

Abstract
The discovery of the microRNA (miRNA) family of small RNAs as fundamental regulators of post-transcriptional gene expression has fostered research on their importance in every area of biology and clinical medicine. In the particular area of liver metabolism and disease, miRNAs are gaining increasing importance. By focusing on two fundamental hepatic biosynthetic pathways, glutathione and methionine, we review recent advances on the comprehension of the role of miRNAs in liver pathophysiology and more specifically of models of hepatic cholestasis/fibrosis and hepatocellular carcinoma.
1. Glutathione synthesis
GSH is synthesized from its constituent amino acids in all mammalian cells in two ATP-requiring enzymatic steps: the first step is the rate-limiting step catalyzed by glutamate-cysteine ligase (GCL), which condenses glutamate and cysteine to form γ-glutamylcysteine, and the second step is catalyzed by GSH synthetase (GS), which condenses glycine and γ-glutamylcysteine to form GSH [1]. GSH synthesis is largely controlled by the activity of GCL, a heterodimeric enzyme composed by a heavy subunit, GCLC (73 kDa) with catalytic activity and a smaller one, GCLM (33 kDa) that has a regulatory role on the other subunit [1]. Under normal physiological conditions GCL activity is regulated mainly by non-allosteric feedback competitive inhibition by GSH (Ki = 2.3 mM) [2] and the availability of the limiting amino acid precursor L-cysteine [3]. Intracellular cysteine concentration is close to the GCL’s Km value for cysteine (0.1–0.3 mM), whereas glutamate concentration is about 10-fold higher than the GCL’s Km value for glutamate (1.8 mM) [2,4]. The cellular concentration of ATP is another limiting variable. Of note, its Km for GCLc is 6 times higher when it is not forming the holoenzyme with the regulatory subunit GCLm [5]. Although GSH synthetase is generally not important in regulating GSH synthesis, it is important in determining overall GSH synthetic capacity in certain tissues (i.e. skeletal muscle) and/or under stressful conditions [6]. One major question that remains largely unresolved is related to the subcellular distribution and compartmentation of GSH. While it is now accepted that GSH is distributed in the cytosol, nucleus and mitochondria, it is still unclear if the regulation of the redox state by GSH in these locations bears differential features among them. The employment of compartment-targeted redox-sensitive fluorescent sensors is progressively allowing to gain insight into this fundamental question [7].
GSH homeostasis in the cell is not only regulated by its de novo synthesis. There are other relevant factors such as utilization, recycling and cellular export, generating a redox cycle, known as the GSH cycle (Fig. 1) [1,8]. These additional pathways incorporate other important antioxidant, redox-related enzymes such as glutathione peroxidase (GPx), which metabolize residual hydrogen peroxide from aerobic respiration by converting 2 GSH molecules to its oxidized form (GSSG). Recent important work has shown how multiple GSSG removal pathways affect GSH/GSSG ratio, subcellular location and homeostasis, at least in yeast [9]. GSSG is recycled back to GSH by the action of glutathione reductase (GR), a reaction that consumes NADPH. GSH works also as cofactor and substrate for the glutaredoxin family (Grx), which catalyze disulfide reductions in the presence of NADPH and glutathione reductase (GR) [10]. Moreover, GSH is also able to react with critical Cys residues in proteins, by forming mixed disulfides (see [11]).
Fig. 1.
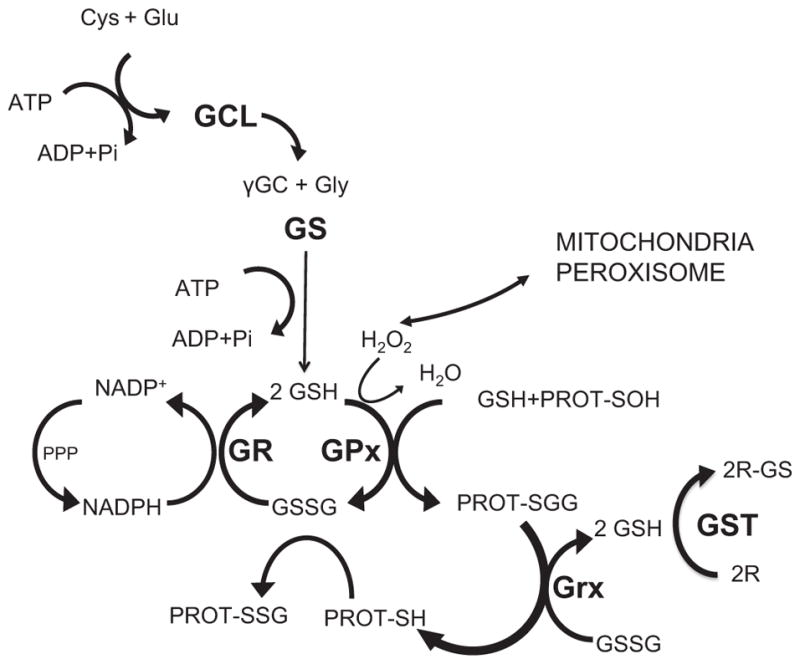
GSH biosynthetic route and GSH cycle. GSH biosynthesis occurs in two different ATP-dependent steps. The first and limiting step is carried out by GCL, formed by two subunits GCLC and GCLM. In this step Glu and Cys react to form γ-glutamylcisteine (γ-GC). GS is responsible of the second step that joins Gly forming γ-glutamylcisteinyl-glycine (GSH). Aerobic respiration or other ROS sources increase H2O2 that should be metabolized, in this case by GPx generating GSSG. This GSSG can be reduced to GSH again with the help of GR, creating a redox cycle, using as a reducing agent NAPDH, from the Penthose Phosphate Pathway (PPP) or folate metabolism (FM). GSH also forms mixed disulfides with proteins through S-Gluthathionylation, protecting redox sensitive Cys residues. Furthermore, glutaredoxins (Grx) can reduce disulfide (S–S) bonds in proteins, and glutathione S transferase (GST) uses GSH to conjugate and detoxify reactive electrophilic compounds (R).
