

Abstract
Metabolite identification is still considered an imposing bottleneck in liquid chromatography mass spectrometry (LC/MS) untargeted metabolomics. The identification workflow usually begins with detecting relevant LC/MS peaks via peak-picking algorithms and retrieving putative identities based on accurate mass searching. However, accurate mass search alone provides poor evidence for metabolite identification. For this reason, computational annotation is used to reveal the underlying metabolites monoisotopic masses, improving putative identification in addition to confirmation with tandem mass spectrometry. This review examines LC/MS data from a computational and analytical perspective, focusing on the occurrence of neutral losses and in-source fragments, to understand the challenges in computational annotation methodologies. Herein, we examine the state-of-the-art strategies for computational annotation including: (i) peak grouping or full scan (MS1) pseudo-spectra extraction, i.e., clustering all mass spectral signals stemming from each metabolite; (ii) annotation using ion adduction and mass distance among ion peaks; (iii) incorporation of biological knowledge such as biotransformations or pathways; (iv) tandem MS data; and (v) metabolite retention time calibration, usually achieved by prediction from molecular descriptors. Advantages and pitfalls of each of these strategies are discussed, as well as expected future trends in computational annotation.
Keywords: Untargeted metabolomics, Annotation
Introduction
The aim of any untargeted metabolomics experiment is to identify and quantify disregulated compounds relevant to a particular disease or stressor. Untargeted data analysis workflow for liquid chromatography electrospray ionization/mass spectrometry (LC/ESI/MS) consist in applying peak-picking algorithms [1–6] to detect peaks associated with metabolites, align those peaks across multiple samples to obtain the so-called peak features (defined as a peak, or a set of aligned peaks across samples with a unique m/z and a specific retention time), and subsequently discover statistically significant variations between experimental groups or conditions. Once features of interest are prioritized, their mass values are searched against metabolite libraries [7–9] to obtain putative metabolite identifications. Next, those features can be identified via fragmentation experiments (tandem MS or MS/MS), usually with quadrupole – Time-of-Flight (q–ToF) instrumentation, by comparing experimental fragmentation patterns with spectral libraries [10]. Ultimately, unambiguous identification, according to the Metabolomics Standards Initiative (MSI) guidelines [11], can only be achieved by comparing the experimental tandem MS spectra with standard materials analyzed under identical conditions.
Although the untargeted metabolomics workflow is well-defined, annotation of metabolites still remains a computational bottleneck due to the nature of LC/ESI/MS data. First, there is a high redundancy of features representing the same metabolite, which can be attributed to to adducts, in-source fragments and isotopes. Library searching of all statistically significant features without prior knowledge of monoisotopic accurate masses of the underlying metabolites might lead to missanotations if adducts or in-source fragments are present [12]. In addition, accurate mass library searches – considering expected adducts – can lead to a large number of potential molecular formulas and thus, molecular entities. Computational feature grouping and annotation is therefore a necessary step to reduce the list of putative identities. Annotation is defined as the process of “noting” each observed feature with a putative identity. Annotation generally refers to assigning each feature with a putative metabolite name or molecular formula, but it also includes assigning each feature with the identity of formed adducts, neutral losses, etc. This, ultimately facilitates the accurate characterization and identification of annotated adduct peaks via tandem mass spectrometry (MS/MS).
This computational annotation process ideally consists in i) grouping features stemming from the same compound – adducts, isotopes and in-source fragments – which gives valuable chemical information for metabolite identification, and ii) determining monoisotopic or neutral molecular mass of each metabolite by annotation of formed adduct peaks and isotopes. Additional strategies have been proposed which attempt to increase the annotation confidence when performing accurate mass search. Those strategies take into account biological information such as pathways, tandem MS/MS data, and retention time calibration or prediction.
This review provides an overview of LC/ESI/MS based un-targeted metabolomics data from a computational and physiochemical perspective, to understand the characteristics of LC/MS data and its challenges for accurate computational annotation. We focused on the occurrence of in-source dissociation phenomena in metabolomics data, and the pitfalls of using accurate mass search without a priori feature annotation. Moreover, we provide an overview of the state of the art computational annotation strategies for LC/MS data. Despite that different computational tools are cited throughout the paper, we focused on their algorithms and strategies rather than providing a comprehensive list of all available annotation tools. For more in depth reviews focused on tools and resources, we encourage the readers to read the following literature [13–15].
Overview of LC/ESI/MS data
LC/ESI/MS data are typically composed of a large amount of features, with a significant redundancy belonging to the same metabolite or chemical entity due to commonly occurring adducts, neutral losses, isotopes and in-source fragments. This section describes how the different levels of redundancy can be used to extract meaningful information and thus converting raw data into biologically interpretable information.
Adducts, neutral losses and isotopes
Under normal conditions, protonated and deprotonated are the most common ion species in LC/ESI/MS metabolomics measurements, however, different molecular adducts are also formed by adduction of ionic species, such as ionic metals (Na+, K+, etc.), halides (Cl−and F−) or additives (acetate, formate, ammonium, etc.) usually found in both solvents and samples or added to improve chromatographic and ionization conditions. Also, features resulting from stable neutral molecules losses such as H2O or CO2 are detected (Figure 1 (c,d)). This yields a feature redundancy, where usually more than one feature per metabolite is observed. Given that the accurate masses of common ion adducts and neutral losses are known, the prediction of m/z distance between two or more adducts and losses is possible. A list of such adducts and neutral losses m/z values is known as annotation rules [16]. Therefore, the presence of two or more adducts together with neutral losses can provide useful information that allows the “triangulation” of the monoisotopic mass by comparing experimental the distance between two features with the theoretical distance between two known adducts. If the experimental distance falls within a user specified window (ppm error) of the theoretical distance, then these two features can be annotated and the neutral mass using the same rules can be calculated. Of note, the relative intensity among adduct peaks as well as the amount of formed adducts will vary from one metabolite to another, as well as for the same metabolite depending on experimental conditions [9]. This can make MS1 spectral information in LC/MS poorly reproducible for annotation purposes. However, some adducts and neutral losses are more frequent than others. As observed from available adduct spectra in MS/MS libraries, protonated and deprotonated are the most frequent adducts, followed by [M+H–H2O]+, [M+Na]+ in positive mode and [M–H–H2O]−, [2M–H]− in negative mode [9].
Fig. 1.
