

SUMMARY
Lung complications are a major cause of rheumatoid arthritis-related mortality. Involvement of gut microbiota in lung diseases by the gut-lung axis has been widely observed, but the underlying mechanism remains mostly unknown. Using an autoimmune arthritis model, we show that a constituent of the gut microbiota, segmented filamentous bacteria (SFB), distantly provoke lung pathology. SFB induce autoantibodies in lung during the pre-arthritic phase, and SFB-dependent lung pathology requires the T helper 17 (Th17) responses. SFB-induced gut Th17 cells are preferentially recruited to lung over spleen due to robust expression in the lung of the Th17 chemoattractant, CCL20. Additionally, we found that in peripheral tissues, SFB selectively expand dual T cell receptor (TCR)-expressing Th17 cells recognizing both an SFB epitope and self-antigen, thus augmenting autoimmunity. This study reveals mechanisms for commensal-mediated gut-lung crosstalk and dual TCR-based autoimmunity.
Graphical Abstract
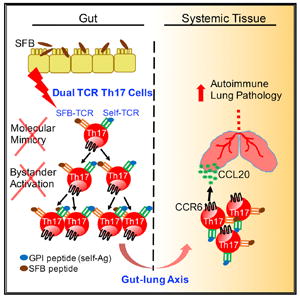
INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune disorder. Lung complications are common (19%–58%) and rank as the second most common cause of death in RA patients (Olson et al., 2011). The low concordance rate of RA in monozygotic twins (~20%) suggests that environmental factors play a key role in RA (Seldin et al., 1999). We have previously demonstrated that the gut microbiota, segmented filamentous bacteria (SFB), act as an environmental cue to enhance autoimmune arthritis by inducing T helper 17 (Th17) and T follicular helper (Tfh) cells (Teng et al., 2016; Wu et al., 2010). A strong interest has emerged in characterizing the role of gut microbiota in lung disease, a gut-lung axis of communication, exemplified by gut microbiota’s impact on diseases including asthma, chronic obstructive pulmonary disease (COPD), and respiratory infections (Budden et al., 2017). However, mechanistically, little is known regarding how gut commensals modulate another mucosal site at the lung.
The K/BxN mice are an autoimmune arthritis model in which transgenic KRN T cells recognize glucose-6-phosphate isomerase (GPI), the self-Ag presented by MHC class II Ag7 molecules. Many autoimmune diseases display ectopic lymphoid tissues (ELTs) in the autoimmune target organs (Neyt et al., 2012). The inducible bronchus-associated lymphoid tissues (iBALT), a type of ELT found in the lung of RA patients, has been shown to correlate with lung tissue damage (Rangel-Moreno et al., 2006). K/BxN mice develop iBALT-like structures characterized by peribronchial and perivascular lymphocytic infiltration (Naskar et al., 2017). Thus, iBALT-like structures provide a clinically relevant index for RA-related lung disease in K/BxN mice.
A long-standing question in the field of host-microbe interactions is how microbes are involved in the development of autoimmunity. Molecular mimicry theorizes that microbes trigger autoimmunity by shared or cross-reactive epitopes between microbes and self-peptides, which activate self-reactive T cells (Albert and Inman, 1999; Münz et al., 2009). A less well-known theory is that dual TCR expression on T cells promotes autoimmunity by allowing autoreactive T cells to escape thymic clonal deletion (Elliott and Altmann, 1995; Ji et al., 2010; Padovan et al., 1995). In both theories, infectious pathogens including viruses and bacteria have been implicated as culprits, and little is known about the molecular mechanism by which commensals could trigger autoimmunity. However, this is an urgent subject, as dysbiosis-related diseases have emerged as new epidemics in the industrialized world (Levy et al., 2017; Yurkovetskiy et al., 2015).
Here, we test whether a gut commensal, SFB, can provoke lung autoimmunity, and if so, what molecular mechanism allows SFB to activate autoimmune T cells. Our results demonstrate that SFB remotely provoke iBALT-like structure formation in lung by upregulating mucosal Th17 cells of the gut-lung axis. We found that SFB boost autoimmunity by expanding a population of dual TCR Th17 cells that sense both SFB and self-Ag.
RESULTS
SFB-Containing Feces Trigger iBALT-like Structures and Robust Autoantibody Production
We first investigated whether microbiota act as an environmental cue to affect lung pathology. We previously established a model to study the effect of SFB in autoimmune development by gavaging SFB-containing (simplified as SFB+ hereafter) feces into SFB negative (SFB−) mice housed in our specific-pathogen-free animal facility (Teng et al., 2016). Arthritis development plateaus on or beyond day 14 post-SFB gavage in this model. We thus examined lung pathology at this arthritic disease phase between 14 and 21 days post-SFB gavage (~5–6.5 weeks old). We found a causative relationship of SFB in triggering lymphocytic infiltration of the lung (Figure 1A). SFB only triggered lung pathology in autoimmune-susceptible animals, as SFB did not induce pathology in B6xNOD (BxN) mice, the non-arthritic control for K/BxN mice (Figure 1A). Just as in RA patients, lymphocytic infiltration in the lung of K/BxN mice is also located in peribronchial and perivascular areas (Figure 1B). SFB-triggered lymphocytic infiltrations contain both T and B cell zones, suggesting an iBALT-like structure (Figure 1C). Next, we examined lung anti-GPI auto-Ab production. Spleen, a systemic lymphoid tissue and the major auto-Ab-producing site in the K/BxN model, was used as a control (Huang et al., 2010). By day 14 post-SFB gavage, K/BxN mice already develop significant ankle thickness, as we reported earlier (Teng et al., 2016 and Figure 1D). At this time point, SFB also induced a strong increase in auto-Ab-secreting cells (ASCs) in both spleen and lung (Figure 1D).
Figure 1. SFB Induce iBALT-like Structures and Auto-Abs in the Lung of Arthritic K/BxN Mice.
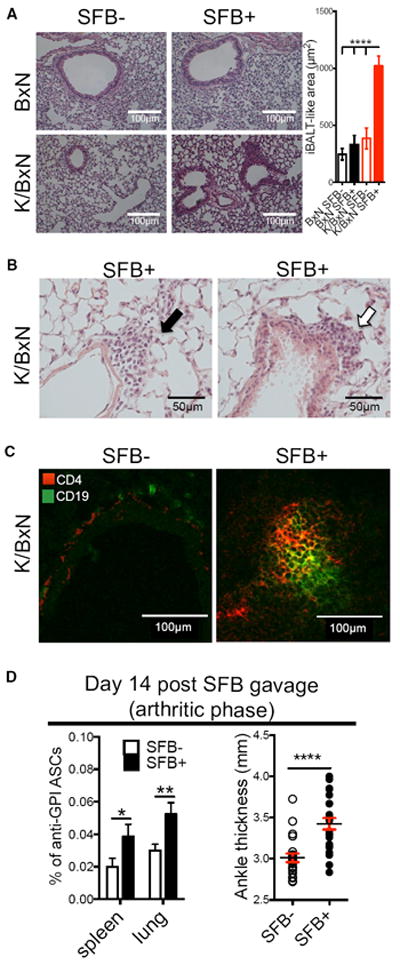
(A) H&E staining of lung from SFB− and SFB+ K/BxN or non-arthritic BxN mice (n = 4/group).
(B) Perivascular (black arrow) and peribronchial (open arrow) lung lymphocytic infiltration (n = 4/group).
(C) T (CD4+) and B cell (CD19+) zone of lymphocyte aggregation (n = 5–7 mice/group).
