

In the nearly 15 years since electron transfer dissociation (ETD) was introduced,1 bioanalytical mass spectrometry (MS) and MS-driven proteomics have continued to develop as the premier tools for characterizing proteome composition, structure, and function.2 The new era of proteomics has focused on throughput, sampling depth, and reproducibility, often relying on traditional collision-based dissociation because of its speed and ease of implementation.3−5 That said, the development of alternative dissociation methods remains an active field in proteome research, offering complementary analyses to collisional activation and valuable insights for analytes that can be intractable with traditional methods.6 Because of its applicability to a wide range of biomolecules and its compatibility with a variety of instrument platforms, ETD is among the most prominent of these alternative approaches. Concentrating on developments in the last 5 years, this review examines the role ETD plays in modern proteomic experiments, including its use in characterizing intact proteins, post-translational modifications (PTMs), and structural aspects of the proteome (Figure 1). We focus on ETD instrumentation and methodological developments and how they are being employed to answer biologically driven problems, and we also include brief discussions about other ion–ion reaction techniques. Finally, we offer our perspective on the future of ETD development and comment on how ETD will continue to be a major contributor to proteome research in years to come.
Figure 1.

Diverse roles of electron transfer dissociation. ETD is a valuable tandem MS method for characterizing many aspects of the proteome and is used in bottom-up, middle-down, and top-down proteomic methods. Structural Proteomics image is reprinted in part by permission from Macmillan Publishers Ltd.: NATURE, Liu, F.; Rijkers, D. T. S.; Post, H.; Heck, A. J. R. Nat. Methods2015, 12, 1179–1184 (ref (255)). Copyright 2015. Deep Proteome Sequencing image is reproduced in part from Proteomics Beyond Trypsin, Tsiatsiani, L; Heck, A.J.R. FEBS J., Vol. 282, Issue 14, pp 2612–2626 (ref (189)). Copyright 2015 Wiley.
Principles of ETD
ETD is an ion–ion reaction involving multiply charged precursor peptide (or protein) cations and singly charged radical reagent anions. An electron from the reagent anion is transferred to the cation, resulting in an odd-electron cation that undergoes free-radical-driven cleavage. Similar to its ion-electron predecessor, electron capture dissociation (ECD),7 this cleavage dissociates N–Cα bonds along the peptide backbone to generate even electron c-type and odd electron z•-type product ions that provide sequence information on the peptide precursor.8 Here we briefly cover basic principles of ETD to provide context for recent developments. For in-depth discussions of radical-mediated gas-phase chemistry and reaction pathways involved in ETD (and related techniques), we direct readers to several excellent recent reviews.9−11 Other reviews have given a more historical perspective on how the introduction of ETD provided new dimensions in proteomic analyses as well.12,13
The success of ETD reactions, i.e., the ability to generate sequence informative fragment ions, is largely dependent on (1) the reagent anion used as an electron donor and (2) the precursor cation charge density. Several small-molecule reagent anions have been explored within the context of ETD reactions, leading to either peptide deprotonation (proton transfer) or deposition of an electron from the reagent anion onto the peptide (electron transfer), the latter of which promotes peptide backbone cleavage.14 Anthracene, 9,10-diphenyl-anthracene, azobenzene, and azulene have demonstrated suitability for ETD reactions,1,14−16 but fluoranthene is generally regarded as one of the more favorable ETD reagents for generating c- and z•-type ions.17−19
In addition to the chemical properties of the reagent anion, precursor cation charge density governs successful generation of product ions in ETD reactions, with higher charge density being much more favorable.20−22 The gas-phase structure of peptide precursor ions is directly related to its charge density: precursors with higher charge density are more linear and those with lower charge density are more compact. When ETD reactions are conducted with lower charge density precursors, peptide backbone cleavage can occur but c- and z•-type product ions are held together by noncovalent interactions present in the more compact structures. Through this process, called nondissociative electron transfer dissociation (ETnoD),23 the radical product ion complex appears in the mass spectrum as an intact species (i.e., [M + nH](n−1)•), yielding no sequence information. One way to combat this challenge involves chemical modification of peptides to increase their charge density and, thus, improve fragmentation.24,25 ETD has also been paired with collision-based fragmentation methods within a given experiment, where ETD is employed for higher charge (z ≥ 3), low m/z precursors and collisional dissociation is used for doubly protonated and higher m/z ions.26,27 These so-called decision-tree methods, where the fragmentation method most suited for a given precursor is chosen in real-time, have proven valuable for increasing identifications and improving confidence of peptide spectral matches in a wide variety of analyses. Indeed, this theme of complementary use of ETD and collision-based fragmentation is present throughout this review and has been discussed previously.8,28
Supplemental Activation for ETD Reactions
Supplemental activation is another approach to increase efficiency of ETD by introducing extra energy into the reaction, and Figure 2 summarizes the most common supplemental activation strategies. Drawing on several years of research in ECD methodology,29−34 the goal of these methods is to disrupt noncovalent interactions that contribute to ETnoD (usually via collisions or photons) to promote product ion generation, especially for low charge density precursors. Supplemental activation schemes that utilize collisions are popular and easily implemented, but they are generally limited to post-ETD activation. Pre-ETD activation is generally ineffective because the bath gas pressures in ion traps where ETD is usually performed are high enough (∼3 mTorr) to promote collisional cooling of activated precursors, which causes them to relax back into more compact structures. Collisional activation during ETD reactions is a nonviable supplemental activation approach because it prevents sufficient overlap of the cation and anion clouds, which is required for the ion–ion reaction to occur.35 Several iterations of post-ETD activation have been explored,36−41 with the most widely adopted being gentle resonant excitation of ETnoD products, a process termed ETcaD.42 Most recently, the Heck group introduced EThcD, which involves broadband activation of all ETD products with high-energy collisional dissociation (HCD).43 Opposed to ETcaD, which produces mainly c/z-type product ions, EThcD generates both c/z- and b/y-type product ions by activating both the ETnoD products and remaining unreacted precursor ions. EThcD produces series of complementary fragments that increase peptide backbone coverage (i.e., the number of inter-residue bonds explained by observed fragments) for improved confidence in peptide identifications, and it boosts the number of peptides that can be identified in LC–MS/MS analyses in complex mixtures compared to ETD and ETcaD.
Figure 2.

Supplemental activation strategies for ETD. ETD spectra from low-charge density precursors can have modest product ion generation (top left), but supplemental activation strategies can increase precursor-to-product ion conversion. In ETcaD (green), ETnoD products are resonantly excited to disrupt noncovalent interactions and release sequence-informative c/z-type product ions. EThcD (blue) uses broadband beam-type collisional activation of all ETD reaction products to generate b/y- and c/z-type fragments. Both ETcaD and EThcD perform supplemental activation steps after the ETD reaction is finished. AI-ETD (red) leverages infrared photoactivation simultaneous to the ETD reaction to promote formation of mostly c/z•-type fragments. Each strategy significantly improves the product ion yield of ETD reactions. Several hybrid MS systems have ETD capabilities and various supplemental activation options (bottom left, supplemental activation options indicated with colored circles). Note, implementations of some methods on the FTICR and TOF systems may differ slightly from the originally described technique, e.g., AI-ETD on the quadrupole-FTICR is only available for postreaction photoactivation.
Two challenges of post-ETD supplemental activation include increased levels of hydrogen migration between ETD product ions and additional time needed to perform the activation, which adds to the overhead time required per MS/MS scan. Hydrogen migrations occur in the radical ETnoD complex, where even electron c-type ions and odd electron z•-type ions are held together for several milliseconds or more by noncovalent interactions. While in this complex, radical z•-type ions can abstract a hydrogen atom from c-type products, resulting in a population of odd-electron c+•-type and even electron z+-type ions.42,44−46 Mixtures of c/z•-type and c+•/z+-type product ions, which are present in ETcaD and EThcD, can complicate spectra and challenge both manual and automated spectral interpretation. The additional scan times required for ETcaD and EThcD differs depending on instrument configuration, but ETcaD often affects scan times more dramatically (tens of milliseconds), while EThcD can add ∼5–15 ms to each MS/MS scan.
