

Abstract
Chronic kidney disease is a major cause of death and renal fibrosis is a common pathway leading to the progression of this disease. Although activated fibroblasts are responsible for the production of the extracellular matrix and the development of renal fibrosis, the molecular mechanisms underlying fibroblast activation are not fully defined. Here we examined the functional role of AMP-activated protein kinase (AMPK) in the activation of fibroblasts and the development of renal fibrosis. AMPKα1 was induced in the kidney during the development of renal fibrosis. Mice with global or fibroblast-specific knockout of AMPKα1 exhibited fewer myofibroblasts, developed less fibrosis, and produced less extracellular matrix protein in the kidneys following unilateral ureteral obstruction or ischemia-reperfusion injury. Mechanistically, AMPKα1 directly phosphorylated cofilin leading to cytoskeleton remodeling and myocardin-related transcription factor-A nuclear translocation resulting in fibroblast activation and extracellular matrix protein production. Thus, AMPK may be a critical regulator of fibroblast activation through regulation of cytoskeleton dynamics and myocardin-related transcription factor-A nuclear translocation. Hence, AMPK signaling may represent a novel therapeutic target for fibrotic kidney disease.
Keywords: AMPK, fibrosis, fibroblast, chronic kidney disease, cofilin, MRTF-A
Introduction
Disease-related injury in organs triggers cellular and molecular responses that can culminate in tissue fibrosis. The dynamic deposition and insufficient resorption of extracellular matrix (ECM) promotes fibrosis and chronic loss of organ function1, which account for an estimated one third of natural deaths worldwide2. As a major fibrotic disorder, renal fibrosis is a hallmark of chronic kidney disease 3,4. Renal interstitial fibrosis is characterized by fibroblast activation and excessive production and deposition of ECM, which leads to the destruction of renal parenchyma and progressive loss of kidney function to end-stage renal disease 5,6.
Activated fibroblasts are the principal cells responsible for ECM production, and their activation is regarded as a key event in the pathogenesis of renal fibrosis 6-8. These cells express α-smooth muscle actin (α-SMA), and are referred to as myofibroblasts. However, the molecular mechanisms underlying fibroblast activation are not fully understood.
Adenosine monophosphate-activated protein kinase (AMPK) is a conserved sensor of cellular energy status and environmental stress, and regulates the activities of a number of enzymes through phosphorylation. Mammalian AMPKs are heterotrimeric complexes containing a catalytic (α) subunit and two regulatory (β and γ) subunits 9, 10. In addition to regulating energy homeostasis and metabolism, AMPK plays important roles in protein synthesis, cell growth, apoptosis 11, cell differentiation 12, actin cytoskeleton reorganization , and gene transcription 14. Recent studies have shown that AMPKα1 participates in the development of renal fibrosis 15, 16. However, how AMPK regulates the development of renal fibrosis is unknown.
To examine the functional role of AMPKα1 in the activation of fibroblasts and the development of renal fibrosis, we generated mice expressing tamoxifen-inducible Crerecombinase (Cre/ESR1) under the control of the chicken beta actin promoter/enhancer coupled with the cytomegalovirus immediate-early enhancer (CAG-Cre) 17 or proα2(I) collagen promoter (Col1-Cre) 18, thus permitting temporally controlled deletion of floxed AMPKα1 gene in a global or fibroblast-specific manner. These transgenic mice were subjected to unilateral ureteral obstruction (UUO) or ischemia-reperfusion injury (IRI) to induce renal fibrosis. We show that AMPKα1 is induced in the kidney during the development of renal fibrosis, and targeted disruption of AMPKα1 inhibits fibroblast activation and attenuates the development of renal fibrosis. Furthermore, we demonstrate that AMPKα1 directly phosphorylates cofilin, which results in actin cytoskeleton reorganization and nuclear translocation of myocardin-related transcription factor-A (MRTF-A; also known as MKL1, MAL, BSAC) leading to fibroblast activation and fibrogenesis.
Results
AMPKα1 is induced during the development of renal fibrosis
We first determined whether AMPKα1 is induced in the kidneys in response to obstructive injury. Western blot analysis showed that the protein levels of AMPKα1 were markedly increased in kidneys after 10 days of UUO compared with the contralateral kidneys (Fig. S1). To characterize the cell types responsible for AMPKα1 induction in the kidney, serial kidney sections were stained with an anti-AMPKα1 antibody by immunohistochemistry. The results revealed that AMPKα1-positive cells increased in the tubulointerstitium of the fibrotic kidneys (Fig. S2). To examine if AMPKα is activated in myofibroblasts of the kidney, kidney sections were stained for p-AMPKα and α-SMA, a myofibroblast marker, by immunofluorescence (Fig. S3). The immunofluorescence results demonstrated that p-AMPKα positive cells are positive for α-SMA indicating that AMPKα is mainly activated in myofibroblasts of the kidney in response to obstructive injury.
Global deletion of AMPKα1 reduces renal fibrosis
To examine whether AMPKα1 regulates renal fibrosis in vivo, mice with loxP-flanked alleles of AMPKα1 were bred with mice harboring a hemizygous CAG-Cre-ER transgene 17. CAG-Cre+AMPKα1flox/flox mice were referred to as GAKO mice and littermate CAG-Cre-AMPKα1flox/flox mice served as controls (Fig. S4). These mice were treated tamoxifen and then subjected to UUO for 10 days. Western blot analysis showed that protein levels AMPKα1 and p-AMPKα were markedly increased in the obstructed kidneys of control mice, which was significantly reduced in the kidneys of GAKO mice with UUO (Fig. 1A). GAKO mice displayed much less collagen deposition in the obstructed kidneys compared with those of their littermate controls (Fig. 1B). We next investigated the effect of AMPKα1 deficiency on the expression of α-SMA, a myofibroblast marker, collagen I and fibronectin, two major ECM proteins. GAKO mice expressed considerably less α-SMA, collagen I, fibronectin in the obstructed kidneys compared with their littermate controls (Fig. 1, C-F). These data indicate that AMPKα1 is indispensable for fibroblast activation and development of interstitial fibrosis induced by UUO.
Fig. 1. Global AMPKαl deficiency attenuates renal fibrosis induced by UUO.
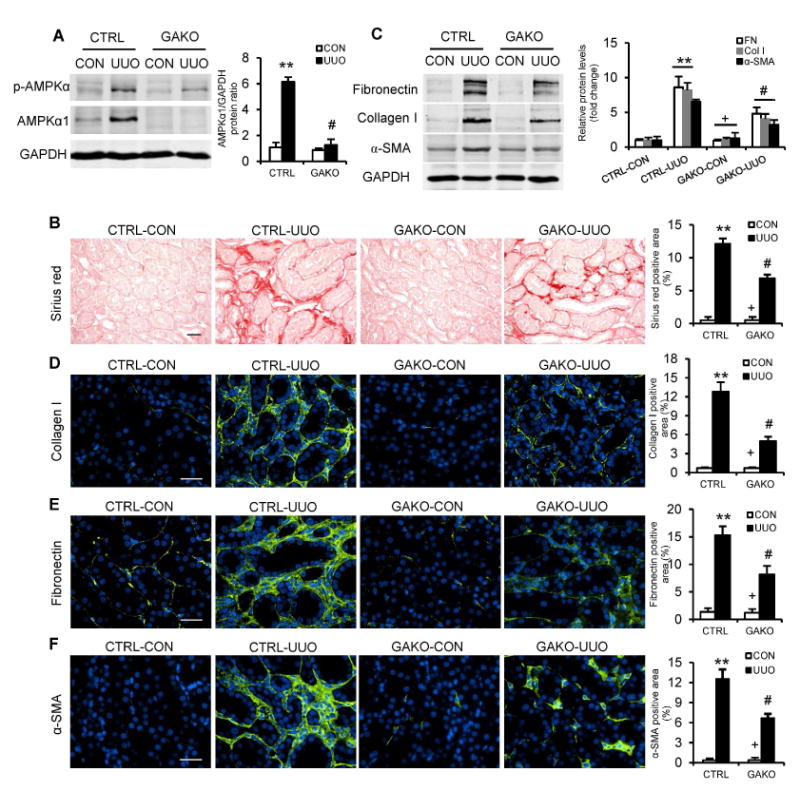
(A) Representative Western blots show the protein levels of AMPKαl and p-AMPKα in the contralateral (CON) and UUO kidneys of GAKO mice and their littermate controls (CTRL). Protein expression was normalized with GADPH. (B) Representative photomicrographs show kidney sections stained with Sirius red for total collagen deposition. Scale bar: 20 μm. The bar graph shows quantitative analysis of renal interstitial collagen content in different groups. (C) Representative Western blots show protein levels of fibronectin (FN), collagen I (Col I), and α-smooth muscle actin (α-SMA) in the kidneys. Representative photomicrographs of the kidney sections stained for collagen I (D), fibronectin (E) and α-SMA (F), and counterstained with DAPI (blue). Scale bar: 30 μm. Bar graphs show quantitative analysis of protein immunostaining in the kidney sections. **P<0.01 compared with the CTRL-CON group, #P<0.05 compared with the CTRL-UUO group, +P<0.05 compared with the GAKO-UUO group. n=6 mice per group.
