

Abstract
Polymers prepared by chemical vapor deposition (CVD) polymerization have found broad acceptance in research and industrial applications. However, their intrinsic lack of degradability has limited wider applicability in many areas, such as biomedical devices or regenerative medicine. Herein, we demonstrate, for the first time, a backbone-degradable polymer directly synthesized via CVD. The CVD co-polymerization of [2.2]para-cyclophanes with cyclic ketene acetals, specifically 5,6-benzo-2-methylene-1,3-dioxepane (BMDO), results in well-defined, hydrolytically degradable polymers, as confirmed by FTIR spectroscopy and ellipsometry. The degradation kinetics are dependent on the ratio of ketene acetals to [2.2]para-cyclophanes as well as the hydrophobicity of the films. These coatings address an unmet need in the biomedical polymer field, as they provide access to a wide range of reactive polymer coatings that combine interfacial multifunctionality with degradability.
Keywords: CVD polymerization, cyclic ketene acetals, free-radical ring-opening polymerization, functional polymers, hydrolytic degradation
Breaking back
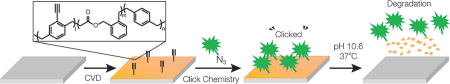
A new class of backbone-degradable polymer coatings was developed via chemical vapor deposition co-polymerization. These polymer coatings address a significant unmet need in the biomedical polymer field, as they provide access to a wide range of reactive polymer coatings that combine interfacial multifunctionality with degradability.
The medical field has increasingly witnessed a shift from permanent implant materials to biodegradable materials that degrade after their intended use.[1, 2] For instance, surgical sutures,[3] controlled drug delivery systems,[4] drug-eluding stent coatings,[5] or tissue engineering scaffolds[6] all benefit from degradable polymers. There exist numerous examples, where surface modification of biodegradable materials is required to introduce functional groups as anchor sites for biomolecule/drug conjugation.[7–9] To date, substrate-independent and widely applicable chemical vapor deposition (CVD) coatings are well established for non-degradable substrates (e.g., metals), but functional, degradable coatings remain elusive. For example, CVD polymerization of [2.2]para-cyclophanes can yield versatile poly(p-xylylene) coatings, which have been successfully applied for a wide range of permanently implanted devices (e.g., stents, pacemakers, or neural probes).[10] Some of these polymers are commercially available and the most widely used member of the family, also known as Parylene C, is an ISO 10993 and United States Pharmacopeia (USP) Class VI (highest biocompatibility class) material. Because of their unique processing through vapor phase polymerization, these polymers feature a range of advantages, such as low-temperature deposition, substrate-independency, absence of process solvents, high conformity, and excellent mechanical properties.[10, 11] CVD-based poly(p-xylylene)s have also been synthesized displaying a wide range of reactive side groups for efficient bioconjugation.[12, 13] While CVD polymers are widely used for functionalization of permanent implants and devices, they are intrinsically not degradable due to the absence of hydrolytically cleavable bonds in their backbone.[14]
Directly addressing this unmet need, we now report a novel class of functionalizable, and hydrolytically degradable polymer coatings made by chemical vapor deposition polymerization. Specifically, we use CVD co-polymerization to prepare degradable co-polymers displaying no functional group (co-polymer 2), hydroxy groups (co-polymer 1), and alkyne groups (co-polymer 3) for further surface modification. Co-polymerization of functionalized [2.2]para-cyclophanes with cyclic ketene acetal (CKA) molecules results in degradable ester linkages inserted into the all-carbon-based poly(p-xylylene) backbone. CKAs fulfill two critical criteria in this context: 1) CKAs polymerize following a radical polymerization mechanism compatible with the CVD polymerization process, while undergoing a rearrangement that can insert ester bonds in the polymer backbone.[15–17] 2) CKAs can sublime under the typical conditions required for CVD polymerization of [2.2]para-cyclophanes.
While a range of different CKAs preferentially undergo ring-opening polymerization,[15] we focused on 5,6-benzo-2-methylene-1,3-dioxepane (BDMO), which was synthesized following a slightly modified literature procedure.[18–20] BMDO features a seven-membered cyclic ketene acetal ring, which has previously been shown to undergo quantitative rearrangement in solution-based reactions.[18, 21] The radical that is generated after rearrangement of BMDO is stabilized by the adjacent benzene ring (Scheme 1), which makes it particularly suitable for CVD polymerization.