Another level of regulation of GSH homeostasis is provided by glutathione S-transferase (GST), that along with other antioxidant enzymes provides the cell with protection against a range of harmful electrophiles produced during oxidative damage to membranes [12]. In humans, GST includes 22 family members, classified according to their structure as cytosolic, mitochondrial and membrane associated. GSTs are dimeric, with both subunits belonging to the same class of GSTs. The activity of GSTs is also dependent upon the presence of GSH, again stressing its importance as an antioxidant molecule [13]. The primary role of GSTs is the detoxification of reactive electrophilic compounds, including environmental toxins, and products of oxidative stress, by conjugation with GSH, forming water-soluble GSH-conjugates that are readily transported out of the cell [14–16]. Due to the action of specific transporters GSH conjugates are removed from the cell, thereby preventing crucial cellular proteins and nucleic acids from the action of reactive electrophilic compounds. The overexpression of GST in mammalian tumor cells has been implicated in the resistance to various anti-cancer agents and chemical carcinogens [17]. Additionally, in acetaminophen-induced hepatotoxicity recent work has shown that downregulation of GST-Pi could prevent toxicity by increasing S-glutathionylation of GCL, protecting its integrity and increasing GSH synthesis [18,19]. For a detailed view of GSH-regulated antioxidant mechanisms and other enzymatic systems related to GSH the readers are referred to Espinosa-Diez et al. [20].
The availability of cysteine, or the pair cysteine/cystine is a limiting step in GSH synthesis. Extracellular cysteine is very unstable as it readily autoxidizes to cystine, the disulfide form of cysteine, which is taken up by many cells and is reduced to cysteine once inside the cell [4]. In hepatocytes, cysteine availability is determined mainly by membrane transport of cysteine (via the ASC system, the ubiquitous Na+-dependent neutral amino acid transporter with preferred substrates alanine, serine and cysteine), cystine (via the Xc− system, the cystine/glutamate antiporter that exchanges extracellular cystine for intracellular glutamate, which is induced under oxidative stress), methionine, (via the L system, the transporter responsible for transport of several large neutral amino acids) and the activity of the transsulfuration pathway, which converts methionine to cysteine [4,21,22]. The transsulfuration pathway is particularly active in the liver, allowing efficient utilization of methionine for the synthesis of GSH [23].
2. The importance of methionine metabolism for glutathione synthesis
The liver plays a central role in both methionine and GSH metabolism. Methionine metabolism in mammals consists of two pathways, the methionine cycle and the transsulfuration pathway, both sharing the first three reactions that convert methionine to homocysteine (Fig. 2) [23]. Up to half of the daily intake of methionine is catabolized in the liver, where methionine adenosyltransferase (MAT) catalyzes the first reaction that converts methionine and ATP to S-adenosylmethionine (SAMe) [24]. SAMe is the principal biological methyl donor and under normal conditions, most of the SAMe formed is used in transmethylation reactions generating S-adenosylhomocysteine (SAH) [24]. SAH is in turn hydrolyzed to homocysteine (Hcy) and adenosine through a reversible reaction catalyzed by SAH hydrolase. SAH is a potent competitive inhibitor of methylation reactions and the thermodynamics of SAH hydrolase favors SAH biosynthesis rather than hydrolysis so prompt removal of Hcy is required to prevent SAH accumulation [25]. The methionine cycle regenerates methionine via remethylation of Hcy, which can occur through a reaction catalyzed by methionine synthase (MS, requires B12 as a cofactor and methyltetrahydrofolate as the methyl donor), and betaine-homocysteine methyltransferase (BHMT, uses betaine, also known as trimethylglycine, as the methyl donor). MS is present in all mammalian cells but BHMT is restricted to liver and kidney [26]. One key function of BHMT is to recover excess methyl groups after, for instance, feeding a high protein diet. The transsulfuration pathway converts Hcy to cysteine via two enzymatic steps. The first step condenses Hcy with serine to form cystathionine in a reaction catalyzed by cystathionine β synthase (CBS, requires vitamin B6). The second step cleaves cystathionine to release free cysteine and α-ketobutyrate in a reaction catalyzed by another vitamin B6-dependent enzyme, γ-cystathionase or cystathionine γ-lyase (CSE). CBS and CSE are known to produce hydrogen disulfide (H2S) from L-cysteine [27]. A third pathway, involves the combined action of cysteine/aspartate aminotransferase (CAT), which produces 3-mercaptopyruvate from L-cysteine and α-ketoglutarate, and 3-mercaptopyruvate sulfurtransferase (MST), which catalyzes the synthesis of H2S from 3-mercaptopyruvate [27]. Recently, a fourth pathway that involves the generation of α-ketoglutarate from D-cysteine (which is formed during food processing), through a reaction catalyzed by the enzyme D-amino acid oxidase (DAO), has also been shown to produce H2S [28]. In the liver, H2S is generated preferentially by CBS, CSE and MST [29]. While at high concentrations H2S inhibits mitochondrial respiration, at lower concentrations H2S acts as a mitochondrial energy substrate, maintaining hepatocyte bioenergetics, and a cytoprotective agent. H2S cytoprotective activity may be explained by its antioxidant and protein S-sulfhydration activities. In this respect, it is interesting to note that H2S stabilizes Nrf2, a transcription factor that regulates antioxidant genes [20], through inhibition of Keap1 via a process that involves the Keap1 sulfhydration [30]. Accordingly, altered H2S content may be involved in numerous liver diseases including ischemia/reperfusion liver injury and liver cirrhosis [27]. The hepatic transsulfuration pathway activity is markedly impaired or absent in the fetus and newborn infant and in cirrhotic patients [1]. This pathway is impaired in cirrhotic patients due to several causes including decreased co-factor availability (such as B vitamins), decreased SAMe content due to reduced MAT activity [24], and reduced CBS activity as CBS is allosterically activated by SAMe [31]. In support of the important contribution of SAMe to hepatic GSH levels is the fact that patients with alcoholic and non-alcoholic fatty liver diseases show increased hepatic GSH levels after six months of treatment with oral SAMe [32].