To further confirm the role of the AMPKα1 in the development of renal fibrosis, we extended our studies to an additional model of renal fibrosis, IRI-induced nephropathy. Western blot analysis showed that protein levels AMPKα1 and p-AMPKα were significantly increased in the kidneys of control mice with IRI, which was substantially reduced in the kidneys of GAKO mice with IRI (Fig. 2A). Global deletion of AMPKα1 markedly reduced interstitial collagen deposition in the IRI-injured kidneys (Fig. 2B). Furthermore, global deletion of AMPKα1 significantly inhibited the expression of α-SMA, collagen I, and fibronectin in kidneys following IRI (Fig. 2, C-F). These data indicate that global deletion of AMPKα1 attenuates IRI-induced fibroblast activation and interstitial fibrosis in the kidney.
Fig. 2. Global AMPKαl deficiency diminishes renal fibrosis induced by IRI.
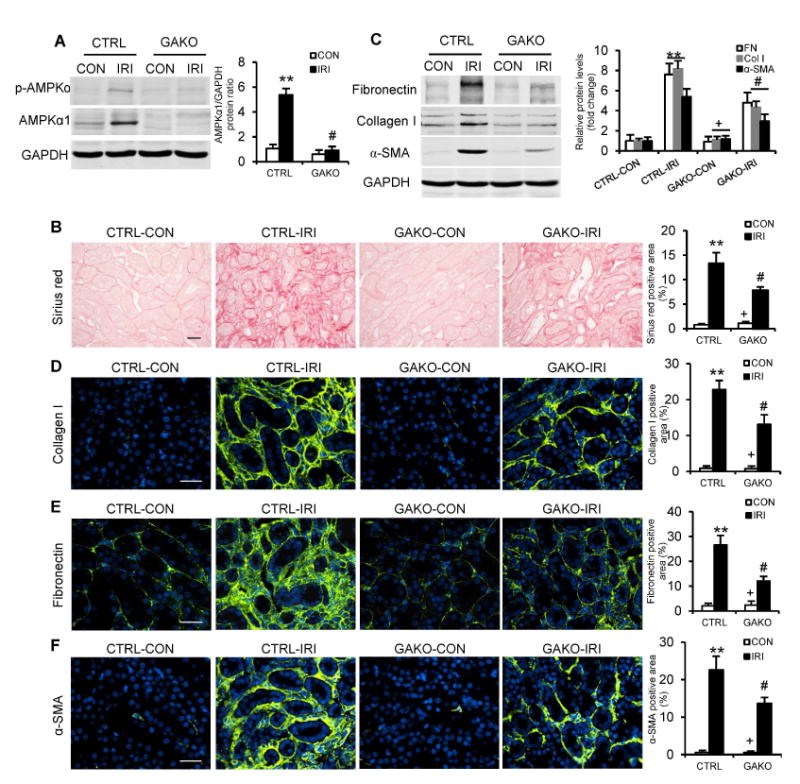
(A) Representative Western blots show the protein levels of AMPKαl and p-AMPKα in the contralateral (CON) and IRI kidneys of GAKO mice and their littermate controls (CTRL). (B) Representative photomicrographs show kidney sections stained with Sirius red for total collagen deposition. Scale bar: 20 μm. (C) Representative Western blots show the protein levels of fibronectin (FN), collagen I (Col I) and α-SMA in the kidneys. Representative photomicrographs of the kidney sections stained for collagen I (D), fibronectin (E) and a-SMA (F), and counterstained with DAPI (blue). Scale bar: 30 urn. **P<0.01 compared with the CTRL-CON group, #P<0.05 compared with the CTRL-IRI group, +P<0.05 compared with the GAKO-IRI group. n=6 mice per group.
Fibroblast-specific deletion of AMPKα1 reduces renal fibrosis
To determine whether the observed effect of AMPKα1 on renal fibrosis was mediated by AMPKα1 in fibroblasts, we generated mice with fibroblast-specific deletion of AMPKα1 by crossing AMPKα1flox/flox mice with tamoxifen-inducible collagen type I promoter/enhancer-driven Cre-ER recombinase transgenic mice (Col1-Cre+)18. AMPKα1fl/fl littermate controls and Col1-Cre+AMPKα1fl/fl mice (referred to as FAKO) were treated with tamoxifen to induce fibroblast-specific deletion of AMPKα1 (Fig. S4). These mice were then subjected to UUO or IRI to induce renal fibrosis. Western blot analysis showed AMPKα1 protein levels were markedly reduced in the kidneys of FAKO mice compared with those of littermate controls (Fig. 3A and 4A). Immunofluorescence staining revealed that AMPKα1 abundance was considerably reduced in the PDGFRβ-positive fibroblasts (Fig. 3C). We then examined the effect of AMPKα1 deficiency in fibroblasts on the development of renal fibrosis. Total collagen accumulation and deposition were significantly attenuated in the injured kidneys of FAKO mice compared with their littermate controls (Fig. 3D and 4B). Furthermore, both Western blot analysis and immunofluorescence staining demonstrated that fibroblast-specific AMPKα1 deficiency attenuated the upregulation of α-SMA, collagen I, and fibronectin in the kidneys with UUO (Fig. 3, E-H) or IRI (Fig. 4, C-F). Collectively, these data indicate that fibroblast-specific deletion of AMPKα1 attenuates renal fibrosis in UUO and IRI models by inhibiting fibroblast activation and ECM protein production.
Fig. 3. Fibroblast-specific depletion of AMPKαl reduces UUO-induced renal fibrosis.