(D) Anti-GPI ASCs of IgG1 isotype in total K/BxN B cells (Avg + SEM) from day 14 post-SFB gavage. Ankle thickness is also shown. Each symbol indicates average ankle thickness from both ankles of each mouse (n = 19/group, six assays combined).
SFB Induce a Strong Lung Th17 Cell Response Essential for RA-Related Lung Pathology Prior to Arthritis Development
Clinical data suggest that lung may be an initiating site for RA (Demoruelle et al., 2014). We therefore measured autoimmune development at the lung mucosal site during the pre-arthritic phase, with spleen as a control. SFB are well known for their ability to induce Th17 differentiation in small intestine lamina propria. Th17 cells are a key cell type in many types of ELT, including iBALT (Pitzalis et al., 2014; Randall and Mebius, 2014). We dissected whether SFB colonization increases lung Th17 cells and, if so, whether SFB-induced Th17 cells are required to prompt iBALT-like structures in lung of K/BxN mice. A strong lung Th17 induction was observed on day 7 after SFB gavage (Figures 2A and S1A), a pre-arthritic disease phase in K/BxN mice (Teng et al., 2016 and Figure 2B). The lung Th17 cell response was much higher than the splenic Th17 response. We also measured the Th1 response using the signature Th1 cytokine, IFN-γ. SFB did not boost the Th1 response (Figure 2A). We recently reported that SFB-induced Tfh cells contribute significantly to K/BxN autoimmune arthritis development (Teng et al., 2016). Here, we chose to focus on Th17 cells because we did not observe a significant (if any) SFB-induced lung Tfh cell population (Figure S1B). In SFB− mice, lungs developed much more robust anti-GPI ASC production than spleen, and SFB further boosted these auto-Ab responses (Figure 2B). Thus, lung has a higher percentage of B cells secreting auto-Ab compared to spleen during the pre-arthritic phase.
Figure 2. SFB Induce a Robust Lung Th17 Response Essential for Lung Pathology.
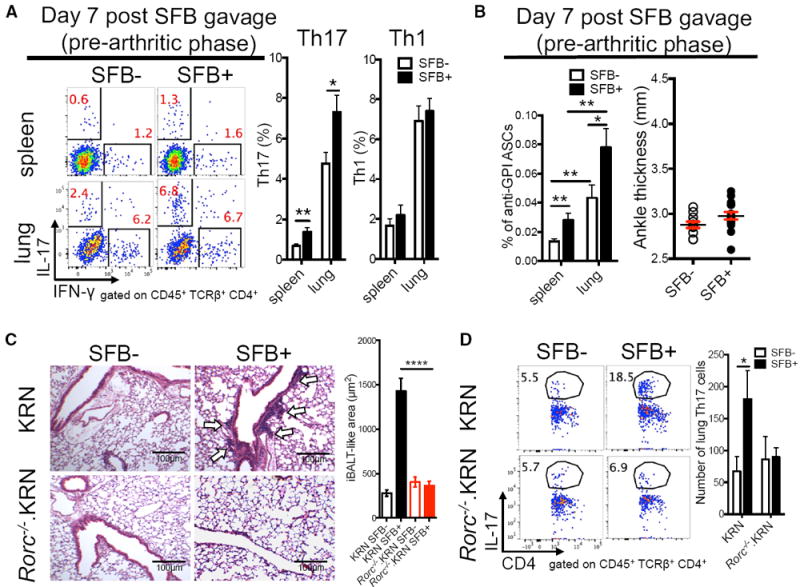
(A) Percentage of IL-17+ (Th17) or IFN-γ+ (Th1) cells in CD4+ T cells isolated from SFB− and SFB+ (day 7 post-SFB gavage) K/BxN mice. Quantitative data are also shown as mean + SEM (n = 10/group, four assays combined).
(B) Anti-GPI ASCs of IgG1 isotype in total B cells (mean + SEM) from experiments in (A) (n = 10/group, data combined from four assays). Ankle thickness of day-7 post-SFB gavage K/BxN mice is shown (n = 10/group, four assays combined).
(C) H&E staining of lung from mice receiving KRN or Rorc−/−.KRN CD4+ T cells (n = 4/group, two assays combined).
(D) Percentage of Th17 cells in CD4+ T cells from the experiment in (C). Quantitative data of Th17 cell numbers are also shown as mean + SEM (n = 6–8/group, three assays combined).
We then asked whether Th17 cells are required for the development of iBALT-like structures by using the K/BxN T cell transfer model, in which arthritis is generated by transferring KRN T cells into T cell-deficient hosts, Tcra−/−.BxN mice, on the B6xNOD background (Monach et al., 2008). Rorc−/−.KRN T cells are defective in mounting a Th17 response because the Rorc gene encodes the Th17 cell master transcription factor, RORγt (Korn et al., 2009). When Rorc−/−.KRN or KRN T cells were transferred into SFB− Tcra−/−.BxN recipients, only minimal iBALT-like structures were observed (Figure 2C). SFB colonization in Tcra−/−.BxN recipient mice strongly enhanced the development of iBALT-like structures in mice receiving KRN but not Rorc−/−.KRN T cells. SFB also boosted the Th17 response when mice received KRN T cells but not Rorc−/−.KRN T cells, despite their SFB+ status (Figure 2D). These results suggested that gut microbiota SFB were able to induce sufficient Th17 cells to cause iBALT-like pathogenesis.
Th17 Cells of Gut-Lung Axis Carry the Crosstalk between Gut Microbiota and Lung
Based on the common mucosal immune system (CMIS), whereby immune cells activated at one mucosal site home to other mucosal tissues via shared homing receptors (Kiyono and Fukuyama, 2004), we hypothesized that the mucosal gut-lung axis provides a communication interface allowing SFB to transfer the initial autoimmune signal from the gut to the lung via SFB-induced Th17 cells. To test this, we determined whether lung Th17 cell induction requires the presence of intestinal SFB by using vancomycin, an antibiotic targeting gram-positive bacteria such as SFB. Vancomycin is poorly absorbed from the gut and thus is non-detectable in the blood when delivered orally (Prasad et al., 2003). Hence, the effect of oral vancomycin is mainly limited to the gastrointestinal tract. SFB were no longer detectable in SFB+ K/BxN mice treated with oral vancomycin (Figure S2A). Concurrently, there was a significant drop in Th17 but not Th1 cells in the spleen and lung of the vancomycin-treated group (Figures 3A and S2B). Thus, Th17 induction in both organs required intestinal SFB, though lung has a much higher Th17 cell percentage than spleen.
Figure 3. SFB Induce Th17 Cells of the Gut-Lung Axis.
