

Abstract
Dendritic cells (DCs) are critical for launching of protective T cell immunity in response to viral infection. Viruses can directly infect DCs, thereby compromising their viability and suppressing their ability to activate immune responses. How DC function is maintained in light of this paradox is not understood. By analyzing the susceptibility of primary human DC subsets to viral infections, we report that CD141+ DCs have an innate resistance to infection by a broad range of enveloped viruses, including human immunodeficiency virus (HIV) and influenza virus. In contrast, CD1c+ DCs are susceptible to infection which enables viral antigen production but impairs their immune functions and survival. The ability of CD141+ DCs to resist infection is conferred by RAB15, a vesicle trafficking protein constitutively expressed in this DC subset. We show that CD141+ DCs rely on viral antigens produced in bystander cells to launch cross-presentation driven T cell responses. By dissociating viral infection from antigen presentation, this mechanism protects the functional capacity of DCs to launch adaptive immunity against viral infection.
Introduction
Dendritic cells (DCs) are a heterogeneous population of antigen-presenting cells essential for the launching of protective T cell immunity. In humans, DCs include three major subsets with different phenotypes, tissue localizations and functions. For example, blood and lung CD1c+ DCs drive the differentiation of mucosal effector CD8+ T cells in response to influenza virus (1); blood and lung CD141+ DCs are the most potent in cross-presentation of antigens from dying cells (2–6); and blood plasmacytoid DCs (pDCs) rapidly produce abundant type I interferons in response to many viruses (7). This functional specialization is determined in part by subset-specific pathogen recognition receptor expression (8) and by spatio-temporal orchestration (9–11). However, the mechanisms that allow DC subsets to develop specialized functions remain incompletely understood (12, 13), especially in the context of viral infection where DCs are both subject to viral infection and key cells to activate and regulate immune response against viruses.
It is well established that DCs respond to viruses and vaccines through their innate sensors, allowing them to initiate adaptive anti-viral effector T cell and B cell responses (12, 14–17). Extracellular viral particles engage sensors facing the extracellular and vesicular space, such as Toll-Like Receptors (TLRs), and the viral antigens are processed for presentation on MHC. Upon infection viruses reach the cytoplasm and start replication, which provides additional antigens for presentation on MHC and activates cytosolic immune sensors (18, 19)(13)(20). However, viral infection also leads to irreversible damage of cellular integrity and enables manipulation of immune responses by the virus (21–23). Viral infection of DCs may also generate inflammatory mediators that can contribute to disease (24). Studies in monocyte-derived DCs (MDCCs) showed that HIV-1 infection is restricted by SAMHD1 (25, 26), which prevents DC activation through the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS) and limits antigen presentation to T cells (19, 27, 28). HIV-2, a virus with reduced pathogenicity compared to HIV-1 (29), abrogates SAMHD1 restriction through its Vpx protein (27). Accordingly, Vpx sensitizes HIV-1 infection in MDDCs and restores the ability of MDCCs to recognize the virus (19, 30). pDCs employ a Vpx-independent mechanism of resistance to HIV-1 infection (31) and respond to viral particles via a TLR7-dependent endosomal pathway (32). In mouse models, lung DCs respond to influenza virus infection (33) and migrate to the draining lymph nodes (34) where they do not appear to be infected (35). Murine pDCs respond to influenza virus through TLR7, even in the absence of viral replication (36–39), while bone-marrow-derived DCs use the cytosolic nucleic acid sensor RIG-I (21). In humans, infection of blood DCs by influenza virus impairs their cross-presentation ability (37, 38) and pDCs resist influenza virus infection through an unknown mechanism (37).
The question thus arises how DCs balance the need to acquire antigen with avoidance of viral targeting and its detrimental consequences to promote protective antiviral immunity. To address this question, we used functional in vitro and genetic approaches in human blood-derived and tissue-resident DCs to evaluate DC-subtype-specific functional responses to two human pathogenic RNA viruses: Human Immunodeficiency Virus (HIV) and influenza virus.
Results
Differential susceptibility of human DC subsets to HIV and influenza virus infection
Blood CD1c+ DCs, CD141+ DCs and pDCs were sorted from healthy donors (Fig. 1A, S1A) and exposed to viruses. Upon infected the cells with titrated doses of a CCR5-tropic HIV-2 reporter virus that encodes GFP instead of Nef (40) (molecular clone JK7312As, HIV-2(JK) herein) (Fig. S1B), CD1c+ DCs became infected with at titers similar to those of susceptible reporter GHOST cells (Fig. S1C). CD141+ DCs and pDCs showed substantially lower frequencies of GFP-expressing cells (4.6-fold and 2-fold, respectively) (Fig. 1B, 1C, S1C, S1D). As expected, an intact Vpx gene was required for infection of all DC subsets by HIV-2, indicating that SAMHD1 is an active restriction factor in all blood DC subsets (Fig. S1E, S1F). All blood DCs were refractory to infection by both CXCR4-tropic HIV-1 (HIV-1(NL4-3)) and CCR5-tropic HIV-1 (HIV-1(BaL)), which lacks Vpx (Fig. 1D, 1E, S1G). Including the Vpx protein in viral particles increased susceptibility of CD1c+ DCs to HIV-1 infection, but CD141+ DCs and pDCs remained resistant (Fig. 1D, 1E, S1G, S1H, S1I).
Fig. 1. Preferential infection of CD1c+ DCs by HIV-1, HIV-2 and influenza virus.
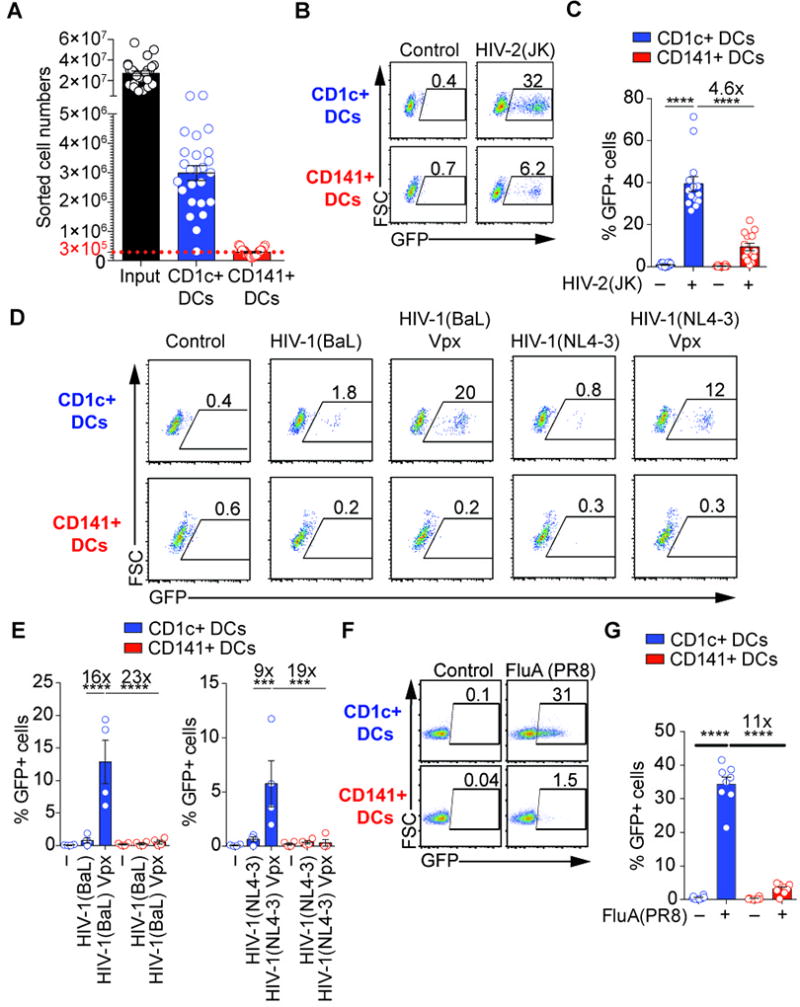
(A) Absolute cell number of sorted DC subsets (n=25 donors). Total DCs were enriched by negative selection with magnetic beads (input) and sorted by FACS
(B) Susceptibility of blood DCs to infection by HIV-2. GFP expression in blood DC subsets that were sorted and infected for 48hr with GFP-coding HIV-2(JK) at MOIGHOST X4R5=0.4.
(C) Quantification as in (B) (n=14 independent donors combined from 7 independent experiments).
(D) Susceptibility of blood DCs to infection by HIV-1 and impact of co-delivered Vpx protein that degrades SAMHD1. GFP expression in blood DC subsets that were sorted and infected for 48hr with GFP-coding HIV-1(BaL) (MOI=0.8), HIV-1(BaL) Vpx (MOI=0.4), HIV-1(NL4-3) (MOI=0.6) or HIV-1(NL4-3) (MOI=0.3). Viruses were not spinoculated in this experiment.