The combination of these challenges associated with post-ETD collision-based supplemental activation led to investigations into photoactivation to improve ETD efficiency. Photoactivation provides greater flexibility for this purpose because it cannot only be used for pre- or postreaction activation, but more importantly, it can also be used to activate ions concurrent to the ion–ion reaction. Pre-ETD photoactivation is rarely used because collisional cooling in the ion trap still occurs, but ideal ion cloud overlap for ETD reactions can be readily achieved while simultaneously irradiating the trapping volume of the reaction cell, meaning photoactivation can happen concomitant with ETD. Activated ion ETD (AI-ETD) uses infrared photoactivation (10.6 μm) concurrent with ETD reactions to disrupt noncovalent interactions and minimize ETnoD, significantly increasing sequence-informative product ion generation.47,48 The concurrent nature of activation in AI-ETD requires no additional time to complete and also minimizes the hydrogen migration seen in ETcaD and EThcD.47 These benefits translate to increased peptide identifications in LC–MS/MS experiments compared to ETD, ETcaD, and EThcD.47−49 In general, AI-ETD produces mainly ETD-specific c/z•-type product ions, although increases in y-type ions (which are generated in standard ETD reactions as well50) are also observed. Furthermore, AI-ETD has been combined with a short (∼4 ms) postactivation using infrared multiphoton activation (IRMPD) to generate spectra rich in both c/z•- and b/y-type fragments, much like EThcD.48 This strategy, termed AI-ETD+, utilizes short settling times built into existing scan sequences to perform its postreaction IRMPD activation following standard AI-ETD, meaning it also adds no additional time to the scan sequence but can further improve peptide backbone coverage and identifications.
Other wavelengths have been explored for ETD supplemental photoactivation to increase product ion yield, too, most notably 193 nm photons used in ultraviolet photodissociation (UVPD). This hybrid reaction scheme is called ETUVPD and can be used for both broadband activation of all ETD products and specific activation of charge reduced species, although activation typically occurs postreaction.51 The 193 nm photons used in ETUVPD are significantly more energetic than the infrared photons used in AI-ETD (6.4 vs 0.12 eV), resulting in UVPD-driven product ion generation in addition to ETD products. Observed fragment ion types included a/x-, b/y-, and c/z-type products, with product ion distributions showing intermediate percentages of fragment types between those seen in ETD and UVPD. Overall, ETUVPD can provide a boost in both product ion generation and sequence coverage over ETD alone but has not been widely implemented.
ETD-Equipped Instrumentation
One of the main drivers for the prevalence of ETD has been its amenability to numerous instrumentation platforms. The development of ETD as an ion–ion reaction that could be performed in rf trapping devices was the technological advance that brought the radically driven fragmentation of ECD to a wider audience. Since its introduction on a quadrupole linear ion trap (QLT), ETD has been incorporated into numerous instrument layouts, and we estimate that well over 1000 ETD-enabled commercial instruments have been installed in various laboratories around the world.
Implementation of ETD on hybrid MS systems that have multiple mass analyzers has greatly improved its utility by offering flexibility in reaction conditions, timing of ion manipulations, and high-resolution mass analysis of precursor and product ions. The first commercially available ETD-enabled hybrid mass spectrometer was a quadrupole linear ion trap-Orbitrap (QLT-OT) system that used a negative chemical ionization (CI) source at the back of the instrument to generate radical anions, similar to the original description of ETD on a standalone QLT system.19 This QLT-OT design served as a major development platform for ETD technologies for years, including decision tree and supplemental activation methods (described above), and was also the first Orbitrap platform to employ the compact, high-field Orbitrap analyzer that decreased the required transient time to achieve a desired resolution,52 a significant benefit for ETD MS/MS spectra of highly charged precursor ions.
Although robust for ETD development, the QLT-OT hybrid system was not necessarily the ideal implementation for ETD. In 2013, our group described a modified system in which the rf quadrupole collision cell used exclusively for HCD was replaced with a multipurpose dissociation cell (MDC) that was specifically designed to improve ETD reactions on the QLT-OT system.53 The MDC had the same dimensions as the HCD collision cell, but it had four independently controllable segments (for simultaneous separate storage of precursor cations and reagent anions) and could perform charge-sign independent trapping for ETD reactions via a secondary rf voltage applied the end lenses of the cell. The net result was an ion–ion reaction cell capable of storing nearly 10 times as many precursor ions than a standard QLT and performing ion–ion reactions at least twice as fast as the QLT due to higher ion cloud densities. Larger precursor ion populations significantly improved product ion signal-to-noise (S/N) in ETD MS/MS spectra, and the faster reaction times increased throughput for LC–MS/MS analyses. Importantly, this provided a direct route for introducing a photons into the ETD reaction cell for AI-ETD54 as well, which is not easily accomplished in the QLT due to instrument geometry. The Heck group also investigated performing ETD reactions in the HCD collision cell itself.55 They created a Z-shaped potential using a static dc gradient, providing two potential minima for different charge signs to allow charge sign independent trapping of cations and anions. In this strategy, increasing the accumulation time (i.e., population size) of reagent anions caused overlap of the cation and anion clouds in the middle of the HCD cell due to space charge, which resulted in ETD. This implementation ultimately proved far less efficient than ETD in the QLT because it required long anion injection times, but it provided a way to increase the size of precursor cation populations by using a bigger reaction cell (similar to the MDC). It also enabled access to photoactivation schemes for ETD and was used for the ETUVPD experiments discussed above.
Another approach to increasing product ion S/N in ETD MS/MS spectra is to perform a single mass analysis on product ions generated from several ETD reactions. This idea of mass analyzing “multiple fills” is prohibited by the position of the reagent anion CI source at the back of the QLT-OT systems because there is no storage region available in the instrument that is not also utilized to transfer reagent anions prior to each ETD reaction. To circumvent this limitation, Hunt and co-workers pioneered the design of a front-end ETD source that permits reagent anion generation near the inlet at the front of the instrument.56 Using the front-end ETD source, both precursor cations and reagent ions can be introduced into the QLT using only the front ion optics of the instrument, allowing products from several ETD reactions to be stored in the C-trap behind the QLT prior to mass analysis for improved spectral quality. Importantly, the front-end ETD source also represents a significant improvement in reagent anion generation over the previously used negative chemical ionization source. The front-end source uses a glow discharge operated in a relatively high pressure region, providing a stable and robust source of electrons for reagent anion generation and eliminating most of the failures seen with filament-based CI sources.
Many of ETD-specific improvements investigated on QLT-OT systems have recently become commercially available on one of the newest generations of Orbitrap hybrid instruments, a quadrupole-Orbitrap-quadrupole linear ion trap (q-OT-QLT) Tribrid MS system.57 Perhaps most notably, the use of the front-end reagent anion source has made ETD much more robust and user-friendly, expanding its use to more nonexpert laboratories. ETD reactions on larger precursor ion populations is now possible as well, even though the QLT is still used as the ETD reaction cell. In an approach called high-capacity ETD, the accumulation of precursor and reagent ions has been modified to allow for reactions on larger precursor ion populations, which has proved especially beneficial for analysis of intact proteins.58 This provides many of the same benefits for product ion S/N seen in the MDC work without requiring specialized hardware modifications. Furthermore, standardization of ETD reactions through calibrated reaction times, also first developed on the QLT-OT platform, has simplified ETD method development and use on the newer system.59 For supplemental activation, the q-OT-QLT is the first system with commercially available EThcD methods (and also has ETcaD). Moreover, the instrument geometry of the q-OT-QLT platform removed many of the constraints that prevented photoactivation in the QLT on the earlier systems, and a simple and robust implementation of AI-ETD was recently described that eliminated many of the challenges of previous AI-ETD setups.48 Overall, the q-OT-QLT system represents one of the most mature ETD-equipped MS platforms with many state-of-the-art ETD technologies.