Fig. 2.
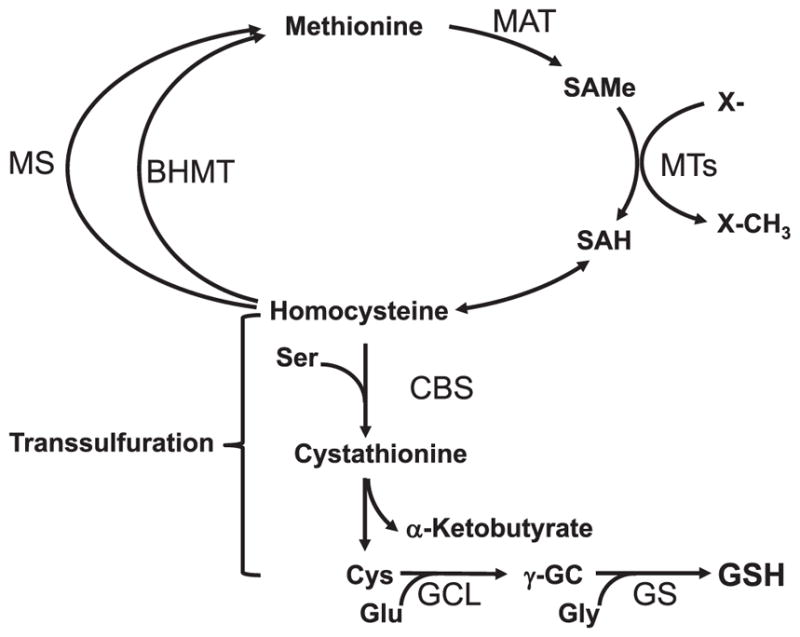
Hepatic methionine metabolism is closely linked to GSH synthesis. Nearly half of the daily intake of methionine is catabolized in the liver via methionine adenosyltransferase (MAT), generating S-adenosylmethionine (SAMe), the principal biological methyl donor that donates its methyl group to a large variety of acceptor molecules in reactions catalyzed by methyltransferases (MTs). Transmethylation produces S-adenosylhomocysteine (SAH), which is a potent inhibitor of all transmethylation reactions. SAH is hydrolyzed by SAH hydrolase, in a reversible reaction generating homocysteine and adenosine. The thermodynamics of SAH hydrolase favors biosynthesis rather than hydrolysis so that prompt removal of homocysteine and adenosine is required to prevent SAH accumulation. Homocysteine can be remethylated to form methionine or converted to cysteine (Cys) via the transsulfuration pathway, a two-step enzymatic process catalyzed by cystathionine β-synthase (CBS) and cystathionase (CSE), both requiring vitamin B6. Remethylation of homocysteine can occur via methionine synthase (MS), which requires folate and vitamin B12 and betaine homocysteine methyltransferase (BHMT), which requires betaine. The liver has the highest transsulfuration activity of all tissues, which allows methionine to be effectively utilized for GSH synthesis.
3. MicroRNAs targeting glutathione synthesis/metabolism
MicroRNAs (miRNAs) are 21–25 nucleotide small noncoding RNAs that regulate gene expression by targeting the 3′UTR region of mRNAs leading to reduce protein translation and/or increased mRNA degradation in the majority of the cases [33]. Recently, it has been shown that microRNAs can also target the mRNA coding sequence and the 5′-UTR. The latter binding has also been suggested to have a different role, related to the enhancement of protein translation [34–38]. MiRNAs were first reported in 1993 and studies over the past two decades have established them as one of the major mechanisms of gene regulation [39].
