

ABSTRACT
Immune responses have been elicited by a variety of cancer vaccines, but seldom induce regressions of established cancers in humans. As a novel therapeutic immunization strategy, we tested the hypothesis that multiple cytokines/chemokines secreted early in secondary responses ex-vivo might mimic the secretory environment guiding new immune responses. The early development of immune responses is regulated by multiple cytokines/chemokines acting together, which at physiologic concentrations act locally in concert with antigen to have non-specific effects on adjacent cells, including the maturation of dendritic cells, homing and retention of T cells at the site of antigen, and the differentiation and expansion of T cell clones with appropriate receptors. We postulated that repeated injections into a metastasis of an exogenous chemokine/cytokine mixture might establish the environment of an immune response and allow circulating T cell clones to self- select for mutant neo-epitopes in the tumor and generate systemic immune responses. To test this idea we injected some metastases in patients with multiple cutaneous melanoma nodules while never injecting other control metastases in the same patient. New immune responses were identified by the development of dense lymphocytic infiltrates in never-injected metastases, and the frequent complete regression of never-injected metastases, a surprising observation. 70% of subjects developed dense infiltrates of cytotoxic CD8 cells in the center and margin of never-injected metastases; 38% of subjects had complete and often durable regressions of all metastases, without the use of check-point inhibitors, suggesting that, as a proof-of-principle, an immunization strategy can control advanced human metastatic melanoma.
KEYWORDS: cancer vaccine, CD8 T cells, cytokines, immunotherapy, metastatic melanoma, tumor infiltrating lymphocytes
Introduction
The induction of effective new immune responses to human cancers has been an elusive goal for many decades.1 Detectable immune responses often have been elicited by therapeutic vaccines of autologous or allogeneic tumor cells, tumor proteins, peptides; expressed by vectors, dendritic cells pulsed with tumor antigens or RNA vaccines encoding tumor epitopes. However responses resulting in clinical regressions in patients have been infrequent. Recently mutant tumor neoantigens have been identified as important targets of nascent anti-cancer immune responses2 that can be enhanced by anti-check point antibodies to CTLA4 or PD-1/L-1, resulting in substantial cancer regressions and prolongation of life in many but not all patients.3–6 The induction of immune responses to additional neo-epitopes and increased lymphocyte infiltration into metastases might enhance the efficacy of anti-checkpoint therapies.7,8 To enhance immune responses, multiple adjuvants have been used including cytokines (CKs) used as proteins or expressed with vectors.9,10 Many preclinical studies using single or limited combinations of three CKs as proteins, plasmids or vaccines of tumor cells expressing CKs, have induced immune responses and restrained the growth of existing cancers, but complete regressions (CRs) occurred in a minority of experimental animals.9 The modification of tumor cells to express a CK revealed that of 30 genes evaluated, GMCSF was optimal in enhancing immune responses to the tumor.11 Clinical approaches using autologous tumor cell lines, DCs or single or combinations of CKs, usually including GMCSF, or CK-expressing cells have led to prolongation of life and partial or occasionally complete responses (CRs).12–17 Several issues may contribute to prior failures of vaccines to induce effective anti-cancer responses: 1, vaccines may not have included sufficient neo-epitopes unique to the patient's tumor; 2, adjuvants or cytokines may not have enhanced immunogenicity; 3, induced T cells were unable to enter the tumor or were inhibited locally; 4, the magnitude and phenotypes of immune responses were insufficient, as compared to effective and durable responses elicited by vaccinia and yellow fever vaccines.18,19
However, single or combinations of 3 recombinant cytokines do not resemble the complex mixture of cytokines and chemokines (CKs) participating in developing immune responses. Cytokines such as colony-stimulating factors can act alone, but the development of immune responses is regulated by multiple cytokines/chemokines from innate and adaptive immune cells acting together, which at physiologic concentrations act locally in concert to have antigen non-specific effects on adjacent cells, including the maturation of dendritic cells, homing and retention of T cells at the site of antigen, the differentiation and expansion of CD4 and CD8 T clones with appropriate receptors,20–22 and potentially increased expression of antigens on the target cells.
Motivated in part by an earlier observation in-vitro that CKs secreted by PBMC responding to a recall microbial antigen enabled PBMC from different donors to respond by proliferation to microbial antigens that did not stimulate these cells in the absence of exogenous, CKs,23 we postulated that a full complement of many (CKs) produced ex-vivo early in a secondary immune response by PBMCs stimulated with a microbial antigen might mimic the secretory environment of a primary response. CKs repeatedly injected into a metastatic nodule might facilitate the development of additional immunity -to metastatic melanoma more effectively than individual cytokines, and constitute an in situ immunization with a “physiological” adjuvant. A mixture of autologous cytokines was used for ethical and safety reasons, and from necessity since many individual CK components were unknown and unavailable when the study was initiated. Would exogenous CKs injected intralesionally modify the microenvironment in injected metastases, but also locally stimulate the development of systemic immune responses affecting distant never-injected metastases? The intralesional provision of a secretory CK environment should expose circulating mononuclear cells to multiple potential neo-epitopes, allowing T cell clone-epitope self-selection including against non-dominant epitopes possibly unique to a patients tumor.
To test our hypothesis in-vivo, prior to the availability of current therapies, we initiated an IRB-approved clinical study in patients with clinically palpable cutaneous or subcutaneous in-transit metastases of an extremity or multiple metastases on the trunk or head. All had developed 2 to 100 metastases during the 2 months prior to entry. They were stages IIIB 2 pts; IIIC 76 pts; and IV-M1a 9 pts . The study brought antigen (melanoma cells), CKs and circulating mononuclear cells together by injecting mixtures of autologous secreted CKs weekly into some cutaneous metastatic melanoma nodules while other metastases in that patient were never injected (Fig. 1). Systemic immune responses would be identified by: 1) the development of new lymphocytic infiltrates in never-injected metastases, 2) an examination of the ability of tumor-infiltrating lymphocytes (TILs) to kill autologous melanoma cells ex-vivo, and 3) an enumeration of any metastases that completely regressed.
Figure 1.
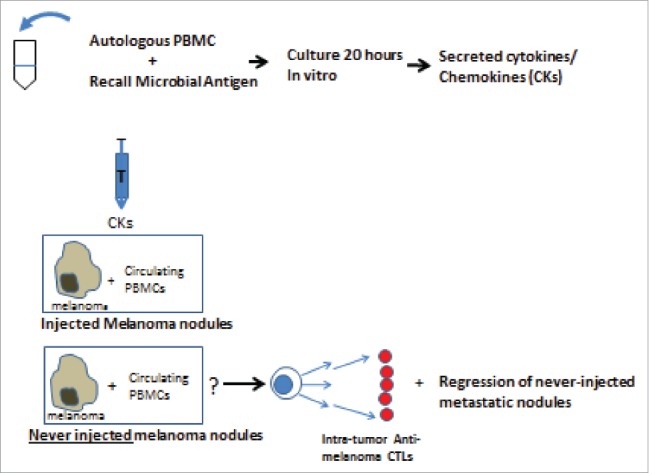
Rationale and design of procedures.
We observed that this immunization strategy resulted in dense lymphocytic infiltrates often into the center of distant never-injected nodules. These responses were associated with CRs in 38% of patients of surprising multi-year durability, and without serious side effects. More recent investigations have provided data on neoantigens and cytokines that may explain the occurrence of the immune responses and CRs observed in our earlier study.