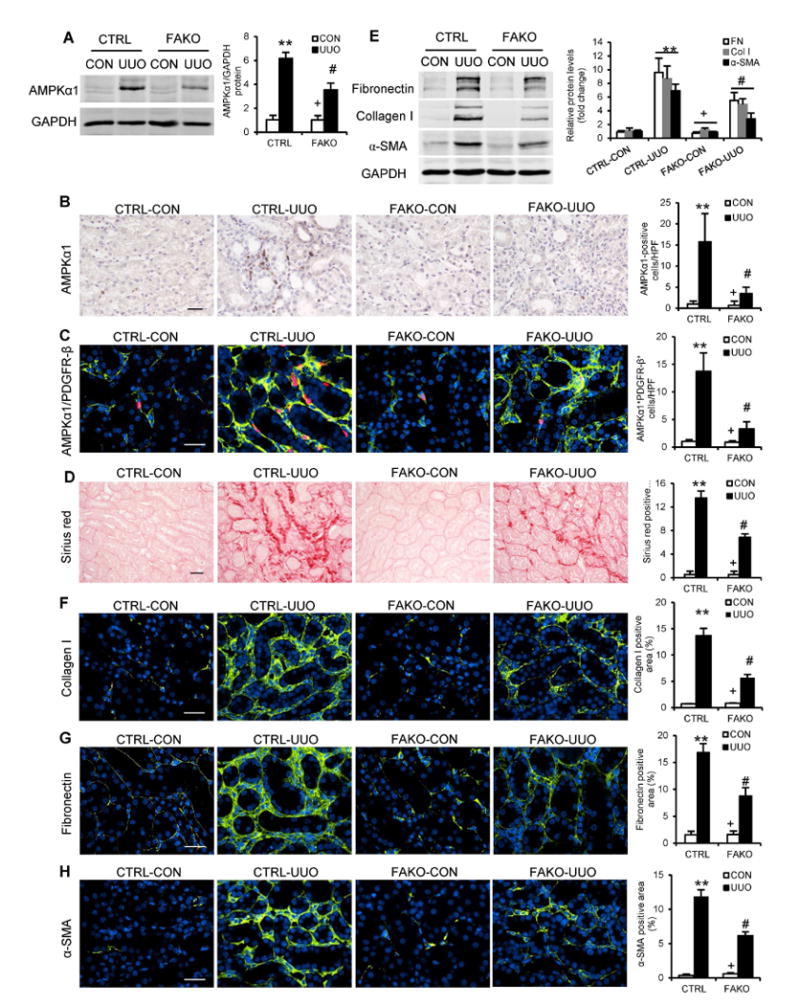
(A) Representative Western blots show the protein levels of AMPKαl in the contralateral control (CON) and UUO kidneys of FAKO mice and their littermate controls (CTRL). (B) Representative photomicrographs of kidney sections stained for AMPKαl (brown) and counterstained with hematoxylin (blue). Bar: 20 μm. (C) Representative photomicrographs of kidney sections stained for AMPKαl (red), PDGFR-β (green), and DAPI (blue). Bar: 30 μm. (D) Representative photomicrographs show kidney sections stained for total collagen deposition. Bar: 20 μm. (E) Representative Western blots show the protein levels of fibronectin (FN), collagen I (Col I) and α-SMA in the kidneys. Representative photomicrographs of the kidney sections stained for collagen I (F), fibronectin (G) and α-SMA (H), and counterstained with DAPI (blue). Bar: 30 μm. **P<0.01 compared with the CTRL-CON group, #P<0.05 compared with the CTRL-UUO group, +P<0.05 compared with the FAKO-UUO group. n=6 mice per group.
Fig. 4. Fibroblast-specific depletion of AMPKαl inhibits IRI-induced renal fibrosis.
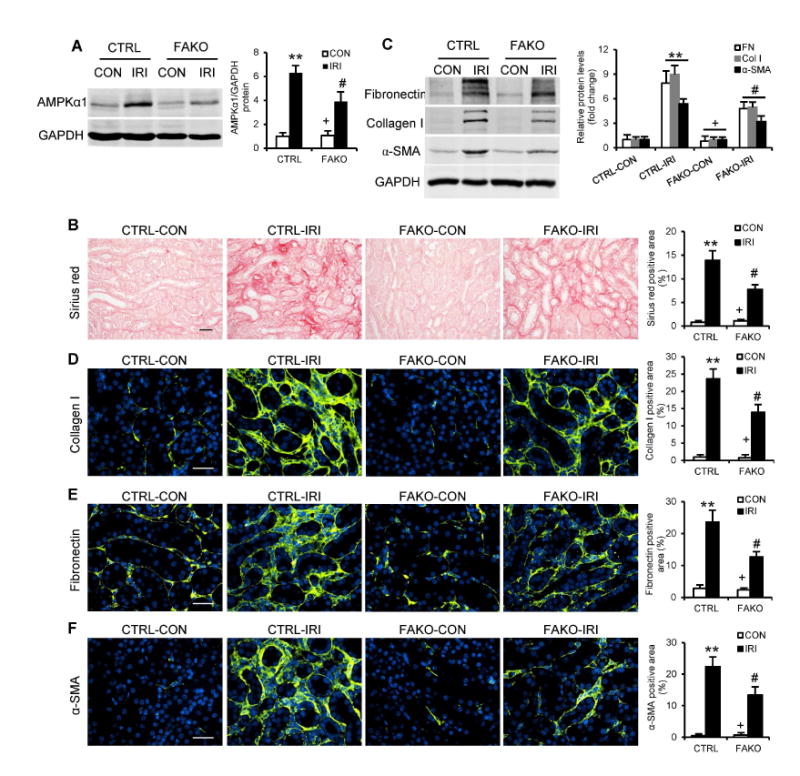
(A) Representative Western blots show the protein levels of AMPKαl in the contralateral (CON) and IRI kidneys of FAKO mice and their littermate controls (CTRL). (B) Representative photomicrographs show kidney sections stained with Sirius red for total collagen deposition. Scale bar: 20 μm. (C) Representative Western blots show the protein levels of fibronectin (FN), collagen I (Col I) and α-SMA in the kidneys. Representative photomicrographs of the kidney sections stained for collagen I (D), fibronectin (E) and α-SMA (F), and counterstained with DAPI (blue). Scale bar: 30 μm. **P<0.01 compared with the CTRL-CON group, #P<0.05 compared with the CTRL-IRI group, +P<0.05 compared with the FAKO-IRI group. n=6 mice per group.
AMPKα1 is required for fibroblast activation
To examine the role of AMPKα1 in activating kidney fibroblasts, the normal rat kidney fibroblasts (NRK-49F), primary kidney fibroblasts and bone marrow-derived monocytes were treated with a direct activator of AMPK, A-769662 10, for 24 hours. A-769662 induced fibroblast activation identified as increased production of α-SMA, fibronectin, and collagen I (Fig. 5, A-C). Compound C, a selective AMPK inhibitor 19, blocked A-769662-induced fibronectin, collagen I, and α-SMA production in NRK-49F cells (Fig. 5A). Furthermore, AMPKα1 deficiency abolished A-769662-induced fibronectin, collagen I, and α-SMA protein production in primary kidney fibroblasts and bone marrow-derived monocytes (Fig. 5, B and C, and Fig. S5). Bone marrow-derived monocytes were also treated with adiponectin, a cytokine that activates AMPK pathway 15, 20, 21. Western blot analysis showed that adiponectin activated AMPKα1 through phosphorylation and stimulated the expression of α-SMA, fibronectin, and collagen I, which was blocked by compound C (Fig. 5D). In addition, ectopic expression of a constitutively active AMPKα1 22, 23 in the human embryonic kidney cells resulted in increased protein levels of α-SMA, fibronectin, and collagen I (Fig. 5E). Collectively, these results indicate that activation of AMPK promotes fibroblast activation and ECM protein production.
Fig. 5. AMPK activation results in fibroblast activation and ECM protein production in the cultured cells.