Hybrid Orbitrap systems are far from the only mass spectrometers to provide ETD capabilities, however, and other platforms offer many benefits not achievable on the q-OT-QLT. ETD was first coupled with Fourier transform ion cyclotron resonance (FTICR) hybrid MS systems in 2008 using a hexapole ion trap to perform ETD reactions,60 which is now available in commercial systems. This implementation uses auxiliary rf potentials on the end lenses of the hexapole ion trap to enable mutual storage of cations and anions, with the duration of mutual storage governing the ion–ion reaction time (similar to work described by McLuckey and co-workers61). Recently, Hendrickson et al. at the National High Magnetic Field Laboratory described the first 21 Telsa (T) FTICR hybrid MS system, which incorporates a QLT at the front of the instrument to enable, among other benefits, ETD reactions.62 This instrument utilizes the front-end ETD reagent anion source described above, has supplemental activation capabilities, and has an external quadrupole trap that allows for multiple fills of MS/MS product ion populations prior to delivery to the ICR cell for mass analysis. The combination of ultrahigh resolution and multiple-fill ETD have shown significant benefits for intact protein analysis, where an impressive ∼90% sequence coverage was achieved for carbonic anhydrase (29 kDa) with ETD alone.63 Furthermore, Weisbrod et al. showed that the number of fills used for ETD mass spectra can be controlled on-the-fly based on precursor ion molecular weight using instrument control software, with the number of fills scaled to appropriately fit smaller or larger protein species.63
ETD has also been implemented on commercially available quadrupole-ion mobility-time-of-flight (q-IM-TOF) MS platforms. Ion mobility spectrometry enables rapid gas-phase separation of ions based on their mobility through a carrier gas and a number of approaches have been described to accomplish mobility separations through various means.64 Coupling ETD with ion mobility spectrometry provides a direct avenue to study detailed higher-order structures of peptides, proteins, and protein complexes. The ETD-capable q-IM-TOF system includes four traveling wave (T-wave) ion guides, with a quadrupole between T-wave cells 1 and 2 and a time-of-flight mass analyzer at the back. Precursor cations and reagent anions are sequentially generated, mass selected using the quadrupole, and stored in T-wave cell 2. Following precursor and reagent ion accumulation, ETD reactions are conducted by propelling precursor cations through the cloud of reagent anions using a traveling wave voltage, the amplitude and velocity of which control the ion–ion reaction time (typically tens of milliseconds).65,66 ETD product ions can then be subjected to traveling wave ion mobility separations and supplemental activation via beam-type collisions prior to TOF mass analysis. Alternatively, broadband ETD activation of all eluting precursor cations (i.e., no mass selection of individual precursors) in a data-independent acquisition fashion, called MSETD, has been demonstrated on this system with relatively simple mixtures, as well.67 The q-IM-TOF platform has been largely utilized for a number of structural proteomics studies discussed further later in this review.
Beyond commercially available instrument platforms, more specialized instruments equipped with ETD have also been developed. Field asymmetric waveform ion mobility spectrometry (FAIMS) and ETD have been combined on a research-grade QLT platform to improve analysis of post-translational modifications (via a device that is available for some commercial systems),68 and Valentine and co-workers recently introduced an ETD-enabled ion trap system modified with a low-pressure drift tube.69 Outside of ion mobility interfaces, two groups have recently coupled matrix-assisted ionization, an ionization technique that can produce multiply charged cations without laser ablation or high voltage, with multiple MS systems to improve characterization of modified peptides and intact proteins generated via ambient ionization using ETD and collisional activation.70,71
Other instrumentation developments have enabled studies designed to provide insight into the fundamentals of ETD reactions and other gas phase chemistries. He et al. described a dual-polarity ion trap to simultaneously manipulate and analyze cations and anions. This system provided a more refined study of ETD reactions by enabling detection of positive and negative mode products from the same reaction to investigate not only peptide dissociation products but also how the reagent anions are modified.72 Oomens and colleagues coupled an ion trap mass spectrometer to a variety of infrared laser sources at the Free Electron Lasers for Infrared eXperiments (FELIX) laboratory, allowing for flexible experimental designs in using infrared ion spectroscopy and IRMPD to characterize biomolecules, including ETD-generated products.73 This instrument has enabled examination of z•-type ETD products with infrared ion spectroscopy, allowing accurate determination of the products and mechanisms associated with ETD-like fragmentation.74 They have also commented on the nature of hydrogen migration in ETD products74 and have identified structures of both the ETD-inducing fluoranthene radical anion in addition to a closed-shell proton-transfer-prone fluoranthene anion, which has provided insights into the reaction mechanism involved in proton transfer.75 Moreover, this instrument setup allows for unique experiments, such as characterization of the degree of activation of small molecules in the active sites of homogeneous catalysts to investigate how ligand environments affect reactivity.76
Clearly the development of ETD instrumentation continues to push the field forward for both fundamental and applied investigations. Improved hardware and modified instrumentation have been a driving force in ETD development since its inception, and we expect future work will focus on both increasing sensitivity in ETD spectra (i.e., product ion S/N) and reducing ion–ion reaction times. Availability of ETD technologies, including both new instrument platforms and strategies like EThcD and AI-ETD, has been key in advancing proteome characterization, and below we discuss how current ETD technologies have enabled analyses of intact proteins, PTMs, cross-linked peptides, and other biomolecules (Figure 1).
ETD and Top-Down Proteomics
ETD is ideally suited for large, highly charged precursor cations, making it a valuable method for top-down proteomics. The top-down approach analyzes intact proteins (rather than proteolytically derived peptides) using tandem MS, aiming to characterize sequence truncations, splice variants, single nucleotide polymorphisms, and combinatorial patterns of PTMs that contribute to proteome complexity. For unambiguous sequence elucidation of proteoforms one must achieve extensive fragmentation of the protein backbone,77 making ETD an attractive method for improving sequence coverage and confidence in proteoform assignments. Advances in top-down proteomics span a diverse swath of technologies, including protein purification, both online and offline protein separations, and data analysis platforms, but ETD remains a critical component in the recent progress of top-down proteomic approaches (along with other alternative dissociation methods).78
ETD provides complementary dissociation of intact proteins compared to collision-based fragmentation,79−81 so the two methods are often used in tandem in top-down experiments. The combination of ETD and collisional dissociation has enabled large-scale, discovery-based profiling of proteoforms, including the study by Tran et al. that identified more than 1000 unique gene products and 3000 proteoforms.82 More recently, Kelleher and colleagues expanded this approach to generate the largest top-down study to date, identifying more than 1220 proteins and 5000 proteoforms from the human lung cancer cell line H1299.83 Despite the clear benefits, using multiple dissociation methods for each precursor ion in these large-scale top-down experiments limits throughput, usually requiring a significant investment in acquisition time. Durbin et al. presented a modified approach to top-down proteomic data acquisition to address this issue by guiding fragmentation of intact protein precursors to limit redundant sampling (Figure 3).84 A valuable extension of decision-tree methods, this automated acquisition scheme, called Autopilot, uses m/z and charge state to judiciously select ETD or collision-based dissociation. It also calculates precursor intact mass and performs online database searches for real-time identifications to prevent sampling of multiple precursor ions from the same protein species, especially those that have already been confidently identified. The Autopilot approach was applied to a large-scale top-down analysis of human fibroblast proteomes, allowing for quantitative comparison of nearly 1600 proteoforms in approximately one-fifth of the total acquisition time of previous experiments, representing a significant advance for top-down analyses of complex systems.85
Figure 3.