(E) Quantification as (D) (n=4 independent donors combined from 2 independent experiments). Viruses were not spinoculated in this experiment.
(F) Susceptibility of blood DCs to infection by influenza virus. GFP expression in sorted CD1c+ and CD141+ DCs pulsed with NS1-GFP H1N1 influenza virus (GFP-tagged FluA(PR8); MOI=2) for 1hr. Analysis was performed 4hr after infection.
(G) Quantification as in (F) in DCs at 24hr post-infection (n=8; 7 donors combined from 7 independent experiments, including 1 donor repeated 2 times; ANOVA).
We next examined infection with influenza A virus. Blood CD1c+ and CD141+ DCs were infected with recombinant live influenza virus H1N1 carrying a GFP reporter gene in the NS1 segment (FluA(PR8) herein) (41). CD1c+ DCs became infected, while CD141+ DCs were comparatively resistant (Fig. 1F, 1G). CD1c+ DCs, but not CD141+ DCs, expressed the non-structural viral protein NS1 (Fig. S1J). Viral titrations confirmed the resistance of CD141+ DCs to influenza virus infection (Fig. S1K). Thus, human DC subsets show differential susceptibility to infection with influenza virus, HIV-2 and HIV-1 with Vpx.
Constitutive inhibition of HIV and influenza virus entry in CD141+ DCs and pDCs
Type I interferons are known to induce a broad antiviral state in cells. Gene expression analysis confirmed earlier studies on expression of interferon-stimulated genes (ISGs) in CD1c+ DCs, and not in CD141+ DCs (Fig. S2A) (42). To test if type I interferon production contributed to the resistance of CD141+ DCs to infection, we used recombinant B18R, a soluble protein encoded by vaccinia virus that neutralizes type I interferon (43) (Fig. S2B, S2C). B18R did not rescue infection of CD141+ DCs with influenza virus or infection of CD141+ DCs and pDCs with HIV-1 (Fig. S2B, S2B). Thus, the resistance of CD141+ DCs was independent of type I interferon-induced antiviral state.
We next examined steps of the viral infection cycle. We measured the ability of HIV to fuse with the cell membrane using the BlaM-Vpr reporter assay (Fig. 2A) (44). HIV-1(BaL) and HIV-1(NL4-3) fused efficiently with CD1c+ DCs, but not with CD141+ DCs and pDCs (Fig. 2A, 2B, S2D). All DC subsets expressed the HIV receptors CD4, CCR5 and CXCR4 (Fig. S2E) at variable levels that did not correlate with the resistance of the cells, suggesting that reduced receptor levels were not responsible for the reduced efficiency of viral fusion in CD141+ DCs and pDCs (Fig. S2E). The quantity of cell-associated HIV-1 p24 protein was also similar between DC subsets at the time of the fusion assay, indicating that lower viral internalization also does not account for reduced fusion (Fig. S2F).
Fig. 2. Resistance of CD141+ DCs to HIV and influenza virus infection at the level of viral fusion.
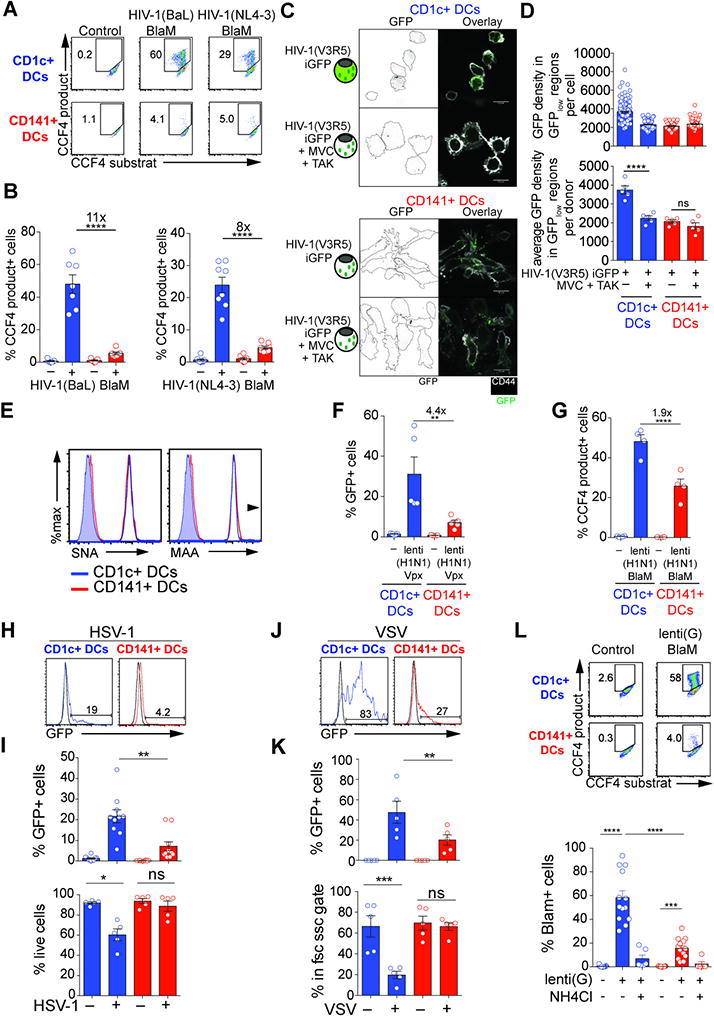
(A) HIV-1 fusion assay in blood DCs. Viral fusion revealed by CCF4 fluorescence in blood DCs after infection with HIV-1(BaL) (MOI=0.8) or HIV-1(NL4-3) (MOI=0.6) containing a BlaM-Vpr fusion protein. Fluorescence of CCF4 product indicates viral fusion with target cells as a result of cleavage of the cell-loaded CCF4 substrate by the viral-contained beta-lactamase (BlaM).
(B) Quantification as in (A) (n=8 donors combined from 4 independent experiments).
(C) Staining of GFP proteins contained in viral particles and CD44 in CD1c+ and CD141+ DCs after infection with HIV-1(V3R5) iGFP containing GAG-iGFP and GFP-Vpr fusion proteins, alone or in the presence of viral entry inhibitors MVC and TAK-779. Scale Bar = 10µm.
(D) Quantification of the GFP density in GFPlow regions as in (C), shown for one representative donor (top) and average for 5 donors (bottom; combined from 2 independent experiments).
(E) Levels of influenza virus receptors on blood DCs. SNA and MAA binding on CD1c+ and CD141+ DCs (representative of 2 independent experiments).
(F) GFP expression in blood DC subsets that were sorted and infected for 48hr with GFP-coding lenti(H1N1) Vpx at MOIGHOST X4R5=1. (n=4 independent donors combined from 2 independent experiments). Viruses were not spinoculated.
(G) Viral fusion revealed as in A by CCF4 fluorescence in blood DCs after infection with lenti(H1N1) (MOI=1) containing a BlaM-Vpr fusion protein. (n=4 donors combined from 2 independent experiments). Viruses were not spinoculated.
(H) GFP expression in blood DC subsets that were sorted and infected for 24hr with HSV-1-GFP at MOI=25. (one representative donor).
(I) Quantification of GFP expression and frequency of live cells in blood DC subsets that were sorted and infected for 24hr with HSV-1-GFP at MOI=25 as in (H). (n=10 combined from 4 experiments).
(J) GFP expression in blood DC subsets that were sorted and infected for 24hr with VSVeGFP at MOI=16. (one representative donor).
(K) Quantification of GFP expression and frequency of live cells in blood DC subsets that were sorted and infected for 24hr with VSVeGFP at MOI=16 as in (J). (n=5 combined from 2 experiments).
(L) Viral fusion revealed as in A by CCF4 fluorescence in blood DCs after infection with lenti(G) (MOI=10) containing a BlaM-Vpr fusion protein. (n=13 combined from 5 experiments).
We next examined the localization of a GFP-labeled virus, HIV-1(V3R5) Gag-iGFP within the cells. In CD1c+ DCs, GFP-enriched puncta and diffuse, low-level staining in the cytosol was observed suggestive of viral fusion that resulted in GFP dispersion from viral particles (Fig. 2C, S2G). Using CCR5 antagonists that prevent viral fusion but preserve incoming viral particles, we confirmed that the diffuse cytosolic staining was abrogated while the GFP-enriched puncta remained, indicating retention of the virus in endocytic compartments (Fig. 2C, 2D, S2H). In CD141+ DCs, the virus exclusively accumulated in puncta while diffuse cytosolic staining was not detectable, consistent with an inhibition of viral fusion in this subset (Fig. 2C, 2D, S2H). In pDCs, the resolution was too low to unequivocally distinguish cytoplasmic staining. We next measured SAMHD1 protein levels after infection, since its degradation induced by Vpx also requires viral fusion. In CD1c+ DCs, SAMHD1 was degraded in the majority of cells (Fig. S2I, S2J). In contrast, most CD141+ DCs and pDCs remained positive for SAMHD1 (Fig. S2I, S2J). We also evaluated progression of the viral cycle by measuring the amount of reverse-transcribed DNA species. All viral DNA species were reduced in CD141+ DCs and pDCs compared to CD1c+ DCs (Fig. S2K). These results indicate that HIV is endocytosed in all DC subsets, but in CD141+ DCs and pDCs viral fusion and entry is inhibited.