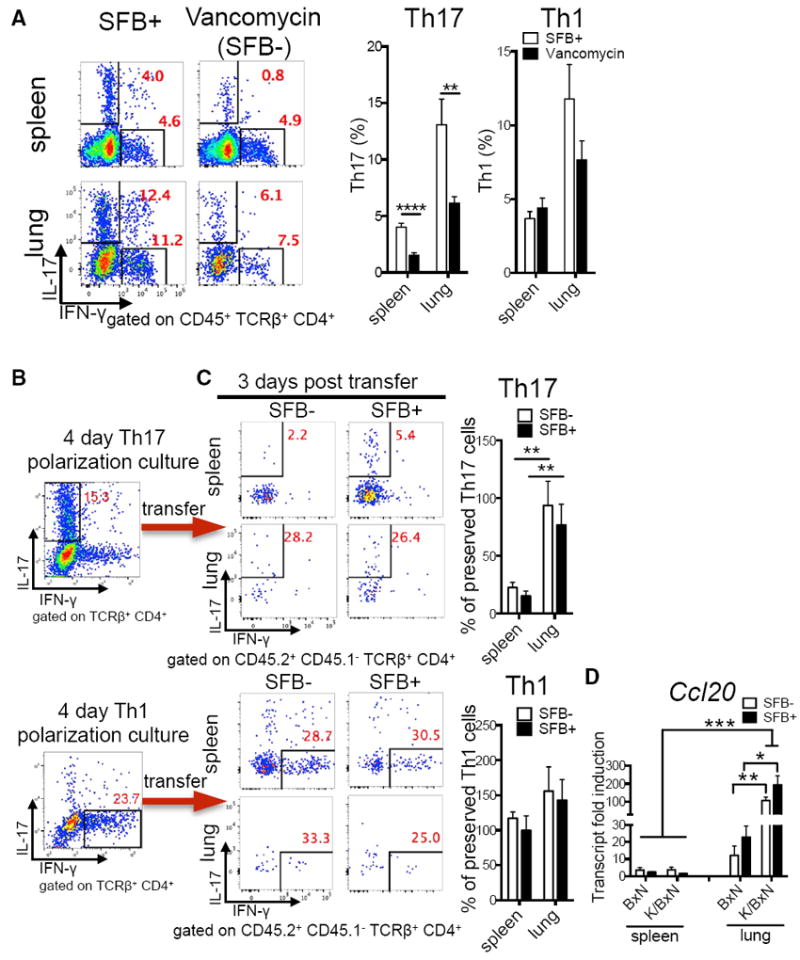
(A) Percentage of lung and splenic Th17 and Th1 cells from littermates of SFB+ K/BxN mice treated with vancomycin or left untreated (n = 8–9/group, two assays combined).
(B) Percentage of Th17 and Th1 cells from Th17- or Th1-polarized KRN T cell cultures before transfer. Representative plots of five assays are shown.
(C) The retention of transferred Th17 polarizing cells in recipient spleen or lung from the experiments in (B) is shown and calculated as percentage of preserved Th17 cells: the post-transfer Th17 percentage in spleen or lung were normalized to the starting Th17 percentage in the polarization culture of each of five experiments (n = 5–8/group, five assays combined). Similar calculations were used for Th1 cells.
(D) Transcript fold induction of CCL20 (n = 6–10/group, two assays combined).
Next, we verified lung as a preferred Th17 trafficking site. In vitro polarized Th1 and Th17 cells were analyzed before transfer into SFB− and SFB+ K/BxN mice (Figure 3B). Host and donor cells were distinguished by CD45.2 (donor) or CD45.1/CD45.2 (host) congenic markers. We avoided transferring Th17 cells into lymphopenic hosts, as that causes rapid phenotype alteration into Th1 cells (Nurieva et al., 2009). Polarized Th1 cells kept their phenotype in both lung and spleen, as we found comparable percentages of IFN-γ-expressing CD4+ T cells before and after transfer in both organs regardless of SFB status (Figures 3B and 3C). In contrast, polarized Th17 cells maintained their phenotype predominantly in the lung, but not spleen, after transfer, and organ-specific IL-17-enrichment occurred regardless of SFB status (Figures 3B and 3C). Th17 cells express the chemokine receptor CCR6. Studies reported a preferential recruitment of Th17 cells to inflamed joints, central nervous system, and intestine by chemokine ligand CCL20, the ligand for CCR6 (Esplugues et al., 2011; Hirota et al., 2007; Reboldi et al., 2009). CCL20 is typically expressed at a low basal level, but can be strongly induced by inflammatory signals (Schutyser et al., 2003). Regardless of SFB status, lung inflammation in K/BxN was sufficient to boost the CCL20 level when compared to non-arthritic BxN mice (Figure 3D). Overall, the CCL20 level was also higher in lung compared to spleen regardless of genetic background. These data imply that the higher Th17 percentage in SFB+ compared to SFB− lung is likely due to a higher output of gut-derived Th17 cells in the SFB+ intestine and suggest that these cells were later recruited to the lung by CCL20 independent of SFB status.
Gut Microbiota Trigger Autoimmunity by Expanding Th17 Cells Co-expressing Microbiota- and Self-Antigen-Specific TCRs
Th17 cells predominantly use TCR Vβ14 to recognize SFB peptide in B6 (MHC class II, H-2b) mice (Yang et al., 2014). Because K/BxN mice express the KRN TCR transgene Vβ6 on a B6xNOD (H-2bxH-2Ag7) background and are Rag competent, we tested the theory of dual TCR-mediated autoimmunity. We hypothesized that SFB preferentially select K/BxN T cells that co-express a second TCR molecule, an endogenous Vβ14 chain recognizing SFB Ag presented by MHC class II H-2b, in addition to their transgenic TCR that recognizes GPI presented by I-Ag7. Although dual TCRs have been considered a culprit in promoting autoimmunity, these studies mostly focus on thymic clonal deletion and rarely investigate how microbiota influence dual TCR T cell selection in the periphery. Studies using both transgenic and wild-type mice indicate that expression of singular or dual TCRs on T cell is controlled beyond the RNA level (Alam and Gascoigne, 1998; Sant’Angelo et al., 2001; Steinel et al., 2010). Thus, we determined TCR expression at the protein level. We found that SFB preferentially skewed KRN (Vβ6+) Th17 cells toward co-expression of dual Vβ segments, Vβ6+Vβ14+, in both lung and spleen (Figures 4A and S2C). Non-Th17 cells did not express dual TCR skewing (Figures 4A and S2C). We asked whether the expansion of Vβ6+Vβ14+ Th17 cells was due to proliferative advantages in SFB+ hosts. SFB indeed increased the ratio of TCR Vβ6+Vβ14+ to total TCR Vβ6+ in EdU+-proliferated Th17 cells, but not in non-Th17 cells (Figure 4B). Isotype controls of TCR Vβ14 staining showed very little background staining (Figures S3A–S3C). SFB did not skew the survival of the Vβ6+Vβ14+ Th17 cell population (Figure S4A). Thus, SFB promote an autoreactive Th17 response by preferentially expanding Vβ6+ KRN T cells that co-express a second TCR Vβ14, known to preferentially recognize SFB.
Figure 4. SFB Trigger Autoimmunity by Expanding Dual TCR-Expressing Autoimmune Th17 Cells.
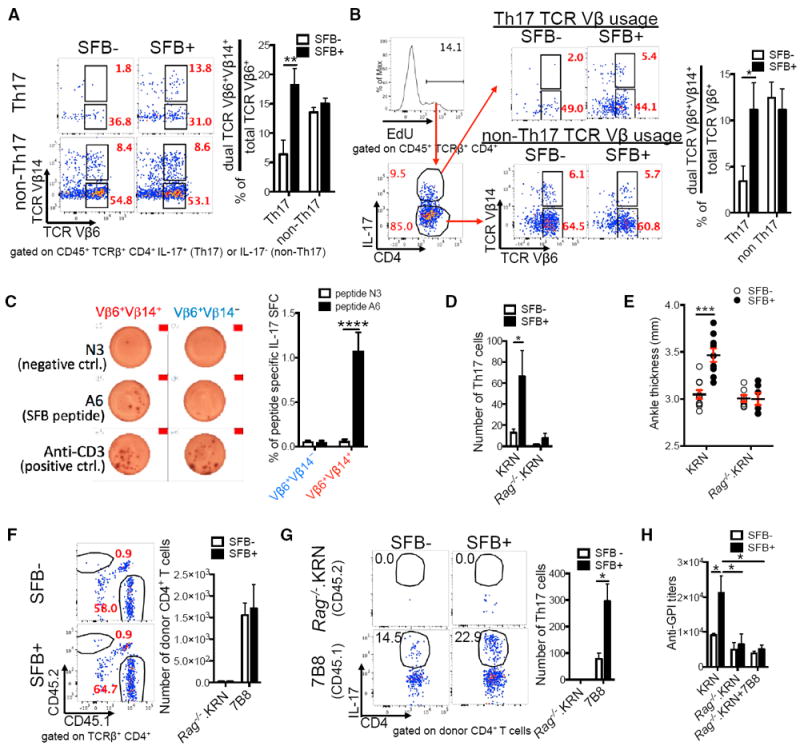
(A) SFB skew dual TCR Vβ6+Vβ14+ usage in lung Th17 cells. Representative plots show TCR Vβ6 versus TCR Vβ14 expression in lung Th17 and non-Th17 cells. Quantitative data showing the percentage of skewed Vβ6+Vβ14+ usage are calculated by dividing the percentage of Vβ6+Vβ14+ Th17 cells by the total Vβ6+ (both Vβ6+Vβ14− and Vβ6+Vβ14+) Th17 cells; similar calculations were applied to non-Th17 cells (n = 10–12/group, four assays combined).