Anti-check point therapy demonstrates that if immunological restraints are diminished, preexisting nascent immune response can control metastatic cancers to some degree in 30% of patients. Clinical responses are predicted by baseline lymphocytic infiltrates at the margin or into the tumor,24–27 and for PD-1 blockade, the number of TIL clones.28 Response rates correlate with the frequency of mutant neo-epitopes, although only a small dominant portion of potential epitopes may contribute to the preexisting immune responses.7 If additional immune responses to neo-epitopes could be induced, preclinical data suggests subsequent anti-check point therapies might result in a higher frequency of CRs.29,30
Results
General responses of injected and never-injected metastases
The study was designed to focus on the responses of never-infected metastatic nodules as evidence of the induction of systemic immunity. During the first 1 to 2 months of weekly injections, new nodules continued to appear and most injected and all never-injected metastases did not become inflamed. After 4 to 6 weeks, in many patients new nodules appeared less frequently, and injected and never-injected nodules often became slightly larger, tender and inflamed. A previously occult subcutaneous nodule occasionally was detected by the development of tenderness and inflammation. Metastases gradually became smaller until undetectable, but occasionally collapsed rapidly. In most responding patients injected nodules began to regress before never-injected nodules, but in 5 patients the first nodule to disappear was never injected. A given metastasis was considered to have regressed if it was undetectable for at least 8 weeks. In 30% of patients no injected or never-injected metastasis regressed completely, although injected nodules became flatter more often than never-injected nodules. Also in 38% of patients all injected and never-injected metastases completely regressed resulting in CRs. In patients with complete regressions of all injected nodules, all never-injected nodules also completely regressed. Because in the majority of patients the number of nodules injected was larger than the number never-injected, we compared the percent of injected and never-injected regressions in each patient. Data from all patient pairs revealed a Spearman correlation of r = 1 with p (two tailed) <0.0001. The high correlation was driven by the many CRs and patients who had no regressions. In patients with many regressions but no CRs, the 3 largest discrepancies by which regressions of injected exceeded never-injected metastases were 65%, 2 at 50%; in one patient never-injected regressions exceeded injected by 30%. The strong correlation between complete regressions of injected and never-injected nodules for most patients suggests that a systemic immune response may be operative in regressions of both categories of metastases. Histologic examination of multiply injected nodules that did not regress revealed central infiltrates less dense than those seen in regressing nodules.
Supplemental Fig. 1 describes the time course of injections in 5 representative patients, the frequent continued development of new metastases initially after starting injections, and the disappearance of many injected and never-injected metastatic nodules in some, but not all patients. After a transition period in which new nodules were appearing while others were regressing, all nodules often regressed over several weeks. Fig. 2 presents photographs of rapidly regressing metastases (Fig. 2A) in patients with local and disseminated (Fig. 2D) cutaneous metastases. In 20% of patients nodules regressed only after 4-6 months of stable disease. A complete regression of all nodules (CR) is defined as no clinically evident disease (NED) by examination and chest x-ray for at least 8 weekly examinations, followed by negative scans. Standard RECIST or WHO criteria for a CR require disappearance for 4 weeks and negative scans.31 Although dimensions of individual metastases were measured, because new nodules often appeared while others in that patient were regressing, categorization by an overall response rate was difficult to calculate. Therefore we enumerated patient responses by the more stringent requirement that all metastases regressed, becoming a CR.
Figure 2.
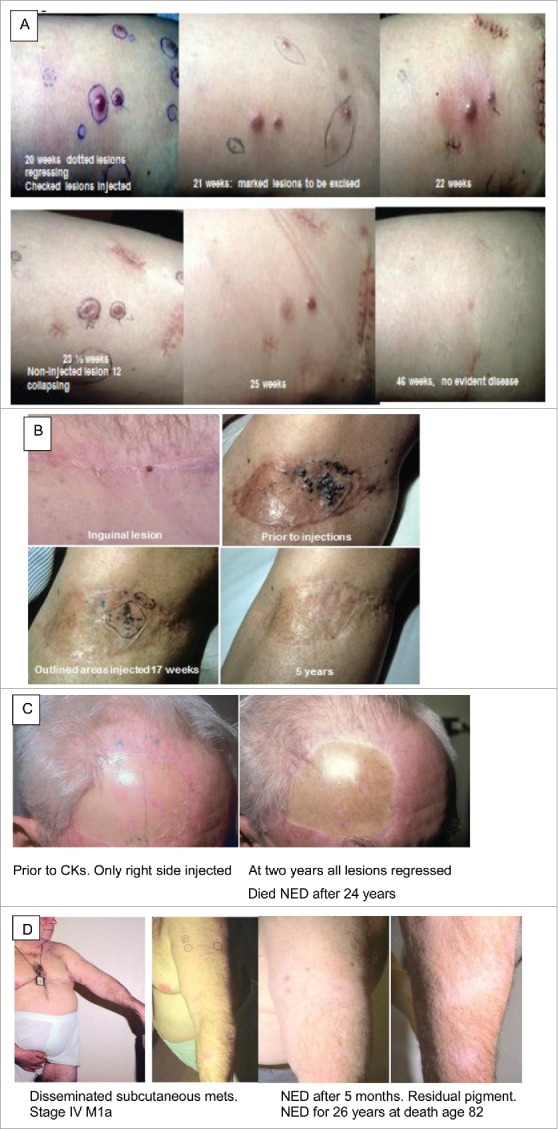
Photographs of patients with cutaneous/subcutaneous metastases experiencing regressions after intralesional CKs. A. 70 year old female: large subcutaneous metastases undergoing rapid rejections. Only checked nodules injected. Dotted nodules are regressing. Collapsing lesion 12 was never injected. Some nodules marked for excision. B. 50 year old female: in transit metastases of left leg and inguinal scar, 4 positive lymph nodes, and no response to chemotherapy. Only circled area injected. Complete regressions and survived disease free for >5 years. Lost to follow up. C. 62 year old male: 6 lesions increased to 57+ small metastases on head and neck. Only lesions on right side injected. All lesions regressed: a 2 × 3 cm mass developed in neck and spontaneously regressed. Needle biopsy showed only residual pigment in melanophages. Became NED after 2 years; died of other causes at age 86, 24 years after entering the study. D. 55 year old male entered with disseminated subcutaneous metastases, face, chest, arms and leg; stage IV-M1a. All lesions including never-injected nodules on arm regressed by 6 months after entry.
Composition of mixtures of autologous CKS
With the goal of mimicking the secretory environment governing the development of a new immune response we stimulated PBMCs with a recall microbial antigen (PPD, Candida, streptokinase) to which the donor was known to respond, and removed the cell-free medium after 20 hours. In order to avoid non-self- proteins, potential infectious agents and for ethical considerations only autologous CK mixtures were employed. Two-D gel separations revealed at least 60 molecules present in the antigen-stimulated supernatants not present in unstimulated cultures (data not shown). Quantitative measurements by Luminex technology of several know cytokines prepared from normal individuals, and CKs prepared from and injected into patients with CRs are presented in Table 1. Individual CKs are present at picogram to nanogram levels. No particular patterns of CKs were associated with CRs. To minimize the level of immunologically detectable microbial antigen carried over in the supernatant media, PBMC were exposed to microbial antigen for only 1 hour washed twice and then cultured for 20 hours. These CK preparations contained no immunologically detectable microbial antigen, but only 10 to 40% of CKs secreted by cells cultured for 20 hours with microbial antigen, and induced fewer regressions. These “minimal antigen” CKs were used for the initial injections in patients and to minimize inflammation occasionally caused in some patients by the presence of microbial antigen.
Table 1.
Concentrations of selected CKs prepared from normal individuals and from and injected into patients who experienced CRs.
| GMCSF | IFN α | IFN λ | IL-12 | IL-2 | IL-15 | IL-7 | TNFα | |
|---|---|---|---|---|---|---|---|---|
| A. Representative cytokine measurements: antigen-stimulated PBMC from 11 normal donors | ||||||||
| Median pgm/ml | 4 | 198 | 77 | 65 | 157 | 500 | 164 | 102 |
| (Range) | (4–150) | (58–662) | (3–2900) | (12–380) | (31–1500) | (54–880) | (47–213) | (2–2380) |
| B. Autologous cytokines injected into 13 patients with complete regressions (stored at minus 800) | ||||||||
| Median pgm/ml | 56 | 55 | 69 | 136 | 105 | 117 | 89 | 23 |
| (Range) | (1–196) | (9–256) | (2–5800) | (3–5250) | (1–2722) | (2–402) | (4–139) | (1–1152) |
Histology and characterization of tumor infiltrating lymphocytes (TILs), and relationship to regressions
In 70% of patients with excisional biopsies of never-injected metastases, examination revealed conspicuous lymphocytic infiltrates in the center or margin of the tumor that were not present in baseline biopsies. The development of dense infiltrates in never-injected nodules is evidence of a new systemic immune response. The density of TIL in never-injected metastases varied between patients, in part as a function of the weeks the nodule had been present while other nodules were being injected, and on the stage of regression. In patients with multiple never-injected metastases who developed a CR, biopsies of an individual nodule revealed moderate to dense lymphocytic infiltrates by 4 to 10 weeks of injections elsewhere (supplemental Fig. 2). After continued injections, infiltrates in never-injected nodules sometimes obscured residual tumor cells, occasionally misdiagnosed as lymphomas by general pathologists. Residual pigmented cutaneous spots revealed only melanophages. All biopsies from patients who experienced CRs contained dense or moderate infiltrates into the center of the metastases. Supplemental Fig. 2A illustrates the absence of any infiltrate in a representative example of the histology of a metastatic nodule before any injections. Supp. Fig. 2B shows a never-injected nodule from the same patient after weeks of injections elsewhere, demonstrating dense infiltrates with lymphocytes contacting tumor cells. Supp. Fig. 2C displays never-injected nodules from 3 additional patients, demonstrating the development of infiltrates restricted to the tumor and not the surrounding tissue, and that enter from the periphery into the center of the metastasis. Baseline infiltrates at the margin of metastases were not required for the development of dense infiltrates and CRs.
