

Abstract
The mechanistic target of rapamycin complex 1 (mTORC1) controls cell growth and metabolism in response to nutrients, energy levels and growth factors. It contains the atypical kinase mTOR and the RAPTOR subunit that binds to the TOS motif of substrates and regulators. mTORC1 is activated by the small GTPase RHEB and inhibited by PRAS40. Here we present the 3.0 Å cryo-EM structure of mTORC1 and the 3.4 Å structure of activated RHEB-mTORC1. RHEB binds to mTOR distally from the kinase active site, yet causes a global conformational change that allosterically realigns active-site residues, accelerating catalysis. Cancer-associated hyperactivating mutations map to structural elements that maintain the inactive state, and we provide biochemical evidence that they mimic RHEB relieving auto-inhibition. We also present crystal structures of RAPTOR-TOS motif complexes that define the determinants of TOS recognition, of an mTOR FKBP12-rapamycin-binding (FRB) domain–substrate complex that establishes a second substrate-recruitment mechanism, and of a truncated mTOR-PRAS40 complex that reveals PRAS40 inhibits both substrate-recruitment sites.
mTORC1 controls multiple aspects of cell growth and homeostasis, including protein synthesis, lipogenesis, glucose metabolism, autophagy, lysosome biogenesis, proliferation and survival, in response to environmental cues ranging from levels of amino acids, glucose, energy and oxygen to growth factors1–3. mTOR and other pathway proteins are frequently mutated in cancer, and mTOR inhibitors are approved for the treatment of cancer4,5. mTORC1 is a ~1 MDa dimeric complex consisting of the phosphoinositide 3 kinase-related protein kinase (PIKK)6 mTOR, and the subunits RAPTOR and mLST87–10. mTORC1 activation requires nutrients, which are sensed as amino acid levels and induce mTORC1 recruitment to lysozomal membranes through RAPTOR2. There, mTORC1 meets its activator, the small GTPase RHEB, which conveys a second set of signals from environmental cues including energy, oxygen levels, and growth factors3,11,12. In metazoan, growth factors also relieve the inhibition of mTORC1 by PRAS4013–16.
The best-studied mTORC1 substrates are the eIF4E-binding protein 1 (4EBP1), whose phosphorylation destabilizes the 4EBP1-eIF4E complex and activates cap-dependent translation17,18, and the ribosomal S6 kinase 1 (S6K1), which promotes multiple aspects of protein synthesis and anabolic pathways1,17,18. mTORC1 exhibits multiple levels of substrate specificity. Like canonical kinases, mTOR has a sequence preference around the phosphorylation site, but it is limited to the P+1 position being a proline, bulky hydrophobic or aromatic residue19,20. It also employs a substrate-recruitment mechanism whereby RAPTOR binds to a ~5 amino acid Tor signaling sequence (TOS) motif present in the 4EBP1 and S6K1 substrates21,22, as well as the PRAS40 inhibitor13–16. In addition, the crystal structure of an N-terminally truncated mTOR kinase complex (mTORΔN-mLST8) suggested the FKBP12-rapamycin binding domain (FRB) recruits the S6K1 substrate into the recessed active site23. The overall architecture of mTORC1 has been described from 5.9 Å and 4.4 Å cryo-EM reconstructions10,24, but the biochemical and structural mechanisms of regulation by RHEB and PRAS40, and of substrate recruitment remain poorly understood. To address these questions, we determined the cryo-EM structures of mTORC1 and RHEB-mTORC1, at 3.0 Å and 3.4 Å, respectively, and the crystal structures of PRAS40-mTORΔN-mLST8, RAPTOR-TOS, and FRB-S6K1 peptide complexes. The crystallographic findings are shown in the context of the mTORC1-RHEB cryo-EM structure in the composite image of Figure 1a.
Figure 1. The FRB domain is a substrate-recruitment site.
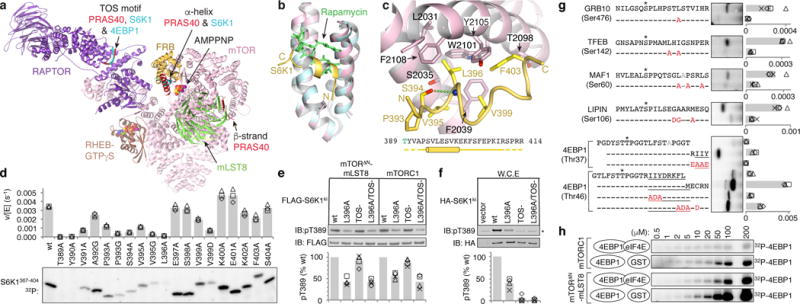
a, Composite image of S6K1 and PRAS40 sub-complex crystal structures superimposed on a RHEB-mTORC1 monomer from the cryo-EM structure. b, The FRB (pink) bound to the S6K1 peptide (yellow) superimposed on the FRB-Rapamycin-FKBP12 complex (blue FRB, green rapamycin; FKBP12 omitted). c, S6K1-FRB interface, S6K1 sequence and secondary structure (cylinder, helix; yellow dashes, unstructured). d, Phosphorylation of w.t. and mutant S6K1367–404 (2 μM) by mTORΔN-mLST8 (30 nM). 32P incorporation plotted as reaction velocity over enzyme concentration (means as columns, three independent experiments with markers). e, In vitro phosphorylation of w.t. and mutant FLAG-S6K1ki (2 μM) by 30 nM mTORΔN-mLST8 or mTORC1. f, Phosphorylation of w.t. and mutant HA-S6K1ki in HEK293 cells (asterisk, endogenous S6K1). In e and f, products detected by pT389-specific antibody and plotted relative to the w.t. reaction of each series (means as columns, four independent experiments with markers). g, Phosphorylation of indicated w.t. and mutant (red mutation) peptides (10 μM) of indicated substrates. Plotted as bars for means and markers for independent experiments (4EBP1, n=3; rest, n=4). 4EBP1 amphipathic helix underlined; asterisk, phosphorylation sites; gray, alanine mutated second phosphorylation sites. h, Phosphorylation of indicated 4EBP1 concentrations by 30 nM mTORΔN-mLST8 or mTORC1 in the presence of 1 molar equivalent eIF4E or GST control (plotted in Extended Data Fig. 1d).
Structure of the S6K1 recruitment peptide bound to the FRB domain
To investigate FRB-mediated substrate recruitment23 by crystallography, we fused a 26-residue S6K1 sequence to the FRB domain due to the low affinity of this interaction (KM= 430 μM, Extended Data Figs 1a, b). In the 1.75 Å crystal structure, a 13-residue S6K1 portion adopts an amphipathic helix and packs with the rapamycin-binding site of the FRB (Fig. 1b; Extended Data Fig. 1e). Central to the interface is the S6K1 Leu396 side chain that extends from the amphipathic helix and inserts into an FRB pocket (Phe2039, Trp2101, Tyr2105 and Phe2108) where rapamycin inserts a key aliphatic group25 (Fig. 1c; Extended Data Fig. 1c). Additional FRB contacts are made by Val395 and Val399 that flank Leu396.
We confirmed the importance of these interactions by alanine-scanning mutagenesis of a 38-residue S6K1 substrate polypeptide (thereafter S6K1367–404). The L396A mutation reduced phosphorylation by the truncated mTORΔN-mLST8 complex to 3 % of the wild type level, and the other mutations had effects commensurate with either their FRB contacts or helix-stabilizing roles (Figs 1c and d; Extended Data Fig. 1b). We also assayed full-length, kinase-inactive S6K1 (thereafter S6K1ki) harboring the L396A mutation and found that its levels of Thr389 phosphorylation by mTORC1 or the truncated mTORΔN-mLST8 complex were 53 % and 37 % of the respective w.t. S6K1ki reactions, irrespective of the TOS motif (Fig. 1e). The L396A mutation also reduced Thr389 phosphorylation of S6K1ki transiently overexpressed in HEK293 cells to 34 % of the w.t. level (Fig. 1f).
The mTORC1 substrates1,4 GRB1019,26, TFEB27, MAF128 and LIPIN29 also contain hydrophobic amino acids within ~15 residues of reported phosphorylation sites (Fig. 1g). We thus mutated one or more hydrophobic residues in each substrate in the context of 20-residue synthetic peptides. The mutations reduced phosphorylation of all four substrates by a factor of 4 to 13, consistent with these substrates utilizing the FRB docking site to enter the catalytic cleft (Fig. 1g).
We also investigated 4EBP1, whose Thr37 and Thr46 phosphorylation sites are followed by an amphipathic helix that binds to eIF4E30. Appreciable Thr37 phosphorylation required extending the peptide to the first turn of the amphipathic helix, which when mutated reduced phosphorylation back to a barely detectable level (Fig. 1g). With the Thr46 site, the 20-residue peptide that reaches partway to the amphipathic helix was phosphorylated ~5-fold less than a peptide encompassing the entire helix. In this longer peptide, mutation of the first and second sets of hydrophobic residues reduced phosphorylation by a factor of ~8 and ~3, respectively (Fig. 1g). Consistent with the amphipathic helix being recruited by the FRB, addition of eIF4E, which sequesters the helix, reduced full-length 4EBP1 phosphorylation by both mTORC1 and mTORΔN-mLST8 by a factor of ~4 (Fig. 1h).
Crystal structures of Raptor-TOS motif complexes
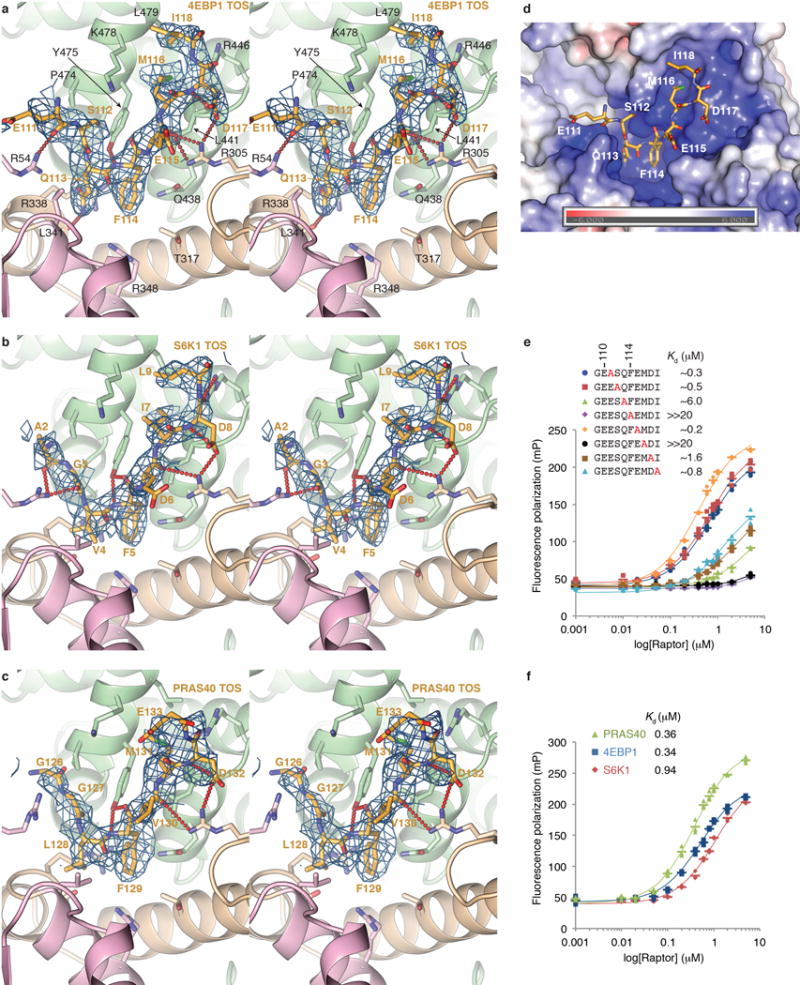
We determined the crystal structures of Arabidopsis thaliana Raptor (atRaptor) bound to TOS-motif peptides from human 4EBP1, S6K1 and PRAS40, at 3.0, 3.1 and 3.35 Å resolution, respectively, and apo-atRaptor at 3.0 Å (Fig. 2a and Extended Data Table). The atRaptor residues that contact the TOS peptides are identical in human RAPTOR, and the atRaptor-4EBP1 TOS interface (Fig. 2b) does not differ discernibly in the 3.0 Å human mTORC1-4EBP1 cryo-EM structure described bellow (Extended Data Fig. 2).
Figure 2. RAPTOR structure and TOS motif recognition.
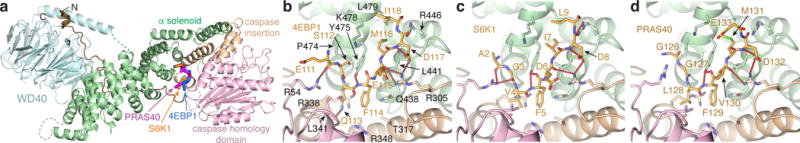
a, atRaptor structure with the three TOS motif co-crystal structures superimposed. atRaptor domains colored as labelled (N-terminal extension dark brown). b–c, Close-up views of the interface between atRaptor, colored as in a, and TOS peptides (yellow sticks) of human 4EBP1 (b), S6K1 (c) and PRAS40 (d). atRaptor side chains shown are identical in human RAPTOR, whose residue numbers are shown (red-dotted lines, hydrogen bonds).