(B) SFB increase the percentage of dual TCR Vβ6+Vβ14+ cells (among total TCR Vβ6+ cells) in proliferated (EdU+) lung Th17 but not non-Th17 cells (n = 11–12/group, three assays combined).
(C) IL-17A ELISPOT assay of both Vβ6+Vβ14− and Vβ6+Vβ14+ lung CD4+ T cell populations from SFB+ K/BxN mice treated with N3 peptide (negative control), SFB A6 peptide, or anti-CD3 (positive control). Left, representative ELISPOT images. Right, data combined from four experiments; each experiment contained pooled lung T cells from 6–8 mice (2–4 ELISPOT replicate wells/group).
(D) Number of Th17 cells in lung of SFB− and SFB+ Tcra−/−.BxN recipients transferred with KRN or Rag−/−.KRN CD4+ T cells (n = 4–9/group, four assays combined).
(E) Ankle thickness of SFB− and SFB+ Tcra−/−.BxN recipients transferred with KRN or Rag−/−.KRN CD4+ T cells from experiments in (D) are shown.
(F) Rag−/−.KRN and 7B8 CD4+ T cells were mixed at 1:1 ratio and co-transferred into SFB− or SFB+ Tcra−/−.BxN recipients. Representative plots indicate the percentage of Rag−/−.KRN (CD45.2+) and 7B8 (CD45.1+) CD4+ T cells in the total lung CD4+ T cells 4 weeks after transfer. The numbers of CD4+ T cells from both donor types in recipients’ lungs are also shown (n = 4–6/group, four assays combined).
(G) Representative plots indicate the percentage of Th17 cells in Rag−/−.KRN and 7B8 lung CD4+ T cells from the experiments in (F). The number of Th17 cells in both Rag−/−.KRN and 7B8 donor cell types are also shown.
(H) Serum anti-GPI titers from the experiments in (D) and (F).
To address whether this second TCR Vβ14 on KRN T cells truly recognizes SFB, we isolated both TCR Vβ6+Vβ14+ and TCR Vβ6+Vβ14− populations from SFB+ K/BxN mice and re-stimulated them with SFB peptide A6 (Yang et al., 2014) plus DCs isolated from Tcra−/−.B6 mice. We used DCs from Tcra−/−.B6 (H-2b) but not K/BxN (H-2b × H-2Ag7) mice so that only the SFB A6 peptide on I-Ab was presented. This avoided presentation of ubiquitously expressed self-antigen GPI by I-Ag7. Neither population responded to the negative control N3 peptide (Figure 4C). The Vβ6+Vβ14+ population responded robustly to the A6 SFB peptide, as shown by a surge of IL-17 spot-forming cells (SFCs), while IL-17 SFCs were at background level in the Vβ6+ Vβ14− group. Both populations responded strongly to the positive control stimulus, anti-CD3. To ultimately confirm the requirement of a second TCR on K/BxN T cells for SFB to boost auto-immunity, we used Rag−/−.KRN mice to exclude the possibility of KRN T cells expressing an endogenous TCR in addition to their transgenic KRN TCR. Rag−/−.KRN T cells reconstituted the recipient mouse T cell populations similarly compared to the KRN T cells in both SFB− and SFB+ conditions (Figure S4B). However, SFB-induced lung Th17 cells and arthritis that developed in mice receiving WT KRN T cells were abolished in mice receiving Rag−/−.KRN T cells (Figures 4D and 4E).
Though our data profoundly support the essential role of dual TCRs in SFB-mediated autoimmunity, another potential mechanism is bystander activation (Münz et al., 2009). We thus tested this model where SFB may trigger autoimmunity by stimulating unrelated (autoreactive) T cells with cytokines such as IL-17 secreted by SFB-specific T cells. 7B8 TCR transgenic T cells express an SFB-specific TCR and preferentially become Th17 cells in SFB+ B6 mice (Yang et al., 2014). We used 7B8 T cells (CD45.1+) as a non-self and SFB-specific enriched source of Th17 cells and co-transferred them with Rag−/−.KRN T cells (CD45.2+) into Tcra−/−.BxN mice. Not only did 7B8 T cells not help autoreactive KRN T cells, but they also out-competed the autoimmune Rag−/−.KRN T cells (Figure 4F). In these co-transfers, SFB enhanced IL-17 expression in 7B8 T cells, but there were hardly any Rag−/−.KRN T cells with or without IL-17 expression (Figure 4G). Thus, the abundant, non-self-reactive Th17 cells induced by SFB could not help the activation of autoimmune T cells. These data indicate that SFB do not activate autoimmune T cells via bystander activation. Finally, since we observed skewed TCR Vβ6+Vβ14+ usage in splenic (systemic) Th17 cells (Figure S2C), we also examined the systemic autoimmune phenotype in the KRN, Rag−/−.KRN, and 7B8+ Rag−/−.KRN T cell transfer recipients. We found that the systemic autoimmune phenotype resembled that of lung, as SFB only increased serum anti-GPI titers in mice receiving KRN, but not Rag−/−.KRN T cells (Figure 4H). Similarly, the robust SFB-mediated Th17 response in 7B8 cells did not increase serum anti-GPI auto-Ab production in SFB+ mice co-transferred with Rag−/−.KRN+7B8 T cells (Figure 4H). Thus, SFB-induced dual TCRs are responsible for both SFB-induced lung and systemic autoimmunity.
DISCUSSION
Environmental factors such as cigarette smoke, allergens, and respiratory infectious agents are known to contribute to iBALT formation (Randall and Mebius, 2014). Our findings point to an unexpected, non-pulmonary contributor, the gut microbiota, as the primary culprit that remotely controls formation of iBALT-like structures in the lung. Parallel to our model, Th17 cells have been shown to play an important role in ELT formation in both infectious and autoimmune models in a variety of tissues, and many human pharmaceuticals in clinical trials are designed to target ELTs by IL-17 depletion (Pitzalis et al., 2014). Thus, our findings have significant implications because gut microbiota, SFB or a human equivalent of Th17-inducing microbiota, may serve as a critical environmental trigger in formations of other ELTs. Recent studies reported a microbiota-induced colonic Treg population that co-expresses Rorγt in addition to Foxp3 and displays an anti-inflammatory effect in colitis models (Sefik et al., 2015; Ohnmacht et al., 2015). It will be interesting to examine whether Rorγt+ Tregs also impact microbiota-dependent lung diseases.