Theoretically the intracellular levels of GSH can be altered by miRNAs that target any of the proteins related to the different steps involved in its enzymatic synthesis [20,40]. In practice, most published work on miRNAs and GSH synthesis has looked at the rate-limiting step catalyzed by GCL. This set of miRNAs may operate either directly by targeting the 3′UTR of GCLc or GCLm, or indirectly by regulating the levels of the essential regulators of GCL subunit expression such as the Nrf2–Keap1 complex [40]. So far, few miRNAs have been declared to pertain to the former set, potential examples being represented by miR-1 and miR-433. miR-1 is one the most relevant miRNAs in cardiovascular pathologies and it is able to enhance cardiac oxidative stress by targeting relevant antioxidant enzymes such as SOD1 and GCLc [41]. Work performed in one of our laboratories showed that miR-433 was able to downregulate the expression of both subunits, GCLC and GCLM, of GCL in an Nrf2-independent manner. Oxidant status regulated miR-433 levels in a reciprocal manner in human endothelial cells and miR-433 increases resulted in reduced GSH/GSSG ratio and diminished eNOS function [42]. With relevance to hepatic dysfunction, we observed that in hepatic cells Huh7 exposed to TGF-β1 GCLC levels were reduced, an effect counteracted by co-transfection with anti miR-433. In the bile duct ligation (BDL) model Gclc and Gclm subunits were significantly downregulated and highly increased levels of miR-433 accompanied this. Studies in renal cells and in the unilateral ureteral obstructive model corroborated the capacity of TGF-β1 to depress GCL levels through the concourse of miR-433, thus providing another mechanistic explanation for the relevance of miR-433 profibrotic action in the kidney [43]. These concepts are summarized in Fig. 3.
Fig. 3.

Glutathione synthesis inhibition regulated by microRNAs. GSH levels can be regulated by miRNAs, either by targeting GCL directly or by indirectly downregulating the levels of Nrf2 (miRs 142,153, 93, 27a/b, 144 and 28, left). MiR-433 was described to downregulate the expression of both GCL subunits in an Nrf2-independent manner. This microRNA is redox-sensitive and responds to situations of either acute (top right) or chronic (bottom right) injury. After a chronic damage as that observed in hepatic or kidney fibrosis, an increase in TGF-β1 induces miR-433 expression inhibiting GCL protein expression, GSH levels and amplifying oxidative stress mediated damage contributing to the fibrotic process.
Indirect regulation by miRNAs of GSH synthesis can occur by targeting key factors that regulate the expression of GCL subunits, such as the expression of Nrf2 and Nrf2-mediated trans-activation of the antioxidant response element (ARE). Nrf2 is a member of the cap ‘n’ collar-basic leucine zipper proteins (CNC-bZIP) that is kept in the cytosol by Keap1 and undergoes proteasomal degradation under non-stressful conditions [20,44,45]. Stress signals cause release of Nrf2 from Keap1 to escape proteasomal degradation and translocate to the nucleus to induce genes involved in defense and survival, such as GSH synthetic enzymes [1,46]. Nrf2 requires heterodimerization with small Maf (MafG, MafK and MafF) and Jun (c-Jun, Jun-D, and Jun-B) proteins in order to bind and activate ARE [46]. Nrf2-mediated activation of ARE is dependent on the abundance of competing proteins and the ability of Nrf2 to form heterodimers to trans-activate ARE. For instance, small Mafs can form homodimers to repress ARE-mediated gene expression [47]. C-Maf, a large Maf protein, can also form heterodimers with small Mafs (but not Nrf2) to repress ARE-mediated gene expression [48]. We have also shown that heterodimerization between Nrf2 and MafG as well as binding to ARE require sumoylation by SUMO-1 at critical sites of Nrf2 and MafG [49]. During hepatic stellate cell activation as well as treatment of macrophages and hepatocytes with lipopolysaccharide (LPS), we observed marked reduction in Nrf2:MafG heterodimer formation and ARE binding due to impaired SUMO-1 sumoylation of Nrf2 and MafG, resulting in reduced GCL expression and lower GSH level [49,50]. Several miRNAs have been reported to target Nrf2 [51]. miR-144 was shown to target Nrf2 directly and higher miR-144 expression resulted in lower Nrf2 expression and lower GSH level [52]. Similarly, miR-28, miR-93, miR-153, miR-27a and miR-142-5p were also shown to directly target Nrf2 3′-UTR in different cell lines and animal models [51]. Recently we described the involvement of miR-27a and miR-27b in downregulating GCLC and GCLM expression during chronic cholestatic liver injury by mechanisms that involve Nrf2 and a novel co-activator of ARE (Fig. 4) [53]. A variety of in vivo and in cellulo models, including bile duct ligation (BDL), in vivo lithocholic acid (LCA) treatment (gavage daily) and Huh-7 cells treated with LCA were employed. We had previously shown that LCA lowers the expression of GCLC and GCLM in Huh-7 cells in a time-dependent manner and consistent with results in the BDL model, this correlated with reduced Nrf2 and higher MafG and c-Maf binding to the ARE. We had speculated that toxic bile acid caused a “displacement” of Nrf2 by MafG and c-Maf. However, we found that following BDL or LCA treatment, Nrf2 and MafG heterodimerization was actually enhanced. This contradictory finding led us to identify the participation of other proteins in this process using biotinylated ARE in pull-down assays, followed by proteomic analysis. C-Myc, which we had shown previously to be induced during BDL [54] and prohibitin 1 (PHB1), a well-known mitochondrial chaperone protein [55], also play important roles in modulating Nrf2-mediated ARE-dependent gene expression. Interestingly, c-Myc is thought to induce PHB1 at the transcriptional level but we found PHB1 expression fell despite the induction of c-Myc during cholestasis [53]. This was a result of c-Myc-mediated induction of miR27a/b, which targets both PHB1 and Nrf2 directly to reduce their expression. Knockdown of c-Myc or miR27a/b attenuated LCA-mediated suppression of Nrf2, PHB1 and GCL subunits expression, whereas overexpression of PHB1 protected against the fall in Nrf2 and GCL subunits. Both c-Myc and PHB1 directly interact with Nrf2 but c-Myc lowers Nrf2 binding to ARE while PHB1 enhances it. Taken together, activation of this c-Myc-miR-27a/b-Nrf2/PHB1 circuit in cholestatic liver injury inhibits GCL expression. Importantly, we found PHB1 expression is reduced in cholestatic liver injury in humans, such as primary biliary cirrhosis and biliary atresia [56]. Whether miR-27 and GCL expression are altered in human chronic cholestatic liver diseases remain to be examined. Another example of a miRNA that may indirectly regulate GCLm through Nrf2 is represented by miR-320 in the context of endothelial toxicity generated by oxidized phospholipids [57]. The above discussion is largely focused on miRNAs that regulate Nrf2. Since Nrf2 ARE trans-activating activity is modulated by many other proteins (such as Keap1 and Maf proteins for instance) and post-translational modifications such as phosphorylation and sumoylation, miRNAs can indirectly regulate GCL through regulation of these other modulatory steps [40,58].