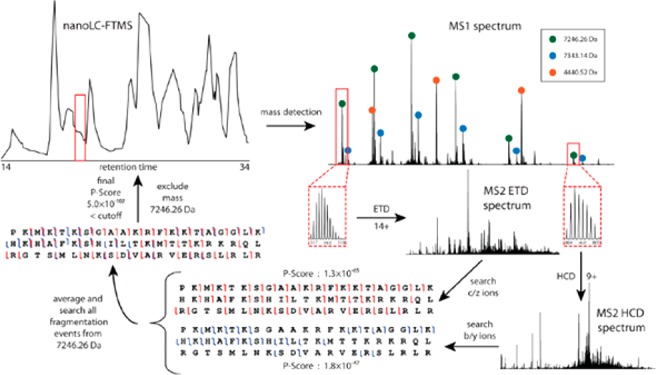
Autopilot data acquisition workflow on an example protein. A full precursor scan is taken, followed by HCD fragmentation of the 9+ charge state on the detected mass 7246.26 Da. After an online search, the software determines more analysis should be performed as the P-score (1.8 × 10–47) is not below the cutoff. An ETD scan of the highest charge state is taken and searched. The fragment ions are combined and the final P-score of 5.0 × 10–102 is below the cutoff. All charge states of the 7246.26 Da species are permanently excluded from further fragmentation and the system goes in search of the next target mass. Reproduced from Durbin, K. R.; Fellers, R. T.; Ntai, I.; Kelleher, N. L.; Compton, P. D. Anal. Chem.2014, 86, 1485–1492 (ref (84)). Copyright 2017 American Chemical Society.
Even though intact proteins tend to carry many charges that, in theory, make them good candidates for electron-driven dissociation, ETD efficiency can still suffer if overall precursor charge density is low. In recent years both EThcD and AI-ETD have been explored for top-down proteomic applications with the goal of increasing the number and S/N of sequence-informative product ions and protein sequence coverage (analogous to peptide backbone coverage).86,87 EThcD often increases the number of sequencing ions over ETD alone, especially for higher m/z precursors, but benefits are typically reduced for high charge density intact protein precursors (low m/z).88,89 That said, EThcD is still a valuable technique to improve intact protein characterization, as shown by Brunner et al. for more confident assignment of phosphosites in a 17.5 kDa phosphoprotein.86 AI-ETD shows promise as a suitable supplemental activation method for top-down proteomics, as it improves ETD-driven activation for precursor ions of all m/z and charge states.87−90 Similar to its performance in peptide dissociation, AI-ETD of intact proteins generates mainly c/z•-type product ions with some y-type fragments (and few b-type fragments), maintaining a similar distribution of product ion types to ETD while increasing the number and S/N of product ions observed.87−89 The similarity in product ion distribution between ETD and AI-ETD shows the additional energy imparted by the concurrent IR photoactivation in AI-ETD serves to disrupt weaker, noncovalent interactions but rarely drives vibrational dissociation of backbone bonds, which differs from EThcD (where the postreaction collisional activation generally increases the number of b/y-type ions observed). Comprehensive sequence coverage (75–100%) of proteins up to ∼20 kDa has been achieved with AI-ETD,88 making it comparable to the extensive top-down fragmentation reported with UVPD.91 Online separations with capillary zone electrophoresis have been coupled with AI-ETD to improve characterization of the Mycobacterium marinum secretome,92 and more thorough high-throughput LC–MS/MS top-down proteomic experiments with AI-ETD are forthcoming.
Even as top-down proteomics continues to progress, current technology has difficulty detecting proteins with molecular weights greater than ∼30 kDa. Raising this size barrier is a current topic in the field. In 2013, Tsybin and co-workers showed that ETD on a TOF MS system could provide extensive sequence information for moderately sized proteins (∼30 kDa) and structural motifs embedded in large proteins (up to ∼80 kDa).93 Just this year, Anderson et al. demonstrated the benefits of the ETD-equipped 21T FTICR hybrid MS system to characterize complex mixtures of human proteins, using a combination of collisional dissociation and multiple-fill ETD.94 ETD spectra typically provided superior confidence scores and proteoform characterization, and they identified 684 unique gene products and 3238 unique proteoforms, a considerable portion of which were >30 kDa. As instrumentation and other offline approaches continue to improve, AI-ETD may also play a role in expanding the molecular weight range that can be characterized in top-down proteomics experiments, as it was recently shown to outperform HCD, ETD, and EThcD for ∼30–70 kDa proteins.89
Other groups have focused on more clinical-based applications of ETD and top-down proteomics. Coelho Graça et al. showed that a targeted method to look for ETD fragments from intact hemoglobin ions from blood samples could provide a simple and flexible methodology that required only hours from sample collection to results, making it suitable for application in clinical laboratories.95 They followed up the proof-of-concept study by comparing rare hemoglobin β chain variants that enabled fast and reliable determination of uncommon hemoglobin proteoforms useful for hemoglobin disorder diagnoses.96 Other groups have worked to bring ETD to spatial analyses of tissues for potential clinical use. Using liquid extraction surface analysis (LESA) MS, Cooper and co-workers used ETD to reliably identify intact proteins in healthy and diseased human liver tissue, helping to distinguish potential protein biomarkers of nonalcoholic liver disease.97,98 Schey et al. combined spatially directed tissue microextraction to analyze intact proteins from specific locations in ocular lens, brain, and kidney tissues using LC–MS/MS with ETD, identifying several proteoforms that were difficult to assign with bottom-up methods.99 From our perspective, ETD offers a number of benefits for intact protein analysis, especially when used in concert with collisional dissociation. We anticipate that top-down proteomics will utilize ETD for years to come and will continue to incorporate new ETD technologies like EThcD and AI-ETD as they become available.
Characterizing Post-Translational Modifications with ETD
Protein post-translational modification is a dynamic and important process that regulates a diverse range of cellular functions.100,101 MS-based approaches are the premier tools for global PTM analysis, boasting high sensitivity, considerable throughput, amenability to diverse classes of PTMs, and the capacity to localize modifications to a single residue. Realization of these benefits, however, requires high-quality MS/MS spectra that have extensive fragmentation along the peptide/protein backbone, in addition to retention of the modifications themselves on the subsequent fragments. ETD has been central in analyses of many PTMs because it drives the radical dissociation of peptide backbone bonds while retaining labile modifications intact at modified residues. The breadth of work surrounding technology development for PTM analysis is far too great to cover here, especially because considerable efforts in these fields concentrate on offline methods, e.g., enrichment strategies, chromatographic separations, and data analysis. In the following section we focus on how ETD methodology specifically has been applied to characterize three specific classes of PTMs in recent years. We direct readers to recent reviews that cover broader technologies for analysis of phosphorylation,102−104 glycosylation,105−107 and PTMs in general100,101 for further insights. We also note that ETD has been valuable for PTM analysis beyond the scope discussed here, including ubiquitylation,108−110 ADP-ribosylation,111−113 and arginine methylation.114−118
Phosphorylation
Protein phosphorylation is a rapid and reversible means to modulate protein activity and transduce signals. Regulation of phosphorylation is a central mechanism in cell health and disease. Phosphorylation most often occurs on serine and threonine residues, with tyrosine phosphorylation also occurring with moderate frequency. The labile nature of phosphoryl groups can limit utility of collision-based dissociation for analysis of phosphorylated peptides and proteins because neutral losses, rather than peptide backbone fragmentation, are energetically favored pathways. Indeed, this shortcoming of collisional activation was a major impetus in the original development of ETD. Modern phosphoproteomic experiments often employ decision tree methods combining collision-based fragmentation and ETD to increase the number and confidence of phosphopeptide identifications,119−123 and more advanced decision tree algorithms have been designed to trigger ETD MS/MS scans based on phosphoric acid neutral losses or evaluation of phosphosite assignments in collisional activation spectra.124−126 These acquisition strategies are often paired with new informatic approaches to integrate ETD spectra into phosphosite assignment algorithms, and analysis of a large synthetic phosphopeptide library with multiple fragmentation methods has aided such developments.127 Neutral loss-triggered decision trees have also been employed for analysis of pyro-phosphorylation, a more rare version of serine and threonine phosphorylation, where EThcD was used to confidently localize pyro-phosphorylation sites and rule out possibilities of multiple standard (e.g., “non-pyro”) phosphosites.128
As expected, EThcD and AI-ETD supplemental activation methods have significantly benefitted ETD-based phosphoproteomic experiments. Increases in the number of sequencing ions and peptide backbone coverage provided by both methods greatly improves the ability to identify and unambiguously localize phosphosites in phosphopeptides, especially for low charge density precursors (which are more prevalent in phosphoproteomic experiments).90,129 Development of EThcD and AI-ETD for phosphoproteomics also led to modifications of the phosphoRS localization algorithm130 that uses observed product ions to statistically evaluate probabilities of phosphosite locations.90,129 Both EThcD and AI-ETD have been used for analyses of intact phosphoproteins,86,90 and AI-ETD has shown superior performance for localizing phosphosites in the multiply phosphorylated protein α-casein (∼23.5 kDa, eight sites). Lössl et al. used combinations of ETD and EThcD methods for integrated bottom-up and top-down phosphoproteomic experiments (among several other MS approaches) to decipher how the number and order of individual phosphorylation events impact protein behavior at a mechanistic level, especially in multiple-protein systems.131 A recent study from Tamara et al. showed that gas-phase phosphate transfer can readily occur between proteins in a complex, enabling high-precision elucidation of binding sites between phosphoproteins and their binding partners, an interesting finding that used EThcD to localize these phosphate-transfer sites.132