Scheme 1.

Proposed mechanism of the CVD synthesis of backbonedegradable polymers. BMDO, a cyclic ketene acetal, was co-polymerized with radicals generated by the pyrolysis of [2.2]para-cyclophanes. BMDO polymerized following a ring-opening radical polymerization mechanism, while undergoing a rearrangement into a polyester.
For CVD co-polymerization, BMDO and [2.2]para-cyclophanes, which act as the radical initiators, were sublimed at 0.07 torr and temperatures above 100°C and transferred in a stream of argon carrier gas into the pyrolysis zone, which was maintained at a temperature of 530°C. After formation of the active intermediates (Scheme 1), the vapor was transferred into the deposition chamber, with the chamber wall temperature set to 120°C and the holder cooled to 15 °C to optimize the deposition. Under these conditions, BMDO underwent molecular rearrangement followed by subsequent co-polymerization with the xylylene moieties. The co-polymerization proceeded with a growth rate of 0.1–0.2 Å s−1 and resulted in well-defined polymers displaying ester bonds in their polymer backbone.
Using this approach, we co-polymerized [2.2]para-cyclophane and BMDO at a molar ratio of 3:5. The resulting co-polymer 2 film was insoluble in common organic solvents, such as acetone, ethanol, or isopropanol. The Fourier transform infrared spectroscopy (FTIR) spectrum of the polymer (Supporting Information) shows a strong band at 1784 cm−1, which is indicative of ester groups. The CVD-based co-polymer 2 degraded in 5 mm KOH/isopropanol solution at room temperature within 12 days, as confirmed by a successive loss of characteristic bands in the FTIR spectra and a continuous decrease in film thickness, as measured by ellipsometry (Figure S2c in the Supporting Information). In addition, the co-polymer film showed slow degradation in an aqueous bicarbonate buffer: At 37 °C, the film thickness of the polymer decreased by 11% after two months. From these results, we concluded that the ester bonds were indeed hydrolytically degradable, albeit the degradation proceeded relatively slowly. Apparently, the hydrophobicity of the polymer films prevented water penetration and slowed down the ester hydrolysis.
To accelerate the degradation in aqueous solution and incorporate functional groups for further surface modification, [2.2]para-cyclophane was replaced with the more polar 4-hydroxymethyl-[2.2]para-cyclophane. Co-polymerization with BMDO (Scheme 2) resulted in polymer films, which featured interfacial hydroxy groups and showed increased hydrophilicity[22] compared to the earlier coatings.
Scheme 2.

Synthesis of co-polymer 1 via CVD polymerization by feeding 1:15 molar ratio of 4-hydroxymethyl[2.2]para-cyclophane (PCP-CH2OH) and BMDO.
As described above, xylylene radicals generated by the pyrolysis of 4-hydroxymethyl[2.2]para-cyclophane initiated the co-polymerization with BMDO to synthesize co-polymer 1. The resulting polymer films were characterized by a combination of surface-sensitive methods, including grazing angle Fourier-transformed infrared reflection absorption spectroscopy (IRRAS), and X-ray photoelectron spectroscopy (XPS). The FTIR spectrum of co-polymer 1 confirmed the existence of ester groups as indicated by a strong band at 1782 cm−1 (Figure 1 a). Moreover, a broad band at 3446 cm−1 confirmed the presence of hydroxy groups in the polymer film. XPS analysis further confirmed the chemical structure of co-polymer 1 (Table 1). The XPS survey spectrum reveals 16.4% oxygen and 83.6% carbon, which compares well to 15.7%oxygen and 84.3%carbon that can be calculated based on the structure of the monomers assuming a monomer feed ratio of 1:15 (PCP-CH2OH: BMDO). The high-resolution C1s-spectrum indicates the presence of carbon in different chemical states (59.5%C–C/C–H, 6.6%C–C=O, 9.7%C–O, 6.5% O–C < C = > O), which is in good agreement with the theoretically calculated values.
Figure 1.

Polymer characterization: a) FTIR spectrum of co-polymer 1; b) TGA traces of the [2.2]para-cyclophane and co-polymer 1 are compared to poly[(hydroxymethyl-p-xylylene)-co-(p-xylylene)] (PPX-CH2OH).