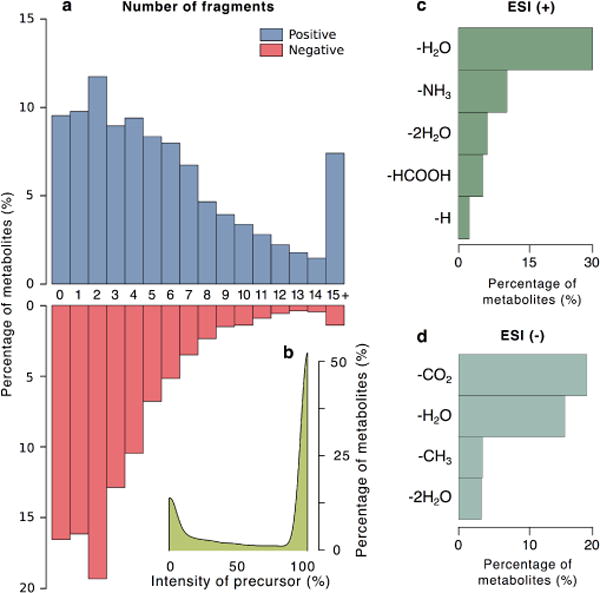
Occurence of in-source fragments. (a) Percentage of metabolites in positive (red) and negative (blue) mode showing a determined number of in-source fragments: from 0 to 14, and 15 or more (15+). (b) Percentage of metabolites versus the intensity of protonated or deprotonaded species. Common neutral losses observed in metabolites for positive (c) and negative (d) modes. Only losses from the precursor ion where considered, i.e., losses from fragments were discarded. The number of fragments, precursor ion intensity and neutral losses descriptive statistics were assessed from METLIN MS/MS database from low collision energy spectra. The number of fragments showing a relative intensity above 5% in a low collision energy (0 V and 10 V) were considered fragments that might appear as in-source fragments. Of note, neutral losses were considered as fragments. The information from ~15,000 experimental MS/MS spectra were used to retrieve these statistics.
Depending on the signal-to-noise ratio, the isotopic envelope of metabolites and their fragments and adducts can increase the feature redundancy. The isotopic pattern (relative 12C/13C isotopomer abundances and their masses) for each molecular formula can be accurately predicted too, but those peaks are characterized by their low intensity, especially for low molecular weight compounds. This is why despite isotopic information has been used to narrow the number of putative molecular formulas (see Section Peak annotation: adducts, neutral losses isotopes and other mass relationships), this approach lacks from suffcient accuracy [17], as even in the cases where peaks are detected by the mass spectrometer and data processing software, the peak intensity precision is too low to provide with accurate results.
In-source fragmentation
ESI is considered to be one of the softest ionization sources, still, in-source fragmentation is a natural phenomenon in LC/MS [18, 19] and, unlike adducts and neutral losses, exact mass of in-source fragments cannot be easily predicted as they are particular of each metabolite. These in-source fragments will usually be the ones observed in low energy MS/MS spectra, with different relative intensities. The occurrence of in-source fragments can be appreciated in Figure 1(a), where only less than 10% of metabolites in METLIN database [7] do not readily dissociate in the source, while ~8% generates more than 15 fragments. Interestingly, the intensity of the protonated and deprotonated species are the most intense signal in more than 50% of the cases. This indicates that assuming that the most intense detected peak is generally a protonated or deprotonated ion can likely lead to misannotations.
Another reason for not relying on accurate mass searches without previous feature annotation is that in-source fragments might match [M+H]+ and [M-H]- species from other metabolites. This might occur when metabolites structural differences differ only in labile chemical moieties and chemical substructures are shared with other metabolites. An example of this occurs with the low energy spectra (0 V or 10 V) of isoxanthopterin and α-guanidinoglutaric acid, where one fragment has the same mass as the protonated ion corresponding to guanine and glutamate respectively. This can be expected due to the structural similarities between pairs of metabolites and it can lead to false annotations and identifications. For instance, if isoxanthopterin is present in a sample and the precursor of guanine, as in-source fragment, is seen, it might be falsely annotated as guanine. In fact, even upon tandem MS experiments to confirm this hypothesis, the similar spectra obtained would lead to an incorrect confirmation of this (false) hypothesis (Figure 2). In those cases, the spectral fragments are usually the same, but the relative intensities might have slight variations. However, these differences could be attributed to different experimental conditions and different instruments from different vendors.
Fig. 2.
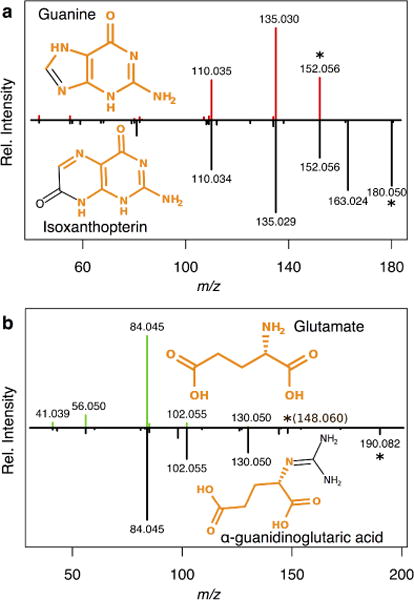
Empirical MS/MS spectra (20 V) of guanine vs. isoxanthopterin (a) and gluatamate vs. α-guanidinoglutaric acid in positive- or negative-ion mode? (b). In an hypothetic case, one of the in-source fragments of isoxanthopterin (low energy spectra not shown) could be mistaken by the protonated peak of guanine. Even in MS/MS fragmentation of this in-source fragment of isoxanthopterin corresponding to the precursor ion of guanine, this would produce similar spectra to guanines. This phenomenon can be explained by the high similarity between metabolite structures (highlited in orange). The precursor ion is marked with * and its m/z between brakets if not observed in the experimental spectra. Isoxanthopterin is a pteridine normally present in plasma, urine, and other bodily fluids. JChem Base 2017 (ChemAxon) was used for structural comparison.
Finally, in-source fragments could be an informative source of molecular identity. For instance, in those cases where only a protonated ion is observed for a certain metabolite, there is no evidence that allows us to assume that the peak is a protonated ion of a molecule and, accurate mass search could lead to incorrect matching for the above-mentioned reasons. In those cases, the presence of in-source fragments might serve as an identity indicator, since each in-source fragment and protonated or deprotonated ion are specific of a small number of compounds.
Computational annotation strategies
The untargeted LC/MS-based metabolomics data processing workflow consist in peak-picking and alignment, provided by widely used tools such as MZmine [1, 2] or XCMS [4, 5], followed by peak annotation. Existing computational annotation tools usually start from the input provided by these peak-picking tools, which output consist of a list of features: a peak, or a set of aligned peaks across samples with a unique m/z and a specific retention time (mzRT).
We classified the existing annotation strategies into five levels. Each level is independent of each other and are usually embedded into computational tools combined or separately. The first level includes peak grouping or MS1 pseudo-spectra extraction, i.e., clustering all the features corresponding to the same metabolite. The second level aims at using ion adducts, in-source or other expected accurate masses to annotate features and thus unravel monoisotopic or neutral masses. These two levels are the most widely used strategies and they are used in almost all computational tools for metabolite annotation. Other complementary strategies make use of biological information (level 3) such as biotransformations or pathways, to increase the annotation confidence, or to use and integrate MS/MS (MS2) data (level 4). A less used but also a valid strategy is considering retention time (level 5), which is usually predicted from molecular descriptors.