RAPTOR has a sausage-like shape, with the N-terminal caspase-homology domain31 at one end, an α–α solenoid of ~8 armadillo repeats in the middle, and a C-terminal WD40 domain at the other end (Fig. 2a). The caspase homology domain can be superimposed on caspase6 with a root-mean square deviation of ~3 Å for 175 Cα atoms, but RAPTOR lacks the caspase Cys-His catalytic dyad31(Supplementary Information discussion). The TOS peptides bind to a groove between the caspase fold and the solenoid, ~65 Å away from the kinase active site (Fig. 1a). One side of this groove is formed by a 4-helix insertion in the caspase fold, and the other side by the first 3 armadillo repeats (Fig. 2a).
All three TOS peptides have an equivalent 8-residue segment ordered in the crystals (Extended Data Figs 3a to d). The key phenylalanine side chain of the TOS consensus21,22 FXΦDΦ (Φ hydrophobic, X any residue) binds to a pocket together with the preceding TOS residue (thereafter F−1 position). In 4EBP1, this unit consists of the Phe114 aromatic ring making π–π stacking interactions with the Gln113 side chain, whereas in S6K1 and PRAS40 the Phe5 and Phe129 aromatic rings make functionally analogous van der Waals contacts with Val4 and Leu128, respectively (Figs 2b to d). In addition, the phenylalanine backbone amide group hydrogen bonds to Tyr475 (human RAPTOR numbering). Alanine mutation of the 4EBP1 Gln113 reduces its affinity for human RAPTOR by a factor of 20, confirming the importance of its stacking with the phenylalanine and recognition of the residue pair as a unit (Kd values in Extended Data Figs 3e and f).
The other conserved TOS residues make overall conserved RAPTOR contacts (Figs 2b to d). The hydrophobic F+2 side chain (Met116, Ile7 and Met131 of 4EBP1, S6K1 and PRAS40, respectively) binds into a tight pocket at the bottom of the groove, and accordingly its alanine mutation reduces binding by nearly two orders of magnitude (Extended Data Fig. 3e). The F+3 aspartic acid side chain forms a hydrogen bond network involving Arg305 and the F+1 backbone carbonyl group, and its mutation increases the Kd 5-fold (Extended Data Figs 3d and e). By contrast, the hydrophobic F+4 side chain (4EBP1 Ile118 and S6K1 Leu9; PRAS40 Glu133 is disordered) binds to a shallow, solvent exposed surface pocket, and its mutation reduces binding only modestly (Extended Data Fig. 3e).
Consistent with the conservation of contacts in the three structures, the PRAS40 inhibitor has a TOS-RAPTOR Kd similar to those of S6K1 and 4EBP1 (Extended Data Fig. 3f). This suggested that PRAS40 has additional mTORC1-interacting elements.
PRAS40 blocks the FRB substrate-recruitment site
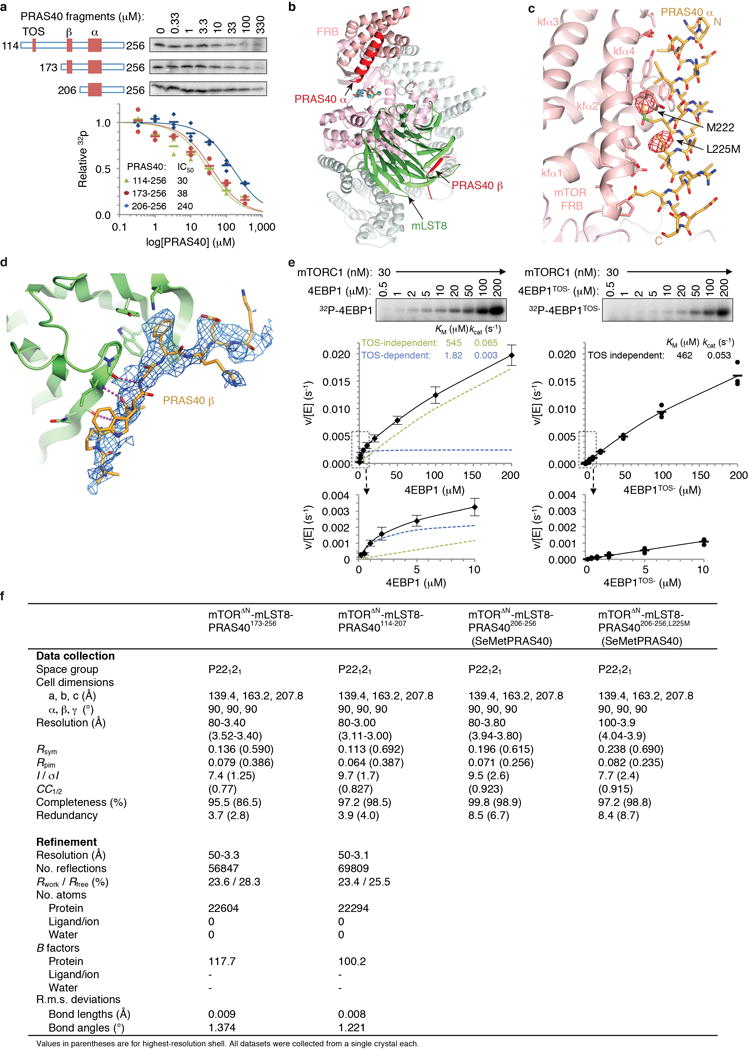
To identify additional mTORC1-binding elements of PRAS40, we first tested whether the PRAS40 segment reported to be necessary for inhibition14 (PRAS40114–256) can also inhibit mTORΔN-mLST8 phosphorylating S6K1367–404. PRAS40114–256 inhibited this TOS-independent reaction with an apparent inhibitor constant (Ki) of ~52 μM (calculated from IC50; Fig. 3a), which is significantly lower than the ~430 μM KM of S6K1367–404 (Extended Data Fig. 1b). Deletion of TOS (PRAS40173–256) had no effect as expected, but deletion of 33 additional residues (PRAS40206–256) reduced inhibition by a factor of ~4 (Fig. 3a). Very similar results were obtained with the 4EBP142–64 substrate (Extended Data Fig. 4a).
Figure 3. PRAS40 blocks the FRB substrate-recruitment site.
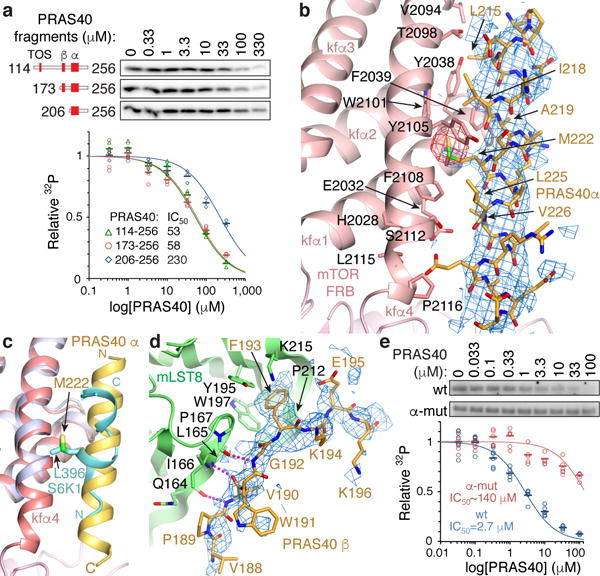
a, Inhibition of mTORΔN-mLST8 (30 nM) phosphorylating S6K1367–404 (10 μM) by indicated PRAS40 fragments (TOS, β strand and amphipathic α helix marked). 32P incorporation plotted as a fraction of the zero PRAS40 reaction of each series, with IC50 curves and values as indicated. Means shown as dashes and values from independent experiments shown with indicated markers and colors (n=3; PRAS40173–256 n=5). b, Close-up view of PRAS40 α helix–FRB interface. 3.4 Å mFo-dFc electron density before PRAS40 was built (blue, 1.8 σ), and anomalous diffraction map (red, 3.5 σ) of SeMet-PRAS40 crystals also shown. c, PRAS40 α helix–FRB interface of b superimposed on FRB (purple)-S6K1 peptide (cyan), highlighting PRAS40 Met222 and S6K1 Leu396. d, Close-up view of PRAS40 β strand–mLST8 interface. 3.4 Å mFo-dFc electron density (blue, 2.0 σ) before PRAS40 was built also shown. e, Inhibition of 20 nM mTORC1 phosphorylating full-length 4EBP1 (5 μM) by w.t. or mutant full-length PRAS40. Quantified and plotted as in a (n=4).
We next determined the 3.4 Å co-crystal structure of PRAS40173–256 bound to mTORΔN-mLST8 (Extended Data Fig. 4f). The structure revealed two PRAS40 anchor points separated by an unstructured segment: an amphipathic α helix (residues 212–232) bound to the FRB domain and a β strand (residues 188–196) bound to the mLST8 WD40 domain (Fig. 1a and Extended Data Fig. 4b).
The amphipathic helix binds to the same FRB site as the S6K1 substrate, but at 5 turns it is substantially longer and makes more extensive contacts than S6K1 (Figs 3b and c; Extended Data Fig. 4c). PRAS40 uses the Met222 side chain to bind to the same rapamycin-binding pocket as the S6K1 Leu396 (Fig. 3c), and five additional hydrophobic side chains (Leu215, Ile 218, Ala219, Leu225 and Val 226) to contact an extended FRB surface.
The PRAS40 β strand has a phenylalanine side chain (Phe193) inserting into a pocket between two mLST8 β propeller blades (Tyr195, Trp197, Pro167, Pro212), while its peptide backbone makes three β sheet hydrogen bonds to the edge of one β propeller (Fig. 3d and Extended Data Fig. 4d). These PRAS40 interactions are consistent with reduced inhibition by PRAS40206–256, which lacks the β strand, compared to PRAS40173–256 (Fig. 3a).
We further confirmed the importance of the amphipathic helix by mutating Met222 and four additional FRB-interacting residues (L215A, I218A, A219G, L225A) in full-length PRAS40. As shown in Figure 3e, the mutations reduced inhibition of mTORC1 phosphorylating full-length 4EBP1 by a factor of ~50.
Cryo-EM structures of active RHEB-mTORC1 and apo-mTORC1
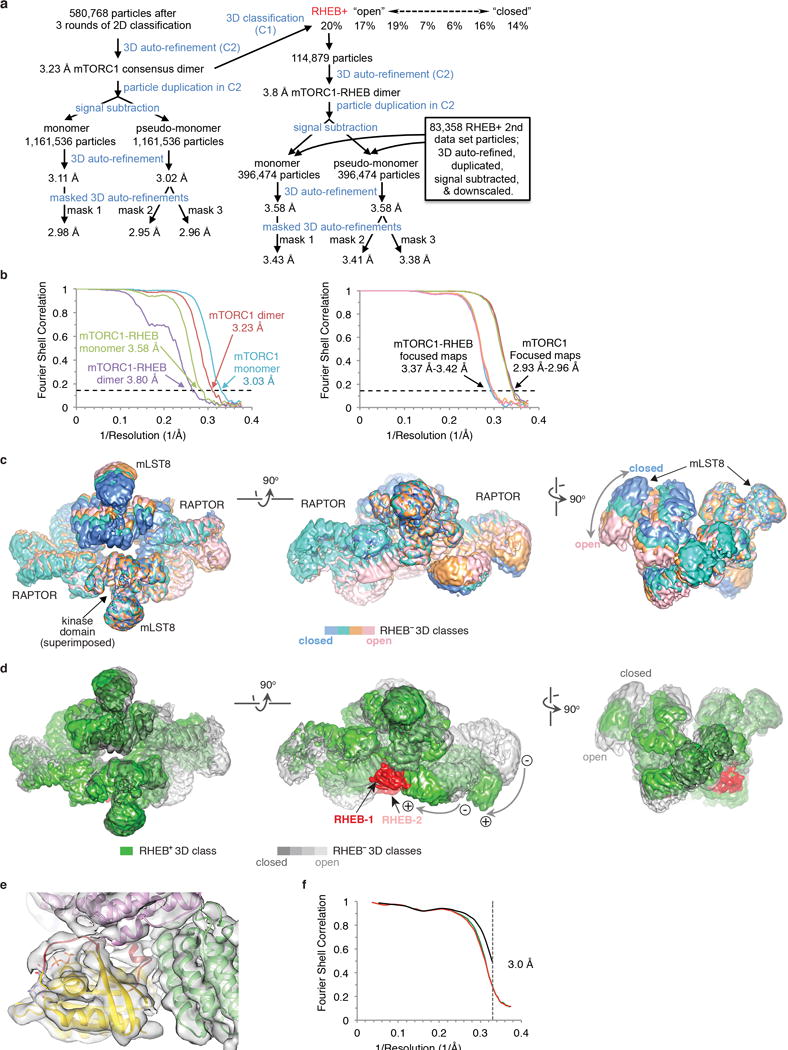
To address how RHEB activates mTORC1, we collected cryo-EM data on mTORC1 that was cross-linked in the presence of excess RHEB-GTPγS and 4EBP1 (Extended Data Fig. 2c). The 3D auto-refinement of 580,768 particles in C2 symmetry led to a consensus reconstruction extending to 3.2 Å resolution, as determined by the gold-standard fourier shell correlation (FSC) procedure32 (Extended Data Figs 5a and b). In subsequent 3D classification in C1, most classes appeared to belong to a continuum of conformational states. One class with ~20 % of the particles had an overall conformation distinct from the ensemble of the other classes, and this was the only class that contained RHEB, one on each mTOR of dimeric mTORC1 (Extended Data Figs 5c to e).