Next, we examined at which step infection by the influenza virus is inhibited in CD141+ DCs. CD1c+ DCs and CD141+ DCs showed similar levels of α2,3- and α2,6-linked sialic acid, which are receptors for influenza virus entry (Fig. 2E). To determine if viral fusion was inhibited, we used H1N1-pseudotyped lentivector (lenti(H1N1)). Infection of CD141+ DCs and pDCs by lenti(H1N1) was reduced compared to CD1c+ DCs, recapitulating our results using FluA(PR8) (Fig. 2F, S2L). Using the BlaM-Vpr assay, we found that CD1c+ DCs fused efficiently with the virus, while viral fusion was reduced in CD141+ DCs and pDCs (Fig. 2G, S2L). Thus, CD141+ DC and pDC resistance to influenza virus can be attributed in part to reduced fusion of the virus with the cell, similar to that observed with HIV.
Broad resistance of CD141+ DCs to infection by enveloped viruses
Since resistance occurs after endocytosis but before viral fusion, we examined how the viral envelope could influence infection of DC subtypes. Vesicular stomatitis virus (VSV) and herpes simplex virus (HSV-1) are enveloped endocytic viruses, modified vaccinia Ankara (MVA) is a membrane-fusing enveloped live attenuated vaccine strain (45, 46) and adenovirus (AdV) is a non-enveloped virus. CD141+ DCs and pDCs were largely resistant to infection-induced cell death by GFP-reporter VSV (47) and HSV, and the fraction of GFP-positive cells was reduced two-fold compared to CD1c+ DCs (Fig. 2H, 2I, 2J, 2K, 2L,S2M). CD141+ DCs and pDCs were also largely resistant to infection by VSV-G-pseudotyped lentivector (Fig. S1E, S1F, S1K) and to viral fusion mediated by VSV-G (Fig. 2L, S2N). In contrast, all DC subsets were readily infected with MVA and GFP-reporter AdV (Fig. S2O, S2P). Thus, CD141+ DCs and pDCs preferentially resist infection by endocytic enveloped viruses.
RAB15 limits viral infection in CD141+ DCs
We next searched for differentially expressed genes that regulate endocytosis in CD141+ DCs and pDCs compared to CD1c+ DCs. RAB proteins are key coordinators of endocytic pathways and vesicular trafficking (48). We found that RAB15 was the only RAB selectively expressed in both CD141+ DCs and pDCs (Fig. 3A, S3A). RNA-seq and RT-qPCR analysis confirmed the selective expression of RAB15 in CD141+ DCs and pDCs and its paucity in CD1c+ DCs (Fig. 3A). We first tested the impact of ectopic expression of RAB15 (Fig. 3B). In CD1c+ DCs, transduction of a GFP-RAB15 lentivector resulted in low expression of GFP-RAB15 and a small but significant decrease in infection with H1N1 and VSV-G-pseusdotyped viruses. To obtain cells with higher levels of RAB15 ectopic expression, we transduced monocytic THP-1 cells with BFP-RAB15 or either BFP alone or the endosomal GTPase BFP-RAB5A as controls and sorted high from low BFP expressers. Strikingly, RAB15 overexpression in THP-1 cells reduced viral infection and infection-induced cell death after exposure to HIV-2(JK), influenza virus, VSV, HSV-1 (Fig. 3B) as well as lentivectors pseudotyped with VSV-G or H1N1 (Fig. S3B). Of note, expression of RAB15 AN1, a splicing variant of RAB15 that lacks the CXC domain required for membrane binding (49), completely and specifically abrogated infection and fusion by influenza virus H1N1 and H1N1-pseudotyped virus (Fig. S3C, S3D, S3E, S3F, S3G, S3H, S3I, S3J).
Fig. 3. RAB15 mediates resistance to HIV and influenza virus entry.
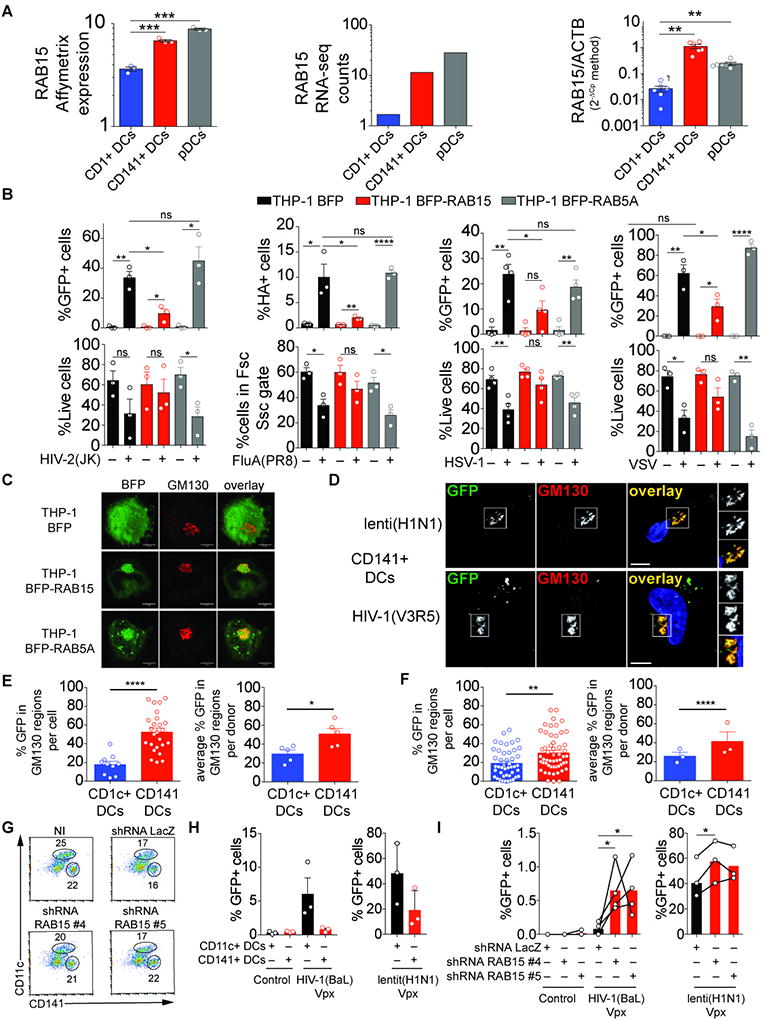
(A) RAB15 expression in blood DCs in the public dataset E-TABM-34 (left panel), measured by RNA-seq (average number of read counts for 3 independent donors; middle panel), and measured by RT-qPCR (n=6 independent donors; right panel).
(B) Viral infection of THP-1 cells over-expressing RAB15 or RAB5A. Viral expression and live cell frequency in THP-1 cells expressing BFP-RAB15, BFP-RAB5A or BFP following infection with GFP-encoding HIV-2(JK) (MOI=1.2) 48hr post infection, GFP-tagged FluA(PR8) (2 µg/ml) 24hr post infection, GFP-encoding HSV-1-GFP (MOI=0.016) 24hr post infection and GFP-encoding VSVeGFP (MOI=25) 24hr post infection.
(C) Localization of BFP, BFP-RAB15 or BFP-RAB5A (green) and GM130 (red) in THP-1 cells. (scale bar=5µm).
(D) Viral particle localization with GM130 in CD141+ DCs. Staining of GFP and GM130 in CD141+ DCs 12hr after infection with lenti(H1N1) iGFP (top) and HIV-1(V3R5) iGFP (bottom) viral particles that contain GFP proteins (scale bar=5µM).
(E) Quantification of HIV-1(V3R5) iGFP viral particles localization with GM130 as in (D). One representative donor (left panel) and average for 5 donors combined from 2 independent experiments (right panel).
(F) Quantification of lenti(H1N1) iGFP viral particles localization with GM130 as in (D). One representative donor (left panel) and average for 5 donors combined from 2 independent experiments (right panel).
(G) Frequencies of CD141+ and CD11c+ cells in CD34+ cells that were expanded, transduced with LacZ or RAB15 shRNA #4 or #5 lentivectors and differentiated (representative of 3 independent donors).
(H) GFP expression in CD34-derived CD141+ DCs and CD11c+ DCs after infection of total CD34-derived cells with GFP-encoding HIV-1(BaL) Vpx or HIV-1(H1N1) Vpx (n=3, combined from 3 independent experiments).