Although gut microbiota may initially seem an unusual suspect for causing lung autoimmune disease, the gut-lung axis has recently emerged as an area of intense interest in other lung diseases such as asthma and COPD, yet little is known regarding the mechanism by which gut microbiota affect lung (Budden et al., 2017). The CMIS such as the one composed of lung and gut tissues, the gut-lung axis, functions as an integrated immune pathway whereby immune cells activated at one mucosal site can home to other mucosal tissues (Kiyono and Fukuyama, 2004). This is supported by an example of nasal vaccination that generates cross-protective immunity in the gut (Kiyono and Fukuyama, 2004). Here, we demonstrate a mechanism by which the gut microbiota SFB remotely condition lung to develop autoimmunity by inducing a gut-lung axis Th17 response. We further reveal that SFB do not alter the ability of the lung to attract or support Th17 cells. Rather, our data suggest that the increase in Th17 cells in the lung of SFB+ compared to SFB− K/BxN mice is due to a larger SFB-induced gut Th17 cell pool, and thus a higher number of gut Th17 cells recruited to the lung in SFB+ mice.
At the molecular level, we demonstrate that SFB selectively expand autoimmune T cells co-expressing SFB-specific TCRs in addition to their self-reactive TCRs. This additional SFB-specific TCR provides a proliferative advantage for autoimmune Th17 cells in SFB+ hosts. Our data suggest that the robust SFB-induced Th17 response of 7B8 T cells could not rescue the autoimmune activity of Rag−/−.KRN T cells through bystander activation. Molecular mimicry is a well-known model for microbe-triggered autoimmunity (Albert and Inman, 1999; Münz et al., 2009). However, the impaired Th17 cell response in mice receiving Rag−/−.KRN T cells despite their SFB+ status and the fact that SFB peptide activates Vβ6+Vβ14+, but not the Vβ6+Vβ14−, KRN T cells both suggest an alternative, dual TCR-based mechanism. Taken together, this study suggests the same T cells need to both recognize self-Ag and be activated by SFB to induce autoimmune Th17 cells to enhance autoimmunity in an SFB-dependent manner. This is supported by previous data showing that in healthy mice, cognate TCR recognition of SFB-Ag is crucial for SFB-mediated Th17 induction (Goto et al., 2014; Yang et al., 2014).
This study demonstrates that a commensal bacterial epitope triggers autoimmunity by skewing dual TCR expression. It has been suggested that dual TCR T cells could contribute to autoimmunity by allowing self-reactive T cells to escape thymic deletion through selection on their non-self-reactive TCR (Auger et al., 2012; Corthay et al., 2001; Elliott and Altmann, 1995; Ji et al., 2010; Padovan et al., 1995). However, studies on this topic have been controversial, and many studies using autoimmune mouse models, such as collagen-induced arthritis or EAE, have failed to show any role of dual TCR T cells in autoimmunity (Corthay et al., 2001; Elliott and Altmann, 1995). Except for one viral infection study (discussed below) (Ji et al., 2010), none of the antigens recognized by the non-self-specific TCR of the dual TCR T cells have been previously identified. Here, identifying the recognition of a commensal epitope by the second (non-self) TCR of dual TCR T cells allows us to elucidate that the discrepancy in previous dual TCR studies may be due in part to an asymptomatic environmental trigger that delivers the last straw required to tip the immune balance toward autoimmunity; i.e., autoreactive T cells co-expressing a second TCR that recognizes gut microbiota preferentially expand to promote autoimmunity only when the gut microbiota Ag is present in the environment.
Importantly, dual TCR expression is not limited to transgenic mouse models. In wild-type mice and in humans, up to 15% and 33%, respectively, of peripheral T cells express dual TCRs, largely due to incomplete allelic exclusion at the Tcra locus (Heath et al., 1995; Padovan et al., 1993). Our animal model thus provides a proof-of-principle study and suggests it is likely that microbiota modulate the dual TCR repertoire in their hosts, which subsequently contributes to autoimmunity. A pioneering study has shown that systemic viral infections following an intravenous injection of vaccinia virus trigger autoimmunity by activating dual TCR T cells (Ji et al., 2010). Here, we report a previously unknown condition, that asymptomatic colonization by commensal bacteria in a local environment, the gut, is capable of triggering systemic autoimmune disease by skewing dual TCR expression in the host. These results have broad and significant implications, as imbalance of gut microbiota has been rising sharply in the industrialized world (Levy et al., 2017; Yurkovetskiy et al., 2015). Future investigation is required to determine whether dual TCRs are involved in other dysbiosis diseases.
STAR☆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents or resources may be directed to and will be fulfilled by Lead Contact Hsin-Jung Joyce Wu (joycewu@email.arizona.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
K/BxN, Tcra−/−.BxN, Rorc−/−.KRN, and Rag−/−.KRN mice were generated as previously described and housed at the University of Arizona (Teng et al., 2016). Briefly, K/BxN mice were generated by crossing KRN TCR transgenic mice on the C57BL/6 (B6) background with NOD mice. Tcra−/−.BxN mice were generated by crossing Tcra−/−.B6 with Tcra−/−.NOD mice. Tcra−/−.BxN mice thus bear NOD MHC class II, I-Ag7, for GPI self-Ag presentation. Non-arthritic control BxN mice were generated by crossing B6 to NOD mice. The 7B8 SFB-TCR transgenic mice on the CD45.1/B6 background were maintained at the New York University. All experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee at both Universities.
K/BxN Transfer Model
Splenic CD4+ T cells for transfer were enriched by CD4-conjugated MACS beads (Miltenyi). KRN and Rorc−/−.KRN CD4+ T cells (5×105) were transferred into SFB- or SFB+ Tcra−/−.BxN recipients for two weeks before analysis. For transfer of polarized Th17 and Th1 cells, 4-day polarized cells (1×106) were transferred into SFB- or SFB+ K/BxN recipients for 3 days before analysis. For KRN and Rag−/−.KRN T cell transfer, an equal number of CD4+ T cells from each mouse strains (5×104 to 2×105) were transferred into SFB- or SFB+ Tcra−/−.BxN recipients for 4 weeks before analysis. For the mixed Rag−/−.KRN + 7B8 transfer, splenic CD4+ T cells from Rag−/−.KRN and 7B8 mice were mixed and co-transferred at a 1:1 ratio (5×104 to 2×105) into SFB− or SFB+ Tcra−/−.BxN recipients for 4 weeks before analysis. SFB status was manipulated by gavaging recipient mice.
METHOD DETAILS
Microbiota Reconstitution and Antibiotic Treatment
Mice in our colony are maintained as SFB− as described previously unless mentioned otherwise (Teng et al., 2016). To obtain SFB+ status, SFB− mice were gavaged with SFB-containing feces collected from the SFB+ mice housed in our colony. For SFB gavage, mice were weaned at 21 days old and rested for one day. Then, mice were orally gavaged with SFB-containing feces collected in-house for 3 consecutive days starting at 23 days old. The SFB− mice were the ungavaged littermate controls. For the antibiotic treatment, the littermates of SFB+ K/BxN mice (maintained as an SFB+ colony by vertical transmission from SFB gavaged moms) were treated with 0.5g/L vancomycin (Hospira) in drinking water containing sweetener (Equal, 2.5 g/300 mL) or left untreated, starting at 23 days old for 2 weeks. For adoptive transfer experiments, SFB status was manipulated by gavaging SFB− recipient mice with SFB prior to cell transfer. The SFB colonization status was routinely examined on day 10 after SFB gavage or on experiment termination day (e.g., day 7 SFB-gavaged experiment and vancomycin experiment) by SFB-specific 16S rRNA quantitative PCR and normalized by the presence of total bacteria (EUB primers). This value was further normalized to Taconic mice (positive control; value set as 1) which allows the comparison of SFB level among experiments (Wu et al., 2010).