Because of the conformational flexibility in between and within the two mTOR-RAPTOR-mLST8 complexes (Extended Data Fig. 5c), we converted the particles to monomers with partial signal subtraction33, and calculated focused reconstructions with three partially overlapping masks (2.98, 2.95 and 2.96 Å; Extended Data Figs 5a and b). Using these three reconstructions with the composite map option of REFMAC534, we refined mTORC1 at 3.0 Å resolution (Extended Data Figs 2c, 5f, 6 and 7a). Using the same procedure, we refined RHEB-mTORC1 at 3.4 Å (Extended Data Figs 2c, 5a and b).
As with the mTORΔN-mLST8 crystal structure23, the PIKK-specific FAT domain adopts a C-shaped solenoid structure that clamps onto the kinase domain (KD), with the start of the solenoid interacting with the KD N lobe, and its end with both the N and C lobes (Fig. 4a). In keeping with the secondary structures of previous mTOR cryo-EM reconstructions24,35, the N-terminal segment missing from mTORΔN starts with an α–α solenoid of 18 HEAT repeats (N-heat), followed by a smaller middle solenoid of 7.5 HEAT repeats (M-heat), and a ~110 residue helical-repeat segment that is structurally contiguous with the subsequent FAT domain and will henceforth be included in the “FAT” descriptor (Extended Data Figs 6, 7b and c). The FAT solenoid acts as an organizing center of mTOR. In addition to its ends clamping onto the KD N and C lobes, the FAT mid-portion packs with the N-heat solenoid end, and the beginning of the FAT packs with the start of the M-heat solenoid, anchoring the two domains (Fig. 4a; Extended Data Figs 6 and 7d; Supplementary Information discussion).
Figure 4. Cryo-EM structure of RHEB-mTORC1.
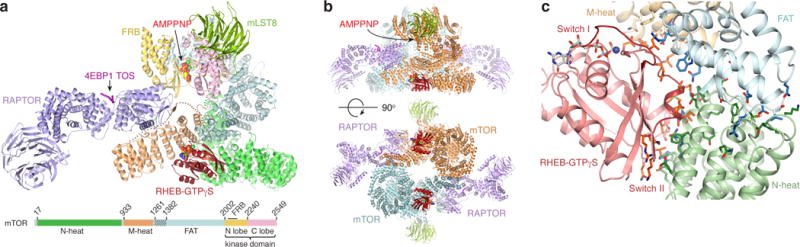
a, One copy of the RHEB-mTOR-RAPTOR-mLST8-4EBP1 complex. mTOR colored according to linear schematic (hatched blue box, extension of conventional FAT boundary), RHEB red, RAPTOR purple, mLST8 green, 4EBP1 TOS magenta. AMPPNP-Mg2+ and GTPγS-Mg2+ in CPK representation (dotted lines, disordered N-heat–M-heat and M-heat–FAT linkers). b, Dimeric mTORC1 oriented similarly to a (top) and rotated as shown (bottom). The two mTOR are cyan and orange, and the other subunits as in a. c, RHEB-GTPγS interface with mTOR N-heat, M-heat and FAT, showing side chains and backbone groups within inter- and intra-molecular contact distance. RHEB is pink, switch regions dark red. FAT residues 1261–1266 omitted for clarity.
The mTORC1 dimer forms through the C-terminal portion of N-heat packing with the M-heat of the second protomer in a reciprocal fashion (Fig. 4b). RAPTOR binds to this interface as well, resulting in a tripartite interface of 5,420 Å2 buried surface area, of which 38 % is from RAPTOR–N-heat, 23 % RAPTOR–M-heat, and 39 % N-heat–M-heat (Extended Data Figs 7e and f).
RHEB binds to the amino-terminal portions of the N-heat, M-heat and FAT domains, forming a 4-way interface (Figs 4a and c). The majority of the contacts are made by the RHEB switch I and switch II regions, which have GTP-dependent conformations as with other small GTPases36. Switch I (residues 33–41) binds to M-heat and FAT, whereas the longer switch II (residues 63–79) interacts with all three mTOR regions (Fig. 4c and Extended Data Fig. 8a).
RHEB induces a conformational change that activates mTORC1 allosterically
In apo-mTORC1, the N-heat RHEB binding site is far away from those on M-heat and FAT, displaced by ~18 Å relative to its position in the RHEB-bound state (Fig. 5a). On RHEB binding, the N-heat solenoid swings in towards M-heat through a ~19° rotation, reconstituting the RHEB-binding site and also inducing new interactions between the N-terminal portions of N-heat and FAT (Figs 4c and 5a; Supplementary Video 1). This causes a conformational change within the FAT domain whose middle portion gets twisted and dragged by the moving N-heat solenoid end that is anchored on it. The intra-FAT conformational change is entirely distinct from the conformational flexibility apo-mTORC1 exhibits (Extended Data Fig. 7f; Supplementary Information discussion). The two conformations are incompatible in a mixed dimer, as this would require a > 20 Å offset in the N-heat portion of the dimerization interface. This explains the lack of single-RHEB 3D classes, and suggests that two RHEB molecules bind to mTORC1 cooperatively. In support, we find that the RHEB-GTPγS response curve of mTORC1 phosphorylating 4EBP1 best fits a Hill slope model with a Hill coefficient of ~2.0 (Fig. 5b; the ~100 μM EC50 in solution likely not reflective of the membrane-surface reaction in vivo2,36).
Figure 5. RHEB allosterically activates the kinase.
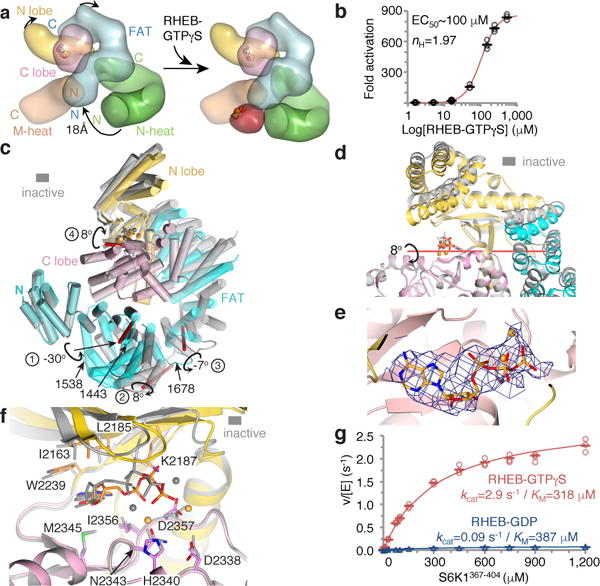
a, Rendering of the inactive (left) and active (right) mTOR structures (colored as in Fig. 4a). Arrows indicate the motions of N-heat, kinase-proximal FAT portion, and N lobe domains. b, Activation of mTORC1 phosphorylating 4EBP1 (10 μM) by RHEB-GTPγS, plotted relative to zero RHEB (means as dashes, three independent experiments as open circles; gel in Extended Data Fig. 8h). Dose-response curve and values also shown. c, FAT-KD portions of active (colored as in a) and inactive (gray) mTOR superimposed on their C lobes. Rotations and their axes (red sticks) and approximate hinge locations (residue numbers) marked. d, Catalytic cleft closure in the inactive-to-active transition. Colored as in c, with orientation rotated ~90° around y (red stick, rotation axis 4 from c). e, Cryo-EM density of the AMPPNP from the 3.4 Å reconstruction of RHEB-mTORC1, looking down vertical axis of f. f, Inactive-to-active transition realigns N lobe residues that contact the ATP relative to the Mg2+ ligands and catalytic residues on the C lobe. g, Steady-state kinetic analysis of mTORC1 phosphorylating S6K1367–404 in presence of 250 μM RHEB-GDP (blue plot) or RHEB-GTPγS (red plot). Data of Extended Data Fig. 8i plotted with means as dashes and values from three independent experiments with indicated markers and colors. KM and kcat values and simulated curves also shown.
The intra-FAT conformational change occurs at hinge regions that allow for relative rotations of flanking segments. One hinge around residue 1443 is associated with a major rotation of 30° between the FAT sub-domains TRD1 and TRD223, while two other hinges exhibit smaller rotations (Fig. 5c). These conformational changes in the FAT are coupled to its C-terminal portion moving away from the kinase, the N lobe of the kinase moving in to the space vacated by the FAT, the FAT–N lobe interface repacking into a looser arrangement, and the catalytic cleft between the N and C lobes closing by 8° (Figs 5c and d; Supplementary Video 2).
The closing of the catalytic cleft changes the relative orientation of the ATP-contacting and catalytic residues from the N and C lobes. This brings the ATP phosphate groups that are bound by the N lobe into closer proximity to critical C lobe residues that include the Mg2+ ligands23 (Asn2343 and Asp2357) and the two catalytic residues23 (Asp2338 and His2340) (Figs 5e and f; Extended Data Figs 8b and c).
This indicates that RHEB activates mTORC1 by allosterically realigning active site residues, bringing them into correct register for catalysis. To confirm this, we compared the steady state kinetic constants of S6K1367–404 phosphorylation by mTORC1 in the presence of 250 μM RHEB-GTPγS or RHEB-GDP. Activation is accounted entirely by a ~30-fold increase in kcat, from 0.09 s−1 to 2.9 s−1, whereas KM values remain essentially unchanged (Fig. 5g). RHEB similarly increased the apparent kcat of full-length 4EBP1 phosphorylation, including under single-turnover conditions, the latter indicating that the kcat effect involves the catalytic step and not a hypothetically rate-limiting product-release step (Extended Data Figs 8d and e). RHEB-GTPγS also accelerated idle ATP hydrolysis, a low-level activity common in protein kinases as well as PI3K37 (Extended Data Fig. 8f).
Cancer-associated hyperactive mTOR mutants
Cancer-associated hyperactivating mutations5,38–40 predominantly involve structure-stabilizing residues. They cluster at the major intra-FAT hinge, the FAT-N lobe packing transition, and the N lobe anchor in a pocket between the C lobe and FAT, suggesting that they act by lowering the barriers to the N lobe adopting the active conformation, mimicking RHEB’s effects (Fig. 6a and Extended Data Figs 9a to c).
Figure 6. Hyperactivating mutations mimic RHEB’s effects.
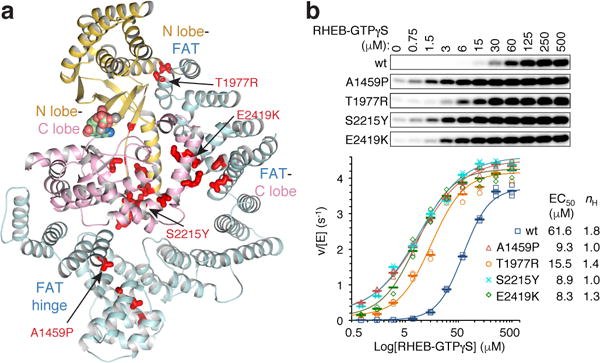
a, Thirty-four cancer-associated hyperactivating mTOR mutations5,38 mapped onto apo-mTORC1 FAT-KD portion, colored as in Figure 4a (thick red sticks, mutated residues; assayed mutants labeled). b, RHEB-GTPγS activating S6K1367–404 (150 μM) phosphorylation by w.t. or indicated mutant mTORC1 (10 nM). Plotted with means as dashes and individual values from three independent experiments with indicated markers and colors. Dose response curves and coefficients also shown.
This hypothesis predicts that the mutations should lower the EC50 of activation by RHEB, as part of the RHEB-mTORC1 binding energy must be used to affect the conformational change, and that they should not synergize with saturating RHEB. To test these predictions, we transiently expressed and purified four representative hyperactive mTORC1 mutants5,38: A1459P in the middle of a helix at the major intra-FAT hinge, T1977R buried at the FAT-N lobe transition, and S2215Y and E2419K at the N lobe-C lobe and juxtaposed C-lobe–FAT interfaces, respectively (Fig. 6a; Extended Data Figs 9a to c).
After confirming that the mutations increase the kcat of mTORC1 phosphorylating S6K1367–404 without affecting the KM (Extended Data Fig. 9d), we assessed their RHEB-GTPγS dose response curves. All four mutations shifted the response curve to lower RHEB concentrations compared to w.t. mTORC1, produced as the mutants (Fig. 6b). A1459P, S2215Y and E2419K reduced the EC50 comparably, by factors of 6.6, 7.0, and 7.4, respectively, whereas T1977R reduced it by a factor of 4.0 (Fig. 6b). Importantly, the mutations did not synergize with RHEB, as at the highest, nearly saturating RHEB concentration, the mutants exhibited S6K1367–404 phosphorylation levels within ~15% of the w.t. control (v/[E] of 3.7 s−1 for w.t., and 4.1–4.4 s−1 for the mutants; Fig. 6b).
The mutations also reduced the cooperativity of RHEB binding, suggesting they allow the formation of single-RHEB containing mTORC1 (Fig. 6b). We presume this is due to the destabilization of the inhibitory FAT clamp, either directly or indirectly at its anchors on the N and C lobes, allowing the FAT-bound N-heat to reach the tripartite dimerization interface of a mixed dimer.