Peak grouping: MS1 pseudo-spectra extraction
A straightforward annotation strategy is to compare expected theoretical distances between well-known ion adduct masses with experimental distances between peaks eluting around a certain retention time. Unfortunately, the high number of peaks in complex metabolomics datasets makes this annotation challenging, as co-eluting peaks originated by other metabolites, in-source fragments, and spurious peaks by the biological matrix or noise due to data processing errors, might lead to an overestimation of adducts and isotopes. Therefore, a complementary strategy, usually applied before taking mass relationships into account, consists in determining which peaks belong to each metabolite. We will refer to this process as the extraction of (MS1) pseudo-spectra (Figure 3(a)). For each metabolite, its pseudo-spectrum should be comprised of its adducts, isotopes, common neutral losses, in-source fragments and any other peaks that, based on a specific metric, are considered to stem from the same molecule. Then, searching for mass relationships among a well-defined group of peaks (pseudospectra) reduces the number of false positive peaks that could lead to false annotations. Specifically, two strategies aimed at extracting pseudo-spectra exist, namely peak-shape and peak-abundance correlation.
Fig. 3.
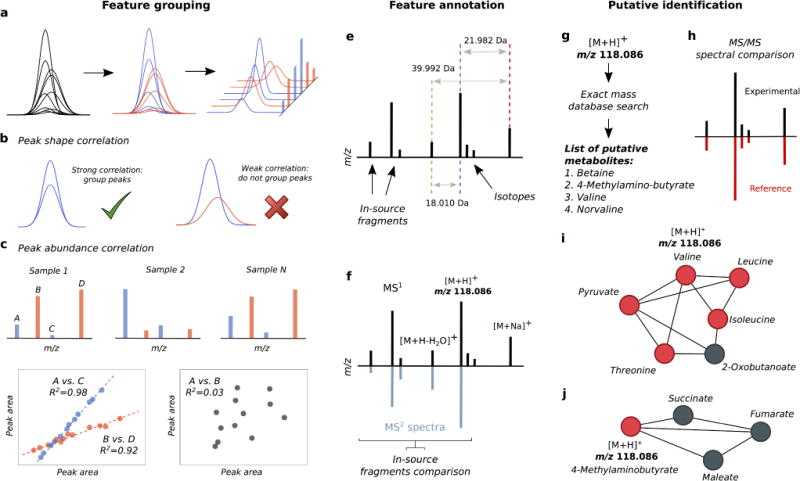
Overview of computational annotation strategies. Annotation comprises feature grouping, annotation and putative identification. Feature grouping (a-c) aims at grouping peaks that belong to each metabolite (a). To do so, if peak shape (b) or peak-abundance (c) correlation between two peaks is above a pre-defined threshold, these two features are considered to belong to the same metabolite. Specifically in peak abundance (c), when the areas of peaks belonging to the same metabolite are compared across samples (A vs. C and B vs. D), a linear relation is expected to be observed, whereas when comparing areas of peaks that do not belong to the same metabolite (A vs. B), no linear relation would be observed. In feature annotation (e, f), expected theoretical distances between known ion adduct masses are compared with experimental distances found among peaks (e). This allows annotating the protonated/deprotonated ion together with adducts and neutral losses (f). For each metabolite, in-source fragments in MS1 data can be characterized by comparison with peaks from low energy MS/MS spectra. After peak annotation, putative identification can be achieved by accurate mass search (g) or by comparison with MS/MS data (h). Finally, pathway biochemical knowledge can be used to increase the annotation confidence (i, j). Statistically significant metabolites retrieved after accurate mass search are projected onto pathway networks. Each metabolite in the pathway is represented by a circle, and those statistically significant are shown in red. From all the hits after accurate mass search (true and false), true identifications are filtered by detecting combinations that have more biochemical sense. Combination of “true” identifications should show an enrichment on local pathway regions (i), whereas false annotations would show a random distribution throughout pathways (j). In this illustrative example, the detected feature m/z 118.0863 has multiple putative hits (g), where only one of the hits correspond the “true” metabolite. When projecting all the putative hits onto pathways, only valine shows a biochemical sense, as other neighboring metabolites in the pathway also show activity (i). Instead, other false hits such as 4-methylaminobutyrate are discarded, as its neighboring metabolites in the pathway show no activity (j).
The first approach to extract MS1 pseudo-spectra from peak-picking strategies is to group peaks based on their chromatographic peak shape. This is to take advantage from the fact that all the peaks with different m/z ratios that belong to the same metabolite elute in the same retention time and, ideally, with a similar peak shape. Therefore, based on the correlation value given by simple Pearson correlation test, we can computationally determine whether two peaks originate from the same metabolite or not, and cluster similar elution profiles together [20] (Figure 3(b)). Different sources of problems hamper the efficiency of this approach. First, the noise introduced by the MS detector, specially at low levels, biological matrix interactions, ion suppression and other chromatographic effects induce changes to the shape of peaks and thus decrease similarities or correlations among shapes of peaks from the same metabolite. Second, coelution with other peaks from different compounds makes the association of those also challenging, i.e., highly co-eluted peaks from two different compounds might show a high similarity/correlation. Interesting examples of poor correlation between related peaks are shown by Mahieu et al [21]. Finally, and due to the aforementioned issues, it is difficult to determine a correlation threshold to define whether two peaks belong to the metabolite or not. A low similarity threshold will lead to an incorrect association of peaks, and peaks from different metabolites will be mixed in the same MS1 pseudo-spectrum, which in turn will obstruct its correct annotation. On the contrary, a high similarity threshold would lead to only consider peaks with highly similar shapes, resulting in important peaks – such as low abundant adducts or isotopes – not being taken into account. Another known pitfall of this approach is the high computational resources needed to perform a clustering based on peak shape, since raw data needs to be re-accessed. Variations on peak-shape approaches have been introduced [22, 23]. Essentially, these variations propose a different metric to measure the similarity between peak shapes, e.g., Ipsen et al. proposed a statistical approach that takes into account the noise of the mass spectrometer.
The second approach to extract MS1 pseudo-spectra is to group peak-features based on their peak-abundance correlation [24–29]. Ideally, when two peaks belong to the same compound, their relative abundance (peak area or intensity) show a strong linear relation across samples - and thus a strong correlation (Figure 3(c)). Therefore peaks are grouped based on their abundance linear relationship or correlation among samples. Comparing the peak abundance relationship is a more robust variable due to the accuracy of current MS detectors, whereas natural occurring differences in the shape of two peaks from the same compound makes the peak-shape strategy more sensible to outliers. This strategy is capable of distinguishing two co-eluting peaks stemming from different metabolites, since their linear relation among samples are likely to be different. Overall, this strategy exploits the joint information provided by all the samples and does not require high computational resources [25]. Pitfalls of this approach are that two co-eluting metabolites could also present a high correlation among samples, i.e., their total concentrations are correlated and, despite being two different metabolites, all their peaks are correlated as well. Another limitation is its sensitivity to outliers, as errors in feature integration one might lead to incorrect peak clustering. Finally, this approach also needs a correlation threshold to be determined. Variations on these approach include RAMClust [28], that uses an hierarchical clustering-based approach to group both MS and MS/MS peaks, or xMSannotator [29], which uses a weighted correlation network analysis and does not require a minimum correlation threshold to be defined.