In patients with some regressing nodules, but the regular development of new nodules and an overall progressive course, biopsies of never-injected progressing nodules revealed moderate to dense infiltrates primarily at the base of the tumor or in short columns into the metastasis (Supp. Fig. 2D). In two patients with dense infiltrates at the base or margin of progressing nodules, lymphocytes from excisional biopsies were able to kill autologous melanoma cells ex vivo even though the nodules were progressing. The lung metastasis in Supp. Fig. 2E, excised while many cutaneous nodules were regressing, was moderately infiltrated with TILs around islands of melanoma cells.
In patients who had no or infrequent shrinking of a nodule and progressive disease, even after several months of injections never-injected nodules revealed only modest often perivascular infiltrates restricted to the margin of the metastases, (Supp. Fig. 2F). Biopsies from multiply injected metastases revealed dense infiltrates in patients who had regressing disease, but infiltrates were minimal in multiply injected nodules of patients who had rare regressions.
The infiltrates were composed of CD8 and smaller numbers of CD4 T cells, and few NK cells, as assessed by immunohistology (Supplemental Fig. 3) and flowcytometry of single-cell suspensions (Supplemental Table 1). Although subject to vagaries of dissociation and timing of the biopsy, single cell suspensions occasionally yielded equivalent numbers of melanoma cells and TILs. TILs were enriched for CD8 T cells and their phenotypes were significantly different from PBMCs obtained at the same time (Supp. Table 1). Needle biopsies of an enlarging 2 × 3 cm mass in the neck of a patient whose scalp metastases regressed, revealed melanophages but no melanoma, and the mass subsequently disappeared.
Ex-vivo function of TILs
Although TILs were retained in melanoma metastases as evidence of immune responses, we wished to examine their functional cytotoxic capacity. In a direct Cr51 release assay, both TILs and blood lymphocytes were able to kill fresh autologous melanoma cells in-vitro without any ex-vivo expansion. In 12 patients for whom TIL and viable melanoma cells were available, TIL from never-injected and injected metastases were equally cytotoxic, and in 18 patients in whom metastases were regressing, blood lymphocytes also were cytotoxic, as illustrated in representative experiments (Fig. 3). In 2 patients with progressive disease enlarging metastases yielded TILs that were cytotoxic to autologous melanoma cells ex vivo even though they were not restraining the growth of the metastasis, suggesting local inhibition of function in the tumor environment. In comparative assays performed at the same time, TIL were 2 to 3 times more cytotoxic than blood lymphocytes (Fig. 3A). TIL and PBMC killed autologous far more than allogeneic melanoma cells in 5 reciprocal donor experiments, suggesting that cytotoxicity was either MHC restricted or directed against unshared, unique epitopes in each case. Depletion of NK cells did not abrogate the killing (Figs. 3B and 3C). TILs did not kill K562 cells, a sensitive NK target. Depletion of CD8 cells markedly decreased cytotoxicity (Fig. 3D). Depletion of CD4 or CD4 plus NK cells decreased the cytotoxicity minimally or not at all. Infiltrates of cytotoxic TILs developed only after the administration of CKs, but insufficient data are available to document any expansion of PBMC cytotoxicity. Sequential cutaneous injections of irradiated autologous melanoma cells with CKs increased the level of PBMC cytotoxicity (Fig. 3E). Note that TIL cytotoxicity was assayed after lymphocytes were removed from the tumor microenvironment.
Figure 3.

Ex-vivo measurements of (TIL) or blood lymphocytes (BL) to kill autologous melanoma cells. Cytotoxicity presented at increasing effector to melanoma target cell ratios. Each figure is representative of 6 to 10 experiments (A) Comparing the same number of lymphocytes, TIL are more lytic than BL. TIL•, BL ▪. (B) TIL preferentially kill autologous melanoma cells as compared to allogeneic cells, consistent with MHC restriction. Pt C TIL•, Pt M TIL▪ (C) Killing is MHC restricted and not due to NK cells. Pt C BL •, Pt C BL minus NK ▪, Pt F BL ▴, Pt F BL minus NK ▾, (D) Depletion of CD8 T cells abrogates the killing. BL •, BL minus CD8 T▪ (E) Repeated injection in 3 patients of CKs mixed with irradiated autologous melanoma cells over many weeks increases the killing ex-vivo.
Clinical patterns and frequencies of regressing metastases
Although clinical outcomes of patients are categorized by the regression of all metastases (CRs), 70% of patients had at least 2 metastases regress, and many had multiple never-injected nodules regress over many months even if they did not have CRs. Metastatic nodules regressed as early as 2 to 6 weeks, but the median time for regressions in all patients was 4 months after entry. 20% of patients experienced multiple regressions by 2 months. In some patients Injected and never-injected nodules regressed over many months of injections. A frequent pattern was for new nodules to appear while never-injected nodules in the same patient were regressing. When all injected nodules in a patient regressed completely all never-injected nodules also regressed completely. 38% of participants had a CR with NED for a median of 59 months (2-334). The time to regression of all metastases (CR) varied from 1 to 48 months after entry (median 15.). Both never-injected and injected nodules in patients receiving CKs from cells pulsed with antigen for only one hour and washed (to minimize microbial antigen) also regressed completely. However these CKs induced fewer regressions than supernatants from cells cultured with antigen for 20 hours, which also contained higher concentrations of CKs. Patients whose disease was progressing rapidly were injected with supernatants from cells cultured with antigen for 20 hours.
A majority of new nodules appearing while others were regressing were injected, with some left as controls. Many new nodules later completely regressed with or without injections. Of 33 patients with NED (>2 months), 11 (33%) developed additional cutaneous/subcutaneous nodules. But 5 of these had a second period of NED, and survived as NED to the end of the study Fig. 4. However, the persistent appearance of new nodules with variable lymphocytic infiltrates in spite of ongoing regressions accounted for the failure to achieve durable CRs in a majority of patients.
Figure 4.
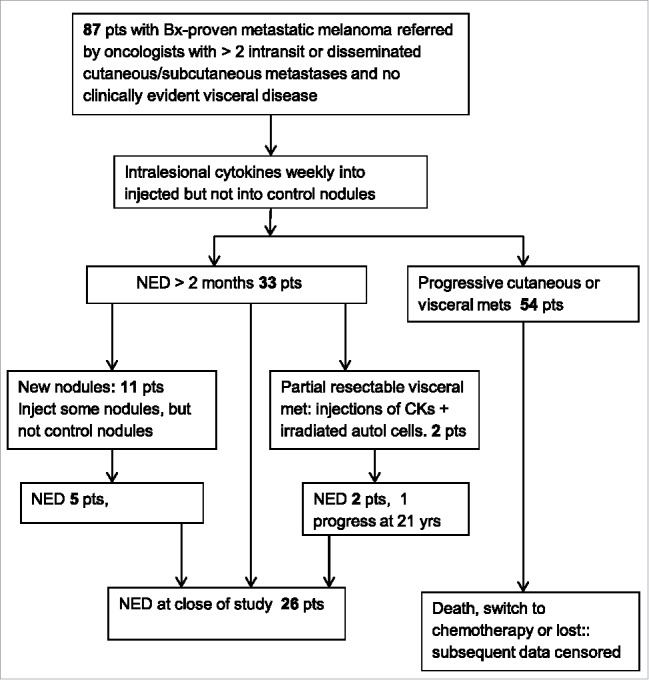
Chart of numbers of patients with regressing and recurrent metastases.
In 3 patients with regressing metastases, benign nevi also injected with cytokines never regressed.