Fig. 4.

Chronic cholestasis turns on c-Myc-miR27a/b circuit to inhibit ARE-dependent GCL expression. Chronic cholestasis induces c-Myc expression, which in turn induces miR27a and miR27b, both of which suppress the inner mitochondrial membrane protein, prohibitin 1 (PHB1) and Nrf2 at the post-transcriptional level directly. In normal liver GCLC and GCLM expression is positively regulated by ARE, which is activated by heterodimer of Nrf2 with small Maf or Jun proteins. In addition, c-Myc and PHB1 also directly interact with Nrf2 but c-Myc serves as a co-repressor while PHB1 acts as a co-activator for Nrf2-driven ARE-dependent gene expression. In normal liver the expression of PHB1 is high and c-Myc is low, allowing for positive Nrf2-driven ARE-dependent GCL expression. In chronic cholestatic liver injury there is induction of c-Myc, leading to down-regulation of both Nrf2 and PHB1. In addition, there is induction of MafG and c-Maf, which can homodimerize (MafG:MafG) and heterodimerize (c-Maf: MafG) to repress ARE-dependent gene expression. These changes all work in concert to bring about down-regulation of GCLC and GCLM expression. Adapted from Yang et al. [53].
Beyond the specific targeting of the GSH enzymatic system and in particular that of GCL, in the past years several miRNAs have been reported to exert effects on GSH levels or on GSH-related metabolic pathways (outside the liver) and as such will be briefly mentioned in this review. One interesting case is exemplified by miR-96-5p, which targeted the cysteine transporter excitatory amino acid carrier 1 (EAAC1) in dopaminergic brain regions and contributed to a reduction in GSH levels [59]. Of interest, the authors showed that the pattern of this miRNA exhibited diurnal rhythmicity, thus contributing to the fluctuation of GSH levels and suggesting that its inhibition could represent a therapeutic avenue for neuroprotection.
The role of GSTs has been explored in the context of several cancer types due to its role in detoxification. An early report described how in a murine model of aging several miRNAs were upregulated in the liver [60]. Of interest, some of them (miR-93, miR-214 and miR-669c) targeted several classes of GSTs. Several groups have also looked at miRNAs targeting the GST system in pathological contexts and for example it has been found that miR-133b may target GST-π and contributed to enhanced sensitivity to chemotherapy in ovarian cancer [61]. A previous report [62] had also shown that miR-133a induced apoptosis in bladder carcinoma cell lines through the targeting of GST-π. Other miRNAs may target GSH levels by interfering with enzymes involved in its recycling, i.e. GR. In this regard miR-214 was also shown to enhance alcohol-induced oxidative stress in liver cells by binding to GR and P450 reductase mRNAs [63]. Overall there is increasing evidence on the functional importance of miRNAs in the regulation of GSH levels, either by directly targeting its synthetic machinery or by interfering with routes related to its recycling or participation in critical GSH-dependent homeostatic pathways, which are essential for the maintenance of the cellular redox state.
4. MicroRNAs targeting methionine metabolism
In the liver, methionine metabolism is tightly regulated in order to keep SAMe within a small concentration range [64]. When the levels of SAMe drop, the methionine regeneration pathway is activated, shunting Hcy away from transsulfuration as CBS activity falls [24]. When SAMe levels are in excess, methyltransferases are induced in order to keep SAMe concentration in check and transsulfuration is activated [24]. Hepatic SAMe levels are controlled by MAT-catalyzed biosynthesis and mainly glycine N-methyltransferase (GNMT)-catalyzed utilization [24]. We have shown that the deficiency in either hepatic MAT or GNMT in mice results in liver injury and spontaneous formation of hepatocellular carcinoma (HCC) (reviewed in [24]). Recent works from our lab and others have identified several miRNAs that target these two major enzymes.