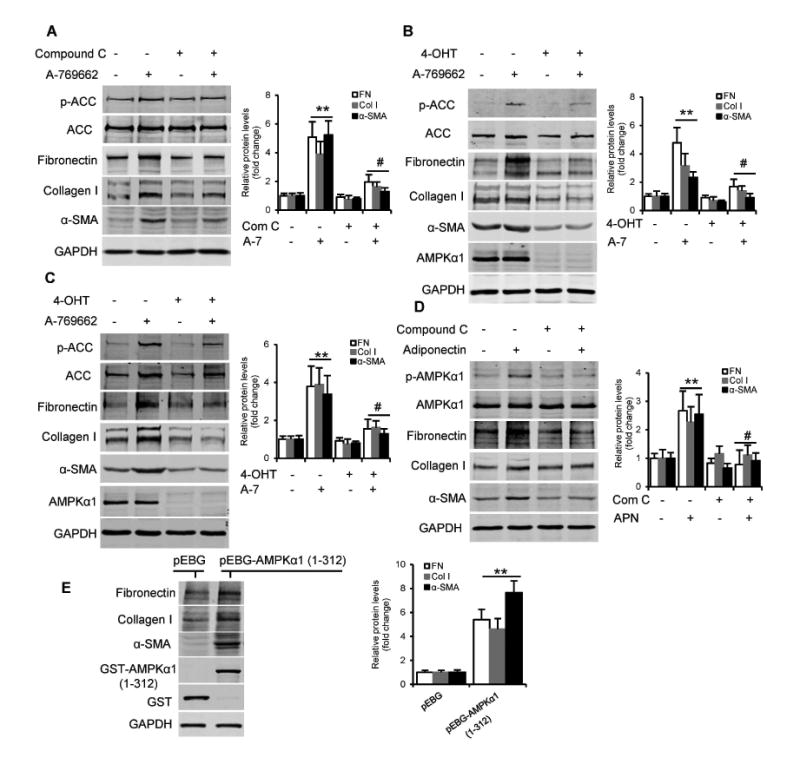
(A) Western blots show A-769662 (A-7, 100 μM for 24 hours) induced the expression of fibronectin (FN), collagen I (Col I), and a-SMA in NRK-49F cells, which was abolished by compound C (Com C, 10 μM). **P<0.01 versus vehicle controls, #P<0.05 versus A-769662 alone. n=3 per group. (B and C) Western blots show A-769662 (100 μM for 24 hours) induced fibronectin, collagen I, and α-SMA expression in primary kidney fibroblasts (B) and bone marrow-derived monocytes (C) from mice, and deletion of AMPKal blocked A-769662-induced fibronectin, collagen I, and α-SMA expression in these cells. AMPKαl was deleted in these cells from CAG-Cre+AMPKa1fl/fl mice by treating with 1 μM 4-hydroxytamoxifen (4-OHT) for 3 days. **P<0.01 versus vehicle controls, #P<0.05 versus A-769662 alone. n=3 per group. (D) Western blotting shows adiponectin (APN, 10 μg/ml for 24 hours) induced the expression of fibronectin (FN), collagen I (Col I), and α-SMA in bone marrow-derived monocytes, which was blocked by compound C. **P<0.01 versus vehicle controls, #P<0.05 versus adiponectin alone. n=3 per group. (E) Ectopic expression of constitutively active AMPKαl increased fibronectin, collagen I, and α-SMA in HEK-293T cells. The cells were transfected with control (pEBG-GST) or pEBG-GST-AMPKal (1-312) plasmids for 24 hours. **P<0.01 versus pEBG plasmid control. n=3 per group. p-ACC, phospho-Acetyl-CoA Carboxylase. ACC, Acetyl-CoA Carboxylase.
MRTF-A mediates AMPKα1-induced fibroblast activation
We then explored the molecular mechanism underlying AMPKα1-induced fibroblast activation. Recent studies have shown that the transcriptional coactivator MRTF-A regulates fibroblast activation and ECM protein production 24-27. To determine whether MRTF-A mediates the effects of AMPKα1 on fibroblast activation, NRK-49F cells and bone marrow-derived monocytes were pretreated with CCG-203971, a selective inhibitor of MRTF-A nuclear localization 27, 28, and then stimulated with A-769662. AMPK-induced fibroblast activation was blocked by CCG-203971 (Fig. 6, A and B). These data indicate that MRTF-A mediates the effect of AMPK on fibroblast activation. Immunofluorescence staining was then performed to determine if AMPK affects MRTF-A nuclear translocation. Activation of AMPK resulted in nuclear accumulation of MRTF-A, which was associated with F-actin formation (Fig. 6C). Western blot analysis confirmed that MRTF-A levels increased in the nuclei following AMPK activation (Fig. 6D). These results indicate that AMPK promotes nuclear translocation and accumulation of MRTF-A leading to fibroblast activation and ECM protein expression.
Fig. 6. AMPKαl activation induces MRTF-A nuclear translocation leading to fibroblast activation and extracellular matrix protein production.
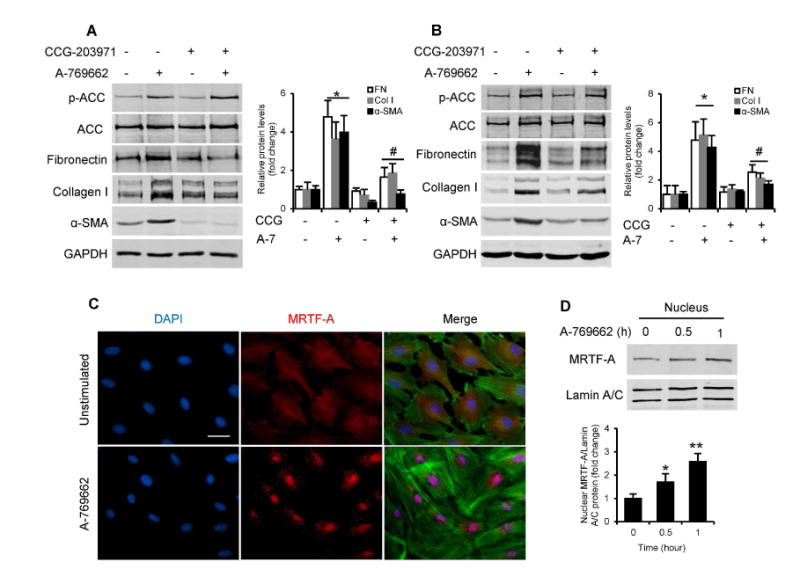
Western blot analyses show inhibition of the MRTF signaling pathway with CCG-203971 (CCG) prevents expression of fibronectin (FN), collagen I (Col I), and α-SMA in NRK-49F cells (A) and bone marrow-derived monocytes (B). The cells were pretreated with vehicle or 10 uM CCG-203971 for 30 minutes and then incubated with vehicle or 100 μM A-769662 (A-7) for 24 hours. *P<0.05 versus vehicle controls, #P<0.05 versus A-769662 without CCG-203971. n=3 per group. (C) NRK-49F cells were serum-starved (0.5 % FBS) for 48 hours, and then stimulated with 200 μM A-769662 for 30 minutes. MRTF-A subcellular localization was determined by immunofluorescence staining for MRTF-A (red), DAPI (blue) and F-actin (green). Scale bar: 30 urn. (D) NRK-49F cells were treated with 200 μM A-769662, harvested at indicated time points (hour). Nuclear translocation of MRTF-A was determined by nuclear extraction and Western blotting. *P<0.05 compared with controls, **P<0.01 compared with controls. n=3 per group. p-ACC, phospho-Acetyl-CoA Carboxylase. ACC, Acetyl-CoA Carboxylase.
AMPK phosphorylates cofilin
We next investigated how AMPK activation resulted in F-actin formation and MRTF-A nuclear accumulation. Recent studies have shown that MRTF-A nuclear localization is regulated by relative amounts of free monomeric globular actin (G-actin) and polymeric filamentous actin (F-actin) 25, 29-31, which are modulated by cofilin 32, 33. To determine whether AMPK acts on cofilin, we first examined whether AMPK activation affects phosphorylation of cofilin (Ser3). NRK-49F cells and bone marrow-derived monocytes were incubated with A762669 for different periods of time, or different concentrations. Activation of AMPK with A-769662 increased cofilin phosphorylation in a dose- and time-dependent manner (Fig. 7, A-D). Furthermore, ectopic expression of constitutively active AMPKα1 increased the levels of cofilin phosphorylation (Fig. 7E). These data indicate that AMPK activation leads to the phosphorylation of cofilin.