Clinical outcomes
The increased sample size of 87 patients enabled us to assess the frequency of clinical benefit of the new induced immune responses. In this analysis, a CR, or no evident disease (NED), required that all metastases disappear by examination and chest x-ray for 8 consecutive weeks with subsequent negative scans (included in RECIST/WHO criteria). Occult visceral metastases usually become evident within two years, and the absence of residual visceral disease in many patients is reflected by many years of disease-free survival. The 38% frequency of (CRs) of all metastases resulting in NED for over 2 months (median 59 months, range 2 to 334 months), and substantial prolongations of disease-free survival were surprising. The frequency and durability of CRs, and overall survival after entry are presented in Table 2. CRs were more frequent in those with fewer metastases on entry. 28%, 24% and 20% of all patients remained free of detectable metastases for cumulative durations of greater than 2, 3, and 5 years. Median overall survival of patients, (progressing and regressing) censored at death on study, when last seen on study prior to switching to chemotherapy or other experimental therapies, or until lost to follow up was 28.5 months (4-348 months), with 54% of patients surviving 2 years, 43% 3 years and 29% 5 or more years. Disease-free (NED) is considered progression-free survival and was achieved in 23% of patients at 1, 2 and 3 years (Table 2). Overall, of 33 patients with NED > 2months, 79% survived with NED for a median of 59 months (2-334). Durations of CRs and overall survivals only reflect months on this study, and are censored when patients died on this study, were last seen, switched to chemotherapy or were lost to follow up.
Table 2.
Clinical Outcomes: Summary of responses and survival. NED = No Evident Disease by examination and chest x- ray for at least 8 weeks followed by negative scans. The table provides median and range of data on all 87 patients for: time to NED, the % of patients achieving NED for greater than 2 months, and the % of patients with a cumulative duration of NED of 2, 3 and 5 years. The table also presents median and range of overall and disease-free survival from entry at different times. Because some patients relapsed after being free of disease for protracted periods, the % of subjects surviving free of disease for 1 to 5 years after entry is less than the cumulative duration of disease free periods. Also a few disease-free patients were lost to follow up.
| Melanoma Clinical Outcomes: Summary Tables | |||||||||
| Time to |
NED Duration in months |
||||||||
| Frequency and durability of responses |
Median |
Range |
% all pts |
Median |
Range |
NED 1 yr |
NED 2 yr |
NED 3 yr |
NED ≥ 5 yr |
| At least 2 completely regressed met | 4 mo | 0–46 | 72% | ||||||
| NED >2 months (CR) | 15 | 0–48 | 38% | 59 | 2–334 | 31% | 28% | 24% | 20% |
| NED when last seen | 25% | 100 | 6–348 | ||||||
| 57 pts (66%) reached stage IV | 11% of stage IV | 100 | 5–334 | ||||||
| Survival in months |
|||||||||
| Survival |
|
|
|
Median |
Range |
Surv 1 yr |
Surv 2 yr |
Surv 3 yr |
Surv ≥ 5 yr |
| All pts overall survival from entry | 28.5 | 4–348 | 70% | 54% | 43% | 29% | |||
| All pts survival from entry, disease free | 25% | 100 | 6–348 | 23% | 23% | 23% | 20% | ||
| Progres. dis. 54 pts, never NED : survival to death or chemo | 18 | 3 to 75 | 56% | 30% | 20% | 9% | |||
Stage IV disease
8 patients entered with stage IV-M1 and 1 with IV-uveal +M1 disease, and 48 additional patients progressed to stage IV during the study. 6 of 57 had a period of NED >5 months after reaching stage IV, although these CRs were often not sustained. Three patients receiving injections into subcutaneous metastases developed pulmonary nodules detected radiographically; in 2 the nodules regressed; in a third the excised nodule revealed a moderate lymphocytic infiltrate (Supplemental Fig. 2 E). In another patient a 6 cm non-resectable adrenal metastasis completely regressed for 21 years leaving only calcified spots.
Injections of CKs with irradiated autologous melanoma cells
Two patients whose cutaneous nodules had regressed completely developed visceral metastases that were only partially resectable. Under a separate IRB-approved protocol, single cell suspensions of tumor cells were obtained, viably frozen, and aliquots of 107 autologous cells irradiated with 20,000 Rads, mixed with autologous CKs were injected intradermally every 2 weeks for 9 months. The two patients became CRs by scans. One additional patient presented with positive inguinal nodes and a subsequent resectable visceral metastasis without evident cutaneous disease. (This patient is not included in response rates.) For these 3 individuals, These 3 patients developed increasing levels of PBMC-mediated cytotoxicity to autologous melanoma ex-vivo (Fig. 3E) and remained free of disease for 18 to 23 years, although one eventually relapsed after 21 years.
The courses of all 87 patients on study are presented in Fig. 5, with the presence of melanoma in black and disease free periods in red. Patients are ranked by duration of follow up, and censored at death on the study, switch to chemotherapy, or when last seen. Many patients who ultimately succumbed to melanoma experienced disease-free periods over one year, and the overall survival with stable disease often exceeded 2 years. Note that clinical metastases recurred after several years of NED in a few patients. Fig. 6 presents a Kaplan-Meier plot of overall survival including deaths after leaving the study in which patients are censored only if free of disease when lost to follow-up or at the end of the study as indicated by a tick mark. Figs. 5 and 6 suggest that survival is prolonged even in subjects without CRs.
Figure 5.

Melanoma status of all patients ranked by the duration of follow up in months and years after entering. The time line for each patient is censored at death, initiation of chemotherapy, or when last examined. Red indicates no clinically evident disease (NED) by examination and chest x-ray for at least 8 weeks, and subsequent negative scans. Several patients had NED* when lost to follow up. Black indicates presence of metastatic melanoma. Note, many patients had prolonged periods of stable or slowly progressive disease. 4 patients who had NED for >2 years experienced a late relapse as indicated. A Patient was experiencing a rapid regression of many metastases after 27 weeks of CK injections (with dense lymphocytic infiltrates in never injected nodules) when she switched to a geographically closer physician, and began receiving injections of an allogeneic melanoma vaccine to enhance immune responses. Within 8 weeks after the last CKs and after 4 weekly injections of vaccine she demonstrated NED. Subsequent indicated recurrences were treated by irradiation or excision.61 B The recurrence of melanoma after 20 years of NED subsequently responded to Ipilimumab.
Figure 6.
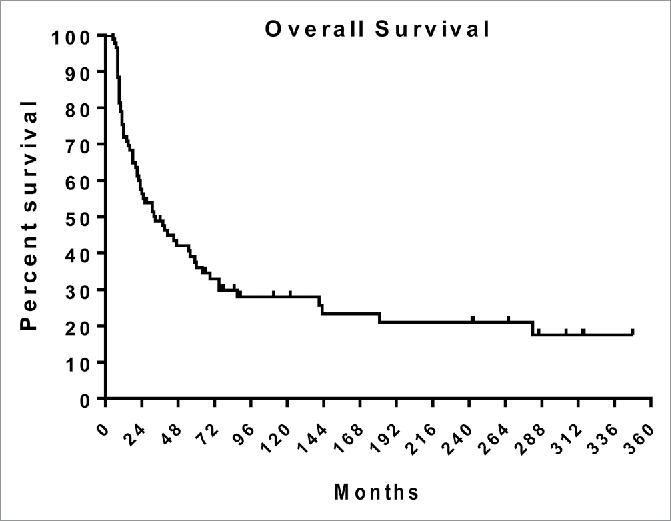
A Kaplan-Meier plot of overall survival on study and after switching to chemotherapy. Tick marks indicate alive at end of study or when lost to follow-up.
Adverse events
Local erythema and tenderness of both injected and never-injected nodules lasting 1 to 5 days occurred in a significant minority of patients. Mild flu-like symptoms in patients undergoing rapid regressions of multiple nodules was the major systemic side effect observed. 15% of subjects experienced local inflammation at the injection site of CKs containing residual microbial antigen, which was avoided by mixing with autologous CKs containing no detectable microbial antigen. Many participants received injections of CKs weekly for several years without adverse events. Local vitiligo of skin where metastases regressed was observed in approximately 20% of patients.
Discussion
This study tested the hypothesis that systemic cellular immune responses to metastatic melanoma could be induced or enhanced by intralesional injections of a mixture of autologous CKs secreted ex-vivo early in a secondary immune response, which may mimic the cytokine environment guiding new immune responses more effectively than individual CKs. The accumulation and retention of new lymphocytic infiltrates in never-injected metastases is consistent with a systemic immune response to melanoma, which may represent the induction of immunity to additional non-dominant epitopes by T cells with appropriate receptors or a marked enhancement of previously undetectable responses. Studies of multiple epitopes were not performed.