The application of these strategies is not exclusive, and both peak shape similarity and peak abundance correlation application can be combined to enhance the efficiency of MS1 pseudospectra extraction [16].
Peak annotation: adducts, neutral losses isotopes and other mass relationships
As previously described, annotation is accomplished by comparing mass distances among experimental peaks to distances among combinations of known adducts, neutral losses, molecular multimers or multiply charged ions (Figure 3(e)). This simple approach, combined with the use of other LC/MS data properties, is the most common strategy used to annotate. For instance, MetAssign [30] uses annotation rules along with peak intensity and retention time information to provide annotations by means of sophisticated Bayesian statistics. MAIT [31] takes into account possible known biotransformations – specific mass differences caused by chemical modifications made by each organism under study – to improve the annotation outcome. Mz.unity [21] aims at expanding the list of annotation rules – which typically cover tens of adduct types –, by taking into account more complex peak mass relationships such as heterodimers, higher complex adducts, distal fragments, relationships between peaks in different polarities, and complex adducts between features and background peaks. Also, RAMSI [32] performs a one-step optimization of chemical rules among observed ions and chemical formula calculation, yielding a convergence that satisfies both criteria. Interestingly, RAMSI does not predefine annotation rules or neutral losses and both positive and negative mode data can be jointly analyzed.
The use of adducts or neutral losses rules alone in peak annotation has its limitations. If for a given pseudo-spectra no adduct is found, no evidence is available which would allow the computation of a monoisotopic mass. Also, due to noise from biological matrix or introduced by peak-picking data processing algorithms, false adducts might be found in the data. Therefore, finding more evidence, e.g., finding more than two adducts that “point” to the same molecular entity, increases the likelihood of those peaks belonging to a real metabolite. In that sense, some studies proposed the use of information from in-source fragmentation as a complementary evidence layer (Figure 3(f)). In-source fragments can be used not only to determine the monoisotopic mass where few or any adducts have been found, but also to provide with a more specific list of candidate metabolites, as each of these fragments are specific of a small number of metabolites. Examples of adoption of this strategy includes RAMClust [28], or the proof of concept by Lynn et al. [33], where they matched in-source fragments with low energy MS/MS spectra in public databases. Similarly, while attempting to predict in-silico in-source fragments and retention time from molecular descriptors, the STOp-1 [34] algorithm uses in-source fragments to rank the list of putative identities.
Alternatively, additional chemical heuristic rules such as hydrogen/carbon ratios, restrictions for the number of elements, or relative isotopic abundances have been proposed to limit the plausible molecular formulas [35–37].
Biochemical knowledge
In cellular metabolism, metabolites are connected among and within pathways through biochemical reactions. That means that a considerable proportion of observed metabolites in biological samples will be related by these chemical reactions, being products or substrates themselves. Different ways to use this biochemical information to narrow down putative identifications based on accurate mass search alone exist. The most simple is to consider only those molecules known to be present in the organism or tissue under study e.g., human serum, mouse urine. Another possibility is to consider putative enzymatic biotransformations among metabolites, by taking advantage of canonical reactions that might occur, e.g., hydrogenation–dehydrogenation (± H2), oxidation (O) or phosphorylation (PO3H) [38]. Each biotransformation corresponds to an addition or subtraction of a known exact mass. As an example, two observed m/z values with a difference of 2.015 Da, are likely to be related by an hydrogenation, and therefore their molecular formula will differ by an addition/removal of H2 [39,40] (Figure 4). Specifically, the proof-of-concept by Rogers et. al. [39] and MAIT [31] take into account these biotransformation relationships, where the presence or absence of related metabolites increase or decrease the likelihood of putative identifications.
Fig. 4.
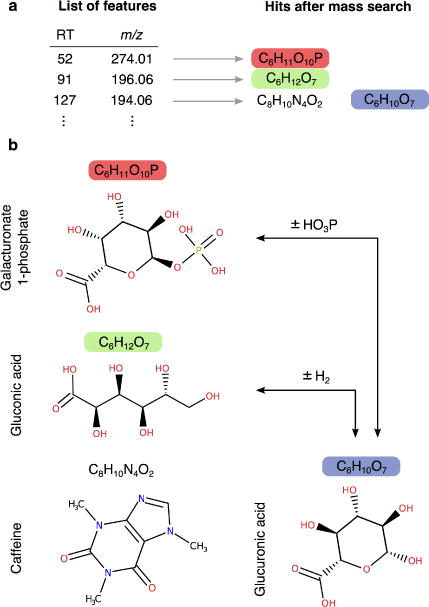
In this hypothetic situation, based on the example by Rogers et al. (2009) three features are observed in a dataset and, for each feature, putative identities (formulas) are assigned after accurate mass search (hits). The third feature (m/z 194.06) might correspond to two compounds (caffeine or glucuronic acid) with two molecular formulas respectively. However, based on mass differences, glucuronic acid might be related to (b) (galacturonate 1-phosphate and gluconic acid) by a phosphorylation (PO3H) and a hydrogenation–dehydrogenation (±H2). Therefore, this supports the evidence that the identity of the third feature (m/z 194.06) might correspond to glucuronic acid instead of caffeine. This is a simplified example and, in fact, the algorithm computes all the formula assignments interdependently, and thus potential assignments are dependent on one another, e.g., glucuronic acid also reinforces the likelihood of gluconic acid being present. Overall, the presence or absence of related metabolites increase or decrease the likelihood of putative identifications.
An extension of this methodology is provided by computational tools such as MI-Pack [41], mummichog [42], ProbMetab [43], xMSannotator [29] or XCMS [44], among others [45, 46]. These tools use biochemical pathways to filter and rank lists of putative identifications obtained after accurate mass search, increasing the likelihood of obtaining correct putative metabolite identifications. Essentially, these algorithms first map putative identifications – from quantitative statistically significant peaks – onto pathway networks. Next, they take into account potential substrate/product biochemical reactions between all possible metabolites combinations (as many metabolites as hits provided by accurate mass matches) and detect the combinations that make more biochemical sense. These approaches take advantage of the fact that if a list of putative identifications reflect biological activity, these should show an enrichment on local pathway regions (Figure 3(i)), whereas false annotations would show a random distribution throughout pathways (Figure 3(j)) [42, 47]. Specifically, mummichog uses a stastistical-based approach to determine the most plausible combinations whereas ProbMetab uses a Bayesian inference approach. At the same time, those algorithms can be used to obtain tentative functional biochemical activities prior to metabolite identification via tandem MS experiments. xM-Sannotator makes use of a correlation-based network analysis to identify related peaks among samples and metabolites within pathways.