Table 1.
XPS analysis results of co-polymer 1. Theoretical values were calculated from the chemical structure of the monomers of co-polymer 1 assuming a ratio of l:m:n = 1:15:1 (i.e., the precursor feed ratio).
| BE [eV][a] | Theoretical [%][b] | Experimental [%][b] | |
|---|---|---|---|
| C–C/C–H | 285 | 61 | 59.5 |
| C–C=O | 285.7 | 7.6 | 6.6 |
| C–O | 286.7 | 8.1 | 9.7 |
| O–C=O | 289.3 | 7.6 | 6.5 |
| π→π* | 291.5 | – | 1.3 |
| O | 533 | 15.7 | 16.4 |
Binding Energy.
Atomic percent.
In addition to the chemical analysis, thermogravimetric analysis (TGA) was performed for co-polymer 1 to confirm successful polymerization (Figure 1 b). The CVD polymer films were compared to the respective monomers used for the CVD polymerization as well as a non-degradable poly[(hydroxymethyl-p-xylylene)-co-(p-xylylene)] (PPX-CH2OH) film (Figure S5), which was also prepared by CVD polymerization. The results show that the different precursors and polymers have distinctly different TGA traces. The volatile BMDO monomer shows a two-step degradation with major weight loss in the temperature range from 67 °C to 151°C and a second step of about 7%weight loss at a higher temperature range (190 °C to 220°C), which is most likely due to a small portion of BMDO that underwent side reactions during storage. We extrapolated onset temperatures in nitrogen of 110 °C, 213°C, 221 °C and 190 °C for BMDO, PCP-CH2OH, PPX-CH2OH and co-polymer 1, respectively. Co-polymer 1 shows a multi-step degradation, which is different from the degradation of the corresponding monomers. Its thermal stability is higher than the stability of the BMDO monomer, but lower than the thermal stability of PCP-CH2OH. In contrast, PPX-CH2OH follows a two-step degradation mechanism. The first infliction point occurs at 209°C and may be due to loss of hydroxy groups, whereas the second step occurs at 470 °C and can be explained by the thermal decomposition of the aromatic ring system. When heated up to 750°C in nitrogen atmosphere, about 16%of carbon residues were still present, while no carbon residues were detected for co-polymer 1 under the same experimental conditions. The thermal stability of co-polymer 1 and PPX-CH2OH is comparable, which confirms the potential for implementation of the hydrolytically degradable co-polymer 1 in various coating applications.
After verification of the chemical composition of co-polymer 1, its degradation behavior was studied at 37 °C in an aqueous sodium carbonate buffer at a pH-value of 10.6 (Figure 2). From the FTIR spectra shown in Figure 2a, we confirmed decreasing intensities of vibrational bands characteristic of ester and hydroxy groups at 1782 and 3446 cm−1. In parallel, the thickness of the co-polymer 1 film (Figure 2b) continuously decreased over time, while the refractive index remained relatively constant around 1.5. Both, FTIR and ellipsometry data, are consistent with a surface erosion mechanism, where the ester groups of the topmost polymer chains are hydrolyzed first, which leads to the successive erosion of the following layers. The surface erosion process of co-polymer 1 appears to be controlled in an aqueous buffer and appears to be similar to the degradation of the polymer of non-functionalized [2.2]para-cyclophane and BMDO (co-polymer 2) that occurred in the 5 mm KOH/isopropanol solution (Supporting Information).
Figure 2.

Polymer degradation: a) FTIR spectra of co-polymer 1 degrading over time. Co-polymer 1 was degraded in a sodium carbonate-sodium bicarbonate aqueous buffer solution with pH value of 10.6 at 37°C; b) thicknesses of co-polymer 1 degrading over time measured by ellipsometry. Co-polymer 2 (co-polymer of [2.2]para-cyclophane and BMDO) degrading in the same aqueous buffer solution (pH 10.6) is shown for the purpose of comparison; c) expanded ESI-mass spectra of the degradation products of co-polymer 1 after totally degradation (in the mass range m/z 200–500) with identified fragments.