Intralesional injections of CK mixtures containing picogram/nanogram levels of individual CKs avoid toxicities and may facilitate development of immunity to multiple melanoma epitopes unique to that patient's cancer. Dense lymphocytic infiltrates in the center of many never-injected nodules and complete regressions of these metastases, accompanied by the presence of CD8 cytotoxic T cells (albeit removed from the tumor microenvironment) suggest that the induction of effective immune responses with combinations of secreted CKs is feasible and can be achieved without significant adverse events. Because it was initiated many years ago, this study demonstrated immunologically mediated CRs some of which resulted in disease-free intervals of over 20 years, and also that late relapses can occur.. Attempts to optimize the composition of CKs, the concentrations and injection schedules have not been undertaken.
The infiltrates were dense and occurred in a larger portion of patients than the nascent infiltrates primarily around the invasive margin and to a smaller extent in the center of tumors that predict favorable responses to checkpoint blockade,24 and most importantly, CK-induced infiltrates often resulted in complete regressions without anti-checkpoint therapy. TILs in the center of the metastases suggest this strategy may often circumvent local inhibitory conditions associated with the tumor.25,32 Complete regressions of nodules were associated with TILs into the center of metastases, but the presence of TILs did not always predict CRs since some nodules with moderately dense TILs progressed even though other nodules in the same patient were regressing. When dense infiltrates were present in progressing never-injected nodules they were usually at the margin with projections into the nodule (Supplemental Fig. 2 D). Although TILs were not always restricting the growth of the tumor in-vivo, in two patients with progressive disease these TILs were able to kill autologous melanoma cells ex-vivo suggesting local inhibition in the tumor microenvironment.
New metastases often regressed with or without being injected, but many patients did not remain free of disease. Only 38% achieved a CR of >2 months duration although many additional patients had protracted periods of stable or slowly progressing disease. New nodules appearing while other never-injected nodules in the same patient were regressing suggests possibilities of antigenic escape,33 and/or local inhibition of lymphocyte function by CTLA-4 or the PD-1/PDL-1 axis,4–6 myeloid-derived suppressor34 or T-regulatory cells,35 or that growth of melanoma cells exceeded the capacity of TIL cytotoxicity.
The TILs were primarily CD8 cells with a smaller variable number of CD4 cells. TILs from never-injected nodules, as well as PBMC from recipients of intralesional CKs, were cytotoxic to fresh autologous melanoma cells in vitro without any pre-stimulation or expansion ex-vivo. Depletion of CD8 cells ex-vivo but not CD4 or NK cells abrogated killing. Preferential cytotoxicity to autologous over allogeneic melanoma cells in several reciprocal experiments is consistent with MHC- restriction or the absence of shared target epitopes. Observations that phenotypes of infiltrating lymphocytes differ from those simultaneously present in the blood (Supplemental Table 1), indicate that cytotoxic CD8 T cells are selectively retained in the tumor.
The accumulation of CD8 cells in never-injected metastases indicates that TILs must be recognizing antigens on at least some of the melanoma cells. The presence of infiltrates in some progressing never-injected nodules implies that not all of the melanoma cells in the nodule can be edited escape mutants. These observations might result from varying expression of melanoma epitopes on different cells within the same metastasis. The ability of TILs from regressing and progressing nodules to kill autologous fresh melanoma cells was demonstrated by removing TILs from the tumor. This suggests local inhibition of T cell function in the microenvironment of some but not all metastases, but does not rule out an additional contribution of escape mutants. The limited phenotypes of tumor-infiltrating cells measured in the present study do not distinguish TILs from nodules that regressed from those that progressed within the same patient. Reagents to assess T regulatory cells, the PD-1/L-1 axis and myeloid-derived suppressor cells were not available when the study was performed. Measurements of the concentrations of a limited number of cytokines in this study (Table 1) were of the same magnitude as CKs from healthy individuals and did not detect patterns distinguishing recipients who did and did not experience CRs (data from progressors not shown).
The multiple exposures to CKs before regressions are observed, the frequent swelling of metastases before regressions and the late appearance of new metastases and regressions are observations similar to those reported for immunological responses revealed by anti-check point therapies.36 CKs obtained after pulsing PBMC with microbial antigen did not contain immunologically detectable antigen but were able to induce infiltrates and complete regressions in some patients, indicating that microbial antigens carried over in the CK mixtures were not essential to induce anti-melanoma responses. If residual microbial antigen were instrumental in inducing the systemic immune responses, then injected nodules, especially in the minority of patients who developed local inflammation due to residual antigen, should have had a greater frequency of CRs, but this did not occur. However, residual microbial antigen might enhance the development of the anti-melanoma responses in the injected nodule. Injected metastases served as the local immunization site but the frequency of regressions in injected and never-injected metastases were similar even though the injected nodules were usually but not always more inflamed. The percent of regressions of injected nodules and never-injected nodules.in each patient were highly correlated for all patient-pairs, primarily because many patients had no complete regressions while others had complete regressions of both injected and never-injected nodules. If all injected nodules regressed in a patient then all never-injected nodules regressed, suggesting that the systemic immune response was operative in CRs of both injected and never-injected metastases.
The observation that repeatedly injected benign nevi never regressed in patients in whom never-injected metastases were regressing suggests that the immune responses were often against epitopes not shared by the benign lesions even though some patients developed local vitiligo at sites of regressions.
In other studies, Injections of dendritic cells pulsed with melanoma antigens have induced cellular immune responses and a modest prolongation of survival, but infrequent clinical responses.12 A gp100 peptide melanoma vaccine administered with high dose IL-2 to patients with advanced melanoma resulted in 9% CRs as compared to 1% CRs with IL-2 alone.14 Recent studies of the intralesional injection of replication competent oncolytic herpes virus expressing GMCSF (talimogene laherparepvec) demonstrated a 16.8% response rate for > 6 months primarily in cutaneous metastases including 10.8% CRs.37 A systemic effect was indicated since non-injected control cutaneous nodules in 5 of 23 patients underwent CRs but only 1 visceral metastasis regressed completely.38 The intralesional injection of IL-2, up to maximally tolerated doses resulted in a high frequency of complete regressions of injected nodules but surprisingly not of non-injected nodules, suggesting that systemic immunity was not induced.39 When IL-2 and TNF conjugated with an antibody to a fibronectin domain was injected into melanoma metastases several noninjected nodules egressed although no patients were reported to have regressions of all nodules.40
We postulate that the intralesional injections of IL-2, herpes virus expressing GMCSF,37 other viruses,29 or toll-like receptor agonists41 may induce systemic immune responses in part by stimulating the local production of CKs. The infusion of IL-2 results in the secondary release of other cytokines,42 although the composition of IL-2- stimulated CKs or those released by other inflammatory stimuli may not mimic that of developing immune responses.
Combinations of 2 or 3 recombinant CKs have been investigated in as therapies for human cancers, usually at maximally tolerated doses. Overall clinical regressions have been no better than the 6% partial or 2% CRs occasionally of multiyear duration seen with infusions of IL-2 alone.14–17
Some cytokines such as colony-stimulating factors have potent biological effects as single proteins. However, during an immune response a complex mixture of CKs is coordinately secreted by activated innate and adaptive immune cells, and act in concert to initiate, modulate and regulate immune responses.20,21 The net effect of a given CK on an immune response when secreted with other CKs may differ from its recognized activity as a single molecule. In mice, different CKs often have overlapping biological activities,43,44 and the effect of a given CK in-vivo may depend on the presence of other CKs.45 IL-2 facilitates the proliferation of lymphocytes but may cause stimulated lymphocytes to die, and enhances inhibitory T regulatory cells.43,46 IL-2 and IL-15 have different effects but share some receptors and signaling pathways and form heterodimers whose functions depend on one another.47,48 The net cellular response to a given CK also depends on the specificity of receptors currently expressed on cells,43 and the concomitant triggering of other linked signaling pathways.49 One cytokine may stimulate the release of additional cytokines.42,45 Multiple CKs may be required to induce and maintain CD8 cell functions.50 CKs produced by CD4 cells are critical for the differentiation of CD8 cells.51 The balance of CKs also is important for induced T regulatory cells.52
The treatment of metastatic melanoma with anti-CTLA-4 monoclonal antibodies was associated with a modest increase in infiltrating CD8 cells.53,54 that did not correlate with clinical responses. Infiltrates seen following ipilimumab appear to be less dense than observed in many never-injected nodules following CKs. Ipilimumab also resulted in increases in activated blood CD4 and CD8 lymphocytes, T regulatory cells, modest increases in interferon-γ-producing T cells, and decreases in myeloid derived suppressor cells.53
The presence in some patients of infiltrates around the margin of a metastasis prior to the administration of anti-PD-1 antibodies predicts a beneficial clinical response, an increase in CD8 cells in the margin and center of the tumor, and the division of CD8 T cells within the metastasis.24 In the current study, preexisting marginal infiltrates were not required for dense TIL to develop in the center of never-injected metastases in patients receiving CKs. However, never-injected nodules in patients who did not develop CRs did develop infiltrates at the tumor margins, suggesting that these metastases might be responsive to subsequent PD-1 blockade.