(I) Role of RAB15 in the resistance of CD141+ DCs to viral infection. GFP expression in CD34-derived CD141+ DCs that were first transduced with the indicated shRNA lentivectors, after infection of total CD34-derived cells with GFP-encoding HIV-1(BaL) Vpx or HIV-1(H1N1) Vpx (n=4, combined from 3 independent experiments).
To understand how RAB15 limits viral infection, we first examined its localization. In THP-1 cells BFP-RAB15 co-localized with the Golgi marker GM130 (Fig. 3C). We then investigated the localization of HIV-1(BaL) and H1N1-pseudotyped lentivector (lenti(H1N1)) in CD1c+ DCs and CD141+ DCs, finding that the two viruses were enriched in GM130-positive compartments specifically in CD141+ DCs (Fig. 3D, 3E, 3F, S3K, S3L). Finally, we sought to test if endogenous RAB15 contributed to the resistance of CD141+ DCs to viral infection. To circumvent the challenge of low numbers of CD141+ DCs that can be isolated from human blood (Fig. 1A), we generated CD141+ DCs ex vivo from CD34+ hematopoietic progenitor cell cultures (50). This system also generates CD11c+ DCs that are similar to monocyte-derived DCs (Fig. 3G). Compared to CD11c+ DCs, CD141+ DCs showed partial resistance to HIV-1(BaL) and lenti(H1N1) infection (Fig. 3H). In contrast, shRNA-mediated knock-down of endogenous RAB15 significantly increased the frequency of infected cells for HIV-1(BaL) and H1N1-pseudotyped lentivector in CD34-derived CD141+ DCs (Fig. 3I, S3M). CD141+ cell differentiation was not affected (Fig. 3G, S3N).
CD141+ DCs sense HIV and influenza in the absence of infection
The resistance of CD141+ DCs to HIV and influenza virus infection prompted us to test the role of innate sensing in the overall response to viral exposure. In response to HIV-2(JK) or HIV-1(BaL) Vpx, CD1c+ DCs upregulated CD86 and produced IP-10, indicating activation of innate immune response in the DCs, and this induction was reduced by inhibitors of viral infection (Fig. 4A, S4A, S4B). CD141+ DCs and pDCs also responded to HIV-2(JK) or HIV(BaL) Vpx as shown by induction of IP-10 in both subsets and CD86 in CD141+ DCs, but the response was not affected by viral infection inhibitors (Fig. 4A, S4A, S4B). These results suggest a functional dissociation of innate sensing from viral infection selectively in CD141+ DCs and pDCs.
Fig. 4. Distinct innate response to HIV and influenza virus determined by viral infection in blood DC subsets.
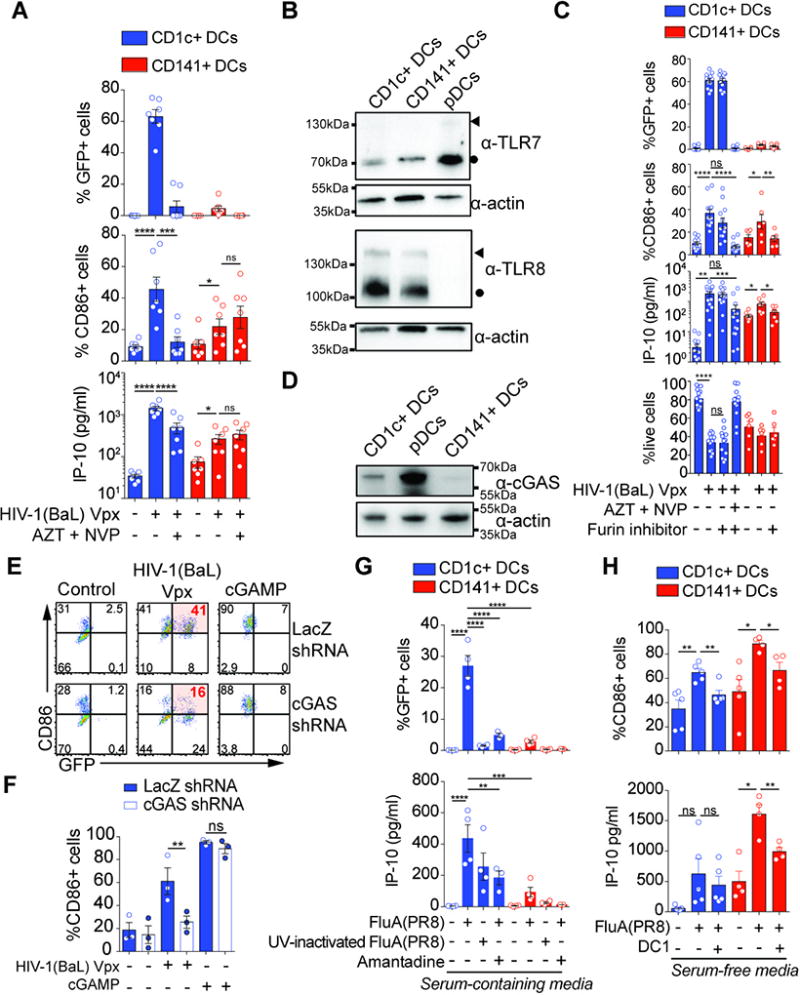
(A) Response of blood DCs to HIV and role of viral replication. GFP and CD86 expression and IP-10 production by sorted blood DCs after infection with GFP-encoding HIV-1(BaL) Vpx for 48hr alone or in the presence of a viral inhibitors AZT and NVP (n=7 donors combined from 4 independent experiments; MOI=0.4).
(B) Expression of TLR7, TLR8 and actin in lysates from blood DC subsets. Blood DC subsets from 3 independent donors were combined at the same ratio and the equivalent of 500,000 cells were loaded. One experiment is shown. Circles indicate protein sizes that match cleaved C-terminal domain of TLR8 or TLR7; arrowheads indicate protein sizes that match full-length TLR8 or TLR7 (53).
(C) Response of blood DCs to HIV-1(Vpx) with or without the furin inhibitor DC1 that prevents maturation of TLR7 and TLR8 proteins. GFP, CD86 and IP-10 expression, and live cells frequency in CD1c+ DCs and CD141+ DCs infected with GFP-encoding HIV-1(BaL) Vpx for 48hr alone or in the presence of AZT and NVP, with or without the furin inhibitor DC1 (MOI=0.8).
(D) Expression of cGAS and actin in lysates from blood DCs subsets. Blood DC subsets from 3 independent donors were combined at the same ratio and the equivalent of 500,000 cells were loaded. Representative of two independent experiments.
(E) Role of cGAS in the response of CD1c+ DCs to HIV infection. Inhibition of cGAS expression in CD1c+ DCs. GFP and CD86 expression in CD1c+ DCs that were transduced with shRNA lentivectors against LacZ or cGAS, and subsequently infected with GFP-encoding HIV-1(BaL) Vpx (MOI=1) or transfected with cGAMP (1.3µg/ml) for 48hr.
(F) Quantification of CD86 expression as in (E) (n=3 donors from 1 experiment representative of 2 independent experiments).
(G) Response of blood DCs to influenza virus. GFP expression and IP-10 production by sorted blood DCs pulsed with GFP-tagged FluA(PR8) or UV-inactivated GFP-tagged FluA(PR8) for 1hr and incubated for 24hr alone or in the presence of Amantadine (n=5 donors for GFP, n=4 donors for IP-10, combined from 2 independent experiments; MOI=2).
(H) Response of blood DCs to GFP-tagged FluA(PR8) with or without the furin inhibitor DC1. CD86 expression and IP-10 production by CD1c+ and CD141+ DCs infected with GFP- tagged FluA(PR8) for 24hr alone or in the presence of the furin inhibitor DC1 (n=5 donors from 2 independent experiments).
We next addressed the contribution of endosomal TLRs to innate sensing of HIV. pDCs respond to HIV-1 RNA through TLR7 (32) and TLR8 can signal in response to HIV-derived RNA (51). We detected expression of TLR8 protein in CD1c+ DCs and CD141+ DCs but not pDCs, while TLR7 protein was expressed in all DC subsets (Fig. 4B, S4C). We used a furin antagonist (DC1) that inhibits TLR7 and TLR8 activity by preventing cleavage (52, 53). DC1 prevented responses of pDCs to CL264, a TLR7 agonist (Fig. S4D). Furin inhibition abrogated the response of CD141+ DCs and pDCs to infection with HIV-1(BaL) Vpx, but not the response of CD1c+ DCs (Fig. 4C, S4E). This prompted us to test the putative role of cGAS in CD1c+ DCs. cGAS was detectable at different levels in all DC subsets (Fig. 4D, S4F). shRNA-mediated knockdown of cGAS (27) (Fig. S4G) prevented CD86 upregulation by CD1c+ DCs after infection by HIV-1(BaL) Vpx (Fig. 4E, 4F). Responses to cGAMP, a product of cGAS, and susceptibility to infection by the virus remained intact upon cGAS knock-down (Fig. 4E, 4F, S4H). Thus, the response of CD1c+ DCs to HIV-1 requires infection for cytosolic sensing, while CD141+ DCs respond to HIV-1 in the absence of infection.
