclusion
In conclusion, ER stress activation contributes to iron damage. In vitro investigation confirmed that ER stress increases NFκB activation, but that this can be prevented by the administration of Sal. Sal reduced exploratory behavior on the EPM. Going forward, we will investigate how tauopathy progresses and contributes to long-term behavior changes. Once this has been characterized, a multi-drug regimen will be established to prevent the onset and duration of neuroinflammation. We propose that this multi-drug regimen may prove beneficial in preventing neurodegeneration following neurotrauma.
Acknowledgments
Aric Logsdon and Brandon Lucke-Wold thank the American Foundation of Pharmaceutical Education for awarding pre-doctoral fellowships. Brandon Lucke-Wold also received funding from Sigma Xi Grants in Aid of Research, Neurosurgery Research and Education Foundation Medical Student Summer Fellowship, American Association of Pharmaceutical Scientists Pre-doctoral Fellowship, and the American Medical Association Foundation Seed Grant.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Doherty C.P., O'Keefe E., Wallace E., Loftus T., Keaney J., Kealy J., Humphries M.M., Molloy M.G., Meaney J.F., Farrell M., and Campbell M. (2016). Blood-brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 75, 656–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shively S.B., Horkayne-Szakaly I., Jones R.V., Kelly J.P., Armstrong R.C., and Perl D.P. (2016). Characterisation of interface astroglial scarring in the human brain after blast exposure: a post-mortem case series. Lancet Neurol. 15, 944–953 [DOI] [PubMed] [Google Scholar]
- 3.Stemper B.D., Shah A.S., Budde M.D., Olsen C.M., Glavaski-Joksimovic A., Kurpad S.N., McCrea M., and Pintar F.A. (2016). Behavioral outcomes differ between rotational acceleration and blast mechanisms of mild traumatic brain injury. Front. Neurol. 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kabu S., Jaffer H., Petro M., Dudzinski D., Stewart D., Courtney A., Courtney M., and Labhasetwar V. (2015). Blast-associated shock waves result in increased brain vascular leakage and elevated ROS levels in a rat model of traumatic brain injury. PLoS One 10, e0127971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan J., Connolly I.D., Dangelmajer S., Kintzing J., Ho A.L., and Grant G. (2016). Sports-related brain injuries: connecting pathology to diagnosis. Neurosurg. Focus 40, E14. [DOI] [PubMed] [Google Scholar]
- 6.Puvenna V., Engeler M., Banjara M., Brennan C., Schreiber P., Dadas A., Bahrami A., Solanki J., Bandyopadhyay A., Morris J.K., Bernick C., Ghosh C., Rapp E., Bazarian J.J., and Janigro D. (2016). Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res. 1630, 225–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner R.C., Lucke–Wold B.P., Robson M.J., Lee J.M., and Bailes J.E. (2016). Alzheimer's disease and chronic traumatic encephalopathy: Distinct but possibly overlapping disease entities. Brain Inj. 11, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucke–Wold B.P., Turner R.C., Logsdon A.F., Nguyen L., Bailes J.E., Lee J.M., Robson M.J., Omalu B.I., Huber J.D. and Rosen C.L. (2016). Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J. Neurosurg. 124, 687–702 [DOI] [PubMed] [Google Scholar]
- 9.Lucke–Wold B.P., Naser Z.J., Logsdon A.F., Turner R.C., Smith K.E., Robson M.J., Bailes J.E., Lee J.M., Rosen C.L., and Huber J.D. (2015). Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl. Res. 166, 509–528 [DOI] [PubMed] [Google Scholar]
- 10.Turner R.C., Naser Z.J., Logsdon A.F., DiPasquale K.H., Jackson G.J., Robson M.J., Gettens R.T., Matsumoto R.R., Huber J.D., and Rosen C.L. (2013). Modeling clinically relevant blast parameters based on scaling principles produces functional & histological deficits in rats. Exp. Neurol. 248, 520–529 [DOI] [PubMed] [Google Scholar]