Conclusion
We show that the TOS motif docking site is ~65 Å from the kinase active site, suggesting that it acts to increase the effective substrate concentration, and we establish a second substrate-docking site corresponding to the rapamycin-binding site at the entrance of the catalytic cleft. PRAS40 binds to both substrate-docking sites and an additional site on mLST8 to achieve inhibition. We also show that the low kinase activity of apo-mTORC1 is due to a misalignment of the kinase N and C lobes and their associated ATP-binding and catalytic residues. The FAT clamp, present in all PIKKs, is a key auto-inhibitory element that keeps the N lobe misaligned. RHEB, binding ~60 Å away from the active site, induces a movement of the N-heat domain, which pulls and twists the FAT clamp, freeing the N lobe to adopt the active conformation. The end result of this process is likely mimicked by cancer-associated mutations that activate mTOR.
METHODS
Protein expression and purification
For the FRB-S6K1389–414 fusion protein, a synthetic gene encoding the FRB domain of human mTOR (residues 2018–2114) followed by a three amino acid linker (SGG) and residues 389–414 of human S6K1αII was cloned into a modified pGEX4T3 vector. The fusion protein was overexpressed in the Escherichia coli strain BL21(DE3), and was purified by glutathione affinity chromatography, removal of the GST tag with TEV protease, and fractionation by ion exchange and size exclusion chromatography. The peak fractions were concentrated to 40 mg ml−1 in 20 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 10% glycerol and 0.5 mM tris-(2-carboxyethyl) phosphine (TCEP).
Human S6K1367–404 wild type and mutant peptides were produced in E. coli as GST-tag proteins using a modified pGEX4T3 vector. Following glutathione affinity chromatography and on-bead cleavage with GST-TEV protease, the recombinant peptides were purified by reversed phase high performance liquid chromatography (HPLC) using a Zorbax 300SB-C3 column (Agilent). All other peptides used for kinase assays were purchased from Bio-Synthesis Inc. or Peptide 2.0 and further purified on HPLC using Zorbax 300SB-C3 or C18 columns when necessary. Molecular weights for all purified peptides were verified using MALDI-TOF mass spectrometry. Peptides used in crystallization experiments were purchased from PEPTIDE 2.0. 4EBP1 and S6K1ki and their mutants were prepared as previously described23. For the TOS-mutated S6K1ki, TOS−, residues 5 to 9 (FDIDL) were mutated to alanine, and for 4EBP1TOS−, residues 114 to 118 (FEMDI) were mutated to alanine.
Human eIF4E was produced by infecting High Five insect cells with a pFastBac1 baculovirus expressing the GST-tagged protein and purified similarly as described above, except that 0.1 mM 7-Methyl-GDP (Sigma) was added to the elution buffer during the glutathione affinity chromatography.
PRAS40 fragments were overexpressed and purified as with the FRB-S6K1389–414 fusion protein. For selenomethionine (SeMet) substituted PRAS40 fragments, E. coli transformants were cultured in LB media at 37°C until the OD600 reached 0.8, pelleted, washed, and resuspended in M9 media containing 50 mg l−1 Leu/Ile/Val, 100 mg L−1 Phe/Lys/Thr, and 90 mg L−1 SeMet. The culture was incubated for 30 minutes to shut down methionine biosynthesis. Protein expression was induced with 1 mM IPTG for 3 hours at 37°C.
Full-length human PRAS40 (PRAS40wt) was produced by infecting High Five insect cells with pFastBac1 baculovirus expressing the GST-tagged fusion protein. It was purified by glutathione affinity chromatography, removal of the GST tag with TEV protease, and by anion exchange and size exclusion chromatography. The peak fraction was concentrated to ~1 mM in 20 mM Tris-HCl, pH 8.0, 400 mM NaCl, 10% Glycerol and 10 mM DTT. PRAS40α-mut containing five mutations on it’s FRB binding α helix (L215A, I218A, A219G, M222A and L225A, generated by QuikChange Lightning Multi Site-Directed Mutagenesis kit, Agilent Technologies) was produced similarly.
Arabidopsis thaliana Raptor (Genebank Q93YQ1) containing an internal deletion (residues 883–942) was produced by infecting High Five insect cells with a pFastBac1 baculovirus expressing the GST-tagged protein. It was purified by glutathione affinity chromatography, removal of the GST tag with TEV protease, and by ion exchange and size exclusion chromatography. The peak fraction was concentrated to 7–9 mg ml−1 in 20 mM Tris-HCl, pH 8.0, 400 mM NaCl, 5% Glycerol, 0.5 mM TCEP.
The mTORC1 complex and mTORΔN–mLST8 complex were prepared from stably-transfected cell as previously described23. The cancer-derived mTORC1 mutants and the matched w.t. control were produced by transient transfection. FLAG-tagged human mTOR wild type and four cancer-derived hyperactivating mutants, A1459P, T1977R, S2215Y, and E2419K (generated with QuikChange Lightning Site-Directed Mutagenesis kit, Agilent Technologies), were cloned into the pcDNA3.1(+) vector. The corresponding mTOR plasmids (40 μg per 15 cm plate) were cotransfected with the pcDNA3.1(+)-human mLST8 plasmid (5 μg per 15 cm plate) using Lipofectamine 2000 into a HEK293-F cell line stably overexpressing the FLAG-tagged human RAPTOR. After incubation for 2 days, ~60 plates of cells per complex were harvested by gentle scraping and lysed in 50 mM Tris-HCl, pH 8.0, 500mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% (v/v) glycerol, 2 mM dithiothreitol (DTT) and protease inhibitors using French Press. After lysate clarification, the Flag-tagged proteins were isolated using anti-Flag M2 agarose beads (Sigma), extensively washed with lysis buffer supplemented with 200 mM Li2SO4, and eluted with 0.2 mg ml−1 flag peptide in lysis buffer. They were then further purified by anion exchange chromatography (MonoQ). Peak fractions containing the wild type or mutant mTORC1 complexes were then concentrated to ~1 mg ml−1 (~2 μM) and stored in small aliquots in −80°C.
For the preparation of nucleotide-bound RHEB, RHEB was overexpressed and purified similarly as FRB-S6K1389–414 fusion protein. To charge RHEB with GDP, the protein was first dialyzed overnight against a buffer containing 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 5% Glycerol, 5 mM EDTA, 0.5 mM TCEP, and 100 mg L−1 acid-washed activated charcoal (Sigma). Next, the protein was incubated with 30-fold molar excess of GDP (Sigma-Aldrich) on ice for 30 minutes followed with addition of 15 mM MgCl2. To charge RHEB with GTPγS, the protein was incubated with 30-fold molar excess of GTPγS (Sigma), 20 mM EDTA, and 10 units of alkaline phosphatase (New England BioLabs) per mg of RHEB at 30°C for 1 hour followed with addition of 40 mM MgCl2. Both states of charged RHEB were then purified by size exclusion chromatography (Superdex 75) in 20 mM Bicine, pH 8.0, 200 mM NaCl, 5% glycerol, 5 mM MgCl2 and 0.1 mM TCEP. The peak fractions were concentrated to 25–40 mg ml−1.
Crystallization and X-ray data collection
FRB-S6K1389–414 crystals were grown by the hanging-drop vapor diffusion method at 16°C from 100 mM Bis-tris propane, pH 7.0, 30% Tacsimate (Hampton Research). Crystals were transferred to 100 mM Bis-tris propane, pH 7.0, 6.3 M NaFormate and flash-frozen in liquid nitrogen.
AtRaptor native crystals were grown at 4°C by the hanging drop vapor diffusion method from a crystallization buffer of 100 mM HEPES, pH 7.5, 50 mM Ca(OAc)2, 4–8% PEG 8000, 0.5 mM TCEP. For heavy atom derivatives, crystals were soaked in 0.4 to 0.8 mM thimerosal for 2 hours before cryo-protection (not shown). The atRaptor-TOS complexes were grown by streak seeding native crystals into atRaptor mixed with a 2-fold molar excess of synthetic TOS peptides corresponding to residues 99–118 of human 4EBP1 (RNSPEDKRAGGEESQFEMDI; only underlined residues are ordered in the crystals), residues 1–14 of human S6K1 (MAGVFDIDLDQPED), and residues 124–139 of human PRAS40 (DNGGLFVMDEDATLQD). Crystals were cryo-protected in crystallization buffer supplemented with 20% PEG 400 and flash-frozen in liquid nitrogen.
For the initial PRAS40–mTORΔN–mLST8 and SeMet-PRAS40 containing complex, 30 μM mTORΔN–mLST8 was incubated with 0.2 mM of truncated PRAS40, followed by crystallization by the hanging-drop vapor diffusion method. Co-crystals of the complex grew in the same condition as the apo mTORΔN–mLST8 crystals23, and subsequent PRAS40 crystals were prepared by soaking apo mTORΔN–mLST8 crystals with 0.5mM PRAS40 fragments. Crystals were cryo-protected as described23.
All diffraction data were collected at −170°C at the ID24C or ID24E beamlines of the Advanced Photon Source. Crystals containing SeMet were collected at the Se edge using the inverse beam strategy. Data were processed with the HKL suite41.
Crystal structure determination and refinement
For the FRB-S6K1 structure, initial phases were obtained by molecular replacement with PHASER42 using the FRB structure (PDB 1FAP) as the search model. The FRB-S6K1 and other crystal structures described in this study were built using O43 and refined with REFMAC542 and PHENIX44. The FRB-S6K1 model has a Molprobity clashscore of 0.36. The Ramachandran plot has 99.8, 0.2, and 0 % of the residues in the favored, allowed and outlier regions, respectively.
For atRaptor, initial phases were calculated from two thimerosal derivatives using both isomorphous and dispersive differences. The atRaptor used in crystallization has an internal deletion of residues 883–942, a region that is highly susceptible to proteolysis. In the crystals the residues 866–882 and 943–956 flanking the internal deletion, and residues 736–830 are disordered. The corresponding regions are also disordered in the 3.0 Å-refined human RAPTOR structure from our cryo-EM reconstruction. The atRaptor model has a Molprobity clashscore of 4.77. The Ramachandran plot has 89.66, 7.75 and 2.59 % of the residues in the favored, allowed and outlier regions, respectively.
The PRAS40–mTORΔN–mLST8 crystal structures were solved by molecular replacement using the mTORΔN–mLST8 structure23 as a search model. The model has a Molprobity clashscore of 1.92, and the Ramachandran plot has 93.74, 5.3 and 0.95 % of the residues in the favored, allowed and outlier regions, respectively.
Cryo-EM sample preparation and data collection
Cryo-EM samples were prepared using two different crosslinking procedures. In the initial, high-salt crosslinking procedure, 0.42 μM mTORC1 was incubated with 250 μM RHEB-GTPγS, 10 μM 4EBP1, 0.2 mM GTPγS, 1 mM AMP-PNP for 10 minutes, and crosslinked with 0.18 % (v/v) glutaraldehyde for 45 minutes on ice in 20 mM Bicine, pH 8.0, 300 mM NaCl, 10% glycerol, 5 mM MgCl2, and 0.5 mM TCEP. The reaction was quenched with 100 mM Tris-HCl, pH 8.0 and the mixture was purified by size exclusion chromatography (Superose 6) in 20 mM Tris-HCl, pH 8.0, 260 mM NaCl, 5 mM MgCl2 and 0.1 mM TCEP. Peak fractions were concentrated by ultrafiltration to 1 mg ml−1 and were supplemented with 100 μM RHEB-GTPγS, 10 μM 4EBP1, 0.2 mM GTPγS and 1 mM AMPPNP. The sample (3 μl) was applied to glow discharged UltrAuFoil 300 mesh R1.2/1.3 grids (Quantifoil). Grids were blotted for 2.2 s at 22°C and ~95% humidity and plunge-frozen in liquid ethane using a FEI Vitrobot Mark IV.
After determining that high ionic strength severely reduced RHEB activation (not shown), we reduced the salt concentration and modified the crosslinking procedure by first reducing the concentrations of mTORC1, RHEB-GTPγS and NaCl to 0.21 μM, 120 μM and 100 mM, respectively, then crosslinking with 0.24 mM BS3 for 45 minutes on ice, followed by the addition of 130 μM RHEB and 160 mM NaCl and further crosslinking with 0.18 % of glutaraldehyde for 45 minutes. Quenching and purification were performed as with the high-salt procedure in 260 mM NaCl. The main data set used for apo-mTORC1 reconstruction was collected at the NYSBC Simons Electron Microscopy Center using LEGINON45 on a Titan Krios microscope operated at 300 kV and equipped with a Gatan K2 Summit camera using defocus values ranging from −1.2 to −3μm. The camera was operated in counting mode with a 1.331 Å pixel size at the specimen level and a dose rate of 8.3 electrons per pixel per second. Each 12 second exposure was dose-fractionated into 60 frames and contained a total dose of ~56 e− per Å2. This dataset, which was acquired over 7 sessions, consisted of 4,502 micrographs from high-salt crosslinked samples and 3740 from lower-salt crosslinked samples. The second data set, which employed only the lower-salt crosslinking condition, was collected at the Sloan-Kettering Institute Titan Krios microscope/Gatan K2 Summit camera operated at 300 kV with a 1.089 Å pixel size and 8.0 electrons per pixel per second. Each 8 second exposure was dose-fractionated into 40 frames and contained a total dose of ~52 e− per Å2. This data set consisted of 8,354 micrographs.