ETD can characterize N-phosphorylation (e.g., lysine and arginine) as well.133−135 Although less common than S/T/Y O-phosphorylation, these alternative phosphorylation events regulate signaling mechanisms in bacterial systems, and more studies are emerging to suggest they may also play a role in eukaryotic signaling. The lability of the phosphoramidate bond leads to neutral losses that considerably increase false localizations in collision-based fragmentation spectra of N-phosphorylated peptides. Conversely, phosphoarginine and phospholysine are sufficiently stable under most ETD conditions, and ETD often enables confident identification and localization of N-phosphorylation. That said, some parameters (e.g., the number of nonmobile protons in the precursor cation) must be considered to account for possible gas-phase rearrangements that can affect reliability of phosphosite localization.136 Clausen and co-workers recently used ETD to characterize the role arginine phosphorylation in protein turnover in Bacillus subtilis, providing new insights into protein degradation pathways in Gram-positive bacteria.137
Glycosylation
Glycoproteomics is one of the arenas of modern proteomics where ETD has the greatest impact. Protein glycosylation is a prevalent, chemically complex, and biologically diverse PTM involved in a wide array of intra- and intercellular functions. It is highly heterogeneous modification that accounts for the greatest proteome diversity over any other PTM. Analysis of intact glycopeptides is imperative to glycoproteome characterization because multiple glycans can modify a given glycosite (i.e., glycan microheterogeneity), which makes glycan identity at a given site crucial to the biological context of the modification. Collision-based fragmentation of intact glycopeptides usually only reveals information about the glycan component of the precursor and offers little sequence information about the peptide backbone. ETD, on the other hand, results in nearly exclusive dissociation of the peptide backbone while leaving the glycan moiety intact, allowing for peptide sequence elucidation and site-specific analysis of the glycan modification.
Consistent with themes discussed throughout this review, ETD is usually paired with collision-based dissociation in glycoproteomic experiments, although we note that this coupling is especially prevalent in glycoproteomics to capitalize on the complementary nature of fragmentation of the two methods for peptide and glycan characterization.138−146 A product ion-dependent decision tree method has become a powerful approach for glycopeptide analyses, triggering ETD scans for precursors when glycan oxonium ions are observed in HCD spectra.147−149 The benefits of this approach are 2-fold: (1) the faster scan rates of HCD MS/MS enable increased sensitivity/greater sampling depth, acquiring slower ETD scans only for precursor ions that have a high likelihood of being glycopeptides (where ETD adds value), and (2) paired HCD and ETD MS/MS spectra for potential glycopeptide ions are automatically acquired, aiding in the interpretation of glycopeptide identifications.
Analysis of N-linked glycosylation, where the glycan is attached at an asparagine residue, is generally more prevalent and somewhat more straightforward. Here, glycans can be readily cleaved off enzymatically for separate analysis of glycans and peptides that can inform intact glycopeptide characterization with ETD. Additionally, modifications happen within the confines of a sequence motif, N-X-S/T (where X is a residue other than proline). O-Glycosylation at serine and threonine residues is more challenging to characterize because cleavage of glycans from the peptide backbone is less straightforward (limiting ability to leverage deglycoproteomic data to inform intact glycopeptide experiments), and the lack of consensus motif makes unambiguous glycosite localization more difficult (i.e., multiple potential modifications sites in a given peptide are more likely). For example, Parker et al. combined intact glycopeptide analysis (using ETD and collisional dissociation) with glycomics of released N-glycans and deglycoproteomic experiments to characterize 863 unique N-glycopeptides, corresponding to 276 N-glycosites on 161 proteins in rat brain lysates.142 Xu et al. identified ∼1150 unique N-linked glycopeptides representing 348 N-glycosites on 270 protein in Arabidopsis influorescence tissue using various ETD-based methods.145 Neither study targeted O-linked glycopeptides. In one of the largest intact glycopeptide characterizations to-date, Trinidad et al. used ETcaD to identify 2100 N-glycopeptides representing ∼700 N-glycosites on 375 proteins.143 This is compared to identification of only 463 O-glycopeptides on 122 proteins, with ∼45% of O-glycosites left ambiguously defined in the same study, highlighting the discrepancy in N- and O-linked glycopeptide analyses.
Several groups have explored strategies to use ETD in concert with other approaches for O-glycopeptide characterization.998 Several years ago, Thaysen-Anderson and co-workers demonstrated the ability of ETD to characterize densely O-glycosylated mucin-type peptides and provided reasonable performance in unambiguous O-glycosite localization.999 Darula et al. used a combination of enrichments (jacalin, a lectin specific for N-acteylgalactosamine seen in O-glycans, ion exchange chromatography, and hydrophilic interaction chromatography) in addition to partial deglycosylation of O-glycans via exoglycosidases to improve ETD fragmentation and glycosite localization of mucin core-1, core-2, and core-3 oligosaccharides.1000 In recent years, the Medzihradszky and Wuhrer groups have followed up with these methods to characterize mucin core-1 type O-glycoproteins in human serum and blood samples with ETD.150,151 Futhermore, removing N-glycans via peptide N-glycosidase F can provide significant benefits for the characterization of O-glycosylation sites with a combination of collisional dissociation and ETD, as underscored by methods investigated by Houcel et al.152 A SimpleCell strategy, which uses genetic engineering to simplify O-glycans to a single truncated O-GalNAc residue, has also been coupled with ETD methods to profile the O-glycoproteome of mammalian cell lines, and similar strategy was used to investigate O-mannosylation in yeast.1001−1004 Windwarer and Altmann used ETD in combination with extensive offline fractionation, generation of large proteolytic glycopeptides, and direct infusion analysis of individual fractions to characterize occupancy of O-glycosylation sites in bovine fetuin, a common glycoprotein standard.153
Beyond general O-glycosylation, ETD has also significantly impacted analysis of O-GlcNAcylation, a common and specific form of O-glycosylation in which a single β-linked N-acetylglucosamine is attached to serine and threonine residues.154 Burlingame and co-workers recently highlighted the essential contribution ETD offers for O-GlcNAc analysis by retaining the labile monosaccharide modification on product ions for unambiguous localization of modification sites.155 Studies of O-GlcNAcylation often examine the modification within the context of cross-talk of between it and phosphorylation, as they both modify serine and threonine residues,156 and ETD plays a central role in the ability to elucidate the interplay between both modifications. Trinidad et al. performed one of the first large-scale characterization of site-specific O-GlcNAc and phosphorylation cross-talk, identifying 1750 and 16 500 sites of O-GlcNAcylation and phosphorylation, respectively, from murine brain tissue.1005 Since then, several studies have expanded those investigations to interrogate O-GlcNAc cross-talk in Alzheimer’s disease,157,158 regulation of circadian clock mechanisms in mice and drosophila,159 and nutrient-sensitive intracellular processes that have significant implications for downstream metabolic regulation.160
As with most applications of ETD, the value of supplemental application cannot be overstated for glycoproteome characterization with ETD. ETcaD has been employed in several large-scale glycopeptide characterization studies,143−145 and the benefits of EThcD and AI-ETD for intact glycopeptide analysis are just beginning to be explored. Yu et al. demonstrated the ability of EThcD to generate both glycan and peptide fragmentation in a single MS/MS spectrum based on the hybrid combination of electron-driven and collision-based dissociation (Figure 4).161 They used EThcD to characterize several glycoproteomic samples, ultimately showing it to be well-suited for large-scale glycopeptide studies by identifying ∼1000–1200 unique N-glycopeptides from rat carotid arteries. A second study by Yu et al. used a combination of approaches, including EThcD, to report the first description of O-linked glycosylation on mouse insulin chains among other proteins and signaling peptides, providing new methods to investigate the role of glycosylation in diabetes.162 Glover et al. used EThcD and HCD-triggered EThcD methods to combine phosphoproteome and glycoproteome analyses, focusing on sialylated and phosphorylated glycopeptides contained in standard phosphopeptide enrichments.163 In all, they characterized ∼4000 phosphopeptides and ∼1000 sialylated glycopeptides from a single enrichment of rat smooth muscle cells, and they also identified glycopeptides with mannose-6-phosphate glycans. Parker et al. noted that EThcD improved glycopeptide spectral quality but commented that further optimizations involving spectral scoring and interpretation were probably needed to maximize its utility.164 Pitteri and co-workers used ETcaD and EThcD-based methods to assess the quality of intact glycopeptide enrichment strategies, ultimately identifying 829 unique glycoforms across 208 N-glycosites in 95 proteins from human plasma.165 Also we recently described the first application of AI-ETD to glycoproteomics,166 showing that AI-ETD enabled the largest intact glycopeptide analysis to date by identifying >7500 localized unique N-glycopeptides. Various pre- and post-ETD supplemental activation schemes characterized the effect of glycosylation on fragmentation of intact proteins, as well, and distinguished high mannose glycans in RNase B (∼14.7 kDa).167
Figure 4.