Anti-CTLA-4 antibodies may facilitate immune responses by enhancing interactions between antigen-presenting cells and T cells rather than solely by removing restraints on effector lymphocytes. New immune responses after ipilimumab were documented in half of recipients by detecting one or more new CD8 T cell clones using a panel of melanoma peptide-specific MHC multimers.55 Ipilimumab increased the number of epitopes recognized but did not increase the number of preexisting CD8 clones. Ipilimumab therapy has resulted in 1.6% CRs but with grade 3 or 4 AEs in 23% of recipients.3 Treatments with antibodies against the PD-1/L-1 axis have caused up to 7% CRs and fewer (15%) adverse events. Nivolumab therapy resulted in a 30% response, a 43% 2 year overall survival, and 1% CRs.56 Ipilimumab and nivolumab administered in combination increased the overall response rate with 22% CRs, but 53% of recipients had serious AEs.57 Most of the anti-checkpoint studies enrolled more stage IV subjects than in the present investigation. In spite of these important and elegant therapeutic gains for 30% of recipients, new strategies are needed to increase the frequency of durable CRs.
At least two aspects of our immunization strategy may account for dense infiltrates in never-injected metastases, the relatively high frequency (38%) of CRs, and the 2 to 20 years of no evident disease. First, the intralesional injections of a secreted combination of CKs may induce new CD4 and CD8 responses against multiple non-dominant mutated epitopes unique to a given patient's tumor, by allowing T cell clone-epitope self-selection, in addition to responses enhanced by anti-check point therapies.58,59 Continued injections into newly appearing metastases while other never-injected nodules are regressing may induce responses to emerging mutated escape epitopes. This is especially important for cancers such as melanoma containing multiple unique mutations.2 A comparison of patients who benefited from ipilimumab with those who did not, revealed that beneficiaries had melanomas with more mutant peptides, and that a subset of epitopes was shared by patients with the best responses.7
Second, the development of effective immune responses in the current study may result from using a combination of secreted CKs at exceedingly low concentrations, mimicking the cytokine environment of a developing immune response, and inducing stronger, more effective immunity. The same effects may not be obtained using single or combinations of 3 CKs. This approach also accounts for the lack of significant side effects.
The efficacy of anti-check point therapies may be limited in part because patients have insufficient nascent anti-melanoma immune responses to be enhanced by tolerable levels of antibodies. If so, the induction of effective anti-melanoma immune responses and dense infiltrates in the tumor and tumor margin using CKs suggests that CKs could be employed prior to the administration of anti-checkpoint therapies. Reciprocally, since many recipients of CKs progressed in spite of developing lymphocytic infiltrates, whose function may be inhibited locally, the administration of PD-1/ L-1 antagonists might increase complete regressions in recipients of CKs. Although autologous secreted CKs resulted in dense infiltrates in never-injected metastases, cytotoxic TILs and many durable CRs, at present injections of CKs cannot be considered a drug, but an investigational procedure or “autologous transplant”. When these studies were conducted, current techniques for studying the tumor microenvironment, epitope specificities and phenotypes of TILs were not available. A full characterization of the required CK components using proteomics, optimization of concentrations and injection schedules, might enable these observations to contribute to a practical therapy. Additional investigations are required to fully characterize the infiltrates and determine why cytotoxic TILs from some metastases do not inhibit tumor cells in-situ. However, these studies do provide an additional and effective strategy for inducing effective immune responses against metastatic melanoma.
We conclude that the intralesional injection of an exogenous and “physiological” mixture of secreted CKs stimulates the development of new systemic and often effective anti-melanoma immune responses to epitopes that intrinsically were not sufficiently immunogenic without CKs, and were not evident prior to injections. The induced immune responses frequently resulted in dense CD8 T lymphocytic infiltrates in the center of never-injected metastases. 38% of patients experienced CRs >2 months, including some with stage IV disease, and 20 % remained disease free for 6 months to over 25 years. These observations demonstrate the feasibility of obtaining complete regressions of advanced metastatic melanoma using a therapeutic vaccine strategy without check-point antagonists, resulting in disease-free periods for many years without significant side effects. The occasional relapse after several disease-free years suggests that continued immunological control may be required. The immunization strategy of using cytokines and chemokines mimicking the secretory environment of an immune response may be more effective than using combinations of 1 to 4 recombinant CKs, and may explain the limited systemic effects of other intralesional inflammatory agents. The study describes an additional strategy to be pursued and combined with anti-checkpoint therapy for the control of metastatic melanoma.
Patients and methods
Study design
This was planned as an immunological study in 20 patients to test the hypothesis that the weekly intralesional injection of autologous secreted CKs was safe and might induce systemic immune responses to melanoma as assessed by the appearance of dense tumor-infiltrating lymphocytes in never-injected metastases (Fig. 1). Patients were in a crescendo phase of their illness for which referring oncologists had no effective therapy. After several patients experienced CRs of all never-injected as well as injected nodules, we expanded the sample size to determine the frequency and durability of disease-free periods. The study was stopped, laboratory investigations limited, and publication efforts suspended due to insufficient funding by the NCI, and strong funding by NCI and NIAID for HIV studies. Fig. 4 presents additional information on patient management. Recently we have obtained follow up on most participants.
Patients participating in this study
87 patients meeting eligibility criteria were referred and monitored by oncologists. They all had clinically palpable cutaneous or subcutaneous in-transit metastases of an extremity or multiple metastases on the trunk or head. All had developed 2 to 100 metastases during the 2 months prior to entry, and 30 had failed to respond to chemotherapy including perfusions ending at least 2 months prior to entry. A majority had positive nodes on elective resection. They were stages IIIB 2 pts; IIIC 76 pts; and IV-M1a 9 pts60 Table 3 tabulates the clinical stages of patients on entry, and the frequency of CRs in different entry groups. Patients with detectable visceral disease on entry were not eligible. There were no other exclusions. 48 patients progressed to Stage IV (2 M1a, 3 M1b; 44 M1c) during the study. 51 patients had excisional biopsies for monitoring, 5 of whom had several nodules resected, but all except one, who had experienced many regressions, had residual disease after biopsies. Patients were studied between 1979 and 1992 when standard chemotherapy was relatively ineffective dacarbazine.
Table 3.
Clinical stages of patients at entry and frequency of CRs. All patients had developed 2 to over 100 palpable cutaneous and subcutaneous metastases during the previous 2 months, and most continued to have new nodules during the first few weeks on study. 30 patients had failed prior chemotherapy ending 2+ months before entry.
| Entry Stage | Prior Chemo | CRs | CRs Progress to IV | CR after stage IV |
|---|---|---|---|---|
| III B, 2 pts | no | 2 of 2 | 0 | |
| III C, 76 pts | yes 29 | 9 of 29 | 5 of 9 | |
| no 47 | 19 of 47 | 6 of 19 | 2 pts after IV-M1b | |
| 1 pt after IV-M1c | ||||
| IV-M1a, 9 pts | yes 1 | 0 | ||
| no 8 | 3 of 8 | 2 pt after IV-M1a | ||
| 1 pt after IV-M1c |
Ethics approvals and consent to participate
Protocols were approved by the New York University School of Medicine IRB (H4356) in accordance with an assurance filed with and approved by U.S. DHHS. Prior to inclusion in the study, patients provided written consent after a discussion with their oncologist, their family and with the principal investigator, and were followed and monitored by their individual oncologists
Injections and monitoring of metastases
As diagramed in Fig. 1, (and supplemental Fig. 1), in patients with cutaneous or subcutaneous metastases, autologous CKs were injected weekly into a majority (50-90%) of nodules, while other control nodules, distal, proximal or contralateral to injected nodules, were never injected at any time. Locations of nodules were identified by plastic-wrap templates, anatomic landmarks and photographs. Nodules began to regress after 2 to 12 weeks with individual nodules regressing completely. Injections were continued at sites of regressed metastases for 8 weeks after the nodule was no longer detectable. Injections into remaining metastases continued until CRs were obtained or until rapid progression mandated that chemotherapy be initiated. Because of the high frequency of disappearances of metastatic nodules and the frequent appearance of new nodules, we present data only on complete regression of a metastasis rather than responses by deceased dimensions. Complete regression of all nodules (CR) is defined as no clinically evident disease (NED) by examination and chest x-ray for at least 8 weekly examinations, followed by negative scans. Standard RECIST or WHO criteria for a CR require disappearance for 4 weeks and negative scans.31 In 2 patients with partially resectable visceral metastases but no remaining cutaneous metastases, fresh autologous melanoma cells were prepared as described below, viably frozen in liquid nitrogen, and 107 cells thawed, washed in autologous serum medium, irradiated with 20,000 Rads, mixed with 0.2 ml of autologous CKs, and injected intracutaneously every 2 weeks for > 9 months.