We note that surface erosion may be more desirable than bulk erosion for some applications like drug delivery, because it can lead to more predictable release kinetics.[23, 24] As shown in Figure 2 a,b, co-polymer 1 is completely degraded after 80 days, with 7.3%of the polymer film being degraded within the first 20 days. After degradation, we extracted the degradation products and analyzed them by positive electrospray ionization (ESI) mass spectrometry.[25] Among the degradation products were small molecule fragments with a mass below 1300 m/z. Some of the prominent fragments had mass-to-charge ratios (m/z) of 267.2 and 409.2 (Figure 2 c). The fragmentation patterns support the assumption of ester bond cleavages further suggesting successive cleavage from both ends of the polymer chains. The fragment at m/z 267.2 corresponds to the terminated carboxylate group and the fragmentation of the ion at m/z 409.2 can be assigned to a product that is terminated by hydroxy and carboxy end groups. Based on this analysis, we concluded that the polymer formed by the CVD polymerization process has a random co-polymer structure. In the past, soluble PPX-films bearing alkyl substituents were characterized using GPC and NMR techniques.[26] The published results clearly confirmed the formation of linear CVD polymers. However, this approach requires the deposition of large polymer quantities, provided that the co-polymers are sufficiently soluble in organic solvents suitable for NMR and GPC. A similar approach was not possible in this case. Instead, we base the preposition of linear co-polymers on the fragmentation patterns obtained by mass spectrometry. While preliminary in nature, these findings are consistent with the earlier findings by Greiner et al. for similar polymer films obtained by NMR spectroscopy and GPC analysis.[26]
To assess the short-term cytotoxicity of these novel polymer coatings, we studied the cell viability of fibroblasts in direct contact with the polymers. Cell growth and confluency were examined under a phase contrast microscope (Supporting Information). As a negative control, a poly(vinyl chloride) containing organo–tin compound (2 wt.% dibutyltin maleate) was included and designed as Ot-PVC. The latter has been used as negative control to generate reproducible cytotoxic responses.[27] Non-coated TCPS (tissue culture polystyrene) was used as positive control. To quantify cell proliferation in response to exposure to the different surfaces, we employed a XTT assay, which is a commonly used colorimetric assay for detecting cell metabolic activities using cells in exponential growth phase.[28]
NIH3T3 fibroblasts were seeded on films comprised of either PPX-CH2OH, co-polymer 1 or partially degraded co-polymer 1. These samples were compared to TCPS and Ot-PVC films. Figure 3 indicates increased spreading of actin filaments after cells were seeded on co-polymer 1 as well as partially degraded co-polymer 1, which are similar to those cultured on TCPS. No statistical differences were observed in the mitochondrial activity of cells seeded on the positive control or either of the CVD polymer coated samples, indicating that the CVD polymer surfaces were non-cytotoxic under these conditions. In contrast, the mitochondrial activity of cells seeded on the negative control was below the detectable limits of the assay. This is consistent with our imaging results, where cells seeded on co-polymer 1 did not exhibit signs of short-term toxicity, neither before nor after degradation.
Figure 3.

Cell viability test of co-polymer 1 before and after degradation. The results from XTT assay are presented as cell viability normalized by positive control, ±the standard deviation on different substrates. The experiments were carried out in triplicates.
To demonstrate surface immobilization onto simultaneously functionalized and degradable CVD coatings, we conducted co-polymerization of alkyne-functionalized 4-ethynyl[2.2]para-cyclophane[30, 31] with BMDO resulting in the novel co-polymer 3 (Figure 4a). Experimentally, the CVD co-polymerization followed the method previously described for the synthesis of co-polymer 1. The FTIR spectrum of co-polymer 3 (Figure 4b) reveals characteristic bands of the ester groups (1780 cm−1) as well as the terminal alkyne groups at 3300 cm−1 and 2100 cm−1, respectively. The FTIR data are further confirmed by the XPS results (Table S1).
Figure 4.

CVD co-polymerization of BMDO and 4-ethynyl[2.2]para-cyclophane: a) chemical structure of the biodegradable co-polymer 3 (polymer of 4-ethynyl[2.2]para-cyclophane and BMDO with the precursor feed ratio 1:5); b) FTIR spectrum of co-polymer 3; c) fluorescence micrograph after microcontact printing (µCP) Alexa Fluor488 azide on the co-polymer 3 surface; d) fluorescence micrograph after µCP biotin-PEG3-azide and streptavidin-Cy3 immobilization.