We next examined DC responses to influenza virus, using UV inactivation or amantadine to inhibit influenza virus entry and infection (54) (Fig. 4G). CD1c+ DCs were susceptible to influenza virus infection while CD141+ DCs were largely resistant. CD1c+ and CD141+ DCs induced IP-10 in response to influenza virus and this was inhibited by UV treatment or amantadine, indicating that an intact virus is required for infection (Fig. 4G). Interferons were induced in CD1c+ DCs and were below limit of detection in CD141+ DCs (Fig. S4I), but the ability of B18R to reduced CD86 induction in response to influenza virus suggests that interferons were indeed also induced in CD141+ DCs (Fig. S2M). To determine the contribution of TLR sensing to DC activation and response, we moved to serum-free medium to limit serum-induced CD141+ DC activation. In these conditions, CD141+ DCs expressed CD86 and induced IP-10 in response to influenza virus, and both were inhibited by DC1 treatment (Fig. 4H). These results show that CD141+ DCs are capable of innate, TLR-mediated sensing of influenza virus despite being resistant to overall infection.
DC infection determines T cell responses to HIV and influenza virus
Since viral infection was essential for innate sensing in CD1c+ DCs but not CD141+ DCs, we examined how infection could determine antigen presentation to T cells by the DCs. For HIV, we utilized a CD8+ T cell line specific to a structural antigen of HIV-1 (SL9 in Gag). CD1c+ DCs activated SL9-specific CD8+ T cells more efficiently than CD141+ DCs after infection by HIV-1 with Vpx (Fig. 5A, S5A). T cell activation was abrogated when viral infection was blocked by RT inhibitors. pDCs did not activate CD8+ T cells after infection (Fig. S5B). All DC subsets activated CD8+ T cells in response to a synthetic SL9 peptide (Fig. S5C). Thus, infection of CD1c+ DCs by HIV is essential for viral antigen presentation to CD8+ T cells.
Fig. 5. Non-redundant antigen-presentation by CD1c+ DCs infected with HIV and influenza virus.
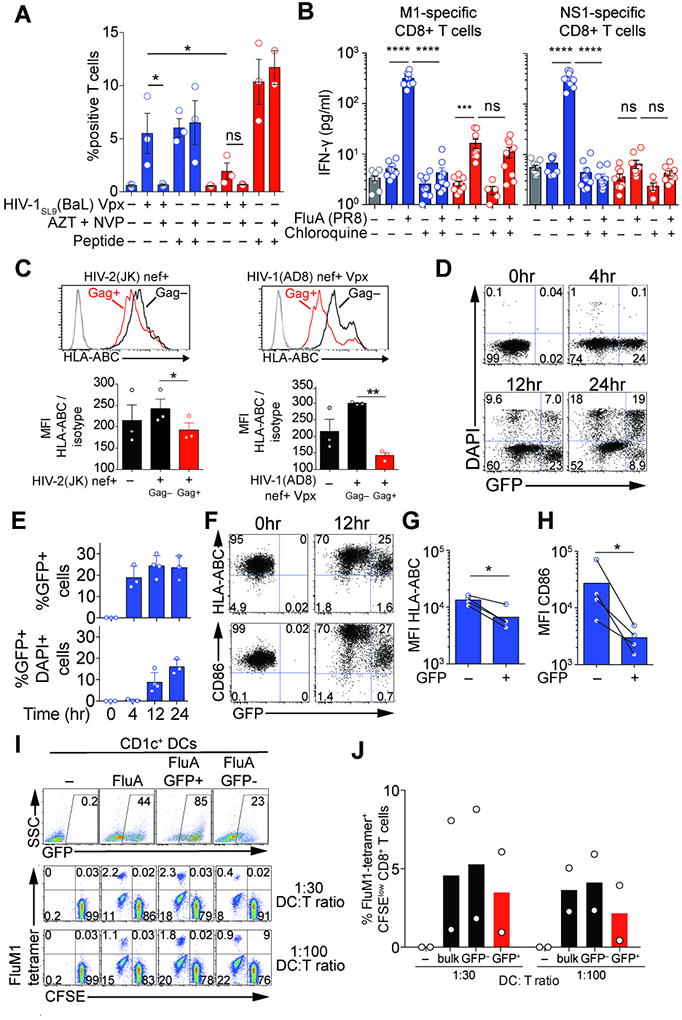
(A) Stimulation of a CD8+ T cell line by infected DCs. Combined intracellular expression of MIP-1β, TNF-α, IFN-γ and IL-2 by a SL9-specific CD8+ T cell line exposed to blood DCs infected for 48hr by HIV-1SL9(BaL) Vpx (MOI=0.8) alone or in the presence of viral inhibitors AZT and NVP (n=3 donors combined from 3 independent experiments).
(B) IFN-γ concentration in culture supernatants of M1- and NS1-specific CD8+ T cell lines exposed for 18hr to DCs that were treated with or without chloroquine for 30min and then pulsed with FluA(PR8) (MOI=2) for 1hr (n=3 donors combined from 3 experiments).
(C) Down-regulation of HLA-ABC in HIV-infected DCs. HLA-ABC expression in Gag+ and Gag- CD1c+ DCs 48h after infection with HIV-2(JK) nef+ (MOI=1) or HIV-1(AD8) nef+ Vpx (MOI=0.8) (n=3).
(D) GFP expression and viability in CD1c+ DCs at 4, 12, 24hr after infection with GFP-encoding influenza virus.
(E) GFP expression and viability as in (D) (n=4; 3 or 4 different donors combined from 3 independent experiments).
(F) Down-regulation of HLA-ABC and CD86 in influenza virus-infected DCs. GFP, HLA-ABC and CD86 expression in DAPI-negative CD1c+ DCs analyzed prior and 12hr after infection with GFP-encoding influenza virus.
(G) HLA-ABC expression as in (F) (n=4; 4 different donors combined from 3 independent experiments).
(H) CD86 expression as in (F) (n=4; 4 different donors combined from 3 independent experiments).
(I) T cell stimulation by infected vs. bystander CD1c+ DCs. GFP expression in bulk CD1c+, sorted GFP+CD1c+ and GFP-CD1c+ DCs 24hr after infection with GFP-encoding influenza virus (top panel). CFSE levels and FluM1-tetramer binding on CD8+ T cells after 7-day cocultures at 1:30 and 1:100 DC:T ratio (bottom panel).
(J) Summary of the percentage of FluM1-tetramer binding CFSElow CD8+ T cells at 7 days after coculture as in (I) (n=2; 2 different donors combined from 2 independent experiments).
To examine antigen presentation by blood DCs in response to influenza virus, we generated CD8+ T cell lines specific to the structural protein M1 and non-structural protein NS1 (Fig. S5D, S5E). M1 is present in viral particles and is expressed in infected cells. CD1c+ DCs induced activated M1-specific CD8+ T cells more efficiently than CD141+ DCs (Fig. 5B). NS1 is present in productively infected cells but not in the viral particles. Only CD1c+ DCs induced IFN-γ in NS1-specific CD8+ T cells (Fig. 5B). To test the requirement for viral infection, we used the endosomal acidification inhibitor chloroquine that inhibits influenza virus fusion. Chloroquine inhibited infection of CD1c+ DCs by FluA(PR8) and the M1- and NS1-specific T cell activation, but it did not inhibit activation of M1-specific T cells by CD141+ DCs in response to viral particles (Fig. 5B). Chloroquine had no effect on peptide presentation by CD1c+ DCs (Fig. S5F, S5G). Thus, viral infection is required in CD1c+ DCs for efficient virus-specific CD8+ T cell activation to both structural and non-structural viral antigens. In contrast, CD141+ DCs are resistant to viral infection but this restricts their presentation on MHC to the antigens present in the viral particles.