Preparation of Single-Cell Suspension from Lung
Lungs were perfused with PBS and finely minced before being placed into digestion buffer containing 1 mg/mL each of Collagenase D (Roche) and MgCl2, and 0.15 mg/mL DNase I (Sigma) in DMEM (HyClone). Lung perfusion is a widely accepted method for the preparation of lung immune cells (Califano et al., 2015; Duerr et al., 2016; von Moltke et al., 2017), which includes cells trapped in the capillaries (Anderson et al., 2012). We opted to use lung perfusion in our study because we intended to include both immune cells from lung parenchyma and also capillaries as tissue capillary-associated immune populations are known to be crucial for participating in immune responses (Rodero et al., 2015; Carlin et al., 2013; Lee et al., 2010). Lungs were digested for 20-25 min at 37°C at 200 RPM then passed through 100 μm cell strainer to prepare single-cell suspension.
Antibodies and Flow Cytometry
For surface staining, fluorophore-conjugated mAbs specific for CD4 (RM4-5), CD19 (6D5), CD45 (30-F11), CD45.1 (A20), CD45.2 (104), PD-1 (RMP1-30) and TCRβ (H57-597) were obtained from BioLegend. Ab recognizing TCR Vβ14 (14-2) and CXCR5 (2G8) were from BD PharMingen. Isotype controls for TCR Vβ14, Rat IgM kappa, were from BD PharMingen. Anti-TCR Vβ6 (RR4-7) was from eBioscience. For intracellular cytokine staining, cells were incubated for 4 hr with BD GolgiPlug (1:1000 dilution), 50 ng/m: phorbol 12-myristate 13-acetate, and 1 μM ionomycin in DMEM (HyClone) supplemented with 10% FCS, 1% nonessential amino acids, penicillin, streptomycin, and glutamine at 37°C. Intracellular cytokine staining was performed with Cytofix/Cytoperm (BD PharMingen). Abs recognizing IFN-γ (XMG 1.2), IL-17 (TC11-18H10.1) and IgG1 (RMG1-1) were obtained from BioLegend. Cells were run on an LSRII (BD Biosciences), and analyses were performed with FlowJo (TreeStar) software. Abs recognizing IgG1 were also used for ELISPOT.
In Vitro Th1 and Th17 Polarization
Splenic KRN CD4+ T cells were enriched with anti-CD4 MACS beads (Miltenyi), and cultured for 4 days in Th1 (anti-CD3ε/CD28 plate coated + 20ng/mL IL-2 + 20ng/ml IL12), or Th17 (anti-CD3ε/CD28 plate coated + 50ng/mL IL-6 + 1ng/mL TGFβ1 + 300nm FICZ) polarization cultures.
ELISA and ELISPOT
ELISA and ELISPOT analyses for GPI-specific IgG1 or GPI specific IgG1 ASCs, respectively were performed as described previously (Teng et al., 2016). Briefly, ELISA plates were coated with recombinant mouse GPI at 5 μg/mL, and diluted mouse sera were added. Subsequently, plates were washed and alkaline-phosphatase (AP)-conjugated anti-mouse IgG1 Abs were added. After the final wash, AP substrate was added and titers were quantified as optical density values via an ELISA reader. For ELISPOT, Multiscreen IP Plates (Millipore) were coated with 10 μg/mL recombinant GPI. B cells were resuspended at 2 × 106 cells/mL in medium, and 100 μL of cell suspension was added to the wells. Cells were serially diluted and incubated for 6 hr at 37°C. Plates were washed, AP-conjugated anti-mouse IgG1 were applied, and the plates were incubated for 1.5 hr at 37°C. Finally, plates were washed and 1-step NBT/BCIP substrate (Pierce) was added at room temperature for spot development (5-10 min). The plates were rinsed with water, dried overnight in the dark, and spots were counted manually using a stereoscopic microscope. IL-17A ELISPOT was performed as previously described (Yang et al., 2014) with a mouse/rat IL-17A ELISPOT Ready-SET-Go! kit (eBioscience). The lung Vβ6+Vβ14+ and Vβ6+Vβ14− CD4+ T cell populations were sorted using a BD FACSAria and co-cultured with Tcra−/−.B6 DCs isolated by CD11c-conjugated MACS beads (Miltenyi). Various stimuli and negative controls including N3 peptide (STGKFNVSGI TAPGIYT; negative control), A6 SFB peptide (DVQFSGAVPNKTD), and α-CD3 (positive control) were used as described in the previous report (Yang et al., 2014). 5.3×103 sorted T cells were cultured with 2×105 DCs on the coated plate for 36 hr at 37°C, 5% CO2 with either 25 μg/mL peptide or 2.5 μg/mL α-CD3 before proceeding with IL-17 detection according to the manufacturer’s instructions.
Proliferation and Apoptosis Assay
K/BxN mice were i.p. injected with 0.2 mg EdU. Twelve hours after injection, cells were processed and stained with surface markers. The samples were then stained with the Click-iT Plus EdU Imaging Kit according to the manufacturer’s instructions. Apoptotic cells were determined by staining cells first with Live/Dead Yellow Dye (LifeTechnologies), and then surface markers. After that, cells were stained with Annexin V (Annexin V/Dead Cell Apoptosis Kit, LifeTechnologies) according to the manufacturer’s instructions.
Quantitative PCR Analysis
Tissues were homogenized in Trizol. RNA were purified and cDNA was synthesized with a Maxima First Strand cDNA Synthesis Kit (Thermo). Quantitative PCR (qPCR) was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems) with TaqMan Gene Expression Assays on CCL20 (Mm01268754) and HPRT (Mm01545399). Reactions were run on a StepOnePlus instrument (Applied Biosystems). The CCL20 Ct values were normalized to HPRT, and further normalized to one of the SFB− BxN spleen samples (value set as 1) in each experiment to allow a comparison between the BxN and K/BxN groups.
Immunohistochemistry, Immunofluorescent and Confocal Microscope
Lung were perfused and fixed with 10% formalin and stained with hematoxylin and eosin (H&E). Lung cryostat sections were stained with Abs against CD4 and CD19. Image analysis was performed using ImageJ software (NIH, Bethesda, MD).