The chemical reactivity of the alkyne-functionalized surfaces was confirmed by copper-catalyzed azide-alkyne cycloaddition (CuAAC), a well-established “click” reaction.[29] Specifically, the surface of co-polymer 3 was patterned via spatially controlled click reaction using microcontact printing (mCP). Microcontact printing is a commonly used method to generate micron-scale patterns[32, 33] and has been used in the past for spatially controlled immobilization of biomolecules onto functionalized poly(p-xylylene)s.[13]
Immobilization of Alexa Fluor488 azide (Figure 4c) was achieved using a microstructured PDMS stamp that was inked with a solution of CuSO4 and brought into contact with the polymer substrate. Next, the covalent binding of a biotin-PEG3-azide followed by streptavidin-Cy3 was used for visualization of the selective modification (Figure 4d). Figures 4c,d and verify the specific reactivity towards the alkyne groups presented on the copolymer surface.
In summary, we have demonstrated the successful synthesis of a novel class of backbone-degradable CVD polymer thin films, which combine the attractive characteristics of both, degradability and chemical functionality for subsequent surface modification. The synthesis was achieved by polymerization of a cyclic ketene acetal, BMDO, with functionalized [2.2]para-cyclophanes via CVD polymerization. The co-polymers were hydrolytically degraded and show no obvious short-term cytotoxicity in a XTT assay. In addition, both, the functionality and the precursor feed ratios, can be controlled to create different combinations of interfacial and bulk properties. This new class of degradable functional thin films has the potential to serve as a widely applicable surface functionalization and coating platform for a broad spectrum of technologies ranging from the life sciences, and medicine to food packaging applications.
Supplementary Material
Acknowledgments
We acknowledge support from the German Science Foundation under the SFB grant 1176. We further acknowledge support the Army Research Office (ARO) under Grant W911NF-11-1-0251.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.201609307.
Contributor Information
Fan Xie, Biointerfaces Institute and Departments of Biomedical Engineering and Chemical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109 (USA); Department of Applied Chemistry, School of Science, Northwestern Polytechnical University, Xi’an 710072 (China).
Dr. Xiaopei Deng, Biointerfaces Institute and Departments of Biomedical Engineering and Chemical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109 (USA).
Dr. Domenic Kratzer, Institute of Functional Interfaces, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldsshafen (Germany)
Kenneth C. K. Cheng, Biointerfaces Institute and Departments of Biomedical Engineering and Chemical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109 (USA)
Dr. Christian Friedmann, Institute of Functional Interfaces, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldsshafen (Germany)
Dr. Shuhua Qi, Department of Applied Chemistry, School of Science, Northwestern Polytechnical University, Xi’an 710072 (China)
Dr. Luis Solorio, Biointerfaces Institute and Departments of Biomedical Engineering and Chemical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109 (USA)
Prof. Dr. Joerg Lahann, Biointerfaces Institute and Departments of Biomedical Engineering and Chemical Engineering, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109 (USA) Institute of Functional Interfaces, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldsshafen (Germany).