Virus infection and antigen presentation are mutually exclusive in CD1c+ DCs
We next sought to understand how CD1c+ DCs could simultaneously activate CD8+ T cells and sustain a viral infection. In HIV-1-infected cells, the Nef protein downregulates class I MHC (22). Using Nef-expressing HIV-1 and HIV-2, we confirmed that class I MHC is downregulated in a cell-intrinsic manner in infected CD1c+ DCs, but not in uninfected bystander CD1c+ DCs (Fig. 5C). We next examined the impact of influenza virus infection on viability and immunostimulatory molecule expression. In response to influenza virus infection, GFP-positive CD1c+ DCs preferentially died after 24 hours (Fig. 5D, 5E). Strikingly, Class I MHC and CD86 expression were downregulated in GFP-positive CD1c+ DCs 12 hours after infection, at a time when most cells were alive (Fig. 5F, 5G, 5H). Thus, influenza virus inhibits immunostimulatory molecule expression in infected CD1c+ DCs in a cell-intrinsic manner and subsequently induces cell death, while non-infected bystander CD1c+ DCs remain unaltered. This raised the possibility that in a culture of CD1c+ DCs partially infected after exposure to the virus, non-infected CD1c+ DCs may also contribute to antigen presentation. To test this idea, we sorted GFP-positive and GFP-negative CD1c+ DCs after infection with influenza virus and measured the expansion of autologous M1-specific CD8+ T cells. Indeed, GFP-negative CD1c+ DCs were more potent at expanding T cells than GFP-positive DCs (Fig. 5I, 5J). Thus, at least in vitro, viral infection in CD1c+ DCs is required for antigen presentation but the infection simultaneously impairs T cell stimulation capacity at the cellular level.
DC subset cooperation for induction of antiviral T cell responses
CD141+ DCs are thought to be specialized in cross-presentation of antigens from necrotic cells, but whether cross-presentation is an essential mode of antigen presentation by these cells is unknown (55). Our findings suggest that since CD141+ DCs resist viral infection, their ability to stimulate viral-specific T cells may require cross-presentation of viral antigens produced in bystander cells. To test this idea, we mixed HLA-A2-negative CD1c+ DCs with HLA-A2-positive CD141+ DCs, infected cells with HIV-1 or influenza virus, and measured T cell responses in the mixed culture (Fig. 6A, 6C). In the mixed culture, viral infection was largely restricted to HLA-A2-negative CD1c+ DCs (Fig. 6B, 6D, S6), recapitulating the situation with purified DC subsets. HLA-A2-positive CD141+ DCs were inefficient at stimulating T cells after infection with influenza virus and HIV. Strikingly, the presence of HLA-A2-negative CD1c+ DCs during infection rescued the ability of HLA-A2-positive CD141+ DCs to efficiently present HIV-1 or influenza virus antigen (Fig. 6B, 6E). These in vitro results support the notion that CD141+ DCs depend upon viral antigen produced in bystander-infected cells such as CD1c+ DCs for efficient T cell stimulation.
Fig. 6. DC subset cooperation for activation of antiviral T cells.
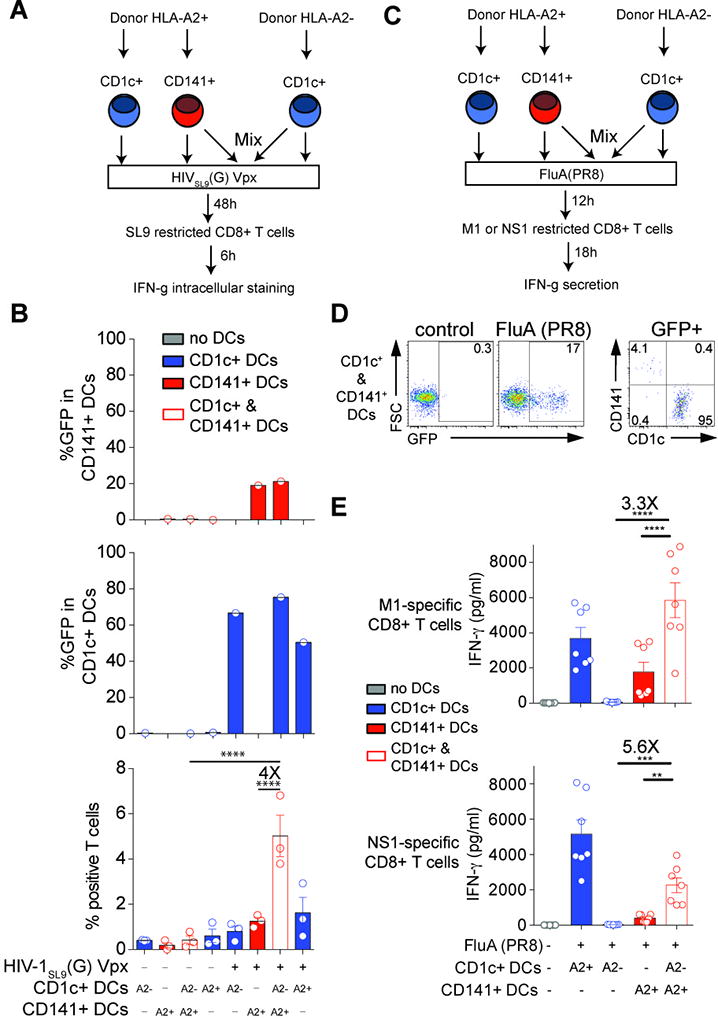
(A) Stimulation of a SL9-specific CD8+ T cell line by HLA-A2- CD1c+ DCs mixed with HLA-A2+ CD141+ DCs after infection with HIV-1, outline of the experiment.
(B) GFP expression in CD1c+ DCs and CD141+ DCs and frequency of CD8+ T cells positive for TNF and IFN-γ expression, 48hr after infection with GFP-encoding HIVSL9(G) Vpx (MOI=1) (n=3 combined from 2 experiments).
(C) Stimulation of M1- and NS1-specific CD8+ T cell lines by HLA-A2- DCs mixed with HLA-A2+ CD141+ DCs after infection with influenza virus, outline of the experiment.
(D) GFP expression in mixed CD1c+ and CD141+ DCs 4hr after infection with GFP-tagged FluA(PR8).
(E) IFN-γ concentration in culture supernatants of M1- and NS1-specific CD8+ T cell lines exposed for 18hr to DCs 12hr after infection with GFP-tagged FluA(PR8) (MOI=2; n=3, combined from 2 experiments).
Resistance of CD141+ to influenza virus infection in vivo
To validate the findings in tissue-resident DCs, we infected sorted human lung DCs with influenza virus. Similar to blood-derived DCs, human lung CD1c+ DCs were susceptible to FluA(PR8) infection ex vivo while lung CD141+ DCs were comparatively resistant (Fig. 7A, 7B). To determine susceptibility to infection in vivo, we infected humanized mice with GFP-reporter influenza virus through the intra-nasal route (Fig. 7C). In the lung, both CD1c+ and CD141+ DCs became GFP-positive over time, likely reflecting productive infection in CD1c+ DCs and antigen capture in CD141+ DCs (Fig. 7D, S7A), although we cannot exclude that CD141+ DCs may carry some undetectable level of intracellular and post-fusion viral material that failed to replicate. In contrast, only CD141+ DCs carried GFP-positive material in the draining lymph nodes (Fig. 7E, S7B). To confirm that CD1c+ DCs were infected while CD141+ DCs were resistant to infection, we analyzed the cell-surface expression of HA on GFP-positive DCs as a measure of cellular viral infection. Lung GFP+HA- cells contained both CD1c+ and CD141+ DCs, while GFP+HA+ cells contained mainly CD1c+ DCs (Fig. 7F). Rare CD141+ positive events were detected among lung GFP+HA+ cells, which may be remaining doublets and are unlikely to represent CD1c+ DCs acquiring CD141 after infection (Fig. 6D). Analysis of HA expression within lung GFP+ CD141+ DCs and GFP+CD1c+ DCs confirmed the much-reduced level of HA expression on CD141+ DCs (Fig. 7G, 7H). In the draining LNs, GFP-positive CD141+ DCs remained uninfected (Fig. 7I, 7J). These results in tissue-resident DCs recapitulate our in vitro observations on differential susceptibility of DC subsets to viral infection, thereby supporting the notion that the functional specialization of DC subsets for viral infection applies to both blood and tissue-resident DCs.
Fig. 7. Resistance of CD141+ DCs to influenza virus in vivo in humanized mice.