In mammals, two different genes encode for the catalytic subunit of MAT – MAT1A that is largely expressed in normal differentiated hepatocytes and MAT2A that is expressed in all extra-hepatic tissues and in the non-parenchymal cells of the liver [24]. Patients with chronic liver disease have reduced hepatic MAT activity because of reduced expression of MAT1A at the mRNA level and inactivation of the MAT1A-encoded enzymes [65]. MAT1A is also often silent in HCC and this is associated with a worse outcome [66]. Importantly, overexpressing MAT1A inhibited growth of HCC in vivo, which was associated with inhibition in angiogenesis and an increase in apoptosis [67]. However, the limited feasibility of this approach led us to search for miRNAs that might regulate MAT1A. We identified three miRNAs whose roles in HCC were unknown: miR-495, miR-664, miR-485-3p. These miRNAs are induced in HCC and suppress MAT1A expression at the mRNA level [68]. We provided evidence that suppressing the expression of these miRNAs resulted in increased MAT1A expression and inhibition of tumor growth, invasion and metastasis in an orthotopic HCC model. We also provided clear evidence that when MAT1A expression increased in HCC, a lot of the MAT1A-encoded protein showed up in the nucleus and this resulted in higher nuclear SAMe level, global DNA hypermethylation and other epigenetic changes [68]. One of the pathways affected by this is the LIN28B–Let-7 pathway. LIN28B is often overexpressed in HCC and plays an important role in tumor invasion and metastasis by down-regulating the expression of let-7, a well-recognized tumor suppressor that targets cell proliferation, angiogenesis and apoptosis [69, 70]. We found that when MAT1A expression increased, the LIN28B promoter region became hypermethylated and LIN28B expression fell. This then led to increased let-7 expression [68]. These findings are summarized in Fig. 5 and show the important role of MAT1A expression in HCC tumorigenesis and the potential of targeting the miRNAs that suppress MAT1A expression in the treatment of HCC. In addition to our report, one other study showed that miR-22 and miR-29b target Mat1a in the rat [71].
Fig. 5.

MiRNAs-mediated down-regulation of MAT1A leads to silencing of let-7 by an epigenetic mechanism. Normal liver expresses high levels of MATα1 and SAMe, which exerts an inhibitory tone on growth factors while being hepatoprotective. In HCC several miRNAs are induced, including miR-664, miR-485-3p and miR-495, and they all suppress MAT1A expression at the mRNA level. This results in reduced SAMe level and global DNA hypomethylation. One of the pathways affected is LIN28B–let-7. Let-7 is a well-known tumor suppressor and when SAMe levels are low, the LIN28B promoter region becomes hypomethylated and LIN28B expression is induced, resulting in suppression of let-7, favoring tumor growth, invasion and metastasis. Taken from Yang et al. [68].
5. Conclusion
We have highlighted the importance of the emerging field of miRNAs as crucial regulatory molecules in the context of two fundamental homeostatic pathways of liver metabolism, which are also interconnected between them, the GSH biosynthetic pathway and the methionine cycle. Both are regulated by specific sets of miRNAs that may act in a concerted action to aggravate previous pathological states or trigger counteracting routes in specific disease settings. In this regard, the liver is an essential organ to scrutinize, as exemplified by the above discussed pathophysiological animal models.
Acknowledgments
Supporting Grants include: Ministerio de Economía y Competitividad (MINECO) SAF 2012-31338 (SL), Instituto de Salud Carlos III REDinREN RD12/0021/0009 (SL), Comunidad de Madrid “Fibroteam” S2010/BMD-2321 (SL) and Fundación Renal “Iñigo Alvarez de Toledo” (SL), all from Spain, NIH Grants R01DK092407 (S. C. Lu), and R01DK51719 (S.C. Lu and J.M. Mato) and European Cooperation in Science and Research COST action BM-1203 (EU-ROS). The CBMSO receives institutional support from Fundación “Ramón Areces”. Cristina Espinosa has been a fellow of the FPI program, MINECO, Spain. Authors thank Dr. Tony Li for help creating some of the figures.
References
- 1.Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830:3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richman PG, Meister A. Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J Biol Chem. 1975;250:1422–1426. [PubMed] [Google Scholar]
- 3.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 4.Bannai S, Tateishi N. Role of membrane transport in metabolism and function of glutathione in mammals. J Membr Biol. 1986;89:1–8. doi: 10.1007/BF01870891. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem. 2005;280:33766–33774. doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- 6.Luo JL, Hammarqvist F, Andersson K, Wernerman J. Surgical trauma decreases glutathione synthetic capacity in human skeletal muscle tissue. Am J Physiol. 1998;275:E359–E365. doi: 10.1152/ajpendo.1998.275.2.E359. [DOI] [PubMed] [Google Scholar]
- 7.Banach-Latapy A, He T, Dardalhon M, Vernis L, Chanet R, Huang ME. Redox-sensitive YFP sensors for monitoring dynamic compartment-specific glutathione redox state. Free Radic Biol Med. 2013;65:436–445. doi: 10.1016/j.freeradbiomed.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 8.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan B, Ezerina D, Amoako TN, Riemer J, Seedorf M, Dick TP. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat Chem Biol. 2013;9:119–125. doi: 10.1038/nchembio.1142. [DOI] [PubMed] [Google Scholar]