Fig. 7. AMPKal directly phosphorylate cofilin.
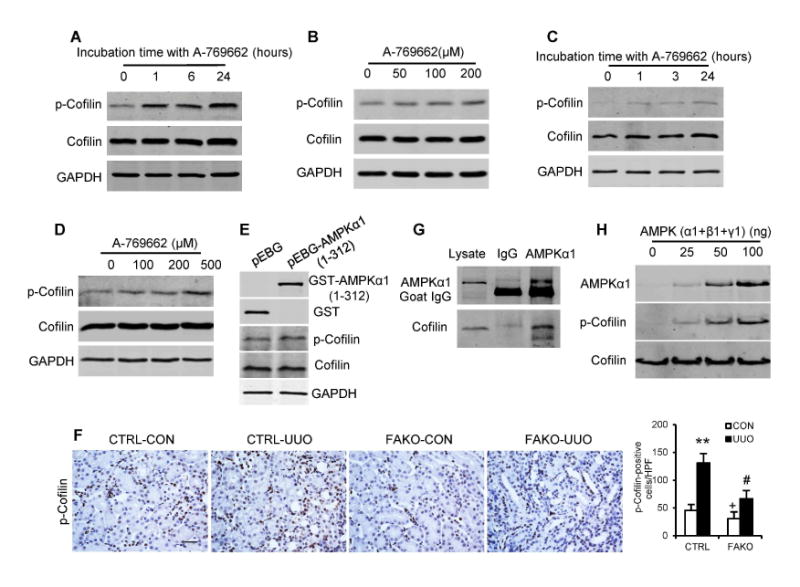
NRK-49F cells were incubated with 100 μM A-769662 for the indicated periods of times (hours) (A), or with various concentrations of A-769662 for 1 hour (B). Bone marrow–derived monocytes were incubated with 100 μM A-769662 for the indicated periods of times (hours) (C), or with various concentrations of A-769662 for 1 hour (D). Cell lysates were immunoblotted with anti-phospho-cofilin (Ser3) and anti-cofilin (A-D). (E) Ectopic expression of constitutively active AMPKαl increased cofilin phosphorylation in HEK-293T cells. FLEK-293T cells were transfected with control (pEBG-GST) or pEBG-GST-AMPKαl (1-312) plasmids for 24 hours. (F) Representative photomicrographs of kidney sections from littermate controls (CTRL) and FAKO mice after 10 days of UUO stained for phosphorylated cofilin (brown) and counterstained with hematoxylin (blue). Scale bar: 20 μm. **P<0.01 compared with the CTRL-CON group, #P<0.05 compared with the CTRL-UUO group, +P<0.05 compared with the FAKO-UUO group, n=6 mice per group. (G) NRK-49F cells were treated with 200 μM A-769662 for 1 hour. Whole cell lysates were used for immunoprecipitation. Western blotting shows the anti-AMPKal antibody pulled down endogenous cofilin. (H) AMPK directly phosphorylates cofilin in vitro. Purified human AMPK (α1+β1+γ1) was incubated with recombinant cofilin protein for 1 hour at 37°C. Cofilin phosphorylation was determined by immunoblotting with a phospho-cofilin (Ser3) antibody.
To examine the effect of AMPKα1 on the phosphorylation of cofilin in the fibrotic kidneys in vivo, kidney sections were stained for phospho-cofilin (Ser3). The positive staining for phosphorylated cofilin increased significantly in the obstructed kidneys after 5 days (Fig. S6) and 10 days (Fig. 7F and Fig. S7) of UUO. AMPKα1 deficiency reduced the numbers of phospho-cofilin-positive cells in the fibrotic kidneys (Fig. 7F and Fig. S7). Similar results were obtained in the IRI model (Fig. S8). Moreover, kidney sections were stained for AMPKα1 and phospho-cofilin (Ser3). AMPKα1-positive cells were also positive for phospho-cofilin in the tubulointerstitium of the injured kidneys (Fig. S9). Finally, kidney sections were stained for phospho-cofilin (Ser3) and α-SMA. AMPK deficiency significantly reduced the number of p-cofilin and α-SMA dual positive cells in the obstructed kidneys (Fig. S10). These results indicate that AMPKα1 induces cofilin phosphorylation in myofibroblasts in the kidney in response to obstructive injury in vivo.
To further determine whether AMPKα1 interacts with cofilin, we performed immunoprecipitation (IP) assay using an anti-AMPKα1 antibody to pull down endogenous proteins in NRK-49F cells. The results showed that the anti-AMPKα1 antibody pulled down cofilin, indicating there is a direct interaction between AMPKα1 and cofilin (Fig. 7G).
Finally, we tested whether AMPKα1 can directly phosphorylate cofilin. We used purified recombinant AMPK kinase containing active human AMPKα1+β1+γ1 proteins as the whole enzyme, and recombinant human cofilin protein as the substrate. Our results showed that AMPK phosphorylated cofilin in a dose-dependent manner (Fig. 7H). These results indicate that AMPK can directly phosphorylate cofilin.
Discussion
Chronic fibrotic diseases account for nearly 45% of deaths in the developed countries 34, 35. However, there is no effective therapy for these devastating disorders. To develop effective therapeutic strategies, it is essential to understand the cellular and molecular mechanisms underlying fibroblast activation and fibrotic development. In the preclinical animal models, we discovered that AMPKα1 significantly induced and activated in the kidney during the development of renal fibrosis. We hypothesize that AMPKα1 may modulate fibroblast activation and fibrogenesis in the kidney. Using the genetic approach, our study identifies AMPKα1 as a critical regulator in fibroblast activation and the development of renal fibrosis. Some studies have reported that pharmacological activators of AMPK may have a protective effect on acute and chronic kidney diseases including possible beneficial effects of metformin in diabetic kidney disease 36-38. However, these pharmacological agents may have AMPK-independent effects. Indeed, it has been reported that the inhibitory effect of metformin gluconeogenesis is independent of AMPK and the renal protective effect of metformin is not dependent on AMPK 39, 40. Utilizing UUO and IRI kidney fibrotic models and conditional AMPKα1 knockout mice, we provide compelling experimental evidence that AMPKα1 contributes to the development of renal fibrosis through activation of fibroblasts (Fig. 1 to 4). We have further demonstrated that either global or fibroblast-specific deletion of AMPKα1 reduces renal fibrosis and the expression of ECM proteins. These results indicated that AMPKα1 has a key role in fibroblast activation and the development of renal fibrosis, which provides a novel link between metabolism and fibroblast activation. It should be emphasized that these two experimental models of renal fibrosis differ considerably in term of etiology and metabolic consequence suggesting that AMPK may play a more general role in fibroblast activation and fibrogenesis. Recently, we have demonstrated that adiponectin, a cytokine that activates AMPK signaling pathway 15, 20, 21, is induced in the kidney following ureteral obstruction or ischemia-reperfusion injury; genetic disruption of adiponectin prevents kidney injury and fibrosis15, 41. The current study suggests that AMPK is at an important nexus for adverse fibrotic remodeling in the kidney.
Our study demonstrates that AMPKα1 deficiency reduces collagen I and fibronectin protein accumulation and deposition in the kidneys 10 days after UUO or 14 days after IRI. Mia et al reported that global deletion of AMPK α1 reduced considerably collage I mRNA expression and slightly, but significantly collagen I protein production in the obstructed kidneys 7 days following UUO16. Furthermore, they did not detect a significant difference in collagen I deposition by immunostaining of the kidneys 7 days after obstructive injury. The discrepancy can be explained that the semi-quantitative method of scoring collagen staining is insensitive and may have failed to detect a reduction in fibrosis that is indicated by other parameters.