Cryo-EM image processing
Motion correction was performed with MOTIONCORR and MOTIONCORR-246 for the first and second data sets, respectively. Contrast transfer function parameters were estimated with CTFFIND447, and subsequent 2D/3D classifications and 3D refinements were carried out with RELION-1.448 and RELION-2.049. All reported resolutions are from gold-standard refinement procedures with the FSC=0.143 criterion32 after post-processing by applying a soft mask, correction for the modulation transfer function (MTF) of the detector, temperature-factor sharpening, and correction of FSC curves to account for the effects of the soft mask as implemented in RELION48,49. Initial references for template-based particle picking were from 2D class averages of manually picked particles. Multiple rounds of 2D and, for some data subsets, 3D classifications were then used to remove false positives and particles that clustered in classes with poorly determined orientations. For the main data set, a total of 580,768 particles were retained (Extended Data Fig. 5a). After an initial 3D auto-refinement with C2 symmetry, the particles were improved by particle-based motion correction and radiation-damage weighting50. The resulting ‘polished’ particles were used for the 3D auto-refinement of a consensus mTORC1 dimer map in C2, yielding a 3.23Å reconstruction (Extended Data Fig. 5a). After 3D classification in point group C1, 114,879 (19.7 %) of the polished particles clustered to a single RHEB-containing class. The fraction of RHEB-containing particles was ~2.5 fold higher in the low-salt crosslinked particles compared to the high-salt ones. The remaining 6 apo-mTORC1 3D classes appeared to sample a continuum of conformational flexibility, both between and within the two mTOR-RAPTOR-mLST8 complexes (Extended Data Fig. 5a). Because of this conformational flexibility, we converted the particles to a “monomeric” form (illustrated in Extended Data Fig. 5a). For this, we duplicated the particle list and advanced the RELION rot angle by 180° to extract the signal of the 2nd copy of the complex superimposed on the first. We then subtracted the signal from the masked map of the second mTOR-RAPTOR-mLST8 complex in the consensus reconstruction (before post-processing) from each particle in the combined set as described33. 3D auto-refinement then yielded a monomeric mTORC1 consensus reconstruction to 3.11 Å. An alternate, “pseudo-monomer” set of particles was calculated by switching the two N-heat domains, as this domain seemed to move more relative to its own mTOR than that of the second protomer. 3D auto-refinement of the “pseudo-monomer” yielded a reconstruction to 3.02 Å. Upon further inspection of the modes of flexibility in 3D classification of the “monomeric” complexes, three partially overlapping soft masks were constructed for masked 3D auto-refinements with local searches of orientation angles. One focused on N-heat (17–903), FAT-KD (1261–2549), the KD-proximal M-heat portion (933–1005), and mLST8 of the actual monomer (mask1 in Extended Data Fig. 5a), and produced a reconstruction to 2.98 Å. The second used the pseudo-monomer focusing on N-heat of protomer 2, RAPTOR, and the RAPTOR-proximal M-heat portion of protomer 1 (1006–1222), and produced a reconstruction to 2.95 Å (mask2 in Extended Data Fig. 5a). The third also used the pseudo-monomer, focusing on N-heat, a smaller portion of the proximal M-heat (1027–1222), and nearby segments of RAPTOR (195–214, 237–255, 274–503), to 2.96 Å (mask3 in Extended Data Fig. 5a). This third reconstruction had the best density for N-heat. The same procedure of particle duplication and signal subtraction was used to convert the 114,879 RHEB-mTORC1 particles to monomer and pseudo-monomer forms (Extended Data Fig. 5a). These two sets of particles were combined with the RHEB-containing monomer and pseudo-monomer particles of the second data set that were processed similarly and which were down-scaled in fourier space with RELION to match the magnification of the first data set. The optimal scale factor was determined by comparing the KD C lobe portion of the refined structures from the two data sets to the crystal structure of mTORΔN–mLST8, minimizing the sum of differences squared in the inter-Cα distances. The combined total of 396,474 RHEB-containing particle sets were then 3D auto-refined as with the consensus structure, except for the inclusion of RHEB bound to M-heat mask 1, and in masks 2 and 3 the inclusion of RHEB and the minimal RHEB-interacting M-heat and FAT elements of protomer 2. The dimeric mTORC1-RHEB reconstruction was to 3.8 Å, the monomers to 3.58 Å, and the three focused 3D refinements to 3.43, 3.41 and 3.38Å (Extended Data Fig. 5a). The inclusion of the down-scaled particles from the second data set improved the resolution limits only marginally, but the maps had better continuity, especially in the relatively less ordered regions.
Cryo-EM structure refinement
Model refinement was done with REFMAC5 modified for cryo-EM34, with a composite map of the three focused 3D reconstructions assigned to the following coordinates: N-heat, and for RHEB-mTORC1, RHEB-GTPγS and minimal RHEB-interacting M-heat and FAT portions (both duplicated) to 3rd focused map; M-heat (961–1222), RAPTOR, TOS, and RAPTOR-bound β strand from M-heat FAT linker to 2nd focused map; M-heat (933–960), FAT-KD (1261–2549), AMPPNP, mLST8, and for RHEB-mTORC1, RHEB-GTPγS and minimal RHEB-interacting regions of N-heat (duplicated) of protomer 2 to 3rd focused map. The three focused maps were aligned on the corresponding regions of the consensus C2 map by first obtaining the rotation-translation matrix with CHIMERA51 and then applying the transformation with CCP442. The resulting composite maps were used for building a model of the monomeric complex using O43, and for refinement with REFMAC542 and PHENIX44. The apo-mTORC1 monomer was refined to 3.0 Å with weak secondary structure restraints (SSR) generated by ProSMART52, and the RHEB-mTORC1 monomer to 3.4 Å with tighter SSRs. RHEB was built based on the published structure36. Validation refinement was done as described34. To refine the dimeric complexes, two copies of the three focused maps of each complex were aligned on the corresponding C2 maps as above. The six resulting maps for each complex were then combined with the composite sfcalc option of REFMAC5 to construct the high-resolution structure factors as described for the monomers. The dimeric apo-mTORC1 and RHEB-mTORC1 models were refined against these structure factors using tight non-crystallographic symmetry (n.c.s.) restraints for the positions and B-factors of the atoms.
In vitro kinase assays
In vitro kinase assays were performed as described23, except reaction duration was 20 minutes. Briefly, reactants were assembled in a buffer of 25 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 2 mM DTT, 5 % (v/v) glycerol and allowed to incubate on ice for 5 minutes. Reactions were started by the addition of cold ATP (0.5 mM final concentration except for Extended Data Fig. 8f, which were added at the indicated concentrations) supplemented with 2 to 4 μCi [γ-32P] ATP (6000 Ci/mmol, Perkin-Elmer) per reaction. Reactions with RHEB-GTPγS or RHEB-GDP were supplemented with 200 μM of the corresponding nucleoside. For reactions with short peptide substrates (Fig. 1g), 0.01% Triton X-100 (Sigma) was added to reduce non-specific interaction of peptides with the test tube. Reactions with peptide substrates were resolved on 16% or 19% Tricine-urea-SDS-PAGE gels53. The Ki values of PRAS40 fragments inhibiting S6K1367–404 phosphorylation shown in Figure 3a were calculated using the IC50 values, the 10 μM S6K1367–404 substrate concentration, and the 430 μM KM of this substrate peptide (Extended Data Fig. 1b) according to the competitive inhibitor equation Ki = IC50/(1+([S]/KM)).
In vivo S6K1 phosphorylation assay
HA-S6K1ki wild type and mutants were cloned into the pcDNA3.1(+) vector. HEK-293F cells (Invitrogen) were maintained in DMEM medium with 10% fetal bovine serum (Sigma) at 37°C and 5% CO2. For transfection, cells were seeded into 6-well tissue culture plates, cultured to 70% confluence and exchanged into fresh media one hour prior to transfection. Cells were transfected with 2 μg each of HA-S6K1ki wild type or mutant plasmids using Lipofectamine 2000 (Invitrogen). 48 hours post transfection, cells were lysed in 50 mM Tris-HCl, 150 mM NaCl, 0.5 mM TCEP, 1% Triton-X100, 2 mM EDTA, 50 mM NaF, 10 mM β-glycerolphosphate and 10 mM Na-pyrophosphate, pH 7.5, and 1 tablet each of cOmplete protease inhibitor and PhosSTOP cocktail (Roche). Whole cell extract (W.C.E.) were adjusted to 2 mg ml−1 with lysis buffer and NuPAGE LDS sample loading buffer (Invitrogen), and boiled for 5 minutes. 20 μg W.C.E. were loaded on gel for immunoblotting with anti-HA antibody (Santa Cruz, SC805) or anti-phospho-S6K1 (T389) antibody (Cell signaling, #9205). The immunoblots were quantified by normalizing the anti-phospho-S6K1 signal to the anti-HA signal of each reaction.
ATPase assay
The ATP hydrolysis assays were set up similarly as the in vitro kinase assays, except without mTOR substrates. To vary the final ATP concentrations, cold ATP was serially diluted and supplemented with [γ-32P] ATP (4 μCi per reaction). The reaction was initiated by mixing the ATP with the enzyme (10 μl total volume), incubated for 20 minutes at 30°C, and stopped by adding 10 μl of 2 M formic acid. 2 μl of each reaction was then spotted on a PEI Cellulose TLC plate (Millipore), developed in 1 M formic acid and 0.5 M LiCl, dried, and quantified by phosphorimaging.
Fluorescence Polarization
FITC-labeled TOS peptides were purchased from Peptide 2.0 Inc. Peptides were quantified by A495 by >20 fold dilution in 10 mM Tris-HCl, pH 8.0, using an extinction coefficient of 75,000 cm−1M−1. A series of 60 μL binding conditions using serially diluted protein with 20 nM FITC-labeled TOS peptides were set up in buffer consisting of 10 mM Tris-HCl, 100 mM NaCl, 2.5% Glycerol and 1 mM TCEP, pH 8.0. Each binding condition was set up in triplicates and equilibrated at room temperature for 15 min. The fluorescence anisotropy measurements were taken with a Cary Eclipse Fluorescence Spectrophotometer with automated polarization accessory (Agilent Technologies), using 485 nm excitation (5 nm slit) and 512 nm emission (10 nm slit) wavelengths, and G factor of 1.5111. The apparent dissociation constants (Kd) values were obtained by fitting the data to a one-site binding model, by minimizing the sum of square of the differences.
Data availability
The cryo-EM maps, including the three focused reconstruction maps and the structure factors of their composite map used in model refinement, and the refined atomic models have been deposited with the Electron Microscopy Data Bank and the Protein Data Bank with accession numbers EMDB-7087 and PDB 6BCX for apo-mTORC1 and EMDB-7086 and PDB 6BCU for RHEB-mTORC1. The coordinates and structure factors of the FRB-S6K1 complex (5WBH), atRaptor (5WBI), atRaptor-4EBP1TOS (5WBJ), atRaptor-S6K1TOS (5WBK), atRaptor-PRAS40TOS (5WBL), mTORΔN-mLST8-PRAS40173–256 (5WBU), mTORΔN-mLST8-PRAS40114–207 (5WBU) have been deposited with the Protein Data Bank.
Extended Data
Extended Data Figure 1. Substrate recruitment by the FRB domain.
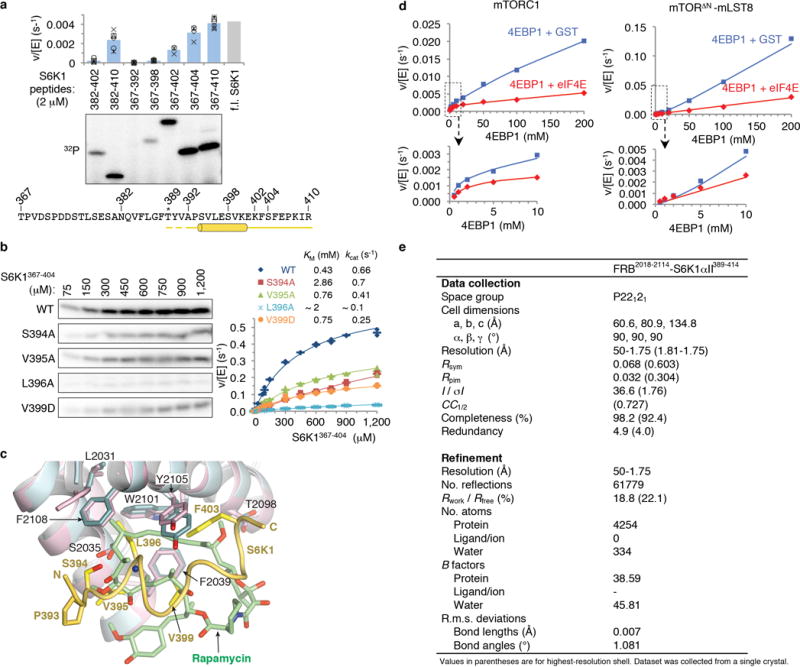
a, Deletion mapping of S6K1 FRB-binding motif polypeptides (2 μM) using phosphorylation by the mTORΔN-mLST8 (30 nM) as the assay, extending previous findings1. Truncation up-to 7 residues N-terminal to the Thr389 phosphorylation site (indicated by asterisk) has minimal effect, whereas C-terminal truncations starting 15 residues from Thr389 successively reduce phosphorylation. The polypeptides were produced as described in Methods for the S6K1367–404 peptide. Column graph shows velocity divided by enzyme concentration from the quantitation of the 32P autoradiogram. Columns show means and markers show values from independent experiments (n=6 for 382–410, 367–392, 367–398 and 367–410 reactions, n=5 for 382–402, n=4 for 367–404, and n=3 for 367–402). The column labeled f.l. S6K1 shows the phosphorylation level of full-length S6K1ki (ki superscript indicates the kinase-inactive K100R mutant) under the same conditions, as reported by Yang et al.1. Truncation of the S6K1 peptide to 20 residues (S6K1382–402), which is the standardized length used in the peptide library of Figure 1g, reduces phosphorylation to ~20 % of S6K1367–404, likely in part because of end-effects destabilizing the helix as well as eliminating some minor contacts. b, Michaelis-Menten steady state kinetic constants for mTORΔN-mLST8 (30 nM) phosphorylating wild type and the indicated mutant S6K1367–404 peptides, quantified as in a. Graph shows means (dashes) and values (markers as indicated) from independent experiments (n=3, except for the 1, 2, 10, 750, 900 and 1,200 μM points which are n=2). Also shown are the KM and kcat values, calculated by non-linear regression fitting of the data, above the graph, and their simulated curves in the graph. Mutations that significantly reduced phosphorylation but do not make substantial direct FRB contacts include S394A, which eliminates a hydrogen bond that stabilizes the helix N-terminus, and the helix breaking V395G mutation that further reduces phosphorylation compared to V395A (Fig. 1d); together, these point to the importance of the helical conformation. Additional mutations include Val391 and Pro393, in the segment between Thr389 and the start of the helix. Pro393 may be important for guiding the FRB-anchored substrate to the kinase active site, and that the P+2 residue Val391 may be involved in contacts to the kinase C lobe, where, by analogy to canonical kinases, the peptide segment of the phosphorylation site and its immediate vicinity are expected to bind to. c, Superposition of the FRB-S6K1 interface onto the FRB-rapamycin-FKBP12 structure2 (FKBP12 is omitted from clarity), highlighting the similarities in the binding of the Leu396 side chain to the same pocket as rapamycin’s key C23 methyl group and flanking portions of its triene arm. The FRB-S6K1 interface is colored as in Figure 1b, rapamycin green and its associated FRB domain cyan. In the crystals of the FRB-S6K1389–414 fusion protein, S6K1 residues 389–391 and 411–414 are disordered, while residues 405–410 are involved in crystal packing. d, Graph with the quantitation of the reactions shown in Figure 1h. e, X-ray data collection and refinement statistics for the FRB2018–2114-S6K1αII389–414 fusion protein structure.