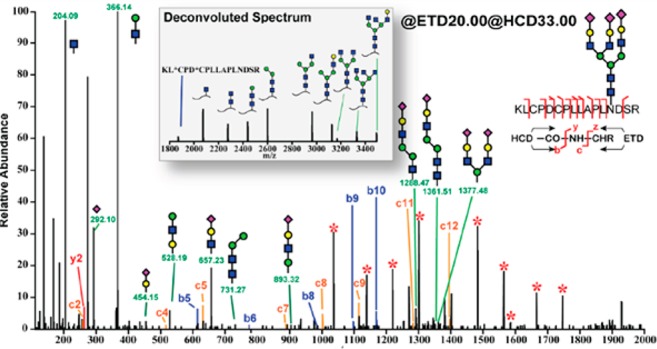
EThcD MS/MS of 3+ charge state precursor ion at m/z 1577.9 of bovine fetuin triantennary N-glycopeptide KLCPDCPLLAPLNDSR (AA 126–141). Starred peaks (*) in the spectra were deconvoluted and annotated in the inset. Reproduced in part from J. Am. Soc. Mass Spectrom., Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization, Vol. 28, 2017, 1751–1764, Yu, Q.; Wang, B.; Chen, Z.; Urabe, G.; Glover, M. S.; Shi, X.; Guo, L. W.; Kent, K. C.; Li, L. (ref (161)) with permission of Springer.
In an effort to enable more quantitative comparisons in large-scale glycoproteomic experiments, Bertozzi and co-workers recently described a new strategy to called isotope-targeted glycoproteomics (IsoTaG), where stable isotopes are incorporated through labeled azido sugars.168 Intact azidosugar-labeled glycopeptides can then be enriched and analyzed via ETD and EThcD based methods. IsoTaG using Ac4GalNAz or Ac4ManNAz sugars enabled identification of 1375 N- and 2159 O-glycopeptides across 15 cell lines,169 and the approach was extended through use of alkynyl sugars as metabolic labels in analysis of the sialylated glycoproteome, identifying 699 intact glycopeptides from 192 glycoproteins in PC-3 cells.170
ETD has enabled characterization of several noncanonical glycosylation modifications as well.143 The Heck group combined high-resolution native mass spectrometry and EThcD-based glycopeptide analyses to study human ethyropoeitn and plasma properdin as models for therapeutic proteins and plasma protein markers.171 They qualitatively and quantitatively monitored coappearing proteoforms in these proteins in addition to revealing PTM localizations, relative abundances, and glycan structures in a site-specific manner. Furthermore, this synergistic approach lead to the discovery of three new C-mannosylation sites, a noncanonical type of glycosylation involving linkage of the glycan through a carbon–carbon bond. In a second study they integrated native mass spectrometry and EThcD-based glycoproteomics workflows to detail the structural microheterogeneity of human complement C9 protein (∼65 kDa), revealing ∼50 distinct signals via native MS and 15 co-occurring proteoforms that included known N- and C-glycosylation and new evidence of O-glycosylation.172 Pronker et al. used ETcaD in combination with X-ray diffraction, structure guided mutations, and several biophysical assays to characterize the structural basis of myelin-associated glycoprotein adhesion and signaling, which included identification of N-glycosylation and a tryptophan C-mannosylation.173 ETD and EThcD revealed that cysteine S-linked N-acetylglucosamine (S-GlcNAcylation) occurs in mammalian systems, as well, including murine and human systems.174 Finally, ETD has been valuable for characterizing sites of glycation (nonenzymatic addition of sugars to amino acids)175−177 in addition to peptidoglycans, which are mostly glycans cross-linked with nonstandard amino acids.178
In terms of scale and scope, glycoproteomic methods have lagged behind analyses of other PTMs, but ETD is quickly making large-scale glycopeptide analysis a realistic endeavor. Methods like EThcD and AI-ETD, which offer complementary dissociation of glycan and peptide components of glycopeptides in a single MS/MS scan, will be essential in enabling global glycoproteomic profiling of complex systems as the field continues to advance.
Disulfide Bonds
Disulfide bonds, naturally occurring intramolecular cross-links between cysteine residues in proteins, play an important role in stabilizing tertiary and quaternary structure, often dictating proper biological function. Disulfide bonds are especially enriched in secreted and membrane proteins and are an important feature in the complexity of the proteome. They represent a challenge for MS/MS approaches, both peptide and intact protein, because both the disulfide and the peptide backbone must be fragmented to access sequence information within the disulfide-enclosed regions. ETD reactions can preferentially cleave disulfide bonds,179,180 lending ETD methods high utility for characterizing this PTM. Often ETD is used to open disulfide bonds181 and subsequent collisional activation provides sequence information on the peptide(s) present;182 however, in many cases ETD alone can generate sufficient c/z•-type product ions through radical cascades initiating at N–Cα bond cleavage that propagates to cleave multiple disulfide bonds.183
Liu et al. developed an EThcD workflow specifically designed to provide a precise, yet generic approach for disulfide bond mapping.184 In EThcD of disulfide-bonded peptides, the ETD reaction preferentially leads to the cleavage of the S–S bonds (in addition to some peptide backbone fragmentation). Supplemental HCD activation then dissociates unreacted and charge-reduced precursor ions of the disulfide-cleaved peptides to provide further peptide backbone fragmentation. They also described a software platform called SlinkS to process the relatively complex spectra generated by this process. Note, pepsin is often the protease of choice in peptide-based disulfide-bond mapping because it is active at low pH, which prevents unwanted disulfide reshuffling during sample preparation.