Myofibroblasts are generally considered to be the main source of increased ECM deposition in renal fibrosis 6-8. Their number correlates closely with the severity of fibrosis and the progression of chronic kidney disease 8, 42, 43. These activated myofibroblasts predominantly originate from resident renal fibroblasts and bone marrow 44-46. We have shown that both accumulation of myofibroblasts identified as α-SMA-positive cells and expression of α-SMA protein are increased in the fibrotic kidneys of WT mice after UUO or IRI, whereas both myofibroblast accumulation and α-SMA expression are significantly reduced in the fibrotic kidneys of Cre-mediated AMPKα1 KO mice. Furthermore, our in vitro studies have shown that AMPK activation induced α-SMA and ECM protein expression; whereas AMPK inhibition or AMPKa1 deletion attenuates expression of α-SMA and ECM proteins (Fig. 5). These results strongly indicate that AMPK plays a pivotal role in fibroblast activation and fibrogenesis.
MRTF-A, a member of the MRTF family of transcriptional coactivators, plays a central role in fibroblast activation 26, 27, 31, 47-49. However, MRTF-A is sequestered in the cytoplasm where it binds to G-actin, and its cytoplasm-nucleus translocation is under the dynamic control of G-actin and F-actin. Polymerization of G-actin into F-actin releases MRTF-A, and enables it to translocate into the nucleus, functioning as coactivator of SRF-mediated transcriptions of CArG box–containing genes 25-27, 29, 30, 49. In the present study, we have shown that AMPK activation results in F-actin formation and MRTF-A nuclear accumulation; inhibition of MRTF-A nuclear translocation with a small molecule inhibitor, CCG-203971, reduced AMPK-induced fibroblast activation and ECM production (Fig. 6). These results suggest that MRTF-A signaling mediates AMPK-induced fibroblast activation and ECM protein production.
Members of the actin-depolymerizing factor (ADF)/cofilin family are conserved small actin-binding protein, and play a pivotal role in actin dynamics. Cofilin binds to F-actin, and severs and depolymerizes F-actin into G-actin. Cofilin is inactivated by phosphorylation at Ser 3 leading to stabilization of F-actin and depletion of G-actin pools 32, 33, which induces G-actin dissociation from MRTF-A and the latter's nuclear translocation. Phosphorylation of cofilin is catalyzed by kinases. Previous study has shown that cofilin can be phosphorylated at Ser 3 by LIM kinases and testicular protein kinase 32, 33. In this study, the most important and unexpected discovery is that AMPK directly phosphorylates cofilin (Fig. 7). For the first time, we identify cofilin-1 protein as a substrate of AMPK. In an in vitro kinase assay, we have demonstrated that AMPK directly phosphorylates cofilin. Furthermore, we have shown that AMPKα1 and cofilin directly interact within cells, and AMPK activation increases cofilin phosphorylation. In addition, the immunofluorescence staining of mouse fibrotic kidneys reveals that AMPKα1 induces cofilin phosphorylation in vivo. Taken together, our study provides compelling experimental evidence that phosphorylation of cofilin by AMPK is the primary trigger for AMPK-induced fibroblast activation and fibrogenesis.
In summary, our study identifies AMPKα1 as a critical regulator of fibroblast activation and renal fibrogenesis. In response to obstructive or ischemia-reperfusion injury, activated AMPK phosphorylates cofilin resulting in F-actin polymerization, MRTF-A nuclear accumulation, which activates fibroblasts and promotes fibrogenesis (Fig. 8). Our findings yield novel insights into the molecular mechanisms of fibroblast activation and fibrogenesis and provide new therapeutic approaches for chronic fibrotic kidney disease.
Fig. 8. A proposed model depicting the molecular mechanism by which AMPK regulates actin dynamics, MRTF-A nuclear translocation, and fibroblast activation.
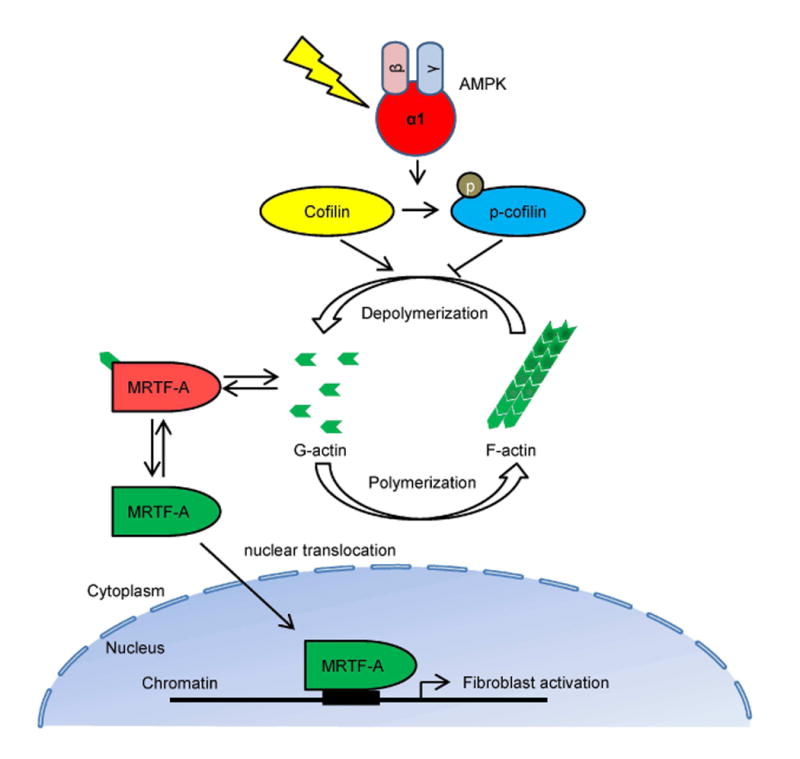
Under normal condition, cofilin severs and depolymerizes F-actin into G-actin, and cytoplasmic G-actin associates and retains MRTF-A in the cytoplasm. In response to injury, activated AMPK directly phosphorylates cofilin, which promotes G-actin to F-actin polymerization. Lower G-actin concentration results in releases of MRTF-A from binding to G-actin, which enters into the nucleus and induces fibroblast activation.
Materials and Methods
Mice
The mice were handled according to NIH Guide for the Care and Use of Laboratory Animals. All experiments were approved by the IACUC of Baylor College of Medicine. The Prkaa1 mutant mice (AMPKα1flox/flox) possess loxP sites flanking exon 3 of the protein kinase, AMP-activated, α1 catalytic subunit (Prkaa1) gene 50. The CAG-Cre+ mice have a tamoxifen-inducible Cre-mediated recombination system driven by the chicken beta actin promoter/enhancer coupled with the cytomegalovirus immediate-early enhancer 17. The Cre/ESR1 protein produced by these transgenic mice was restricted to the cytoplasm, and can only gain access to the nuclear compartment after exposure to tamoxifen. These mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). The Col1-Cre+ mice carrying the tamoxifen-inducible Cre-recombinase (Cre/ESR1) under the control of fibroblast-specific regulatory sequence from the proα2(I) collagen gene, were kindly gift from Binoit de Crombrugghe 18. The AMPKα1flox/flox mice mated with CAG-Cre+ mice and Col1-Cre+ mice, respectively. The offspring were used for generating tamoxifen-induced, Cre-mediated AMPKα1flox/flox gene deletions. Genotyping was performed by PCR of tail biopsies (see Fig. S4C for primer sequences).