Generally, biochemical/pathway knowledge-based strategies facilitate further identification steps, providing also preliminary mechanistic insights of the biological system under study. However, it should be noted that these approaches are limited by the fact that they assume hypothesis (e.g., local enrichments) that are not always met in real studies, and even when those are met, it is difficult to correctly discover the identity of peaks based on mass search alone and pathway activity. Also, it is well known that pathways are not completely annotated and not all links between them have been discovered. On the computational side, a limiting aspect is that pathway databases such as KEGG [48], Cytoscape [49], MetaCyc [50], are generally commercial, non-downloadable or non-redistributable. Only Reactome [51] has a Creative Commons license which allows developers to easily integrate it with their own computational tools.
Use and integration of tandem MS data
Tandem mass spectrometry (MS/MS) is employed to generate MS/MS spectra by fragmentation of ions of interest. Those contain specific fragments that, in most cases, allow the differentiation among molecules with the same neutral mass with the exception of stereoisomers. In fact, a level 2 identification, according to the MSI guidelines [11], can be achieved by comparing experimental MS/MS spectra with reference libraries. Thus, MS/MS data is a rich source of information that some tools have taken advantage of. As described before, an example of integration of MS/MS information to annotate is considering in-source fragments, which can be retrieved from low energy MS/MS spectra. For example, RAMClust [28], reduces false positive annotations by jointly processing both MS1 and data-independent MS2 data, whereas Scheubert et. al. [52] propose a false discovery rate of these annotations to filter “significative” annotations by considering the effect of different spectrum-spectrum match criteria on the number and the nature of the molecules annotated.
Traditionally, tandem MS has been used in a targeted fashion – where only selected ions in an inclusion list are fragmented –, or via data-dependent acquisition modes, such as “auto MS/MS”, a less time consuming mode in which fragmentation is triggered on precursor ions that meet user-defined criteria of intensity and charge state [53]. Limited metabolite coverage is obtained when using data-dependent modes since only intense ions are fragmented and many artifact ions will be detected and fragmented due to the inherent presence of isotopes, in-source fragments or high abundance ions from contamination or chemical noise [53]. Resources to annotate MS/MS spectra by comparison with experimental [53] or in-silico spectral libraries have been proposed, including MolFind [54], MetFrag [60], MetFusion [55], MyCompoundID [56, 57], CFM-ID [58] and MS-FINDER [59]. Alternatively, data-independent acquisition (DIA) modes such as MSE [62] or SWATH [63], in which all fragment ions for all precursors are simultaneously acquired, allows an increased MS/MS spectral coverage and reinforces the annotation confidence [64]. However, processing DIA data is more challenging, since the direct link between the fragmented precursor ion and its specific fragments is lost [65]. Algorithms to specifically process DIA MS/MS data include MS-DIAL [64] or MetDIA [65].
High-throughput identification of tandem MS can be achieved by comparing acquired spectra with reference libraries. However, only ~10% of known metabolites in databases such as Human Metabolome Database (HMDB) [8] or METLIN [7] have experimental spectral data [9]. In that sense, in-silico prediction of MS/MS spectra has gained a substantial interest, and multiple studies report different algorithms to provide MS/MS data not yet available in databases [66–69]. Conversely, inverse solutions have also been proposed where experimental spectra, that cannot be attributed to any metabolite when compared to available experimental or even in-silico libraries, is interpreted to provide putative structural annotations [70].
In comparison to peptides and proteins, which are composed of sequences of amino-acids and thus, their MS/MS spectra can be much more easily predicted, in-silico prediction of MS/MS spectra from metabolite structures still represents a challenging process. Defined fragmentation rules are used in proteomics which allows accurate in-silico MS/MS predictions, but due to the high chemical diversity of metabolites, existing rules to model CID behavior are not sufficiently accurate [71]. In-silico MS/MS data in metabolomics are usually generated from heuristic approaches that predict fragmentation routes by indirectly estimating bond dissociation energies. Since all possible fragmented substructures of a given metabolite can be relatively easily determined, in-silico prediction is characterized by a high probability of detection – recall – but a low specificity. This means that whereas most of the in-silico predicted fragments are going to match those in the experimental spectra, more in-silico false fragments are going to be predicted that actually are detected in empirical spectra [68]. As an example of in-silico strategy, a competitive fragmentation modeling (CFM) was proposed [68], where a model was built to determine the likelihood of particular bond breaking under specific conditions (Figure 5 (a)). CFM takes into account the presence of atoms adjacent to a broken bond, formation of specific neutral losses and molecular rearrangements. Machine learning algorithms are used to build the model. Bond strength might dramatically vary with different adjacent chemical structures, which potentially leads to low performance of in-silico predictions if it is not correctly modeled [72]. An extended review of algorithms for in-silico MS/MS prediction can be found elsewhere [73].
Fig. 5.
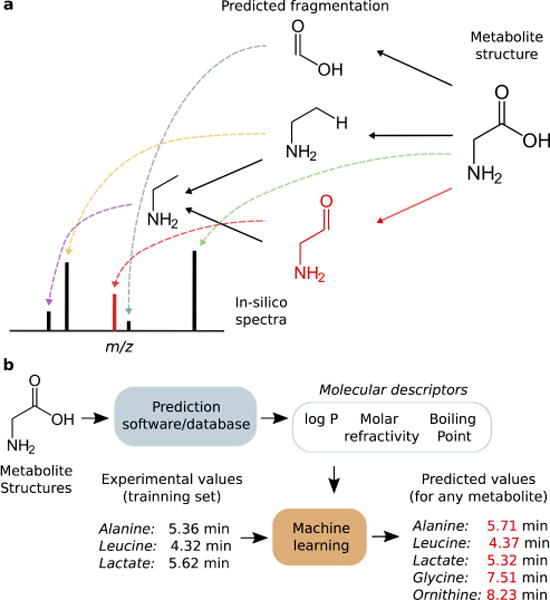
In-silico MS/MS and retention time prediction. In (a), in-silico predictions are based on inferring resulting substructures after fragmentation for a given collision energy. Each of these substructures correspond to a m/z ratio. If substructures are not correctly predicted, false peaks will be generated in the in-silico spectra (red). Of note, each of these substructures or neutral losses can undergo through rearrangements and neutral losses. In (b), from a given metabolite structure, softwares or databases are used to predict molecular descriptors. From these predicted descriptors, a machine learning algorithm is trainned with experimental values. Machine learning then builds a model that relates molecular descriptors with retention time. This allows computing predicted retention times for any metabolite not initially included in the tranning set.