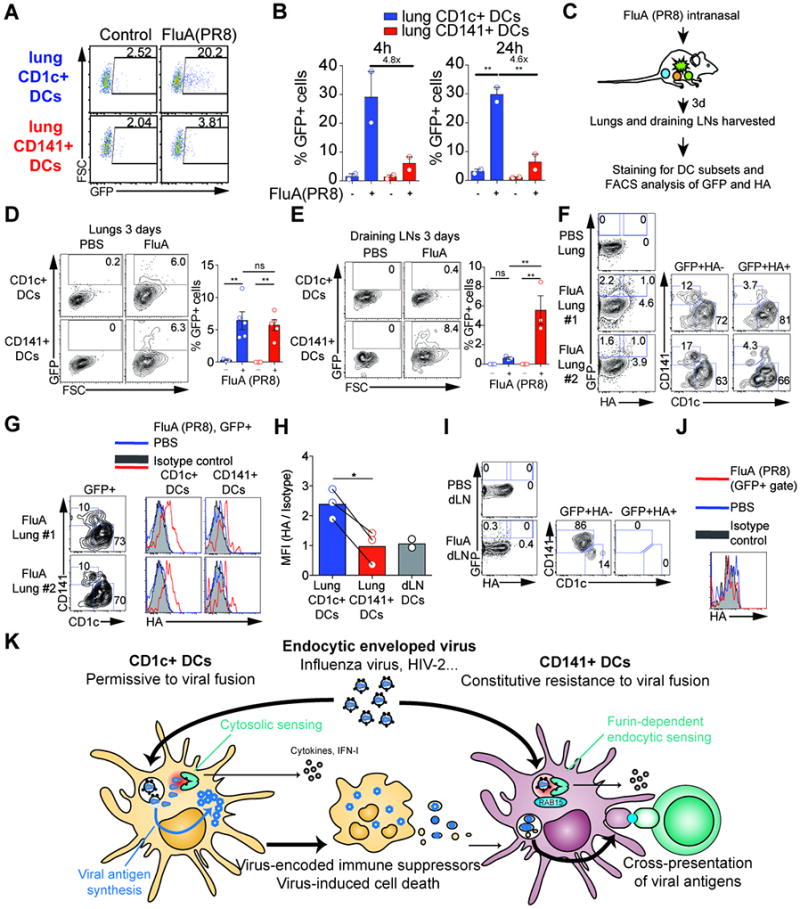
(A) Infection of human lung DC subsets. GFP expression in CD1c+ and CD141+ DCs sorted from human lungs and pulsed with GFP-tagged FluA(PR8) (MOI=2) for 1hr. Analysis was performed at 4hr after infection.
(B) Quantification as in (A) at 4, 24hr after infection (n=2 donors from 2 independent experiments).
(C) Intranasal infection of humanized mice by GFP-tagged FluA(PR8), outline of the experiment.
(D) Detection of GFP in CD1c+ DCs and CD141+ DCs in the lungs of infected humanized mice as in C, one representative sample and combined data (n=5 from 3 independent experiments).
(E) Detection of GFP in CD1c+ DCs and CD141+ DCs in the draining lymph nodes of infected humanized mice as in C, one representative sample and combined data (n=3 from 3 independent experiments).
(F) FACS plots illustrating the GFP and surface HA staining on lung DCs from PBS treatment or GFP-tagged FluA(PR8) infection of humanized mice as in C. GFP+ HA- and GFP+HA+ cells were examined for the expression of CD1c and CD141. (Representative of two independent experiments)
(G) Total GFP+ DCs as gated in panel (F) were further defined as CD1c+ and CD141+ DCs and analyzed for the expression of surface HA (red line). DC subsets from PBS-treated lungs (grey shaded) were stained with anti-HA as the control. (Each sample was a pool of 3 mice. Representative of two independent experiments)
(H) The same gating as in (F) on draining LN DCs from PBS treatment or GFP-tagged FluA (PR8) infection. (Each sample was a pool of 3–6 mice. Representative of two independent experiments)
(I) The same gating as in (F) on draining LN DCs from PBS treatment or GFP-tagged FluA (PR8) infection (each sample was a pool of 3–6 mice; representative of two independent experiments).
(J) HA expression in draining LN DCs. Total GFP+ DCs as gated in panel (I) were analyzed for the expression of surface HA (red line). Isotype staining (grey shaded) and HA staining from PBS-treated LN DCs (blue line) as the control (representative of two independent experiments).
(K) Model for T cell activation by CD141+ DCs that depends on productive infection of bystander CD1c+ DCs for the endocytic enveloped viruses tested.
Discussion
Infection of DCs with viruses can be considered a dilemma for the immune system: it allows activation of the innate and adaptive immune response, but it simultaneously facilitates manipulation by the viruses of the immune system itself. We show that this paradox is resolved by dissociation of viral infection from viral antigen presentation to T cells across DC subsets. Blood and lung DC subsets display differential susceptibility and selective resistance to infection with influenza virus (blood and lung DCs), as well as HIV, VSV and HSV (blood DCs). CD1c+ DCs are permissive to viral fusion by influenza virus and HIV, support viral infection and cytosolic sensing, and generate viral antigen that is needed to launch T cell responses, but also rapidly die due to the infection. In contrast, CD141+ DCs resist viral entry and rely on virus-infected CD1c+ DCs (and other cell types) as source of antigen for cross-presentation to promote activation of T cells (Figure 7K). We propose that this resistance of CD141+ DCs to viral entry to be a mechanism to avoid the deleterious effects of viral infection while preserving the functional capacity of DCs to launch T cell stimulation and subsequent cell-mediated immunity.
Given the critical role of DCs in the induction of antiviral immunity, it is remarkable that CD141+ DCs resist such a broad range of viruses. Nonetheless, our observations do not preclude the possibility of variable infectivity of individual viruses towards CD141+ DCs, given that additional virus-specific factors and mechanisms could play an additive role. Influenza virus and VSV are pH-dependent and require viral internalization followed by acidification in the endosomes (56). Entry of HIV-1 is pH-independent (57), but endocytosis prior to viral fusion has been extensively documented in primary CD4+ target cells (58–60). HIV receptor levels are reduced on CD141+ DCs compared to CD1c+ DCs, but receptor levels did not correlate with the pattern of resistance among subsets and thus are not the dominant factor explaining the broad resistance of CD141+ DCs.
Our results demonstrate an essential role for RAB15 in the resistance of CD141+ DC (and pDC). RAB15 appears to define a distinct antiviral resistance pathway based on constitutive and selective expression in CD141+ DCs and pDCs. HIV-1 and influenza-pseudotyped virus accumulated in GM130-positive compartments in CD141+ DCs. This correlates with the localization of RAB15 after ectopic expression, suggesting that viral particles undergo a retrograde transport and/or accumulation into the Golgi that impairs their fusogenic capacity in a RAB15-dependent manner. The transit and maturation of viral envelope glycoproteins and cellular receptors during their biogenesis, in the absence of recognized Golgi-to-Golgi fusion, supports the idea that the Golgi apparatus may be a privileged compartment with intrinsic resistance to viral envelope protein-mediated fusion. It is possible that other endocytic enveloped viruses, such as Respiratory Syncytial Virus, evade this mechanism of antiviral resistance (61, 62). Interestingly, neutralization of type I interferon had a marginal ability to rescue infection in pDCs, which contrasts with the prevailing view that the resistance of pDCs to viral infection would be mediated by autocrine type I interferon production.
Although we have identified a key role for RAB15 in promoting resistance of CD141+ DCs to viral infection, how resistance is achieved at the biochemical level remains to be determined. It is possible that CD1c+ DCs express specific gene programs that render them susceptible to viral infection in a way that overcomes RAB15 activity. An interesting scenario is that interferon-stimulated gene expression in CD1c+ DCs is not sufficient to impose an antiviral state and that CD1c+ DCs are equipped with specific programs enabling viral replication. A similar concept has been proposed for marginal zone macrophages (63). Alternatively, RAB15 might depend on specific effectors and co-factors that are also expressed selectively in CD141+ DCs that remain to be identified. How RAB15 may increase virus localization in GM130-positive compartments, and how the Golgi apparatus may limit virus-mediated fusion thus needs further study. Other RAB GTPases previously linked to RAB15 activity and to HIV and influenza virus infection could also be implicated (64–66). In addition, we find that the vaccine strain MVA poxvirus infects all DC subsets, presumably through efficient fusion at the cell surface (46). The ability to bypass RAB15-mediated resistance may be a desirable property for the immunogenicity of live attenuated vaccines such as MVA, since it would maximize production of viral antigens and triggering of cytosolic sensors within the APCs.
How DCs deal with viral infection and perform antigen presentation is highly organized in space and time (1, 9, 10, 55). Here, we have demonstrated a mechanism enabling this dual function is a dissociation of these two events. To ensure that antigen is presented, CD141+ DCs are protected from viral infection and utilize cross-presentation. This paradigm may allow a better understanding of the induction of protective immunity against viruses and live-attenuated vaccines, and sheds light on the existence of constitutive mechanisms of resistance to viral infection.
Supplementary Material
Acknowledgments
We thank Anna Lisa Lucido, Sebastian Amigorena and Philippe Benaroch for critical reading of the manuscript. Some reagents were provided by the NIH AIDS Reagent Program. We thank our healthy volunteers; Dr. Joseph Fay; Patrick Metang; the Clinical Core, the Apheresis Core, the Flow Cytometry Core, the Imaging Core, and the Genomic Core at BIIR; the Flow Cytometry facility, Vincent FRAISIER from the Cell and Tissue Imaging (PICT-IBiSA) at Institut Curie, member of the French National Research Infrastructure France-BioImaging (ANR10-INBS-04). Sean Whelan initially provided VSVeGFP to MI.