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of mice per group is indicated in the corresponding figure legends. In all figures, the mean value is depicted + SEM. Differences were considered significant when p < 0.05 by Student’s t test (two-tailed, unpaired; used when comparing between 2 groups) or two-way analysis of variance (ANOVA; used when comparing between 3 or more groups) (Prism 6, Graph-Pad Software). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Littermate mice were randomly assigned into experimental groups after matching for gender. Investigators remained unblinded to group assignments. No data points were excluded from statistical analysis.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| Anti-mouse CD4 Antibody, clone RM4-5, BV421 conjugated | BioLegend | Cat#100544; RRID: AB_11219790 | ||
| Anti-mouse TCR beta antibody, clone H57-597, APC-eFluor780 conjugated | eBioscience/ThermoFisher Scientific | Cat#47-5961-82; RRID: AB_1272173 | ||
| Anti-mouse CD45 antibody, clone 30-F11, PE/Dazzle 594 conjugated | BioLegend | Cat#103146; RRID: AB_2564003 | ||
| Anti-mouse IL-17A antibody, clone TC11-18H10.1, PE/Cy7 conjugated | BioLegend | Cat#506922; RRID: AB_2125010 | ||
| Anti-mouse TCR V beta 6 antibody, clone RR4-7, PerCP-eFluor710 conjugated | eBioscience/ThermoFisher Scientific | Cat#46-5795-82; RRID: AB_11150054 | ||
| Anti-mouse TCR V beta 14 antibody, clone 14-2, Biotin conjugated | BD Biosciences | Cat#553257; RRID: AB_394737 | ||
| Anti-mouse TCR V beta 14 antibody, clone 14-2, FITC conjugated | BD Biosciences | Cat#553258; RRID: AB_394738 | ||
| Anti-mouse CD44 antibody, clone IM7, Alexa Fluor 700 conjugated | BioLegend | Cat#103026; RRID: AB_493713 | ||
| Anti-mouse CD45.1 antibody, clone A20, FITC conjugated | BioLegend | Cat#110705; RRID: AB_313494 | ||
| Anti-mouse CD45.2 antibody, clone 104, Alexa Fluor 700 conjugated | BioLegend | Cat#109822; RRID: AB_493731 | ||
| Anti-mouse PD-1 antibody, clone RPM1-30, PE conjugated | BioLegend | Cat#109104; RRID: AB_313421 | ||
| Anti-mouse CXCR5 antibody, clone 2G8, Biotin conjugated | BD Biosciences | Cat#551960; RRID: AB_394301 | ||
| Anti-mouse IFN-γ antibody, clone XMG1.2, PE conjugated | BioLegend | Cat#505808; RRID: AB_315402 | ||
| Anti-mouse CD4 antibody, clone RM4-5, PE conjugated | BioLegend | Cat#100512; RRID: AB_312715 | ||
| Anti-mouse CD19 antibody, clone 1D3, FITC conjugated | BD Biosciences | Cat#553785; RRID: AB_395049 | ||
| Anti-mouse CD19 antibody, clone 6D5, PerCP/Cy5.5 conjugated | BioLegend | Cat#115534; RRID: AB_2072925 | ||
| Alkaline Phosphatase AffiniPure Goat Anti-Mouse IgG, Fcγ fragment specific | Jackson ImmunoResearch Laboratories | Cat#115-055-008; RRID: AB_2338530 | ||
| Anti-mouse IgG1 antibody, clone RMG1-1, biotin conjugated | BioLegend | Cat#406604; RRID: AB_315063 | ||
| Rat IgM, κ Isotype Control, clone R4-22, biotin conjugated | BD Biosciences | Cat#553941; RRID: AB_10053773 | ||
| Rat IgM, κ Isotype Control, clone R4-22, FITC conjugated | BD Biosciences | Cat#553942; RRID: AB_479631 | ||
| Biological Samples | ||||
| SFB feces | Laboratory of H.-J.J.W.; Teng et al., 2016 | N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||||
| A6 peptide (DVQFSGAVPNKTD) | GenScript; Yang et al., 2014 | N/A | ||
| N3 peptide (STGKFNVSGITAPGIYT) | GenScript; Yang et al., 2014 | N/A | ||
| GPI protein | Laboratory of H.-J.J.W.; Monach et al., 2008 | N/A | ||
| CD4 (L3T4) MicroBeads, mouse | Miltenyi Biotec | Cat#130-049-201 | ||
| CD11c MicroBeads UltraPure, mouse | Miltenyi Biotec | Cat#130-108-338 | ||
| Vancomycin | Hospira | Cat#CA-2581 | ||
| Collagenase D | Roche | Cat#11088882001 | ||
| Deoxyribonuclease I from bovine pancreas | Sigma | Cat# DN25-1G; CAS: 9003-98-9 | ||
| Anti-CD3 | BioLegend | Cat#100314 | ||
| Anti-CD28 | BioLegend | Cat#102112 | ||
| Recombinant mouse IL-2 | Peprotech | 212-12 | ||
| Recombinant mouse IL-12 | Peprotech | 210-12 | ||
| Recombinant mouse IL-6 | Peprotech | Cat#216-16 | ||
| recombinant human TGF-β1 | Peprotech | Cat#100-21 | ||
| 6-Formylindolo(3,2-b)carbazole (FICZ) | Enzo Life Sciences | BML-GR206-0100 | ||
| Tripure Isolation Reagent | Roche | 11667165001 | ||
| Streptavidin, APC conjugated | BioLegend | Cat#405207 | ||
| Streptavidin, BV605 conjugated | BioLegend | Cat#405229 | ||
| Phosphatase Substrate Tablets | Sigma | Cat#S0942-200TAB | ||
| Critical Commercial Assays | ||||
| Click-iT Plus EdU Imaging Kit | ThermoFisher Scientific | C10637 | ||
| Dead Cell Apoptosis Kit with Annexin V Alexa Fluor 488 | ThermoFisher Scientific | V132415 | ||
| LIVE/DEAD Fixable Yellow Dead Cell Stain Kit | ThermoFisher Scientific | Cat#L34968 | ||
| Mouse/rat IL-17A ELISPOT Ready-SET-Go! Kit | eBioscience | 88-7370-21 | ||
| CCL20 TaqMan Gene Expression Assay | ThermoFisher Scientific | Cat#4453320; Assay ID: Mm01268754_m1 | ||
| HPRT TaqMan Gene Expression Assay | ThermoFisher Scientific | Cat#4453320; Assay ID: Mm01545399 | ||
| TaqMan Fast Advanced Master Mix | ThermoFisher Scientific | Cat#4370074 | ||
| Maxima First Strand cDNA Synthesis Kit | ThermoFisher Scientific | Cat#K1642 | ||
| SYBR Select Master Mix | ThermoFisher Scientific | Cat#4472908 | ||
| Experimental Models: Organisms/Strains | ||||
| Mouse: K/BxN (KRN transgenic on C57BL/6J background × NOD) | Laboratory of H-J.J.W; Monach et al., 2008 | N/A | ||
| Mouse: BxN (B6 × NOD) | Monach et al., 2008 | N/A | ||
| Mouse: Tcra−/−.B6: B6.129S2-Tcratm1Mom/J | Jackson Laboratories | JAX: 002116 | ||
| Mouse: Tcra−/−.BxN (Tcra−/−.B6 × Tcra−/−.NOD) | Laboratory of H-J.J.W; Teng et al., 2016 | N/A | ||
| Mouse: Rorc−/−.KRN | Laboratory of H-J.J.W; Teng et al., 2016 | N/A | ||
| Mouse: Rag−/−.KRN | Laboratory of H.H. and H-J.J.W; Teng et al., 2016 | N/A | ||
| Mouse: 7B8 SFB-TCR CD45.1/B6 | Laboratory of D.R.L; Yang et al., 2014 | N/A | ||
| Oligonucleotides | ||||
| Primer: SFB Forward: GAC GCT GAG GCA TGA GAG CAT | Eurofins Genomics; Teng et al., 2016 | N/A | ||
| Primer: SFB Reverse: GAC GGC ACG GAT TGT TAT TCA | Eurofins Genomics; Teng et al., 2016 | N/A | ||
| Primer: EUB Forward: CGG CAA CGA GCG CAA CCC | Eurofins Genomics; Teng et al., 2016 | N/A | ||
| Primer: EUB Reverse: CCA TTG TAG CAC GTG TGT AGC C | Eurofins Genomics; Teng et al., 2016 | N/A | ||
| Software and Algorithms | ||||
| FlowJo v10.0.8 | FlowJo | http://www.flowjo.com | ||
| Prism (v7) | GraphPad | http://www.graphpad.com | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ | ||
Highlights.
Gut commensal SFB trigger lung autoimmune pathology in the pre-arthritic phase
SFB aggravate lung autoimmunity by inducing Th17 cells of the gut-lung axis
SFB do not rely on molecular mimicry or bystander activation to induce Th17 cells
SFB expand Th17 cells co-expressing dual TCRs for SFB epitope and self-antigen
Acknowledgments
This work was supported by grants from the R01AI107117 to H.-J.J.W., U01 HL121831-01 to K.S.K., R01AI087645 to H.H., R01DK103358 to D.R.L., and from the Southwest Clinic and Research Institute Fund to H.-J.J.W. K.E.B. was partially supported by NIH grant 2T32AI007090.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.chom.2017.10.007.