Extended Data Figure 2. RAPTOR-4EBP1 TOS interface and density in the Cryo-EM structure of mTORC1.
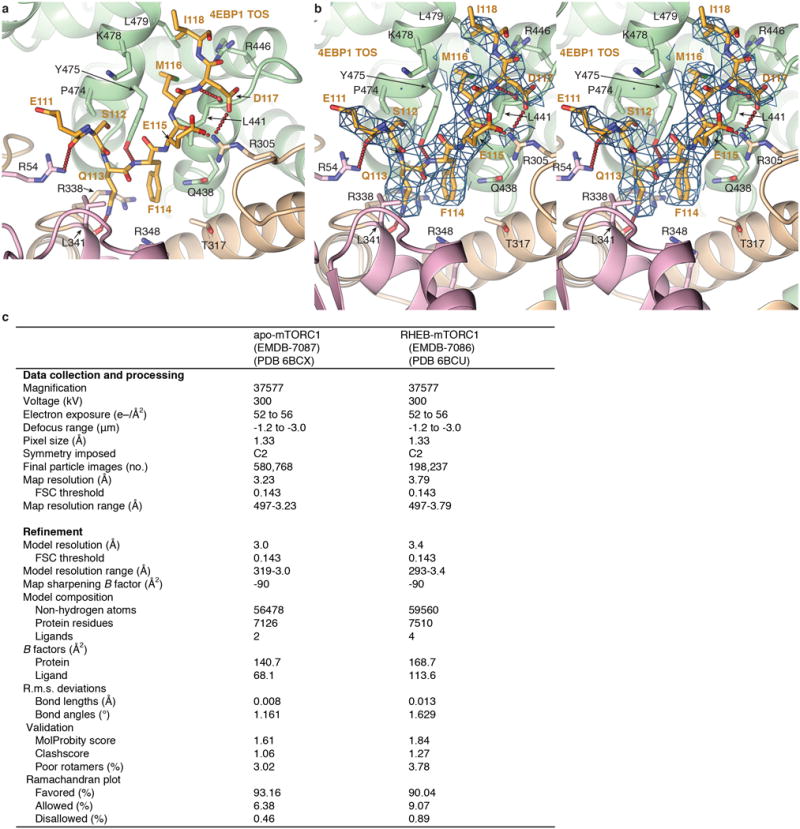
a, Human RAPTOR-4EBP1 TOS interface from the 3.0 Å refined human mTORC1 cryo-EM structure, colored as in Figure 2b. Only the RAPTOR side chains that make hydrogen bond (red dotted lines), electrostatic or van der Waals contacts to 4EBP1 are shown. b, Stereo view of the cryo-EM density of the human RAPTOR-4EBP1 TOS interface. Although the complex contained full-length 4EBP1, only an 8-residue segment is ordered in the cryo-EM reconstruction (also see Extended Data Fig. 7a). c, Cryo-EM data collection and refinement statistics.
Extended Data Figure 3. AtRaptor-TOS motif crystal structures and human RAPTOR-TOS motif dissociation constants.
a, Stereo view of the mFo-dFc electron density of the atRaptor-4EBP199–118 co-crystals, calculated with phases from atRaptor before any 4EBP1 was built into the model. The structure is colored as in Figure 2b, and the map, calculated at 3.0 Å and contoured at 2.2 σ, is shown as blue mesh. Of 20-residues in the 4EBP1 peptide in the crystals, only the 8-residue segment shown is ordered. b, Stereo view of the mFo-dFc electron density, calculated as in a, of the atRaptor-S6K11–14 co-crystals. The 3.1 Å map is contoured at 2.2 σ. As with the 4EBP1 co-crystals, only 8 of the 14 S6K1 residues are ordered. c, Stereo view of the mFo-dFc electron density, calculated as in a, of the atRaptor-PRAS40124–139 co-crystals. The 3.35 Å map is contoured at 1.9 σ. Only 8 of the 16 PRAS40 residues are ordered. In addition, side chain of Glu133 has poor density and is only tentatively built. d, Molecular surface representation of the atRaptor TOS-binding groove, colored according to the electrostatic potential (–6 to +6 kT) calculated without the 4EBP1 peptide (shown as sticks) using APBS3 and illustrated with PyMOL. The TOS-binding groove has an overall basic electrostatic potential owing to five arginine and one lysine residues, explaining the tendency of acidic residues to be present at the non-conserved and flanking positions of the TOS motif. e, Binding of the indicated human 4EBP1 TOS mutant peptides (mutation in red) to human RAPTOR measured by fluorescence polarization anisotropy. Graph shows means as dashes and values from three independent experiments with the indicated markers and colors. Dissociation constants from the non-linear regression fitting of the data are also shown, and simulated binding curves are overlaid on the data. f, Binding of the TOS motif peptides of human 4EBP1 (blue), S6K1 (red) and PRAS40 (green) to human RAPTOR measured by fluorescence polarization anisotropy. Graph shows means as dashes and values from three independent experiments with the indicated markers and colors. Also shown are the dissociation constants from the non-linear regression fitting of the data and the simulated binding curves.
Extended Data Figure 4. PRAS40 is a competitive inhibitor of the FRB substrate-recruitment site and additionally binds to mLST8.
a, Inhibition of mTORΔN-mLST8 (30 nM) phosphorylating the TOS-less 4EBP142–64 (10 μM) by indicated PRAS40 fragments (red rectangles mark TOS, β strand and amphipathic α helix). 32P incorporation is plotted as a fraction of the zero PRAS40 reaction of each series, with means as dashes and values from independent experiments with the indicated markers and colors (n=3 for PRAS40206–256; n=2 for PRAS40114–256 and PRAS40173–256). IC50 values from the non-linear regression fitting of the data and their simulated curves are also shown. b, Co-crystal structure of the PRAS40173–256–mTORΔN–mLST8 complex. The mTOR FAT domain is colored cyan, FRB salmon, kinase pink, mLST8 green and PRAS40 red. c, Anomalous diffraction fourier map (red mesh) of crystals containing the SeMet-substituted L225M mutant of PRAS40206–256 bound to mTORΔN-mLST8 showing two SeMet peaks that confirm the direction of the PRAS40 helix. The 5.4 Å map is contoured at 3.5 σ and superimposed on the structure of the wild type complex, which has a slightly different unit cell from the SeMet crystals. The anomalous diffraction map of wild type seleno-methionine substituted PRAS40 co-crystals is shown in the main text (Fig. 3b). d, mFo-dFc electron density of PRAS40114–207, which contains only one of the three phenylalanine residues present in the PRAS40173–256 polypeptide, bound to mTORΔN-mLST8 confirming the sequence assignment of the PRAS40 beta strand. Map was calculated with phases from before PRAS40 was built into the model. The structure and map, calculated at 3.0 Å and contoured at 2 σ, are colored as in Figure 3d. e, Phosphorylation of full-length 4EBP1 by apo-mTORC1 (left panel) does not obey Michaelis-Menten kinetics, and in the absence of a single substrate KM value we cannot calculate the Ki of full-length PRAS40 for the reaction of Figure 3e. Left panel shows the 32P incorporation data, plotted as velocity over enzyme concentration (means ±s.d., n=11 except for the 10, 50 and 200 μM reactions that are n=6), for the curve of apo-mTORC1 phosphorylating full-length 4EBP1. The curve of reaction velocity/[enzyme] versus substrate concentration is parabolic, with product levels comparatively higher at low substrate concentration (up-to around 10 μM) and lower at higher substrate concentration than a Michaelis-Menten type response. The two substrate concentration ranges display very different KM and kcat values. The entire substrate range can thus be modeled (black curve) as the sum of two reactions, one having a tight KM of ~2 μM but very slow kcat of 0.003 s−1 (blue dashed curve), and another reaction of a weak KM of 545 μM but a faster kcat of 0.065 s−1 (green dashed curve). The tight KM reaction is dependent on the TOS motif, as its mutation (right panel; graph shows means as dashes and values from three independent experiments) results in a reaction that obeys Michaelis-Menten kinetics, with KM and kcat values of 462 μM and 0.053 s−1, respectively, that are very similar to the values of the weak KM faster kcat curve of w.t. 4EBP1. We presume that the TOS-independent weak KM reaction reflects, in part, substrate interactions with the FRB. It is possible that the non-Michaelis-Menten behavior is due to the presence of multiple, likely non-equivalent phosphorylation sites on 4EBP1. We also cannot reliably measure the KM value of full-length S6K1, as this substrate aggregates and then precipitates at concentrations higher than ~50 μM, before reaching saturation (not shown). f, X-ray data collection and refinement statistics for the structures of mTORΔN-mLST8 bound to PRAS40 fragments.
Extended Data Figure 5. Cryo-EM reconstruction of mTORC1 and RHEB-mTORC1 complexes.
a, Flow chart of single particle cryo-EM data processing. Details are described in Methods. b, Left graph shows gold-standard FSC plots between two independently refined half-maps for the consensus mTORC1 C2 reconstruction (red curve), the masked monomer with signal subtraction (blue curve), the RHEB-mTORC1 C2 reconstruction (purple curve), the masked RHEB-mTORC1 monomer with signal subtraction containing additional signal-subtracted particles from a second, downscaled data set (green curve). The FSC cutoff of 0.143 and associated resolution for each plot are marked. Right graph shows gold-standard FSC plots for the three focused refinements of the consensus mTORC1 C2 reconstruction with mask 1 (purple curve), mask 2 (red curve) and mask 3 (green curve) described in Methods. Also shown are the corresponding curves for RHEB-mTORC1 masks 1, 2, 3 in cyan, orange and pink. The FSC cutoff of 0.143 and associated resolution ranges of the three masked refinements for consensus mTORC1 and RHEB-mTORC1 are marked on each plot. c, The four largest RHEB-less 3D classes of the flowchart (a) superimposed on the C-lobe of the kinase domain (marked by arrow) of one mTOR protomer. The closest approach of the two mLST8 subunits ranges from 123 Å (“closed” conformation in light blue) to 145 Å (“open” conformation in pink). The RHEB-containing class and two minor classes of suboptimal density are omitted for clarity. The four classes shown appear to represent samples along a continuum of conformations between the open and closed states, as more intermediate states get populated in a 3D classification with a larger number of classes (not shown). The 3D classes shown are from a calculation with a partial data set ~50% the size of the final data set. d, Comparison of the RHEB-containing class (green, with the RHEB density in red) with the RHEB-less classes (colored light gray to dark gray). The maps are superimposed as in c. The RHEB-containing class has two RHEB molecules with very similar density, even though the 3D classification was done in C1. In the figure, one of the two RHEBs is occluded in the left and right panels, and is in lighter background (labeled as RHEB-2) in the middle panel. None of the RHEB-less classes has any significant density at either of the two RHEB-binding sites. The RHEB-containing class has an inter-mLST8 distance intermediate of the open and closed conformations of the RHEB-less classes, but the relative positions of N-heat and its associated RAPTOR are distinct from the apparent continuum of conformational states of the RHEB-less classes. Curved arrows indicate the transitions from the RHEB-less classes (“minus” sign) to the RHEB-containing (“plus” sign) class. e, Cryo-EM density of the RHEB-containing particles from the 3D classification showing cartoon representations of the refined RHEB-GTPγS structure (yellow, its switch I and II segments in red,) and the RHEB-interacting portions of the mTOR N-heat (green) M-heat (pink) and FAT (pink) segments. f, FSC plots of the final model versus the composite cryo-EM map from REFMAC5 (black), and of a model validation protocol4 refining against one of two half maps after an initial random displacement of atoms is applied to the model to remove model bias (FSCwork in red), and cross-validating the same model against the other half map (FSCfree green).