Male mice homozygous for AMPKα1flox/flox with hemizygous Cre (Cre+AMPKα1flox/flox) were administered tamoxifen, to delete AMPKα1 gene. Male littermate mice homozygous for AMPKα1flox/flox without Cre (Cre-AMPKα1flox/flox) were used as controls. Both cre and control mice aged 8–10 weeks (weighing 20–30 g) were given daily intraperitoneal injections of 0.1 ml of 10 mg/ml tamoxifen (Sigma-Aldrich, St. Louis, MO) in corn oil for 10 days before surgeries. Daily tamoxifen treatment was continued until the day before sacrifice because fibroblasts continue to be activated during the development of renal fibrosis.
Mouse models
Male mice were subjected to UUO or IRI surgery after 10 days' tamoxifen treatment. The UUO procedure was performed as described15, 46, 51, 52. To induce unilateral IRI, left kidneys were exposed through flank incision and were subjected to ischemia by clamping renal pedicles with nontraumatic microaneurysm clamps, as described15, 52, for 60 minutes. Kidneys were harvested 2 weeks after IRI. Unless otherwise specified, 6 mice per group were used in each experiment.
Cell culture and treatment
All cells were cultured in a humid incubator at 37°C and 5% CO2. The normal rat kidney fibroblasts NRK-49F (ATCC CRL1570) and the human embryonic kidney cells HEK-293T (ATCC CRL-3216) were obtained from the American Type Culture Collection and cultured according to manufacturer's protocols. NRK-49F cells were starved overnight and stimulated in Dulbecco's modified Eagle's medium (DMEM) with 1 g/L glucose containing 0.5% fetal bovine serum (FBS) and 1% penicillin and streptomycin.
Primary kidney fibroblasts were isolated from mice as described 46. Briefly, mouse kidney was decapsulated, minced, and digested with Liberase (Roche, Indianapolis, IN) for 30 minutes at 37°C. Cells were filtered through a 40 urn strainer, centrifuged, and cultured in DMEM containing 10% FBS and 1% penicillin and streptomycin. After subculture, more than 98%) of adherent cells were kidney fibroblasts as identified by positive staining for type I collagen and vimentin.
Mouse bone marrow-derived monocytes were isolated and cultured as described 15,46, 53. Briefly, bone marrow-derived monocytes were isolated from 8- to 10-week-old mice, and cultured in RPMI-1640 medium containing 10% FBS, 10% L929-conditioned medium, and 1% penicillin/streptomycin. After 5 days, cells were serum-starved and stimulated in RPMI-1640 medium containing 2% FBS and 2% L929-conditioned medium.
4-hydroxytamoxifen (Sigma-Aldrich) at 1 μM was used to delete AMPKal in primary kidney fibroblasts and bone marrow-derived monocytes from CAG-Cre+AMPKα1fl/fl mice54.
The pEBG-AMPKαl (amino acids 1-312) and pEBG (empty backbone of GST fusion vector) plasmids were obtained from Addgene (plasmid no 27632 and 22227). Cells were transfected with plasmid DNAs using Neon Transfection System (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol, or using X-tremeGENE HP DNA transfection reagent (Sigma-Aldrich, St. Louise, MO).
In vitro AMPK kinase assay
Active human AMPKαl + AMPKβi + AMPKγl full length protein (ab79803 Abcam, Cambridge, MA) was used as AMPK kinase, and recombinant human cofilin full length protein (ab62958 Abcam) was used as the substrate. Various concentrations of AMPK kinase and 1 μg of cofilin were incubated for 1 hour at 37°C in a kinase buffer containing 20 mM HEPES (pH 7.4), 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.2 mM AMP, 0.1 mM ATP, 0.05 mM DTT, and 50 ng/μl BSA. Reactions were terminated with SDS sample buffer, and subjected to SDS-PAGE. Phosphorylation of cofilin was determined by Western blots.
Renal morphology
Mice were sacrificed and perfused with PBS to remove blood. One portion of the kidney tissue was fixed in 10% buffered formalin and embedded in paraffin, cut at 4-μm thickness, and stained with Sirius red to identify collagen fibers. The Sirius red-stained sections were scanned using a microscope equipped with a digital camera (Nikon Instruments, Melville, NY), and quantitative evaluation was performed using the NIS-Elements Br 3.0 software (Nikon Instruments) 15,51,55,56.
Immunohistochemistry
Immunohistochemical staining was performed on paraffin sections which were processed as described 15, 46. Antigen retrieval was performed with antigen unmasking solution (Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched with 3% H2O2. Vectastain Elite ABC kits (Vector Laboratories) were used for the staining according to manufacturer's instructions. The reaction was visualized by incubation with diaminobenzidine solution (ImmPACT DAB, Vector Laboratories). Slides were then counterstained with hematoxylin. The images were acquired with a microscope equipped with a digital camera.
Immunofluorescence
Immunofluorescence staining of kidney sections was performed on paraffin sections as described 15, 51, 52, 57. After antigen retrieval, nonspecific binding was blocked with protein block (Dako, Carpinteria, CA). Kidney sections were incubated with primary antibodies overnight, followed by appropriate Alexa Fluor 488- or 594-conjugated secondary antibodies (Life Technologies). Cell staining was performed according to the Immunocytochemistry (ICC) protocol 58. Cells were grown on glass coverslips. After washing with PBS, the samples were fixed in 4% paraformaldehyde (Alfa Aesar, Ward Hill, MA), and permeabilized with 0.5% Triton X-100 in PBS. Cells were stained with anti-MRTF-A (Santa Cruz Biotechnology), followed by Alexa Fluor 594-conjugated secondary antibody. Alexa Fluor 488 conjugated Phalloidin (Cell Signaling Technology) was used to stain F-actin. Slides were mounted with medium containing DAPI (Vector Laboratories). Fluorescence intensity was visualized using a Nikon microscope image system. Quantitative analysis of sections was performed using NIS-Elements Br 3.0 software.
Coimmunoprecipitation
Cells were harvested in Pierce IP Lysis Buffer containing cocktail proteinase and phosphatase inhibitors (Thermo Fisher Scientific) after washing in PBS. Protein A/G PLUS-agarose (Santa Cruz Biotechnology) was used for immunoprecipitation, according to the manufacturers' protocols. Beads were washed 5 times with lysis buffer. Eluted proteins were analyzed by Western blotting. In this study, goat IgG was used as a negative control.
Western blot analysis
Renal tissues or cells were homogenized and lysed in radioimmunoprecipitation (RIPA) buffer containing cocktail proteinase and phosphatase inhibitors (Thermo Fisher Scientific). Protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Nuclear extracts were prepared as described 59. Equal amounts of protein were separated on SDS-polyacrylamide gels in a Tris/glycine buffer system, transferred onto nitrocellulose membranes, and blotted according to standard procedures 15, 46, 55, 56, 60, 61. The specific bands of target proteins were analyzed using an Odyssey IR scanner (LICOR Bioscience, Lincoln, NE), and signal intensities were quantified using NIH ImageJ software.
Statistical analysis
Data were represented as mean ± SEM. Multiple group comparisons were performed by ANOVA followed by the Bonferroni procedure for comparison of means. Comparisons between two groups were analyzed by the two-tailed t test. P values of less than 0.05 are considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Binoit de Crombrugghe at University of Texas M. D. Anderson Cancer Center for providing collagen I promoter/enhancer driven Cre recombinase transgenic mice. This work was supported by the National Institutes of Health grants – R01DK095835 (to Yanlin Wang), R01AR63686 (to Z Hu), R01HL089792 (to ML Entman), and R37DK37175 (to WE Mitch), and the U.S. Department of Veterans Affairs grant – I01BX02650 (to Yanlin Wang). The contents of the article do not represent the views of the U.S. Department of Veterans Affairs or the United States government.