- 10.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 11.Mieyal JJ, Chock PB. Posttranslational modification of cysteine in redox signaling and oxidative stress: focus on s-glutathionylation. Antioxid Redox Signal. 2012;16:471–475. doi: 10.1089/ars.2011.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye ZW, Zhang J, Townsend DM, Tew KD. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim Biophys Acta. 2015;1850:1607–1621. doi: 10.1016/j.bbagen.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazzetti AP, Fiorile MC, Primavera A, Lo Bello M. Glutathione transferases and neurodegenerative diseases. Neurochem Int. 2015;82:10–18. doi: 10.1016/j.neuint.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Board PG, Menon D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim Biophys Acta. 2013;1830:3267–3288. doi: 10.1016/j.bbagen.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Giffen PS, Pick CR, Price MA, Williams A, York MJ. Alpha-glutathione S-transferase in the assessment of hepatotoxicity – its diagnostic utility in comparison with other recognized markers in the Wistar Han rat. Toxicol Pathol. 2002;30:365–372. doi: 10.1080/01926230252929945. [DOI] [PubMed] [Google Scholar]
- 16.Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Common structural features of MAPEG – a widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci: Publ Protein Soc. 1999;8:689–692. doi: 10.1110/ps.8.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIlwain CC, Townsend DM, Tew KD. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGarry DJ, Chakravarty P, Wolf CR, Henderson CJ. Altered protein S-glutathionylation identifies a potential mechanism of resistance to acetaminophen-induced hepatotoxicity. J Pharmacol Exp Ther. 2015;355:137–144. doi: 10.1124/jpet.115.227389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci USA. 2000;97:12741–12745. doi: 10.1073/pnas.220176997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sanchez-Perez P, Cadenas S, Lamas S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015;6:183–197. doi: 10.1016/j.redox.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilberg MS. Amino acid transport in isolated rat hepatocytes. J Membr Biol. 1982;69:1–12. doi: 10.1007/BF01871236. [DOI] [PubMed] [Google Scholar]
- 22.Takada A, Bannai S. Transport of cystine in isolated rat hepatocytes in primary culture. J Biol Chem. 1984;259:2441–2445. [PubMed] [Google Scholar]
- 23.Finkelstein JD. Methionine metabolism in liver diseases. Am J Clin Nutr. 2003;77:1094–1095. doi: 10.1093/ajcn/77.5.1094. [DOI] [PubMed] [Google Scholar]
- 24.Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–1542. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tehlivets O, Malanovic N, Visram M, Pavkov-Keller T, Keller W. S-adenosyl-l-homocysteine hydrolase and methylation disorders: yeast as a model system. Biochim Biophys Acta. 2013;1832:204–215. doi: 10.1016/j.bbadis.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 27.Szabo C, Ransy C, Modis K, Andriamihaja M, Murghes B, Coletta C, Olah G, Yanagi K, Bouillaud F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2014;171:2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N, Kimura H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun. 2013;4:1366. doi: 10.1038/ncomms2371. [DOI] [PubMed] [Google Scholar]
- 29.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hourihan JM, Kenna JG, Hayes JD. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid Redox Signal. 2013;19:465–481. doi: 10.1089/ars.2012.4944. [DOI] [PubMed] [Google Scholar]
- 31.Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Nat Acad Sci USA. 2006;103:6489–6494. doi: 10.1073/pnas.0509531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vendemiale G, Altomare E, Trizio T, Le Grazie C, Di Padova C, Salerno MT, Carrieri V, Albano O. Effects of oral S-adenosyl-L-methionine on hepatic glutathione in patients with liver disease. Scand J Gastroenterol. 1989;24:407–415. doi: 10.3109/00365528909093067. [DOI] [PubMed] [Google Scholar]
- 33.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 34.Tsai NP, Lin YL, Wei LN. MicroRNA mir-346 targets the 5′-untranslated region of receptor-interacting protein 140 (RIP140) mRNA and up-regulates its protein expression. Biochem J. 2009;424:411–418. doi: 10.1042/BJ20090915. [DOI] [PubMed] [Google Scholar]
- 35.Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA. 2010;16:2493–2502. doi: 10.1261/rna.2384610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci USA. 2007;104:9667–9672. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M. MicroRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valinezhad Orang A, Safaralizadeh R, Kazemzadeh-Bavili M. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int J Genom. 2014;2014:970607. doi: 10.1155/2014/970607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic Biol Med. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Yuan Y, Li J, Ren H, Cai Q, Chen X, Liang H, Shan H, Fu ZD, Gao X, Lv Y, Yang B, Zhang Y. MicroRNA-1 aggravates cardiac oxidative stress by post-transcriptional modification of the antioxidant network. Cell Stress Chaperones. 2015;20:411–420. doi: 10.1007/s12192-014-0565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espinosa-Diez C, Fierro-Fernandez M, Sanchez-Gomez F, Rodriguez-Pascual F, Alique M, Ruiz-Ortega M, Beraza N, Martinez-Chantar ML, Fernandez-Hernando C, Lamas S. Targeting of gamma-glutamyl-cysteine ligase by miR-433 reduces glutathione biosynthesis and promotes TGF-beta-dependent fibrogenesis. Antioxid Redox Signal. 2015;23:1092–1105. doi: 10.1089/ars.2014.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li R, Chung AC, Dong Y, Yang W, Zhong X, Lan HY. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-beta/Smad3-Azin1 pathway. Kidney Int. 2013;84:1129–1144. doi: 10.1038/ki.2013.272. [DOI] [PubMed] [Google Scholar]