Extended Data Figure 6. Secondary structure and conservation of mTOR.

Human mTOR sequence showing conservation from yeast to man (blue column graph above sequence) and secondary structure elements in the refined model. Helices are indicated as rectangles, β strands as arrows, segments lacking regular secondary structure as solid lines, and disordered regions as dashed lines. N-heat secondary structure is colored green, M-heat orange, FAT in cyan (including helices fα1 to fα6 that are continuous with the FAT structure, even though they are outside the FAT boundary defined by sequence conservation in PIKKs), the kinase domain N-lobe in yellow (FRB helices are name kfα1 to kfα4 for consistency with the mTORΔN-mLST8 paper) and C-lobe in pink. The three hatched N-heat helices have not been assigned a sequence, and their boundaries are indicated tentatively. The dashed-lines indicate disordered regions.
Extended Data Figure 7. Apo-mTORC1 cryo-EM density, interfaces and conformational flexibility.
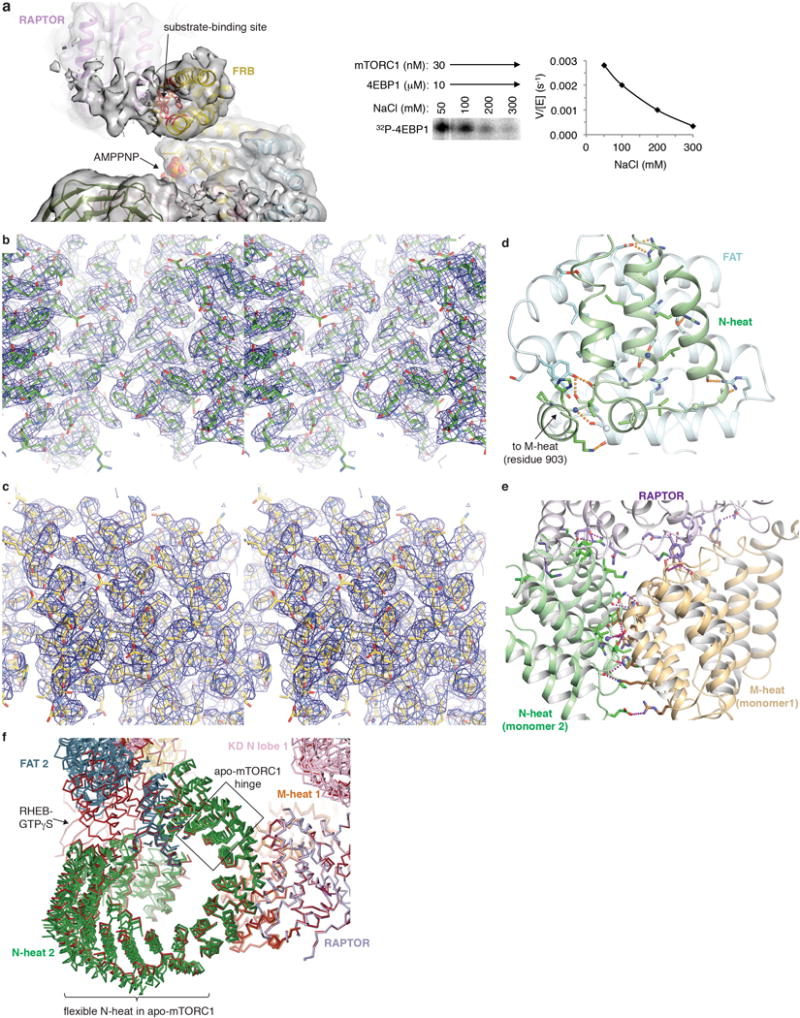
a, The 4EBP1 amphipathic helix density on the FRB does not have interpretable cryo-EM density. The map shown is of a 3D class (~30 % of particles) with the highest relative level of density; the FRB side chains that contact the S6K1 substrate in Figure 1c are shown as red sticks to map the site, the rest of mTOR is colored as in Figure 4a, and AMPPNP is in space-filling representation. Unlike the 4EBP1 TOS, which has a level of density comparable to that of its binding site (Extended Data Fig. 2b), the putative density of the 4EBP1 amphipathic helix is much weaker than that of the FRB, suggestive of partial occupancy. We presume this is due, in part, to our cryo-EM samples containing 260 mM NaCl, which substantially reduces 4EBP1 phosphorylation and thus likely further weakens substrate-FRB association (right panel). b, Stereo view of the 3.0 Å cryo-EM density of the consensus apo-mTORC1 reconstruction, showing mTOR N-heat (residues ~650–850 shown in stick representation colored half-bonded green, red and blue for C, O, N atoms, respectively). c, Stereo view of the 3.0 Å cryo-EM density of the consensus apo-mTORC1 reconstruction showing mTOR M-heat (residues ~960–1105 shown in stick representation colored half-bonded sand, red and blue for C, O, N atoms, respectively). d, The end of the N-heat solenoid (residues 848–898; green) is anchored on the middle of the FAT domain (residues 1565–1627; light cyan). Close-up view showing side chains (glycine Cα atoms as spheres) and backbone groups (blue spheres for amide and sticks for carbonyl groups) involved in intra-molecular van der Waals or hydrogen bond (yellow dotted lines). For clarity, only N-heat residues 836–903 and FAT residues 1537–1664 are shown. e, The mTOR binding elements of RAPTOR (purple) are encompassed within its conserved RNC5, with the caspase domain contacting M-heat of one mTOR protomer (sand), the caspase insertion contacting both M-heat and the N-heat (green) of the other mTOR protomer, and the first three armadillo repeats of its solenoid contacting N-heat. Side chains are shown as in d. Hydrogen bond contacts are shown as red dotted lines. N-heat and M-heat structural elements above the plane of the figure are omitted for clarity. f, The conformational flexibility of apo-mTORC1 is associated with bending at a major hinge region of three heat repeats (indicated by a box) in the N-heat solenoid, in between its FAT and RAPTOR-M-heat interacting segments. Figure shows Cα trace of the tripartite interface between N heat of protomer 2, M heat of protomer 1 and RAPTOR of the four apo-mTORC1 3D classes of Exteded Data Figure 5c (colored as in Figure 4a) and RHEB-mTORC1 (all red). The four apo-mTORC1 classes were refined to 4.5 Å or better and together with RHEB-mTORC1 were superimposed on RAPTOR (residues 52–422). Figure also highlights the flexibility on the N-terminal half of the N-heat solenoid in apo-mTORC1 (see Supplementary Information discussion).
Extended Data Figure 8. RHEB-induced conformational change in mTORC1.
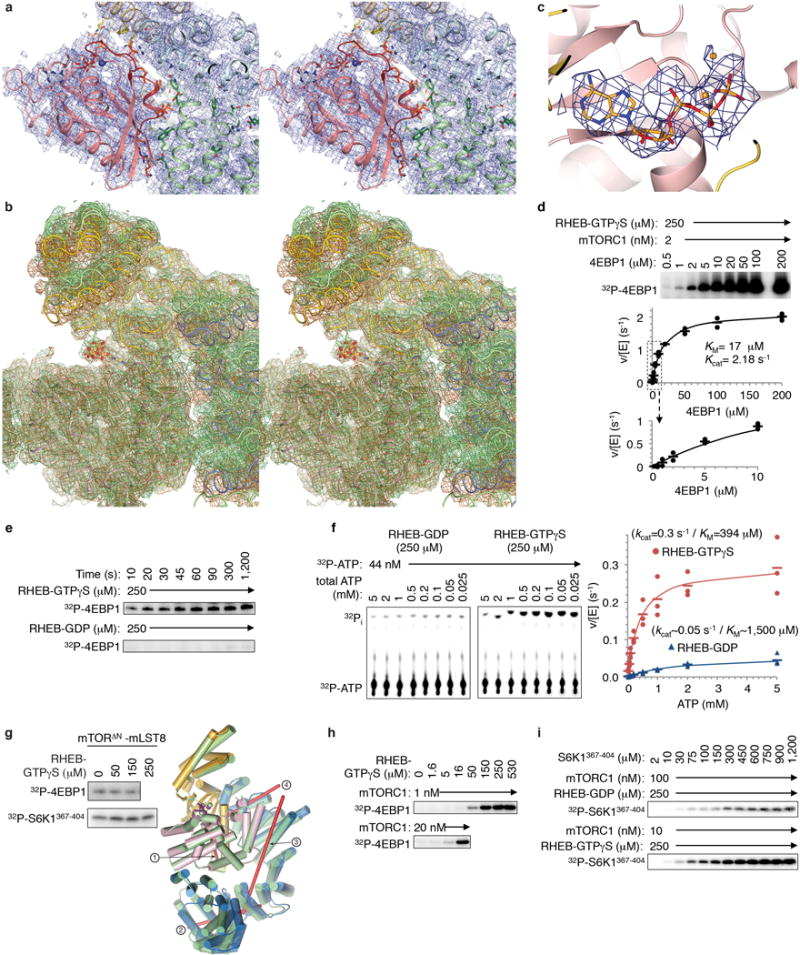
a, Stereo view of the cryo-EM density from the 3.4 Å RHEB-mTORC1 reconstruction, showing the RHEB-mTOR interface in the same orientation and coloring as in Figure 4c. The RHEB-interacting segments of mTOR are nα3 to nα7 of N-heat, mα2-mα4 of M-heat and fα2-fα3 of FAT. The majority of the contacts made by RHEB are from its switch I and switch II regions, with a small number of additional contacts to N-heat and M-heat contributed by the nearby segments of residues 5 to 7 and 106 to 111. b, Stereo view of cryo-EM density from RHEB-mTORC1 (sand) and the 3.0 Å apo-mTORC1 (green), with the two structures and maps superimposed on the C lobes as in Figure 5d. c, AMPPNP (orange) cryo-EM density of apo-mTORC1. d, Steady state kinetic analysis of mTORC1 phosphorylation of intact 4EBP1 in the presence of 250 μM RHEB-GTPγS. Reactions quantified by 32P incorporation and plotted as velocity over enzyme concentration (means as dashes and values from two independent experiments as filled circles). The KM and kcat values, calculated by non-linear regression fitting of the data, and simulated curves are also shown. Note that in contrast to the reaction in the absence of RHEB shown in Extended Data Figure 4e, the curve of reaction velocity versus 4EBP1 concentration obeys Michaelis-Menten kinetics. e, RHEB-GTPγS activation of 4EBP1 phosphorylation by mTORC1 under single-turnover conditions. A master mix of excess mTORC1 (500 nM) over 4EBP1 substrate (100 nM) was incubated with 250 μM RHEB-GTPγS or 250 μM RHEB-GDP in the standard kinase buffer on ice for 5 minutes. Reactions were started by the addition of a mixture of cold ATP (50 μM final) and [γ -32P] ATP (8 μCi per reaction time point). The reactions were done on ice to slow down the reaction. At indicated time points, an aliquot of the reaction was drawn, stopped, and analyzed as described in Methods. The experiment was repeated three times with very similar results. f, ATP steady-state kinetic parameters of ATP hydrolysis by mTORC1 in the presence of 250 μM RHEB-GDP (left, blue plot) or RHEB-GTPγS (right, red plot). Reactions quantified by 32P incorporation as in d. Graph shows means as dashes and values from three independent experiments with the indicated markers and colors. The steady-state kinetic constants of the RHEB-GDP containing reaction are approximate owing to the weak signal of these reactions. g, As expected, RHEB-GTPγS did not activate the truncated mTORΔN-mLST8 complex phosphorylating 4EBP1 or S6K1367–404 (10 μM both; mTORΔN at 20 and 30 nM, respectively) (left panel; experiments were repeated two times with very similar results). mTORΔN-mLST8 has an intermediate kcat of 0.66 s−1 (Extended Data Fig. 1b) compared to the 0.09 and 2.9 s−1 kcat values of apo-mTORC1 and RHEB-mTORC1, respectively (Fig. 5g). mTORΔN-mLST8 has a distinct FAT conformation likely due to the absence of N-heat. Right panel shows superposition of the FAT plus kinase domain portions of inactive apo-mTORC1 on the crystal structure of mTORΔN-mLST8 done by aligning their C lobes. Apo-mTORC1 is in green and mTORΔN is colored blue for FAT, yellow for N-lobe and pink for C-lobe. The rotation axes (red lines) are numbered according to the hinges of Figure 5c. Compared to the inactive to active transition, the comparison of the mTORΔN FAT conformation to that of the inactive state exhibits bigger changes around the major hinge with a rotation in the opposite direction and a different rotation axis far from the hinge axis (labeled “1”). The rotations around the two minor hinges are comparably modest although distinct, with the rotation axes nearly orthogonal to those of the inactive to active transition. h, Autoradiogram showing activation of mTORC1 phosphorylating 4EBP1 (10 μM) by RHEB-GTPγS, repeated three times. Gel quantification is shown in Figure 5b. i, Steady-state kinetic analysis of mTORC1 phosphorylating S6K1367–404 in presence of 250 μM RHEB-GDP (top row) or RHEB-GTPγS (bottom row). 32P incorporation data is plotted as velocity over enzyme concentration in Figure 5g (n=3).
Extended Data Figure 9. Hyperactivating mTOR mutations cluster in three regions of the mTOR structure.