Footnotes
Conflicts of interest: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 2.Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol. 2013;304:C216–225. doi: 10.1152/ajpcell.00328.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schainuck LI, Striker GE, Cutler RE, et al. Structural-functional correlations in renal disease. II. The correlations. Hum Pathol. 1970;1:631–641. doi: 10.1016/s0046-8177(70)80061-2. [DOI] [PubMed] [Google Scholar]
- 4.Nath KA. The tubulointerstitium in progressive renal disease. Kidney Int. 1998;54:992–994. doi: 10.1046/j.1523-1755.1998.00079.x. [DOI] [PubMed] [Google Scholar]
- 5.Eddy AA. Progression in chronic kidney disease. Adv Chronic Kidney Dis. 2005;12:353–365. doi: 10.1053/j.ackd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Neilson EG. Mechanisms of disease: Fibroblasts--a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101–108. doi: 10.1038/ncpneph0093. [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 8.Asada N, Takase M, Nakamura J, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121:3981–3990. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 10.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010;1804:581–591. doi: 10.1016/j.bbapap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 11.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 12.Zhao JX, Yue WF, Zhu MJ, et al. AMP-activated protein kinase regulates beta-catenin transcription via histone deacetylase 5. J Biol Chem. 2011;286:16426–16434. doi: 10.1074/jbc.M110.199372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miranda L, Carpentier S, Platek A, et al. AMP-activated protein kinase induces actin cytoskeleton reorganization in epithelial cells. Biochem Biophys Res Commun. 2010;396:656–661. doi: 10.1016/j.bbrc.2010.04.151. [DOI] [PubMed] [Google Scholar]
- 14.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J, Lin SC, Chen G, et al. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol. 2013;24:1644–1659. doi: 10.1681/ASN.2013030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mia S, Federico G, Feger M, et al. Impact of AMP-Activated Protein Kinase alpha1 Deficiency on Tissue Injury following Unilateral Ureteral Obstruction. PLoS One. 2015;10:e0135235. doi: 10.1371/journal.pone.0135235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 18.Zheng B, Zhang Z, Black CM, et al. Ligand-dependent genetic recombination in fibroblasts : a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol. 2002;160:1609–1617. doi: 10.1016/S0002-9440(10)61108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heiker JT, Kosel D, Beck-Sickinger AG. Molecular mechanisms of signal transduction via adiponectin and adiponectin receptors. Biol Chem. 2010;391:1005–1018. doi: 10.1515/BC.2010.104. [DOI] [PubMed] [Google Scholar]
- 21.Shehzad A, Iqbal W, Shehzad O, et al. Adiponectin: regulation of its production and its role in human diseases. Hormones (Athens) 2012;11:8–20. doi: 10.1007/BF03401534. [DOI] [PubMed] [Google Scholar]
- 22.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crute BE, Seefeld K, Gamble J, et al. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- 24.Penke LR, Huang SK, White ES, et al. Prostaglandin E2 inhibits alpha-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J Biol Chem. 2014;289:17151–17162. doi: 10.1074/jbc.M114.558130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riches DW, Backos DS, Redente EF. ROCK and Rho: Promising therapeutic targets to ameliorate pulmonary fibrosis. Am J Pathol. 2015;185:909–912. doi: 10.1016/j.ajpath.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luchsinger LL, Patenaude CA, Smith BD, et al. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J Biol Chem. 2011;286:44116–44125. doi: 10.1074/jbc.M111.276931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sisson TH, Ajayi IO, Subbotina N, et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol. 2015;185:969–986. doi: 10.1016/j.ajpath.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell JL, Haak AJ, Wade SM, et al. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett. 2013;23:3826–3832. doi: 10.1016/j.bmcl.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esnault C, Stewart A, Gualdrini F, et al. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014;28:943–958. doi: 10.1101/gad.239327.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald ME, Li C, Bian H, et al. Myocardin-related transcription factor A regulates conversion of progenitors to beige adipocytes. Cell. 2015;160:105–118. doi: 10.1016/j.cell.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25:457–469. doi: 10.1016/j.cellsig.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman SL, Sheppard D, Duffield JS, et al. Therapy for fibrotic diseases: nearing the starting line. Science translational medicine. 2013;5:167sr161. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 36.Lempiainen J, Finckenberg P, Levijoki J, et al. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br J Pharmacol. 2012;166:1905–1915. doi: 10.1111/j.1476-5381.2012.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cavaglieri RC, Day RT, Feliers D, et al. Metformin prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. Mol Cell Endocrinol. 2015;412:116–122. doi: 10.1016/j.mce.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Ravindran S, Kuruvilla V, Wilbur K, et al. Nephroprotective Effects of Metformin in Diabetic Nephropathy. J Cell Physiol. 2017;232:731–742. doi: 10.1002/jcp.25598. [DOI] [PubMed] [Google Scholar]
- 39.Foretz M, Hebrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christensen M, Jensen JB, Jakobsen S, et al. Renoprotective Effects of Metformin are Independent of Organic Cation Transporters 1 &2 and AMP-activated Protein Kinase in the Kidney. Sci Rep. 2016;6:35952. doi: 10.1038/srep35952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin X, Chen J, Hu Z, et al. Genetic deficiency of adiponectin protects against acute kidney injury. Kidney Int. 2013;83:604–614. doi: 10.1038/ki.2012.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol. 2009;5:319–328. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- 44.Lebleu VS, Taduri G, O'Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broekema M, Harmsen MC, van Luyn MJ, et al. Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol. 2007;18:165–175. doi: 10.1681/ASN.2005070730. [DOI] [PubMed] [Google Scholar]
- 46.Yan J, Zhang Z, Yang J, et al. JAK3/STAT6 Stimulates Bone Marrow-Derived Fibroblast Activation in Renal Fibrosis. J Am Soc Nephrol. 2015;26:3060–3071. doi: 10.1681/ASN.2014070717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rahaman SO, Grove LM, Paruchuri S, et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest. 2014;124:5225–5238. doi: 10.1172/JCI75331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velasquez LS, Sutherland LB, Liu Z, et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A. 2013;110:16850–16855. doi: 10.1073/pnas.1316764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Small EM, Thatcher JE, Sutherland LB, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen G, Lin SC, Chen J, et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol. 2011;22:1876–1886. doi: 10.1681/ASN.2010080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia Y, Yan J, Jin X, et al. The chemokine receptor CXCR6 contributes to recruitment of bone marrow-derived fibroblast precursors in renal fibrosis. Kidney Int. 2014;86:327–337. doi: 10.1038/ki.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot5080. pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 54.Fuhrmann-Benzakein E, Garcia-Gabay I, Pepper MS, et al. Inducible and irreversible control of gene expression using a single transgene. Nucleic Acids Res. 2000;28:E99. doi: 10.1093/nar/28.23.e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia Y, Entman ML, Wang Y. CCR2 Regulates the Uptake of Bone Marrow-Derived Fibroblasts in Renal Fibrosis. PLoS One. 2013;8:e77493. doi: 10.1371/journal.pone.0077493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia Y, Entman ML, Wang Y. Critical Role of CXCL16 in Hypertensive Kidney Injury and Fibrosis. Hypertension. 2013;62:1129–1137. doi: 10.1161/HYPERTENSIONAHA.113.01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen J, Xia Y, Lin X, et al. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab Invest. 2014;94:545–556. doi: 10.1038/labinvest.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haudek SB, Cheng J, Du J, et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Keulenaer GW, Wang Y, Feng Y, et al. Identification of IEX-1 as a biomechanically controlled nuclear factor-kappaB target gene that inhibits cardiomyocyte hypertrophy. Circ Res. 2002;90:690–696. doi: 10.1161/01.res.0000012922.40318.05. [DOI] [PubMed] [Google Scholar]
- 60.Yang J, Chen J, Yan J, et al. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PLoS One. 2012;7:e52415. doi: 10.1371/journal.pone.0052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu J, Lin SC, Chen J, et al. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol. 2011;301:H538–547. doi: 10.1152/ajpheart.01114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.