References
- 1.Grafahrend D, Heffels K-H, Beer MV, Gasteier P, Mçller M, Boehm G, Dalton PD, Groll J. Nat. Mater. 2011;10:67–73. doi: 10.1038/nmat2904. [DOI] [PubMed] [Google Scholar]
- 2.Jeong B, Bae YH, Lee DS, Kim SW. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 3.Vert M. Biomacromolecules. 2005;6:538–546. doi: 10.1021/bm0494702. [DOI] [PubMed] [Google Scholar]
- 4.Jin Q, Maji S, Agarwal S. Polym. Chem. 2012;3:2785–2793. [Google Scholar]
- 5.Takahashi A, Palmer-Opolski M, Smith RC, Walsh K. Gene Ther. 2003;10:1471–1478. doi: 10.1038/sj.gt.3302010. [DOI] [PubMed] [Google Scholar]
- 6.Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biomaterials. 2006;27:3413–3431. doi: 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 7.Kim TG, Park TG. Biotechnol. Prog. 2006;22:1108–1113. doi: 10.1021/bp060039t. [DOI] [PubMed] [Google Scholar]
- 8.Lim Y-b, Kim S-M, Lee Y, Lee W-k, Yang T-g, Lee M-j, Suh H, Park J-S. J. Am. Chem. Soc. 2001;123:2460–2461. doi: 10.1021/ja005715g. [DOI] [PubMed] [Google Scholar]
- 9.Murphy WL, Mooney DJ. J. Am. Chem. Soc. 2002;124:1910–1917. doi: 10.1021/ja012433n. [DOI] [PubMed] [Google Scholar]
- 10.Hsu JM, Rieth L, Normann RA, Tathireddy P, Solzbacher F. IEEE Trans. Biomed. Eng. 2009;56:23–29. doi: 10.1109/TBME.2008.2002155. [DOI] [PubMed] [Google Scholar]
- 11.Rodger DC, Fong AJ, Li W, Ameri H, Ahuja AK, Gutierrez C, Lavrov I, Zhong H, Menon PR, Meng E, Burdick JW, Roy RR, Edgerton VR, Weiland JD, Humayun MS, Tai Y-C. Sens. Actuators B. 2008;132:449–460. [Google Scholar]
- 12.Deng X, Lahann J. J. Appl. Polym. Sci. 2014;131:1–9. [Google Scholar]
- 13.Chen H-Y, Lahann J. Langmuir. 2010;27:34–48. doi: 10.1021/la101623n. [DOI] [PubMed] [Google Scholar]
- 14.Tian H, Tang Z, Zhuang X, Chen X, Jing X. Prog. Polym. Sci. 2012;37:237–280. [Google Scholar]
- 15.Agarwal S. Polym. Chem. 2010;1:953–964. [Google Scholar]
- 16.Hiraguri Y, Tokiwa Y. J. Polym. Environ. 2010;18:116–121. [Google Scholar]
- 17.Sanda F, Endo T. J. Polym. Sci. Part A. 2001;39:265–276. [Google Scholar]
- 18.Bailey WJ, Ni Z, Wu SR. Macromolecules. 1982;15:711–714. [Google Scholar]
- 19.Wickel H, Agarwal S. Macromolecules. 2003;36:6152–6159. [Google Scholar]
- 20.d’Ayala GG, Malinconico M, Laurienzo P, Tardy A, Guillaneuf Y, Lansalot M, D’Agosto F, Charleux B. J. Polym. Sci. Part A. 2014;52:104–111. [Google Scholar]
- 21.Bailey WJ, Ni Z, Wu S-R. J. Polym. Sci. Polym. Chem. Ed. 1982;20:3021–3030. [Google Scholar]
- 22.Lahann J, Klee D, Pluester W, Hoecker H. Biomaterials. 2001;22:817–826. doi: 10.1016/s0142-9612(00)00244-1. [DOI] [PubMed] [Google Scholar]
- 23.Gçpferich A. Biomaterials. 1996;17:103–114. doi: 10.1016/0142-9612(96)85755-3. [DOI] [PubMed] [Google Scholar]
- 24.von Burkersroda F, Schedl L, Gçpferich A. Biomaterials. 2002;23:4221–4231. doi: 10.1016/s0142-9612(02)00170-9. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee S, Mazumdar S. Int. J. Anal. Chem. 2012;40 doi: 10.1155/2012/282574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bier AK, Bognitzki M, Mogk J, Greiner A. Macromolecules. 2012;45:1151–1157. [Google Scholar]
- 27.Schutte RJ, Xie L, Klitzman B, Reichert WM. Biomaterials. 2009;30:160–168. doi: 10.1016/j.biomaterials.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. J. Immunol. Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- 29.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001;113:2056–2075. [Google Scholar]
- 30.Nandivada H, Chen H-Y, Bondarenko L, Lahann J. Angew. Chem. Int. Ed. 2006;45:3360–3363. doi: 10.1002/anie.200600357. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006;118:3438–3441. [Google Scholar]
- 31.Deng X, Friedmann C, Lahann J. Angew. Chem. Int. Ed. 2011;50:6522–6526. doi: 10.1002/anie.201101581. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123:6652–6656. [Google Scholar]
- 32.Wilbur JL, Kumar A, Kim E, Whitesides GM. Adv. Mater. 1994;6:600–604. [Google Scholar]
- 33.Chen HY, Lahann J. Adv. Mater. 2007;19:3801–3808. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.