AUTHOR CONTRIBUTIONS
C.P.B. and H.-J.J.W. conceived the study. C.P.B., F.T., and K.M.F. designed and performed most experiments and analyzed the data. T.S. and D.R.L. provided 7B8 spleens and contributed to the design of 7B8- and SFB peptide-related experiments. D.N. performed ELISPOT analysis. K.E.B. and H.H. provided the initial Rag−/−.KRN spleens and contributed to Rag−/−.KRN-related experimental design. K.S.K. contributed to lung histology analysis. H.-J.J.W. designed the study and wrote the manuscript.
References
- Alam SM, Gascoigne NR. Posttranslational regulation of TCR Valpha allelic exclusion during T cell differentiation. J Immunol. 1998;160:3883–3890. [PubMed] [Google Scholar]
- Albert LJ, Inman RD. Molecular mimicry and autoimmunity. N Engl J Med. 1999;341:2068–2074. doi: 10.1056/NEJM199912303412707. [DOI] [PubMed] [Google Scholar]
- Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol. 2012;189:2702–2706. doi: 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger JL, Haasken S, Steinert EM, Binstadt BA. Incomplete TCR-β allelic exclusion accelerates spontaneous autoimmune arthritis in K/BxN TCR transgenic mice. Eur J Immunol. 2012;42:2354–2362. doi: 10.1002/eji.201242520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, Hansbro PM. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15:55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, Li H, Avram D. Transcription Factor Bcl11b Controls Identity and Function of Mature Type 2 Innate Lymphoid Cells. Immunity. 2015;43:354–368. doi: 10.1016/j.immuni.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, Geissmann F. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153:362–375. doi: 10.1016/j.cell.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthay A, Nandakumar KS, Holmdahl R. Evaluation of the percentage of peripheral T cells with two different T cell receptor alpha-chains and of their potential role in autoimmunity. J Autoimmun. 2001;16:423–429. doi: 10.1006/jaut.2001.0504. [DOI] [PubMed] [Google Scholar]
- Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr Opin Rheumatol. 2014;26:64–71. doi: 10.1097/BOR.0000000000000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, Eva MM, Gauchat JF, Qureshi ST, Mazer BD, et al. Type I inter-feron restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 2016;17:65–75. doi: 10.1038/ni.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JI, Altmann DM. Dual T cell receptor alpha chain T cells in autoimmunity. J Exp Med. 1995;182:953–959. doi: 10.1084/jem.182.4.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O’Connor W, Jr, Rongvaux A, Van Rooijen N, Haberman AM, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, Ivanov II. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath WR, Carbone FR, Bertolino P, Kelly J, Cose S, Miller JF. Expression of two T cell receptor alpha chains on the surface of normal murine T cells. Eur J Immunol. 1995;25:1617–1623. doi: 10.1002/eji.1830250622. [DOI] [PubMed] [Google Scholar]
- Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc Natl Acad Sci USA. 2010;107:4658–4663. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol. 2010;11:628–634. doi: 10.1038/ni.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyono H, Fukuyama S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat Rev Immunol. 2004;4:699–710. doi: 10.1038/nri1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, Chaconas G, Kubes P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol. 2010;11:295–302. doi: 10.1038/ni.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. 2017;17:219–232. doi: 10.1038/nri.2017.7. [DOI] [PubMed] [Google Scholar]
- Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. 2008;Chapter 15:22. doi: 10.1002/0471142735.im1522s81. [DOI] [PubMed] [Google Scholar]
- Münz C, Lünemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naskar D, Teng F, Felix KM, Bradley CP, Wu HJ. Synthetic Retinoid AM80 Ameliorates Lung and Arthritic Autoimmune Responses by Inhibiting T Follicular Helper and Th17 Cell Responses. J Immunol. 2017;198:1855–1864. doi: 10.4049/jimmunol.1601776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 2012;33:297–305. doi: 10.1016/j.it.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Chung Y, Dong C. Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J Immunol. 2009;182:2565–2568. doi: 10.4049/jimmunol.0803931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORγt+ T cells. Science. 2015;349:989–993. doi: 10.1126/science.aac4263. [DOI] [PubMed] [Google Scholar]
- Olson AL, Swigris JJ, Sprunger DB, Fischer A, Fernandez-Perez ER, Solomon J, Murphy J, Cohen M, Raghu G, Brown KK. Rheumatoid arthritis-interstitial lung disease-associated mortality. Am J Respir Crit Care Med. 2011;183:372–378. doi: 10.1164/rccm.201004-0622OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor alpha chains: dual receptor T cells. Science. 1993;262:422–424. doi: 10.1126/science.8211163. [DOI] [PubMed] [Google Scholar]
- Padovan E, Casorati G, Dellabona P, Giachino C, Lanzavecchia A. Dual receptor T-cells. Implications for alloreactivity and autoimmunity. Ann N Y Acad Sci. 1995;756:66–70. doi: 10.1111/j.1749-6632.1995.tb44482.x. [DOI] [PubMed] [Google Scholar]
- Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14:447–462. doi: 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
- Prasad YV, Puthli SP, Eaimtrakarn S, Ishida M, Yoshikawa Y, Shibata N, Takada K. Enhanced intestinal absorption of vancomycin with Labrasol and D-alpha-tocopheryl PEG 1000 succinate in rats. Int J Pharm. 2003;250:181–190. doi: 10.1016/s0378-5173(02)00544-6. [DOI] [PubMed] [Google Scholar]
- Randall TD, Mebius RE. The development and function of mucosal lymphoid tissues: a balancing act with micro-organisms. Mucosal Immunol. 2014;7:455–466. doi: 10.1038/mi.2014.11. [DOI] [PubMed] [Google Scholar]
- Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. 2006;116:3183–3194. doi: 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Rodero MP, Poupel L, Loyher PL, Hamon P, Licata F, Pessel C, Hume DA, Combadière C, Boissonnas A. Immune surveillance of the lung by migrating tissue monocytes. Elife. 2015;4:e07847. doi: 10.7554/eLife.07847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sant’Angelo DB, Cresswell P, Janeway CA, Jr, Denzin LK. Maintenance of TCR clonality in T cells expressing genes for two TCR heterodimers. Proc Natl Acad Sci USA. 2001;98:6824–6829. doi: 10.1073/pnas.121179998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. 2015;349:993–997. doi: 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seldin MF, Amos CI, Ward R, Gregersen PK. The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum. 1999;42:1071–1079. doi: 10.1002/1529-0131(199906)42:6<1071::AID-ANR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Steinel NC, Brady BL, Carpenter AC, Yang-Iott KS, Bassing CH. Posttranscriptional silencing of VbetaDJbetaCbeta genes contributes to TCRbeta allelic exclusion in mammalian lymphocytes. J Immunol. 2010;185:1055–1062. doi: 10.4049/jimmunol.0903099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, Tran NL, Umesaki Y, Wu HJ. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer’s Patch T Follicular Helper Cells. Immunity. 2016;44:875–888. doi: 10.1016/j.immuni.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, O’Leary CE, Barrett NA, Kanaoka Y, Austen KF, Locksley RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. 2017;214:27–37. doi: 10.1084/jem.20161274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkovetskiy LA, Pickard JM, Chervonsky AV. Microbiota and autoimmunity: exploring new avenues. Cell Host Microbe. 2015;17:548–552. doi: 10.1016/j.chom.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.