Cytokines/chemokines (CKs)
To obtain a complete complement of CKs secreted in immune responses, and eliminate potential transmission of infectious agents, autologous CKs were prepared by stimulating autologous peripheral blood mononuclear cells (PBMC) one million cells/ml in 10% autologous plasma-containing tissue culture medium (modified Eagles medium) for 20 hours. PBMC were prepared from fresh peripheral heparinized blood by centrifugation over Ficol-Hypaque, placed in culture, and stimulated with sterile microbial antigens (1 to10 µg/ml) to which the patient's PBMC respond as assessed by previous proliferation assays. Commercially available microbial antigens used include: tuberculin PPD skin test antigen, streptokinase for injection, candida albicans skin test antigen. Alternatively, the cytokines were prepared by pulsing the cells briefly (1 hour) with the antigen, washing the cells, and collecting the supernatant medium after 20 hours. Supernatants from PBMC pulsed with an antigen to which the donor is not sensitive do not stimulate the PBMC of a donor exquisitely sensitive to that antigen and thus do not contain immunologically detectable microbial antigen. CKs were passed through a 0.2 micron filter before injection. Complete regressions were seen in recipients of CKs prepared with each of the microbial antigens. The cytokine mixtures contain a variable range of low concentrations of cytokines as quantified by a Luminex method using Life Sciences reagents. 4 to 6,000 picograms /ml of an individual cytokine were present in the mixtures. Table 1 present examples of concentrations of 8 cytokines from a probable >100 proteins secreted by antigen-stimulated PBMC from healthy individuals, and from frozen (minus 800) supernatants from stimulated PBMC of melanoma patients who experienced complete regressions. In measuring only 30 cytokines, no pattern seems to be associated with complete regressions
An individual patient received between 0.05 and 2.0 ml CKs intralesionally each week as a function of the number of metastases.
Monitoring of infiltrates
Excisional biopsies of never-injected and injected metastases were prepared and examined by a dermatopathologist with an estimate of the extent of lymphocytic infiltration, and compared with biopsies from the patient prior to injections. The composition of tumor-infiltrates lymphocytes (TILs) also was examined by immunohistology using immunoperoxidase kits with anti- CD4, CD8 and CD56 monoclonal antibodies, and examination of single-cell suspensions by flowcytometry (Supplemental Fig. 3 and Supplemental Table 1).
The tumor was not examined for other phenotypes, myeloid-derived suppressor cells, T-regulatory cells, CTLA4 or the PD-1/PDL-1 axis since reagents were not available at the time.
Cell preparations and ex-vivo cytotoxicity measurements
Excised metastatic melanoma was cut into 1-2 mm cubes, digested with collagenase and DNAse, gently vortexed, and every 10 minutes released cells separated and washed. Tumor cells and TIL were separated by repeated centrifugations on discontinuous Ficol-Hypaque (PBMC at 100%; tumor cells at 75% interface) or percoll gradients, followed by a 2 hour incubation on plastic. Melanoma target cells were used after a 16 hour incubation to allow damaged cells to die, or suspended in serum-DMSO and stored over liquid nitrogen. Tumor cells were labeled with 500 µCi Cr,51 and 1,000 viable tumor cells washed in 10% autologous serum, and added to wells as targets. TIL or PBMC effector cells, at indicated effector (E) to target (T) cells ratios, were directly incubated with target cells for 16 hours without any in vitro “priming”. Assays were conducted in autologous serum. Total Cr51 incorporated in target cells was measured by detergent release. % cytotoxicity was calculated as % of total cellular Cr51 released after incubation with effector cells, corrected for spontaneous release (<22%). Depletion of CD8, CD4 T cells or NK cells from PBMC or TIL was accomplished using beads conjugated with anti- CD4, CD8 or CD56 antibodies or anti CD56 plus complement. NK depletion was monitored by loss of ability to kill K562 cells. Probable MHC restriction of cytotoxicity or the absence of shared target epitopes were assessed by multiple comparisons of the reciprocal killing of autologous and allogeneic melanoma targets by cells from pairs of patients. Note that TIL cytotoxicity was assayed after lymphocytes were removed from the tumor microenvironment.
Supplementary Material
Disclosures of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Laboratory experiments and preparation of patient cytokineswere conducted by Jin-Sheng Yao, Anna Maria Paolino and Vincenza Itri. Histopathology for this study was performed by Dr. A. Bernard Ackerman. We thank Drs. James Speyer and Ruth Oratz, Medical Oncologists, who also followed patients during the study and cared for the many patients who did not respond to cytokines. We also thank Dr. Speyer for suggestions on the design of the manuscript. We are grateful to the patients who participated in this study, often over several years
Funding
This work was initially supported by the National Cancer Institute CA 19529 (FTV), and also supported by the Fan Fox and Leslie Samuels Foundation, Friends of Caroline, and the Cancer Surgery Research Fund. Clinical visits were conducted in the General Clinical Research Center: current grant UL1TR000038.
Author's contributions
Conception and design: F.V, F.G, M.H., D.R.
Director of Study: F.V.
Acquisition of patient data and biopsies: F.G, D.R, M.H.
Laboratory studies: F.V.
Analysis and interpretation of data: F.V, F.G, D.R
Writing of manuscript: F.V.
Review of manuscript: F.V, D.R, M.H, F.G
References
- 1.Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, Gilboa E, Hammond SA, Hershberg R, Korman AJ, et al.. The human vaccines project: A roadmap for cancer vaccine development. Sci Transl Med. 2016;8:334ps9–ps9. doi: 10.1126/scitranslmed.aaf0685. PMID:27075624 [DOI] [PubMed] [Google Scholar]
- 2.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al.. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. PMID:23945592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Thomas L, Bondarenko I, O'Day S, M DJ, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al.. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. PMID:21639810 [DOI] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. PMID:22658127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. PMID:22658128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamid O, Robert C, Daud A, Hodi F. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:133–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al.. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. PMID:25409260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, et al.. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3:399–411. doi: 10.1158/2326-6066.CIR-14-0215. PMID:25678581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agha-Mohammadi S, Lotze MT. Immunomodulation of cancer: Potential use of selectively replicating agents. J Clin Invest. 2000;105:1173–6. doi: 10.1172/JCI10026. PMID:10791989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barouch DH, Letvin NI, Seder RA. The role of cytokine DNAs as vaccine adjuvants for optimizing cellular immune responses. Immunol Rev. 2014;202:266–74. doi: 10.1111/j.0105-2896.2004.00200.x. [DOI] [PubMed] [Google Scholar]
- 11.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539–43. doi: 10.1073/pnas.90.8.3539. PMID:8097319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dillman RO, Cornforth AN, DePriest C, McClay EF, Amatruda TT, de Leon C, Ellis RE, Mayorga C, Carbonell D, Cubellis JM. Tumor stem cell antigens as consolidative active specific immunotherapy: A randomized phase II trial of dendritic cells versus tumor cells in patients with metastatic melanoma. J Immunother. 2012;35:641–9. doi: 10.1097/CJI.0b013e31826f79c8. PMID:22996370 [DOI] [PubMed] [Google Scholar]
- 13.Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K, et al.. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–71. doi: 10.1200/JCO.2009.24.3675. PMID:19884534 [DOI] [PubMed] [Google Scholar]
- 14.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, et al.. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27. doi: 10.1056/NEJMoa1012863. PMID:21631324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vacchelli E, Eggermont A, Fridman WH, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Immunostimulatory cytokines. Oncoimmunology. 2013;2:e24850. doi: 10.4161/onci.24850. PMID:24073369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Gast G, Klümpen H-J, Vyth-Dreese F, Kersten M-J, Verra N, Sein J, Batchelor D, Nooijen WJ, Schornagel JH. Phase I trial of combined immunotherapy with subcutaneous granulocyte macrophage colony-stimulating factor, low-dose interleukin 2, and interferon alpha in progressive metastatic melanoma and renal cell carcinoma. Clin Cancer Res. 2000;6:1267–72. PMID:10778950 [PubMed] [Google Scholar]