Retention time prediction
The comparison of two orthogonal pysicho-chemical properties such as neutral mass or MS/MS spectral pattern with retention time (RT) is a robust metric to assess the identity of a metabolite. Retention time can differentiate between multiple isomeric candidates obtained by accurate mass search or even tandem MS. Unlike gas chromatography – mass spectrometry, where the robustness of capillary columns allow the standardization of RT into library-available retention index, analysis of pure standards are needed to obtain empirical metabolite RT in LC/MS. To overcome this problem, RT prediction has been proposed as an alternative to the use of standard materials. These strategies propose the use of a series of theoretical molecular descriptors obtained from molecular structures, and correlate these descriptors with empirical retention time of a “training set” of metabolites. Once a statistical model is created, it is possible to infer the RT for any existing metabolite not included in the “training set”, greatly improving metabolite annotation. Those models are known as quantitative structure–retention relationships (QSRR) models [74]. As an example, partition-coefficient (log P) is known to show a relevant correlation with experimental RT in reverse phase chromatography [75]. Different studies proposed (QSRR) models to predict metabolite RT [54,76–84] . For example, Creek et al. [76] performed a multiple linear regression among molecular descriptors and RT in hydrophilic interaction liquid chromatography (HILIC) chromatography. Wolfer et al. [82] combined artificial neural networks to select the best combination of molecular descriptors to subsequently predict RT in reversed-phased (RP) chromatography by support vector machine and random forest learning methods. As commented before, the 1-SToP algorithm [34] also uses QSSR models to predict both in-source fragments and retention time.
QSRR models performance largely depend on the “training set” used, and therefore they need a wide representative set of empirical retention times to be able to effectively predict RT for any other metabolite, which molecular predictors have never been “seen” before by the QSRR model. While a small set of training metabolites will not be able to accurately predict RT, a large number of training metabolites requires several experimental assays. QSRR models performance also depend on the limited accuracy of molecular descriptors [75]. Moreover, those models are going to be unique for the chromatographic method used (e.g., type of column, solvent), which makes them non-extensible to other configurations.
An interesting concept was recently introduced by Stanstrup et al. [85], a collaborative web-based platform for RT prediction named PredRet. Users upload their empirical RT along with the description of the chromatographic method used. PredRet then constructs a model by projecting users’ RT into a similar chromatographic system in the database. The RT of the metabolites in the database can then be projected back onto the user’s chromatographic system, allowing a larger number of metabolites RT to be obtained. This approach is however limited by the number of chromatographic systems and metabolite RT available in the database.
Conclusion and perspective
Despite the fair range of algorithms, tools and resources to annotate data derived from complex experiments in untargeted metabolomics studies, users still have to go through a tiresome process of annotation, which involves manual data curation. This is attributed to the rich and complex nature of LC/MS-based untargeted metabolomics samples, which makes the computational translation of raw signals into interpretable biological knowledge difficult. In gas chromatography – mass spectrometry (GC–MS) for instance, the highly reproducible electron impact (EI) ionization source together with the robustness of capillary columns allow a more straightforward identification of metabolites, using both spectral and standardized retention time comparison with longstanding libraries such as NIST. Specifically, and compared to GC–MS, while the application of more advances multivariate techniques for high-throughput spectral extraction in GC–MS have been used [86, 87], the application of those approaches in LC/MS data is still a proof-of-concept [88]. In GC–MS, these multivariate strategies have allowed to replace features as the analysis entity, and provide a single metabolite quantitative value per sample. In LC/MS, an effective extraction of MS1 pseudospectra could replace the feature for the metabolite as the analysis entity, thus reducing the number of features and facilitating the quantitative and annotative tasks in metabolomics. Developing smarter and more accurate tools for LC/MS based metabolomics studies is of great interest to the entire community, given the broader metabolome coverage and simpler sample preparation steps compared to GC–MS based experiments.
Whereas genes or proteins are composed of well-defined sequences of amino acids, metabolite structures are highly diverse, which makes spectral libraries a necessary resource in metabolomics research. However, only ~10% of known metabolites in databases such as Human Metabolome Database (HMDB) [8] or METLIN [7] have experimental spectral data [9]. On the other hand, in-silico MS/MS or retention time predictions are still far from having the necessary accuracy to serve as gold standard reference for metabolite identification. Advanced models based perhaps on first-principle CID fragmentation, or other innovative resources have to be designed to provide accurate layers of complementary – or orthogonal – information that help address these issues.
Overall, the available tools and methods for metabolite annotation now partially enable the broad utility of metabolomics. However, ultimately this field will depend on new computational resources, algorithms and instrumental advances to facilitate the complete interpretation of complex mass spectrometry data.
Acknowledgments
This work was partially supported by Ecosystems and Networks Integrated with Genes and Molecular Assemblies (ENIGMA), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory for the US Department of Energy, Office of Science, Office of Biological and Environmental Research under contract number DE-AC02-05CH11231 (G.S.); and National Institutes of Health grants R01 GMH4368 (G.S.) and PO1 A1043376-02S1 (G.S.).
References