- 11.Lucke–Wold B.P., Logsdon A.F., Smith K.E., Turner R.C., Alkon D.L., Tan Z., Naser Z.J., Knotts C.M., Huber J.D., and Rosen C.L. (2015). Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol. Neurobiol. 52, 1119–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nisenbaum E.J., Novikov D.S., and Lui Y.W. (2014). The presence and role of iron in mild traumatic brain injury: an imaging perspective. J. Neurotrauma 31, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logsdon A.F., Turner R.C., Lucke–Wold B.P., Robson M.J., Naser Z.J., Smith K.E., Matsumoto R.R., Huber J.D., and Rosen C.L. (2014). Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front. Cell Neurosci. 8, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucke-Wold B.P., Logsdon A.F., Smith K.E., Turner R.C., Alkon D.L., Tan Z., Naser Z.J., Knotts C.M., Huber J.D., and Rosen C.L. (2014). Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol. Neurobiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner R.C., Naser Z.J., Bailes J.E., Smith D.W., Fisher J.A. and Rosen C.L. (2012). Effect of slosh mitigation on histologic markers of traumatic brain injury: laboratory investigation. J. Neurosurg. 117, 1110–1118 [DOI] [PubMed] [Google Scholar]
- 16.Chen–Roetling J., Liu W., and Regan R.F. (2011). Iron accumulation and neurotoxicity in cortical cultures treated with holotransferrin. Free Radic. Biol. Med. 51, 1966–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Logsdon A.F., Lucke–Wold B.P., Nguyen L., Matsumoto R.R., Turner R.C., Rosen C.L., and Huber J.D. (2016). Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res. 1643, 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tucker L.B., Burke J.F., Fu A.H., and McCabe J.T. (2016). Neuropsychiatric symptom modeling in male and female C57BL/6J mice after experimental traumatic brain injury. J Neurotrauma. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harch P.G., Andrews S.R., Fogarty E.F., Amen D., Pezzullo J.C., Lucarini J., Aubrey C., Taylor D.V., Staab P.K., and Van Meter K.W. (2012). A phase I study of low-pressure hyperbaric oxygen therapy for blast-induced post-concussion syndrome and post-traumatic stress disorder. J. Neurotrauma 29, 168–185 [DOI] [PubMed] [Google Scholar]
- 20.Turner R.C., Lucke–Wold B.P., Logsdon A.F., Robson M.J., Dashnaw M.L., Huang J.H., Smith K.E., Huber J.D., Rosen C.L., and Petraglia A.L. (2015). The quest to model chronic traumatic encephalopathy: a multiple model and injury paradigm experience. Front. Neurol. 6, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerson J., Castillo–Carranza D.L., Sengupta U., Bodani R., Prough D.S., DeWitt D.S., Hawkins B.E., and Kayed R. (2016). Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in Htau mice. J. Neurotrauma. 33, 2034–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tweedie D., Rachmany L., Rubovitch V., Li Y., Holloway H.W., Lehrmann E., Zhang Y., Becker K.G., Perez E., Hoffer B.J., Pick C.G., and Greig N.H. (2016). Blast traumatic brain injury-induced cognitive deficits are attenuated by preinjury or postinjury treatment with the glucagon-like peptide-1 receptor agonist, exendin-4. Alzheimer's Dement. 12, 34–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daneshvar D.H., Goldstein L.E., Kiernan P.T., Stein T.D., and McKee A.C. (2015). Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol. Cell. Neurosci. 66, 81–90 [DOI] [PubMed] [Google Scholar]
- 24.Galgano M.A., Cantu R. and Chin L.S. (2016). Chronic traumatic encephalopathy: the impact on athletes. Cureus 8, e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H., Constantini S., and Chen Y. (2015). A two-model approach to investigate the mechanisms underlying blast-induced traumatic brain injury. In: Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Kobeissy F.H. (ed). Boca Raton, FL, Chapter 17 [PubMed] [Google Scholar]
- 26.Logsdon A.F., Lucke–Wold B.P., Nguyen L., Matsumoto R.R., Turner R.C., Rosen C.L. and Huber J.D. (2016). Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res. 1643, 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faden A.I., Wu J., Stoica B.A., and Loane D.J. (2016). Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 173, 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanaan N.M., Cox K., Alvarez V.E., Stein T.D., Poncil S., and McKee A.C. (2016). Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 75, 19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]