Funding: AS was successively supported by a Ministère de l’Education Nationale de la Recherche et de Technologie fellowship and by Sidaction. SC was supported by Sidaction. This work was supported by ATIP-Avenir program, Ville de Paris Emergence program, European FP7 Marie Curie Actions, LABEX VRI (ANR-10-LABX-77), LABEX DCBIOL (ANR-10-IDEX-0001-02 PSL* and ANR-11-LABX-0043), ACTERIA Foundation, Fondation Schlumberger pour l’Education et la Recherche (FSER) and European Research Council grant 309848 HIVINNATE for NM, ANRS (France REcherche Nord & Sud Sida-hiv Hépatites) for NM and AM, Baylor Research Institute (BRI; AKP), NIH grants U19AI057234 and U19AI089987 (AKP), R01DA033773 (AG-S), 1U19 AI 118610-01 (MM), U19AI117873 (MM, RAA and AG-S), UO1AI095611 (MM, AG-S and AKP), and Center for Research on Influenza Pathogenesis (NIAID CEIRS HHSN272201400008C).
Footnotes
The authors declare no conflict of interest.
Author contributions: AS & CIY performed most experiments. XL, FI and SC performed parts of HIV experiments. CB performed experiment with lung DCs. JBB performed parts of RAB15 experiments. WHK performed parts of influenza virus experiments. CC provided technical help. MM developed image analysis scripts. CG, SML & YW performed bioinformatics analysis. VP and EP directed RNAseq experiments. RAA contributed to influenza virus experiments. MI provided provided reagents and expertise in experiments involving VSV. AGS provided influenza virus reagents and expertise. BG provided expertise on RAB proteins. MD directed HIV experiments with FI. AM directed HIV experiments with SC. MM designed parts of the study. JI, AS, AFS and MM contributed to manuscript writing. AKP and NM designed the study and wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The data for this study have been deposited in the database NCBI with accession number.
References and Notes
- 1.Yu CI, et al. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity. 2013;38:818–830. doi: 10.1016/j.immuni.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachem A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. The Journal of Experimental Medicine. 2010;207:1273–1281. doi: 10.1084/jem.20100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crozat K, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. The Journal of Experimental Medicine. 2010;207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jongbloed SL, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. The Journal of Experimental Medicine. 2010;207:1247–1260. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulin LF, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. The Journal of Experimental Medicine. 2010;207:1261–1271. doi: 10.1084/jem.20092618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haniffa M, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. doi: 10.1016/j.immuni.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 8.Buschow SI, Figdor CG. Dendritic cell subsets digested: RNA sensing makes the difference! Immunity. 2010;32:149–151. doi: 10.1016/j.immuni.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Hor JL, et al. Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates CD4+ and CD8+ T Cell Activation to Localized Viral Infection. Immunity. 2015;43:554–565. doi: 10.1016/j.immuni.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 10.Eickhoff S, et al. Robust Anti-viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions. Cell. 2015;162:1322–1337. doi: 10.1016/j.cell.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 12.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 13.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;(54 Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Reis e Sousa C. Dendritic cells as sensors of infection. Immunity. 2001;14:495–498. doi: 10.1016/s1074-7613(01)00136-4. [DOI] [PubMed] [Google Scholar]
- 15.Pichlmair A, Reise e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Silvin A, Manel N. Interactions between HIV-1 and innate immunity in dendritic cells. Adv Exp Med Biol. 2013;762:183–200. doi: 10.1007/978-1-4614-4433-6_7. [DOI] [PubMed] [Google Scholar]
- 17.Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;43:336–341. doi: 10.1016/j.cyto.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Buseyne F, et al. MHC-I-restricted presentation of HIV-1 virion antigens without viral replication. Nat Med. 2001;7:344–349. doi: 10.1038/85493. [DOI] [PubMed] [Google Scholar]
- 19.Manel N, et al. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 23.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillai PS, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laguette N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hrecka K, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahaye X, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Gao D, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowland-Jones SL, Whittle HC. Whittle, Out of Africa: what can we learn from HIV-2 about protective immunity to HIV-1? Nat Immunol. 2007;8:329–331. doi: 10.1038/ni0407-329. [DOI] [PubMed] [Google Scholar]
- 30.Manel N, Littman DR. Hiding in plain sight: how HIV evades innate immune responses. Cell. 2011;147:271–274. doi: 10.1016/j.cell.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bloch N, et al. HIV type 1 infection of plasmacytoid and myeloid dendritic cells is restricted by high levels of SAMHD1 and cannot be counteracted by Vpx. AIDS Res Hum Retroviruses. 2014;30:195–203. doi: 10.1089/aid.2013.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lepelley A, et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011;7:e1001284. doi: 10.1371/journal.ppat.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao X, Kim TS, Braciale TJ. Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol. 2008;82:4908–4919. doi: 10.1128/JVI.02367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 35.Helft J, et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest. 2012;122:4037–4047. doi: 10.1172/JCI60659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 37.Smed-Sorensen A, et al. Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog. 2012;8:e1002572. doi: 10.1371/journal.ppat.1002572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fonteneau JF, et al. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 2003;101:3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- 39.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 40.Gao F, et al. Human infection by genetically diverse SIVSM-related HIV-2 in west Africa. Nature. 1992;358:495–499. doi: 10.1038/358495a0. [DOI] [PubMed] [Google Scholar]
- 41.Manicassamy B, et al. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robbins SH, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biology. 2008;9:R17. doi: 10.1186/gb-2008-9-1-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Symons JA, Alcami A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81:551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 44.Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 45.Chang SJ, Chang YX, Izmailyan R, Tang YL, Chang W. Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions determine strain-specific virus entry pathways into HeLa CHO-K1 and L cells. J Virol. 2010;84:8422–8432. doi: 10.1128/JVI.00599-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol. 2005;86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 47.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 49.Pham TV, et al. Rab15 alternative splicing is altered in spheres of neuroblastoma cells. Oncol Rep. 2012;27:2045–2049. doi: 10.3892/or.2012.1731. [DOI] [PubMed] [Google Scholar]
- 50.Balan S, et al. Human XCR1+ dendritic cells derived in vitro from CD34+ progenitors closely resemble blood dendritic cells, including their adjuvant responsiveness, contrary to monocyte-derived dendritic cells. J Immunol. 2014;193:1622–1635. doi: 10.4049/jimmunol.1401243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 52.Hipp MM, et al. Processing of human toll-like receptor 7 by furin-like proprotein convertases is required for its accumulation and activity in endosomes. Immunity. 2013;39:711–721. doi: 10.1016/j.immuni.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishii N, Funami K, Tatematsu M, Seya T, Matsumoto M. Endosomal localization of TLR8 confers distinctive proteolytic processing on human myeloid cells. J Immunol. 2014;193:5118–5128. doi: 10.4049/jimmunol.1401375. [DOI] [PubMed] [Google Scholar]
- 54.Budowsky EI, Bresler SE, Friedman EA, Zheleznova NV. Principles of selective inactivation of viral genome. I. UV-induced inactivation of influenza virus. Arch Virol. 1981;68:239–247. doi: 10.1007/BF01314577. [DOI] [PubMed] [Google Scholar]
- 55.Segura E, Amigorena S. Cross-Presentation in Mouse and Human Dendritic Cells. Adv Immunol. 2015;127:1–31. doi: 10.1016/bs.ai.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stein BS, et al. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 1987;49:659–668. doi: 10.1016/0092-8674(87)90542-3. [DOI] [PubMed] [Google Scholar]
- 58.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marechal V, et al. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol. 2001;75:11166–11177. doi: 10.1128/JVI.75.22.11166-11177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu HJ, Reuter MA, McDonald D. HIV traffics through a specialized surface-accessible intracellular compartment during trans-infection of T cells by mature dendritic cells. PLoS Pathog. 2008;4:e1000134. doi: 10.1371/journal.ppat.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta MR, Kolli D, Garofalo RP. Differential response of BDCA-1+ and BDCA-3+ myeloid dendritic cells to respiratory syncytial virus infection. Respir Res. 2013;14:71. doi: 10.1186/1465-9921-14-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson TR, Johnson CN, Corbett KS, Edwards GC, Graham BS. Primary human mDC1, mDC2 and pDC dendritic cells are differentially infected and activated by respiratory syncytial virus. PLoS One. 2011;6:e16458. doi: 10.1371/journal.pone.0016458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Honke N, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol. 2011;13:51–57. doi: 10.1038/ni.2169. [DOI] [PubMed] [Google Scholar]
- 64.Zuk PA, Elferink LA. Rab15 differentially regulates early endocytic trafficking. J Biol Chem. 2000;275:26754–26764. doi: 10.1074/jbc.M000344200. [DOI] [PubMed] [Google Scholar]
- 65.Sieczkarski SB, Whittaker GR. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic. 2003;4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 66.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.