- 1.Katajamaa M, Jarkko M, Matej O. Bioinformatics. 2006;22:634–636. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- 2.Pluskal T, Castillo S, Villar-Briones A, Oresic M. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lommen A, Harrie JK. Metabolomics. 2012;8:719–726. doi: 10.1007/s11306-011-0369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 5.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. Anal Chem. 2012;84:5035– 5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia J, Wishart DS. Curr Protoc Bioinformatics. 2016;55:14.10.1–14.10.91. doi: 10.1002/cpbi.11. [DOI] [PubMed] [Google Scholar]
- 7.Smith CA, O’Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G. Ther Drug Monit. 2005;27:747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 8.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, Scalbert A. Nucleic Acids Res. 2013;41:801–807. doi: 10.1093/nar/gks992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, Yanes O. Trends Analyt Chem. 2016;78:23–35. [Google Scholar]
- 10.Patti GJ, Yanes O, Siuzdak G. Nat Rev Mol Cell Biol. 2012;13:63–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR. Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kind T, Fiehn O. BMC Bioinformatics. 2006;7:234. doi: 10.1186/1471-2105-7-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misra BB, van der Hooft JJJ. Electrophoresis. 2015;37:86–110. doi: 10.1002/elps.201500417. [DOI] [PubMed] [Google Scholar]
- 14.Misra BB, Fahrmann JF, Grapov D. Electrophoresis. 2017;38:2257–2274. doi: 10.1002/elps.201700110. [DOI] [PubMed] [Google Scholar]
- 15.Spicer R, Salek RM, Moreno P, Cañueto D, Steinbeck C. Metabolomics. 2017;13:106. doi: 10.1007/s11306-017-1242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhl C, Tautenhahn R, Bottcher C, Larson TR, Neumann S. Anal Chem. 2012;84:283–289. doi: 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda F, Shinbo Y, Oikawa A, Hirai MY, Fiehn O, Kanaya S, Saito K. PLOS ONE. 2009;4:e7490. doi: 10.1371/journal.pone.0007490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown LJ, Smith RW, Toutoungi DE, Reynolds JC, Bristow AW, Ray A, Sage A, Wilson ID, Weston DJ, Boyle B, Creaser CS. Anal Chem. 2012;84:4095–4103. doi: 10.1021/ac300212r. [DOI] [PubMed] [Google Scholar]
- 19.Xu YF, Lu W, Rabinowitz JD. Anal Chem. 2015;87:2273–2281. doi: 10.1021/ac504118y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou B, Xiao JF, Tuli L, Ressom HW. Mol Biosyst. 2012;8:470–481. doi: 10.1039/c1mb05350g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahieu NG, Spalding JL, Gelman SJ, Patti GJ. Anal Chem. 2016;88:9037–9046. doi: 10.1021/acs.analchem.6b01702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ipsen A, Want EJ, Lindon JC, Ebbels TMD. Anal Chem. 2010;82:1766–1778. doi: 10.1021/ac902361f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Chang J, Lei Z, Huhman D, Sumner LW, Zhao PX. Anal Chem. 2014;86:6245–6253. doi: 10.1021/ac501162k. [DOI] [PubMed] [Google Scholar]
- 24.Draper J, Enot DP, Parker D, Beckmann M, Snowdon S, Lin W, Zubair H. BMC Bioinformatics. 2009;10:227. doi: 10.1186/1471-2105-10-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alonso A, Julià A, Beltran A, Vinaixa M, Díaz M, Ibañez L, Correig X, Marsal S. Bioinformatics. 2011;27:1339–1340. doi: 10.1093/bioinformatics/btr138. [DOI] [PubMed] [Google Scholar]
- 26.Tikunov YM, Laptenok S, Hall RD, Bovy A, de Vos RC. Metabolomics. 2012;8:714–718. doi: 10.1007/s11306-011-0368-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu H, Gowda GA, Neto FC, Opp MR, Raftery D. Anal Chem. 2013;85:10771–10779. doi: 10.1021/ac4019268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broeckling CD, Afsar FA, Neumann S, Ben-Hur A, Prenni JE. Anal Chem. 2014;86:6812–6817. doi: 10.1021/ac501530d. [DOI] [PubMed] [Google Scholar]
- 29.Uppal K, Walker DI, Jones DP. Anal Chem. 2017;89:1063–1067. doi: 10.1021/acs.analchem.6b01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daly R, Rogers S, Wandy J, Jankevics A, Burgess KE, Breitling R. Bioinformatics. 2014;30:2764–2771. doi: 10.1093/bioinformatics/btu370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernàndez-Albert F, Llorach R, Andrés-Lacueva C, Perera A. Bioinformatics. 2014;30:1937–1939. doi: 10.1093/bioinformatics/btu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baran R, Northen TR. Anal Chem. 2013;85:9777–9784. doi: 10.1021/ac402180c. [DOI] [PubMed] [Google Scholar]
- 33.Lynn KS, Cheng ML, Chen YR, Hsu C, Chen A, Lih TM, Chang HY, Huang CJ, Shiao MS, Pan WH, Sung TY, Hsu WL. Anal Chem. 2015;87:2143–2151. doi: 10.1021/ac503325c. [DOI] [PubMed] [Google Scholar]
- 34.Broeckling CD, Ganna A, Layer M, Brown K, Sutton B, Ingelsson E, Peers G, Prenni JE. Anal Chem. 2016;88:9226–9234. doi: 10.1021/acs.analchem.6b02479. [DOI] [PubMed] [Google Scholar]
- 35.Kind T, Fiehn O. BMC Bioinformatics. 2007;8:105. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ipsen A, Want EJ, Ebbels TM. Anal Chem. 2010;82:7319–7328. doi: 10.1021/ac101278x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treutler H, Neumann S. Metabolites. 2016;6:E37. doi: 10.3390/metabo6040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breitling R, Ritchie S, Goodenowe D, Stewart ML, Barrett MP. Metabolomics. 2006;2:155–164. doi: 10.1007/s11306-006-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers S, Scheltema RA, Girolami M, Breitling R. Bioinformatics. 2009;25:512–518. doi: 10.1093/bioinformatics/btn642. [DOI] [PubMed] [Google Scholar]
- 40.Dunn WB, Erban A, Weber RJM, Creek DJ, Brown M, Breitling R, Hankemeier T, Goodacre R, Neumann S, Kopka J, Viant MR. Metabolomics. 2013;9:44–66. [Google Scholar]
- 41.Weber RJM, Viant MR. Chemometr Intell Lab. 2010;104:75–82. [Google Scholar]
- 42.Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, Pulendran B. PLoS Comput Biol. 2013;9:e1003123. doi: 10.1371/journal.pcbi.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva RR, Jourdan F, Salvanha DM, Letisse F, Jamin EL, Guidetti-Gonzalez S, Labate CA, Vencio RZ. Bioinformatics. 2014;30:1336–1337. doi: 10.1093/bioinformatics/btu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huan T, Forsberg EM, Rinehart D, Johnson CH, Ivanisevic J, Benton HP, Fang M, Aisporna A, Hilmers B, Poole FL, Thorgersen MP, Adams MWW, Krantz G, Fields MW, Robbins PD, Niedernhofer LJ, Ideker T, Majumder EL, Wall JD, Rattray NJW, Goodacre R, Lairson LL, Siuzdak G. Nat Meth. 2017;14:461–462. doi: 10.1038/nmeth.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaquerel E, Kuhl C, Neumann S. Metabolomics. 2013;9:904–918. [Google Scholar]