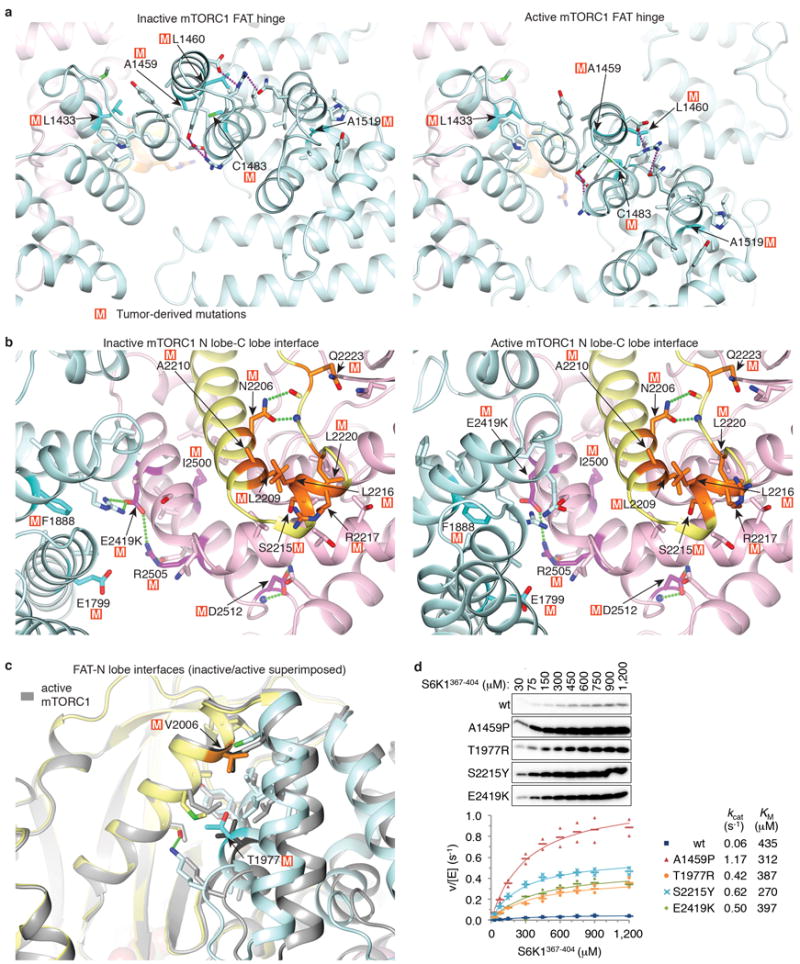
a, Hyperactivating mutations that cluster at the major hinge region of the FAT domain. The mutations, which occur at residues with structure-stabilizing roles, likely disrupt the structural integrity of the FAT clamp and thus its ability to block the movement of the N lobe into the active position. Cancer-genome mutations shown experimentally to be hyperactivating6 are mapped onto the apo-mTORC1 (left) and RHEB-mTORC1 (right) structures. The two structures are aligned on the C lobes of their kinase domains to highlight the different packing arrangements at the hinge. The side chains of mutated residues are indicated with the letter “M” and colored dark cyan, the side chains that they interact with are in light cyan, while the rest of the structural elements are colored as in Figure 4a. Red dotted lines indicate groups within hydrogen bond distance. b, The largest cluster of hyperactivating mutations is centered on an N lobe helical extension (kα3, kα3b) that is anchored in a pocket between the C lobe and FAT domains. Here, the structural mutations either at the N lobe kα3-kα3b helices or at their binding site on the C lobe and the adjacent FAT domain would weaken the structural coupling of the N lobe with the C lobe, possibly allowing the N lobe to assume conformations closer to its active state. Mutations mapped onto the apo-mTORC1 (left) and RHEB-mTORC1 (right) structures. c, Hyperactivating mutations that cluster at where the FAT transitions into and packs with the N lobe. Mutations here likely destabilize the structural elements and their packing that prevents the N lobe from moving into its active position, mimicking the RHEB-induced conformational change and the associated looser FAT-N lobe interface. Mutations are mapped onto apo-mTORC1 (FAT in cyan, N lobe yellow, C lobe pink) and RHEB-mTORC1 (all in gray). The two structures are superimposed on their N lobe domains to facilitate comparison. d, Steady-state kinetic analysis of S6K1367-404 phosphorylation by 30 nM mTORC1 containing w.t. or the indicated hyperactive mTOR mutants. 32P incorporation was quantified and velocity over enzyme concentration values were plotted as means (dashes) and values from two independent experiments with the indicated markers and colors. Dissociation constants from the non-linear regression fitting of the data are also shown, and simulated binding curves are overlaid on the data.
Extended Data Table.
Data collection and refinement statistics for the atRaptor-TOS and apo-atRaptor crystal structures.
| atRaptor | atRaptor-4EBP199–118 | atRaptor-S6K11–14 | atRaptor- PRAS40124–139 | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 89.1, 112.6, 134.1 | 89.1, 113.1, 153.5 | 89.1, 113.1, 152.9 | 89.1, 113.1, 151.8 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50-3.00 (3.11-3.00) | 80-3.00 (3.11-3.00) | 80-3.10 (3.21-3.10) | 80-3.35 (3.47-3.35) |
| Rsym | 0.086 (0.533) | 0.082 (0.800) | 0.084 (0.819) | 0.088 (0.758) |
| Rpim | 0.057 (0.362) | 0.035 (0.307) | 0.035 (0.331) | 0.042 (0.377) |
| I/σI | 19.8 (2.6) | 19 (1.53) | 19.2 (1.75) | 22.3 (2.3) |
| CC-1/2 | (0.713) | (0.801) | (0.846) | (0.770) |
| Completeness (%) | 97.4 (97.3) | 98.7 (99.6) | 99.2 (99.4) | 98.5 (97.6) |
| Redundancy | 3.1 (3.0) | 6.2 (7.2) | 6.5 (6.9) | 5.1 (4.8) |
| Refinement | ||||
| Resolution (Å) | 20-3.0 | 20-3.0 | 20-3.11 | 20-3.35 |
| No. reflections | 24264 | 27720 | 25050 | 19535 |
| Rwork/Rfree (%) | 24.4/27.8 | 22.8/27.4 | 21.6/25.6 | 21.0/26.5 |
| No. atoms | ||||
| Protein | 8310 | 8378 | 8332 | 8331 |
| Ligand/ion | 0 | 0 | 0 | 0 |
| Water | 0 | 0 | 0 | 0 |
| B factors | ||||
| Protein | 75.3 | 102.4 | 104.4 | 115.3 |
| Ligand/ion | – | – | – | – |
| Water | – | – | – | – |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.008 | 0.008 | 0.008 | 0.008 |
| Bond angles (°) | 1.356 | 1.371 | 1.348 | 1.355 |
Values in parentheses are for highest-resolution shell. All datasets were collected from a single crystal each.
Supplementary Material
Supplementary Video 1: A morph from the inactive to the active mTOR conformation.
The roundtrip morph from the inactive (at movie start) to the active (movie middle) monomeric mTORC1 structures superimposed on the KD C lobes (mLST8 not shown). Orientation and coloring similar to Figure 4a.
Supplementary Video 2: Close-up view of the catalytic cleft in the morph from the inactive to active mTOR conformation.
Close-up of Supplementary Video 1 focusing on the relative orientation of the kinase N and C lobes. View is similar to Figure 5d looking into the kinase catalytic cleft.
Acknowledgments
We thank the staff of the Advanced Photon Source, the NYSBC Simons Electron Microscopy Center, the HHMI Cryo-EM facility, the MSKCC Cryo-EM facility and Subangstrom LLC for help with data collection. Supported by HHMI and National Institutes of Health grant CA008748.
Footnotes
Author Contributions: H.Y., B.L. and N.P.P. designed the experiments, determined the mTOR structures and wrote the manuscript. N.P.P. and X.J. determined the Raptor crystal structures and biochemical constants. H.Y., H.J.Y., M.M., A.Y. and A.D. carried out the mTOR enzyme assays and biochemical experiments.
Author Information: The authors declare no competing financial interests.
SI Guide
Supplementary Information: Supplementary Discussion; Supplementary Data
This file contains the Supplementary Discussion sections about steady state kinetic constants, about other mTORC1 substrates in Figure 1g, about Raptor structure, about mTORC1 structure, and about apo-mTORC1 conformational flexibility. The file also contains Supplementary Figure 1 showing the source data (autoradiograms and immunoblots of gels) for the Main text and Extended Data figures.
References
- 1.Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Current opinion in cell biology. 2017;45:72–82. doi: 10.1016/j.ceb.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends in cell biology. 2014;24:400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends in cell biology. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grabiner BC, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer discovery. 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 7.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 8.Hara K, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 9.Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 10.Aylett CH, et al. Architecture of human mTOR complex 1. Science. 2016;351:48–52. doi: 10.1126/science.aaa3870. [DOI] [PubMed] [Google Scholar]
- 11.Saucedo LJ, et al. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 12.Stocker H, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–565. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 13.Sancak Y, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282:20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 16.Oshiro N, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem. 2007;282:20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 19.Hsu PP, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang SA, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13:797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 22.Nojima H, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278 doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 23.Yang H, et al. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, et al. 4.4 A Resolution Cryo-EM structure of human mTOR Complex 1. Protein & cell. 2016;7:878–887. doi: 10.1007/s13238-016-0346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–242. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 26.Yu Y, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roczniak-Ferguson A, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michels AA. MAF1: a new target of mTORC1. Biochemical Society transactions. 2011;39:487–491. doi: 10.1042/BST0390487. [DOI] [PubMed] [Google Scholar]
- 29.Peterson TR, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell. 1999;3:707–716. doi: 10.1016/s1097-2765(01)80003-4. [DOI] [PubMed] [Google Scholar]
- 31.Ginalski K, Zhang H, Grishin NV. Raptor protein contains a caspase-like domain. Trends Biochem Sci. 2004;29:522–524. doi: 10.1016/j.tibs.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nature methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheres SH. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods in enzymology. 2016;579 doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Brown A, et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr D Biol Crystallogr. 2015;71:136–153. doi: 10.1107/S1399004714021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baretic D, Berndt A, Ohashi Y, Johnson CM, Williams RL. Tor forms a dimer through an N-terminal helical solenoid with a complex topology. Nature communications. 2016;7:11016. doi: 10.1038/ncomms11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Y, et al. Structural basis for the unique biological function of small GTPase RHEB. J Biol Chem. 2005;280:17093–17100. doi: 10.1074/jbc.M501253200. [DOI] [PubMed] [Google Scholar]
- 37.Klink TA, Kleman-Leyer KM, Kopp A, Westermeyer TA, Lowery RG. Evaluating PI3 kinase isoforms using Transcreener ADP assays. Journal of biomolecular screening. 2008;13:476–485. doi: 10.1177/1087057108319864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagle N, et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer discovery. 2014;4:546–553. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohne Y, et al. Isolation of hyperactive mutants of mammalian target of rapamycin. J Biol Chem. 2008;283:31861–31870. doi: 10.1074/jbc.M801546200. [DOI] [PubMed] [Google Scholar]
- 40.Urano J, et al. Point mutations in TOR confer Rheb-independent growth in fission yeast and nutrient-independent mammalian TOR signaling in mammalian cells. Proc Natl Acad Sci U S A. 2007;104:3514–3519. doi: 10.1073/pnas.0608510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta crystallographica Section A, Foundations of crystallography. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suloway C, et al. Automated molecular microscopy: the new Leginon system. Journal of structural biology. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 46.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of structural biology. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of structural biology. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimanius D, Forsberg BO, Scheres SH, Lindahl E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife. 2016;5 doi: 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scheres SH. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife. 2014;3:e03665. doi: 10.7554/eLife.03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Nicholls RA, Fischer M, McNicholas S, Murshudov GN. Conformation-independent structural comparison of macromolecules with ProSMART. Acta Crystallogr D Biol Crystallogr. 2014;70:2487–2499. doi: 10.1107/S1399004714016241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schagger H. Tricine-SDS-PAGE. Nature protocols. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
Extended Data References
- 1.Yang H, et al. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–242. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 3.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown A, et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr D Biol Crystallogr. 2015;71:136–153. doi: 10.1107/S1399004714021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 6.Grabiner BC, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer discovery. 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1: A morph from the inactive to the active mTOR conformation.
The roundtrip morph from the inactive (at movie start) to the active (movie middle) monomeric mTORC1 structures superimposed on the KD C lobes (mLST8 not shown). Orientation and coloring similar to Figure 4a.
Supplementary Video 2: Close-up view of the catalytic cleft in the morph from the inactive to active mTOR conformation.
Close-up of Supplementary Video 1 focusing on the relative orientation of the kinase N and C lobes. View is similar to Figure 5d looking into the kinase catalytic cleft.
Data Availability Statement
The cryo-EM maps, including the three focused reconstruction maps and the structure factors of their composite map used in model refinement, and the refined atomic models have been deposited with the Electron Microscopy Data Bank and the Protein Data Bank with accession numbers EMDB-7087 and PDB 6BCX for apo-mTORC1 and EMDB-7086 and PDB 6BCU for RHEB-mTORC1. The coordinates and structure factors of the FRB-S6K1 complex (5WBH), atRaptor (5WBI), atRaptor-4EBP1TOS (5WBJ), atRaptor-S6K1TOS (5WBK), atRaptor-PRAS40TOS (5WBL), mTORΔN-mLST8-PRAS40173–256 (5WBU), mTORΔN-mLST8-PRAS40114–207 (5WBU) have been deposited with the Protein Data Bank.