- 44.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1–Nrf2–ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki T, Yamamoto M. Molecular basis of the Keap1–Nrf2 system. Free Radic Biol Med. 2015;88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 47.Dhakshinamoorthy S, Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 48.Dhakshinamoorthy S, Jaiswal AK. c-Maf negatively regulates ARE-mediated detoxifying enzyme genes expression and anti-oxidant induction. Oncogene. 2002;21:5301–5312. doi: 10.1038/sj.onc.1205642. [DOI] [PubMed] [Google Scholar]
- 49.Ramani K, Tomasi ML, Yang H, Ko K, Lu SC. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J Biol Chem. 2012;287:36341–36355. doi: 10.1074/jbc.M112.370775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomasi ML, Ryoo M, Yang H, Iglesias Ara A, Ko KS, Lu SC. Molecular mechanisms of lipopolysaccharide-mediated inhibition of glutathione synthesis in mice. Free Radic Biol Med. 2014;68:148–158. doi: 10.1016/j.freeradbiomed.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ayers D, Baron B, Hunter T. miRNA influences in NRF2 pathway interactions within cancer models. J Nucl Acids. 2015;2015:143636. doi: 10.1155/2015/143636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–4348. doi: 10.1182/blood-2009-04-214817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang H, Li TW, Zhou Y, Peng H, Liu T, Zandi E, Martinez-Chantar ML, Mato JM, Lu SC. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid Redox Signal. 2015;22:259–274. doi: 10.1089/ars.2014.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang H, Li TW, Ko KS, Xia M, Lu SC. Switch from Mnt-Max to Myc-Max induces p53 and cyclin D1 expression and apoptosis during cholestasis in mouse and human hepatocytes. Hepatology. 2009;49:860–870. doi: 10.1002/hep.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ko KS, Tomasi ML, Iglesias-Ara A, French BA, French SW, Ramani K, Lozano JJ, Oh P, He L, Stiles BL, Li TW, Yang H, Martinez-Chantar ML, Mato JM, Lu SC. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology. 2010;52:2096–2108. doi: 10.1002/hep.23919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barbier-Torres L, Beraza N, Fernandez-Tussy P, Lopitz-Otsoa F, Fernandez-Ramos D, Zubiete-Franco I, Varela-Rey M, Delgado TC, Gutierrez V, Anguita J, Pares A, Banales JM, Villa E, Caballeria J, Alvarez L, Lu SC, Mato JM, Martinez-Chantar ML. Histone deacetylase 4 promotes cholestatic liver injury in the absence of prohibitin-1. Hepatology. 2015;62:1237–1248. doi: 10.1002/hep.27959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrottmaier WC, Oskolkova OV, Schabbauer G, Afonyushkin T. MicroRNA miR-320a modulates induction of HO-1, GCLM and OKL38 by oxidized phospholipids in endothelial cells. Atherosclerosis. 2014;235:1–8. doi: 10.1016/j.atherosclerosis.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 58.Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic Biol Med. 2015;89:548–556. doi: 10.1016/j.freeradbiomed.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinoshita C, Aoyama K, Matsumura N, Kikuchi-Utsumi K, Watabe M, Nakaki T. Rhythmic oscillations of the microRNA miR-96-5p play a neuroprotective role by indirectly regulating glutathione levels. Nat Commun. 2014;5:3823. doi: 10.1038/ncomms4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maes OC, An J, Sarojini H, Wang E. Murine microRNAs implicated in liver functions and aging process. Mechan Ageing Dev. 2008;129:534–541. doi: 10.1016/j.mad.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 61.Chen S, Jiao JW, Sun KX, Zong ZH, Zhao Y. microRNA-133b targets glutathione S-transferase pi expression to increase ovarian cancer cell sensitivity to chemotherapy drugs. Drug Des, Dev Therapy. 2015;9:5225–5235. doi: 10.2147/DDDT.S87526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uchida Y, Chiyomaru T, Enokida H, Kawakami K, Tatarano S, Kawahara K, Nishiyama K, Seki N, Nakagawa M. MiR-133a induces apoptosis through direct regulation of GSTP1 in bladder cancer cell lines. Urol Oncol. 2013;31:115–123. doi: 10.1016/j.urolonc.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 63.Dong X, Liu H, Chen F, Li D, Zhao Y. MiR-214 promotes the alcohol-induced oxidative stress via down-regulation of glutathione reductase and cytochrome P450 oxidoreductase in liver cells. Alcohol Clin Exp Res. 2014;38:68–77. doi: 10.1111/acer.12209. [DOI] [PubMed] [Google Scholar]
- 64.Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- 65.Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, Prieto J, Lu SC, Caballeria J, Rodes J, Mato JM. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 66.Frau M, Tomasi ML, Simile MM, Demartis MI, Salis F, Latte G, Calvisi DF, Seddaiu MA, Daino L, Feo CF, Brozzetti S, Solinas G, Yamashita S, Ushijima T, Feo F, Pascale RM. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology. 2012;56:165–175. doi: 10.1002/hep.25643. [DOI] [PubMed] [Google Scholar]
- 67.Li J, Ramani K, Sun Z, Zee C, Grant EG, Yang H, Xia M, Oh P, Ko K, Mato JM, Lu SC. Forced expression of methionine adenosyltransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am J Pathol. 2010;176:2456–2466. doi: 10.2353/ajpath.2010.090810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang H, Cho ME, Li TW, Peng H, Ko KS, Mato JM, Lu SC. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J Clin investig. 2013;123:285–298. doi: 10.1172/JCI63861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70–80. doi: 10.3747/co.v17i1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang YC, Chen YL, Yuan RH, Pan HW, Yang WC, Hsu HC, Jeng YM. Lin-28B expression promotes transformation and invasion in human hepatocellular carcinoma. Carcinogenesis. 2010;31:1516–1522. doi: 10.1093/carcin/bgq107. [DOI] [PubMed] [Google Scholar]
- 71.Koturbash I, Melnyk S, James SJ, Beland FA, Pogribny IP. Role of epigenetic and miR-22 and miR-29b alterations in the downregulation of Mat1a and Mthfr genes in early preneoplastic livers in rats induced by 2-acetylaminofluorene. Mol Carcinog. 2013;52:318–327. doi: 10.1002/mc.21861. [DOI] [PubMed] [Google Scholar]