- 46.Uppal K, Soltow QA, Promislow DEL, Wachtman LM, Quyyumi AA, Jones DP. Front Bioeng Biotechnol. 2015;3:87. doi: 10.3389/fbioe.2015.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deo RC, Hunter L, Lewis GD, Pare G, Vasan RS, Chasman D, Wang TJ, Gerszten RE, Roth FP. PLoS Comput Biol. 2010;6:e1000692. doi: 10.1371/journal.pcbi.1000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Karp PD. Nucleic Acids Res. 2016;44:D471–480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, Matthews L, May B, Milacic M, Rothfels K, Shamovsky V, Webber M, Weiser J, Williams M, Wu G, Stein L, Hermjakob H, D’Eustachio P. Nucleic Acids Res. 2016;44:D481–487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheubert K, Hufsky F, Petras D, Wang M, Nothias L, Duehrkop K, Bandeira N, Dorrestein P, Boecker S. bioRxiv. 2017:109389. doi: 10.1038/s41467-017-01318-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benton HP, Ivanisevic J, Mahieu NG, Kurczy ME, Johnson CH, Franco L, Rinehart D, Valentine E, Gowda H, Ubhi BK, Tautenhahn R, Gieschen A, Fields MW, Patti GJ, Siuzdak G. Anal Chem. 2015;87:884–891. doi: 10.1021/ac5025649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menikarachchi LC, Cawley S, Hill DW, Hall LM, Hall L, Lai S, Wilder J, Grant DF. Anal Chem. 2012;84:9388–9394. doi: 10.1021/ac302048x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerlich M, Neumann SJ. Mass Spectrom. 2013;48:291–298. doi: 10.1002/jms.3123. [DOI] [PubMed] [Google Scholar]
- 56.Li L, Li R, Zhou J, Zuniga A, Stanislaus AE, Wu Y, Huan T, Zheng J, Shi Y, Wishart DS, Lin G. Anal Chem. 2013;85:3401–3408. doi: 10.1021/ac400099b. [DOI] [PubMed] [Google Scholar]
- 57.Huan T, Tang C, Li R, Shi Y, Lin G, Li L. Anal Chem. 2015;87:10619–10626. doi: 10.1021/acs.analchem.5b03126. [DOI] [PubMed] [Google Scholar]
- 58.Allen F, Pon A, Wilson M, Greiner R, Wishart D. Nucleic Acids Res. 2014;42:W94–99. doi: 10.1093/nar/gku436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsugawa H, Kind T, Nakabayashi R, Yukihira D, Tanaka W, Cajka T, Saito K, Fiehn O, Arita M. Anal Chem. 2016;88:7946–7958. doi: 10.1021/acs.analchem.6b00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruttkies C, Schymanski EL, Wolf S, Hollender J, Neumann S. Journal of Cheminformatics. 2016;8:3. doi: 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witting M, Ruttkies C, Neumann S, Schmitt-Kopplin P. PLoS ONE. 2017;12:e0172311. doi: 10.1371/journal.pone.0172311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-Perez JM, Nicholson JK. Rapid Commun Mass Spectrom. 2006;20:1989–1994. doi: 10.1002/rcm.2550. [DOI] [PubMed] [Google Scholar]
- 63.Zhu X, Chen Y, Subramanian R. Anal Chem. 2014;86:1202–1209. doi: 10.1021/ac403385y. [DOI] [PubMed] [Google Scholar]
- 64.Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, Arita M. Nat Meth. 2015;12:523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, Cai Y, Guo Y, Chen F, Zhu Z. Anal Chem. 2016;88:8757–8764. doi: 10.1021/acs.analchem.6b02122. [DOI] [PubMed] [Google Scholar]
- 66.Wolf S, Schmidt S, Mller-Hannemann M, Neumann S. BMC Bioinformatics. 2010;11:148. doi: 10.1186/1471-2105-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heinonen M, Shen H, Zamboni N, Rousu J. Bioinformatics. 2012;28:2333–2341. doi: 10.1093/bioinformatics/bts437. [DOI] [PubMed] [Google Scholar]
- 68.Allen F, Greiner R, Wishart D. Metabolomics. 2015;11:98–110. [Google Scholar]
- 69.Dührkop K, Shen H, Meusel M, Rousu J, Böcker S. PNAS. 2015;112:12580– 12585. doi: 10.1073/pnas.1509788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aguilar-Mogas A, Sales-Pardo M, Navarro M, Guimerà R, Yanes O. Anal Chem. 2017;89:3474–3482. doi: 10.1021/acs.analchem.6b04512. [DOI] [PubMed] [Google Scholar]
- 71.Blaženović I, Kind T, Torbašinović H, Obrenović S, Mehta SS, Tsugawa H, Wermuth T, Schauer N, Jahn M, Biedendieck R, Jahn D, Fiehn O. J Cheminform. 2017;9 doi: 10.1186/s13321-017-0219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Wang X, Zeng X. Metabolomics. 2017;13:116. [Google Scholar]
- 73.Hufsky F, Scheubert K, Böcker S. TrAC Trends in Analytical Chemistry. 2014;53:41–48. [Google Scholar]
- 74.Put R, Heyden YV. Anal Chim Acta. 2007;602:164–172. doi: 10.1016/j.aca.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 75.Stanstrup J, Gerlich M, Dragsted LO, Neumann S. Anal Bioanal Chem. 2013;405:5037–5048. doi: 10.1007/s00216-013-6954-6. [DOI] [PubMed] [Google Scholar]
- 76.Creek DJ, Jankevics A, Breitling R, Watson DG, Barrett MP, Burgess KEV. Anal Chem. 2011;83:8703–8710. doi: 10.1021/ac2021823. [DOI] [PubMed] [Google Scholar]
- 77.Hall LM, Hall LH, Kertesz TM, Hill DW, Sharp TR, Oblak EZ, Dong YW, Wishart DS, Chen M, Grant DFJ. Chem Inf Model. 2012;52:1222–1237. doi: 10.1021/ci300092s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goryński K, Bojko B, Nowaczyk A, Buciński A, Pawliszyn J, Kaliszan R. Anal Chim Acta. 2013;797:13–19. doi: 10.1016/j.aca.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 79.Eugster PJ, Boccard J, Debrus B, Bréant L, Wolfender J, Martel S, Carrupt P. Phytochemistry. 2014;108:196–207. doi: 10.1016/j.phytochem.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 80.Cao M, Fraser K, Huege J, Featonby T, Rasmussen S, Jones C. Metabolomics. 2015;11:696–706. doi: 10.1007/s11306-014-0727-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aicheler F, Li J, Hoene M, Lehmann R, Xu G, Kohlbacher O. Anal Chem. 2015;87:7698–7704. doi: 10.1021/acs.analchem.5b01139. [DOI] [PubMed] [Google Scholar]
- 82.Wolfer AM, Lozano S, Umbdenstock T, Croixmarie V, Arrault A, Vayer P. Metabolomics. 2016;12:8. [Google Scholar]
- 83.Bruderer T, Varesio E, Hopfgartner GJ. Chromatogr B. 2017 doi: 10.1016/j.jchromb.2017.07.016. Just Accepted. [DOI] [PubMed] [Google Scholar]
- 84.Randazzo GM, Tonoli D, Strajhar P, Xenarios I, Odermatt A, Boccard J, Rudaz SJ. Chromatogr B. 2017 doi: 10.1016/j.jchromb.2017.04.032. Just Accepted. [DOI] [PubMed] [Google Scholar]
- 85.Stanstrup J, Neumann S, Vrhov sek U. Anal Chem. 2015;87:9421–9428. doi: 10.1021/acs.analchem.5b02287. [DOI] [PubMed] [Google Scholar]
- 86.Domingo-Almenara X, Brezmes J, Vinaixa M, Samino S, Ramirez N, Ramon-Krauel M, Lerin C, Díaz M, Ibáñez L, Correig X, Perera-Lluna A, Yanes O. Anal Chem. 2016;88:9821–9829. doi: 10.1021/acs.analchem.6b02927. [DOI] [PubMed] [Google Scholar]
- 87.Domingo-Almenara X, Brezmes J, Venturini G, Vivó-Truyols G, Perera A, Vinaixa M. Metabolomics. 2017;13:93. [Google Scholar]
- 88.Gorrochategui E, Jaumot J, Lacorte S, Tauler R. Trends Analyt Chem. 2016;82:425–442. [Google Scholar]