Other approaches involve more nuanced tactics to study disulfide bonds, including targeted methods to dissociate intrachain disulfide bond containing peptides with ETD based on prior analysis of reduced/alkylated samples and assigning disulfide bond connectivity via extracted ion chromatograms of disulfide bonded peptide pairs that are generated together following ETD reactions.185,186 In all, ETD-based analyses of disulfide bonds have been used in recent studies to investigate neuropeptides,185 secreted proteins,182 HIV envelope proteins,186 and protein storage stability,187 among other things.
Alternative Proteases, Middle-Down Proteomics, and Peptidomics
The vast majority of proteomics experiments utilize trypsin to proteolytically cleave proteins C-terminal to lysine and arginine residues, generating peptides for further analysis with LC–MS/MS. This bottom-up, or shotgun, approach with trypsin is particularly well-suited for ubiquitously available, high-throughput collision-based dissociation methods; reliance on characterization of exclusively tryptic peptides, however, limits the depth and coverage of the proteome that can be sampled. Use of alternative proteases (e.g., LysC, GluC, ArgC, AspN, and chymotrypsin) in a complementary fashion with trypsin can significantly increase sequence coverage across the proteome, but many proteases generate peptides that are not conducive to collision-based dissociation. Thus, ETD has directly benefited deep sequencing efforts and has contributed to much greater proteome coverage via multiple-protease approaches. Two recent reviews provide a broader perspective of the field beyond ETD.188,189
In the first large-scale multiple protease study to leverage ETD, Swaney et al. showed that ETD methods could offer improved characterization of ArgC, AspN, GluC, and LysC peptides when compared with collisional dissociation, and the combination of identifications from these complementary proteases with tryptic peptide identifications enabled a 3-fold increase in protein sequence coverage across the yeast proteome.190 In 2015, Nardiello et al. followed-up on this idea by using collisional activation and ETD to characterize peptides derived from digestions of standard proteins using trypsin and chymotrypsin. They showed that the combination of both dissociation methods and proteases could increase sequence coverage and aid in identification of PTMs, species-specific residues, and single-point amino acid modifications in natural protein variants.191 Trypsin and chymotrypsin were also used in tandem by Somasundaram et al. to identify protein C-termini, where they derivatized carboxylic acids and used collisional dissociation and ETD to improve C-termini characterization.192 Multiple proteases (trypsin, chymotrypsin, subtilisin, and AspN) and combinations of collisional activation and ETD were used to improve characterization of the density and complexity of glycosylation in polymeric mucin MUC2, which has a highly O-glycosylated mucin domain.193 One of the more robust applications of multiple protease characterization with ETD in recent years came from Guthals et al.194 Using overlapping peptides from multiple digests and corroborating b/y/c/z•-type fragments from collisional activation and ETD, they de novo assembled peptide sequences averaging nearly 70 amino acids in length at 99% sequencing accuracy.
Tsiatsaiani et al. investigated the chromatographic separation properties, ETD and HCD fragmentation behavior, and (phospho)proteome sequence coverage of LysargiNase, a protease that mirrors the proteolytic activity of trypsin by cutting N-terminal to lysine and arginine residues with high specificity.195 Interestingly, LysargiNase peptides fragment as near mirror images to tryptic peptides, with LysargiNase peptides generating predominantly N-terminal (c- and b-type) product ions using ETD and HCD, respectively (Figure 5). ETD of LysargiNase peptides provided especially informative sequence ladders (although HCD performance was worse than for trypsin); and overall, analysis of LysargiNase generated peptides provided complementary characterization, adding coverage in both proteome and phosphoproteome analysis. In a similar vein, Aebersold and co-workers introduced arginyl-tRNA protein transferase (ATE)-mediated LysC/AspN proteolysis that generates arginylated peptides with basic amino acids on both termini.196 Dissociation of these peptides generates near complete sequence ladders from both N- and C-terminal ends, and ETD generates complete c- and y-type fragments in addition to other product ion types (e.g., z-type).
Figure 5.

Fragmentation characteristics of proteolytic K/R(X)n and (X)nK/R peptides. N-terminal or C-terminal protons drive the formation of opposite but complementary ion patterns for the alike peptides during ETD, with basic residues depicted in red. (a) ETD MS/MS spectra of LysargiNase (top) and tryptic peptides (bottom) with a single basic residue or with multiple basic residues. b) Fragmentation heat maps based on ion intensities for K/R(X)n and (X)nK/R peptides during HCD and ETD. Normalized relative intensity values were calculated for peptides 6–20 amino acids long. Series numbers are matched to the sequence orientation. Reproduced in part from Tsiatsiani, L.; Giansanti, P.; Scheltema, R. A.; van den Toorn, H.; Overall, C. M.; Altelaar, A. F. M.; Heck, A. J. R. J. Proteome Res.2017, 16, 852–861 (ref (195)) Copyright 2017 American Chemical Society.
Middle-down proteomics is an extension of the alternative protease approach, with the goal of generating large peptides that cover the middle mass range (∼3–10 kDa) between tryptic peptides and intact proteins. In theory, the middle-down approach should allow for the detection of some PTM combinations and provide more extensive proteome coverage not achievable with shotgun approaches. Either nontraditional proteases or limited trypsin digestion can be used to accomplish this goal.197 Whichever approach is leveraged, ETD is well-suited to sequence these large peptides. In 2012, Kelleher and colleagues described a method for restricted enzymatic proteolysis using the outer membrane protease T (OmpT), a member of the novel omptin protease family derived from the Escherichia coli K12 outer membrane.198 OmpT produced peptides with >6.3 kDa mass on average, and ETD showed highly complementary dissociation behavior to collisional dissociation for these peptides. In all, the OmpT middle-down workflow enabled identification of 3697 unique peptides from 1038 proteins, including those with PTMs, and closely related protein isoforms could be readily differentiated. Cristobal et al. recently focused on optimizing several aspects of middle-down workflows, including proteolysis with GluC and AspN (and a nonenzymatic formic acid induced cleavage), chromatographic separations with larger pore-sized particles and peptide dissociation with HCD, ETD, and EThcD.199 They found the generation of larger peptides suitable for true middle-down analysis was not readily achievable with GluC or AspN, which highlights the value of new middle-down proteases like OmpT and other limited proteolysis approaches. Regardless, EThcD performed best for the analysis of midsized peptides, providing high MS/MS success rates and higher peptide sequence coverage (up to 95% peptide backbone coverage) compared to HCD and ETD.
Peptidomics likewise involves analysis of nontryptic peptides, as it focuses on endogeonously produced protein fragments. Opposed to the other methods described in the section, proteases are rarely used to prepare peptidomic samples, so a wide range of endogenous, nontryptic protein fragments are present that require a combination of dissociation methods to properly characterize.200 Collision-based dissociation and ETD are used together in a wide range of peptidomic experiments, including large-scale identification of secretory peptides in human endocrine cells,201 microproteins and endogenous peptides in murine brain tissues,202,203 and native peptides present in urine of healthy women during pregnancy.204 Peptidomics also enables screening of natural peptide products that have medicinal or therapeutic uses. Gucinski and Boyne used ETD to identify multiple forms of protamine sulfate, a complex peptide drug product with multiple basic peptides.205 Juba, Bishop, and co-workers used ETD-based methods to screen Komodo dragon (Varanus komodoensis) and American alligator (Alligator mississippiensis) plasma for cation antimicrobial peptides (CAMPs), potential targets for development of future antibacterial theurapeutics.206,207 ETD has been used in characterizations of venoms, as well, which are a rich source for discovering new peptide and protein products with biotechnological applications.208
Cases Where ETD is Particularly Helpful
Major Histocompatibility Complex/Human Leukocyte Antigen Peptides
The major histocompatibility complex (MHC), referred to in humans in the human leukocyte antigen complex (HLA), is a group of proteins that are central to immune response. They function by presenting peptides at the cell surface for recognition by different T-cells for recognition. The MHC peptides presented are generated via proteolysis and can be derived from cytosolic proteins (MHC Class I proteins) or from antigens that are internalized into antigen presenting cells via phagocytosis (MHC Class II proteins). Class I peptides are typically 8–11 residues in length, while Class II peptides range from 10 to 25 amino acids; much like the challenges seen in peptidomics, these endogeonously derived MHC peptides can be difficult to characterize, especially because they can have PTMs that play important roles in their recognition. MS-based approaches to study MHC peptides were pioneered by Hunt and co-workers more than 2 decades ago,209 but the introduction of ETD and related techniques has greatly aided discovery of new properties of MHC peptides in recent years.