- 17.Lotze MT. Cytokines in the treatment of cancer In: Caligiuri MA, Lotze MT, editors. Cytokines in the genesis and treatment of cancer. Totowa: (New Jersey: ): Humana Press; 2007. p. 307–471. [Google Scholar]
- 18.Sivapalasingam S, Kennedy JS, Borkowsky W, Valentine F, Zhan MX, Pazoles P, Paolino A, Ennis FA, Steigbigel NH. Immunological memory after exposure to variola virus, monkeypox virus, and vaccinia virus. J Infect Dis. 2007;195:1151–9. doi: 10.1086/512161. PMID:17357051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, et al.. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–22. doi: 10.1016/j.immuni.2008.02.020. PMID:18468462 [DOI] [PubMed] [Google Scholar]
- 20.Yamane H, Paul WE. Cytokines of the gamma(c) family control CD4+ T cell differentiation and function. Nat Immunol. 2012;13:1037–44. doi: 10.1038/ni.2431. PMID:23080204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–52. doi: 10.1016/j.immuni.2012.01.002. PMID:22265676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281. PMID:19293190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valentine FT, Lawrence HS. Lymphocyte stimulation: Transfer of cellular hypersensitivity to antigen in vitro. Science. 1969;165:1014–6. doi: 10.1126/science.165.3897.1014. PMID:5804724 [DOI] [PubMed] [Google Scholar]
- 24.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. doi: 10.1038/nature13954. PMID:25428505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. PMID:25970248 [DOI] [PubMed] [Google Scholar]
- 26.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. doi: 10.1016/j.immuni.2013.07.008. PMID:23890060 [DOI] [PubMed] [Google Scholar]
- 27.Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al.. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827–37. doi: 10.1158/2159-8290.CD-15-1545. PMID:27301722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, Gopalakrishnan V, Wang F, Cooper ZA, Reddy SM, et al.. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9:eaah3560. doi: 10.1126/scitranslmed.aah3560. PMID:28251903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. PMID:24598590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al.. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–30. doi: 10.1016/j.celrep.2015.04.031. PMID:25959818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, et al.. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. PMID:10655437 [DOI] [PubMed] [Google Scholar]
- 32.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al.. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–16. doi: 10.1158/2159-8290.CD-15-0283. PMID:26645196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. PMID:21436444 [DOI] [PubMed] [Google Scholar]
- 34.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22. doi: 10.1038/ni.2703. PMID:24048123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin Cancer Res. 2012;18:3022–9. doi: 10.1158/1078-0432.CCR-11-3216. PMID:22510350 [DOI] [PubMed] [Google Scholar]
- 36.Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G, et al.. Guidlines for the evaluation of immune therapy in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. PMID:19934295 [DOI] [PubMed] [Google Scholar]
- 37.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al.. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8. doi: 10.1200/JCO.2014.58.3377. PMID:26014293 [DOI] [PubMed] [Google Scholar]
- 38.Kaufman HL, Amatruda T, Reid T, Gonzalez R, Glaspy J, Whitman E, Harrington K, Nemunaitis J, Zloza A, Wolf M, et al.. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J Immunother Cancer. 2016;4:12. doi: 10.1186/s40425-016-0116-2. PMID:26981242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weide B, Eigentler TK, Pflugfelder A, Leiter U, Meier F, Bauer J, Schmidt D, Radny P, Pföhler C, Garbe C. Survival after intratumoral interleukin-2 treatment of 72 melanoma patients and response upon the first chemotherapy during follow-up. Cancer Immunol Immunother. 2011;60:487–93. doi: 10.1007/s00262-010-0957-3. PMID:21174093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Byers BA, Temple-Oberle CF, Hurdle V, McKinnon JG. Treatment of in-transit melanoma with intra-lesional interleukin-2: A systematic review. J Surg Oncol. 2014;110:770–5. doi: 10.1002/jso.23702. PMID:24996052 [DOI] [PubMed] [Google Scholar]
- 41.Sagiv-Barfi I, Kohrt HE, Burckhardt L, Czerwinski DK, Levy R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood. 2015;125:2079–86. doi: 10.1182/blood-2014-08-593137. PMID:25662332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mier JW, Vachino G, Vandermeer JWM, Numerof RP, Adams S, Cannon JG, Bernheim HA, Atkins MB, Parkinson DR, Dinarello CA. Induction of circulating tumor necrosis factor (Tnf-Alpha) as the mechanism for the febrile response to interleukin-2 (Il-2) in cancer-patients. J Clin Immunol. 1988;8:426–36. doi: 10.1007/BF00916947. PMID:3265420 [DOI] [PubMed] [Google Scholar]
- 43.Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, Yamauchi A, Bloom ET, Mietz J, John S, et al.. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331. doi: 10.1016/1074-7613(95)90141-8. PMID:7719938 [DOI] [PubMed] [Google Scholar]
- 44.Prlic M, Lefrancois L, Jameson SC. Multiple choices: Regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. J Exp Med. 2002;195:F49–52. doi: 10.1084/jem.20020767. PMID:12070294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton SE, Prlic M, Jameson SC. Environmental conservation: Bystander CD4 T cells keep CD8 memories fresh. Nat Immunol. 2004;5:873–4. doi: 10.1038/ni0904-873. PMID:15334079 [DOI] [PubMed] [Google Scholar]
- 46.Shrikant P, Mescher MF. Opposing effects of IL-2 in tumor immunotherapy: Promoting CD8 T cell growth and inducing apoptosis. J Immunol. 2002;169:1753–9. doi: 10.4049/jimmunol.169.4.1753. PMID:12165496 [DOI] [PubMed] [Google Scholar]
- 47.Arneja A, Johnson H, Gabrovsek L, Lauffenburger DA, White FM. Qualitatively different T cell phenotypic responses to IL-2 versus IL-15 are unified by identical dependences on receptor signal strength and duration. J Immunol. 2014;192:123–35. doi: 10.4049/jimmunol.1302291. PMID:24298013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ring AM, Lin JX, Feng D, Mitra S, Rickert M, Bowman GR, Pande VS, Li P, Moraga I, Spolski R, et al.. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat Immunol. 2012;13:1187–95. doi: 10.1038/ni.2449. PMID:23104097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bezbradica JS, Rosenstein RK, Demarco RA, Brodsky I, Medzhitov R. A role for the ITAM signaling module in specifying cytokine-receptor functions. Nat Immunol. 2014;15:333–42. doi: 10.1038/ni.2845. PMID:24608040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, et al.. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201:139–48. doi: 10.1084/jem.20041057. PMID:15630141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamane H, Paul WE. Cytokines of the gamma(c) family control CD4+ T cell differentiation and function. Nat Immunol. 2012;13:1037–44. doi: 10.1038/ni.2431. PMID:23080204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wan YY, Flavell RA. The roles for cytokines in the generation and maintenance of regulatory T cells. Immunol Rev. 2006;212:114–30. doi: 10.1111/j.0105-2896.2006.00407.x. PMID:16903910 [DOI] [PubMed] [Google Scholar]
- 53.Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, Sander C, Yin Y, Holtzman M, Johnson J, et al.. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One. 2014;9:e87705. doi: 10.1371/journal.pone.0087705. PMID:24498358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, Sazegar H, Seja E, Villanueva A, Gomez-Navarro J, et al.. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res. 2011;17:4101–9. doi: 10.1158/1078-0432.CCR-11-0407. PMID:21558401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJ, van der Burg S, Kapiteijn E, Michielin O, et al.. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6:254ra128. doi: 10.1126/scitranslmed.3008918. PMID:25232180 [DOI] [PubMed] [Google Scholar]
- 56.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al.. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. PMID:24590637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al.. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. doi: 10.1056/NEJMoa1414428. PMID:25891304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, et al.. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31:e439–42. doi: 10.1200/JCO.2012.47.7521. PMID:24043743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al.. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81. doi: 10.1038/nature13988. PMID:25428507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balch CM, Gershenwald JE, Soong SJ, Thompson JF. Update on the melanoma staging system: The importance of sentinel node staging and primary tumor mitotic rate. J Surg Oncol. 2011;104:379–85. doi: 10.1002/jso.21876. PMID:21858832 [DOI] [PubMed] [Google Scholar]
- 61.Mitchell MS, Kan-Mitchell J, Kempf RA, Harel W, Shau HY, Lind S. Active specific immunotherapy for melanoma: Phase I trial of allogeneic lysates and a novel adjuvant. Cancer Res. 1988;48:5883–93. PMID:3262416 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.