The majority of work has focused on the Class I immunopeptidome. Combinations of ETD and collisional dissociation are leveraged to study phosphorylation of HLA-I peptides,210,211 and Mommen et al. showed in 2014 that EThcD could significantly improve both sequence coverage and PTM characterization, including phosphorylation, for global analyses of HLA-I peptides. A follow up study of HLA-II peptides also demonstrated the value of ETD-based approaches in combination with other dissociation methods to characterize the complex nature of the HLA class II system.212 Further studies with EThcD have indicated new PTMs on HLA-I peptides, including O-GlcNAc,213 extended O-GlcNAc structures,214 and dimethylated arginine,215 and Liepe et al. shed new insight into the frequency and abundance of proteasome-catalyzed peptide splicing events in Class I peptides.216 ETD is also leveraged for the study of drug binding interactions with Class I and II peptides,217 and Malaker et al. recently showed that Class II peptides have 17 different glycoforms, indicating a role for diverse and complex glycosylation patterns in MHC recognition.218
Histones
Histones are fundamental protein components of chromatin, which is the structural framework for chromosomal DNA in the cell nucleus. The modification states and sequence variants of histones are directly involved in gene expression, making histones supremely important in epigenetic regulation. Bottom-up analyses of histones can be challenging since N-terminal regions are lysine and arginine rich (resulting in small, difficult to analyze tryptic peptides) and combinatorial patterns of PTMs on histone tails (i.e., the “histone code”) are important to understanding regulation states, meaning that middle-down and top-down methods are particularly valuable for histone characterizations.219−221 ETD thus provides obvious benefits for histone analysis, and indeed, it has been highly utilized for histone studies.
Bottom-up and middle-down proteomic analysis of histones with ETD are used to study histones in a variety of capacities, including cellular reprogramming of histone H4 in induced pluripotent stem cells,222 the role of H2A modifications in vertebrate embryo development,223 histone ADP-ribosylation in DNA damage response,224 and histone modification states in transformed cell line vs primary cell monocyte-derived macrophages.225 Schriemer and co-workers recently described a new alternative protease called neprosin that is well-suited for middle-down histone analysis.226 Neprosin cleaves C-terminal to proline residues, making relatively large peptides that depend more heavily on ETD for characterization. Importantly, it also creates relatively large peptides from histone tails (1–38 for H3 and 1–32 for H4) for analysis of co-occurring PTMs. Various separation methods have been tested for middle-down workflows for histone characterization with ETD fragmentation, with weak cation exchange chromatography,227,228 capillary electrophoresis,229 and ion mobility230 all being demonstrated as compatible online separation techniques. Data-independent ETD methods have been investigated with some success for middle-down histone characterization as well.231 Quantitative comparisons of histones and their modifications can be accomplished through several strategies enabled by bottom-up and middle-down approaches.232−234
The top down approach offers some advantages for histone analysis because intact characterization best captures the biological information present in histone PTM patterns and provides insight into the complexity of the histone proteome; that said, technology development is still needed to make top-down methods more accessible.235 Advances in ETD-based methods are making significant inroads toward this goal, and three recent studies, all using front end ETD sources in various capacities, have noticeably improved the ability to analyze intact histones on chromatographic time scales.236−238 Furthermore, Molden et al. showed how combinations of bottom-up and top-down approaches can comprehensively profile histone changes.239
Antibody Characterization
Monoclonal antibodies (mAbs) and related biological molecules are an important and growing class of human therapeutics; more than 30 immunoglobulins (Igs) have been approved for the treatment of cancer, immunological diseases, and infectious diseases.240 The specificity and affinity of mAbs are among their key advantages, but engineering these molecules to have the desired traits requires thorough characterization. Their large size (two heavy chains at ∼50 kDa and two light chains at ∼25 kDa for a total mass of ∼150 kDa), number of disulfide bonds (which are critical for structure and activity), sequence differences in key variable regions, and presence of PTMs (e.g., glycosylation) make comprehensive characterization challenging, but as discussed above, ETD-based methods offer several benefits for many of these features.
Bottom-up, middle-down, and top-down methods have all been explored for mAb characrterization, and ETD plays a role in each approach. Extending their de novo sequencing approach to mAb analysis, Guthals et al. recently showed that a combination of multiple proteases and multiple fragmentation methods could sequence mAb from human serum with no sequence database required, enabling discovery of new mAbs.241 Hunt and others have used online proteolysis with pepsin to analyze mAbs in bottom-up and middle-down approaches,242−244 using ETD to characterize large peptides derived from limited proteolysis, including those with intact disulfide bonds. Several laboratories have explored using alternative proteases (including immunoglobulin G-degrading enzyme of Streptococcus pyogenes [IdeS] and secreted aspartic protease 9 [Sap9]) or simple reduction of disulfide bonds to characterize subunits and intact chains of IgG with a variety of fragmentation methods, often employing ETD for large subunits that require extensive fragmentation.245−249 Others have investigated top-down methods to characterize fully intact mAbs (i.e., no proteolysis) with some success (Figure 6), and they rely on ETD to derive sequence information from the ∼150 kDa precursor ions, although sequence coverage has been limited to ∼30%.250−253
Figure 6.

Intact mAb analysis with ETD. Product ion abundance analysis of ETD top-down MS of adalimumab with 10 ms ETD duration and trastuzumab with 10 and 25 ms duration. In the case of trastuzumab, the color-coded histogram demonstrates the improvement in sequence coverage obtained through the combination of 10 ms (magenta) and 25 ms (cyan) ETD MS/MS data. Whereas medium and large product ions (e.g., z117 and z63) are generally produced using 10 ms duration, smaller ions such as z16, z17, and z18 are detected only in the case of a longer ETD duration, presumably as the result of secondary fragmentation (i.e., refragmentation of larger product ions). Note the ability of ETD to generate fragments within a disulfide bridge region. Reprinted from J. Proteomics, Vol. 159, Fornelli, L.; Ayoub, D.; Aizikov, K.; Liu, X.; Damoc, E.; Pevzner, P. A.; Makarov, A.; Beck, A.; Tsybin, Y. O. Top-down analysis of immunoglobulin G isotypes 1 and 2 with electron transfer dissociation on a high-field Orbitrap mass spectrometer, pp 67–76 (ref (253)). Copyright 2017, with permission from Elsevier.
Structural Characterization Using ETD
Structural characterization of the proteome is a natural extension from recent advances in instrumentation, top-down proteomics, and PTM analyses. Beyond providing information about the presence and abundance of proteins and their various proteoforms, MS contributions to molecular and structural biology are gaining momentum and offering new ways to examine protein–protein interactions, small molecule–protein interactions, and other studies of large biomolecular assemblies (recently reviewed comprehensively by Lössl et al.254). ETD offers several benefits to this effort, including analysis of cross-linked peptides, peptides and proteins labeled via hydrogen–deuterium exchange, and proteins ionized via native electrospray ionization.
Cross-Linked Peptides
Chemical cross-linking proteomics provides an avenue to investigate protein–protein interactions (i.e., the “interactome”) and protein conformations. Cross-linking reagents have two reactive groups directed toward chemical reactivity with specific amino acid residues, and the reactive groups are connected by a spacer arm that can vary in length. Cross-linking reagents are introduced prior to proteolysis to introduce intra- and interprotein cross-links that help discern structural information. Following proteolysis, cross-linked peptides are analyzed with tandem MS (sometimes via multiple stages) to elucidate which sequences exist within a given proximity in the proteome. Note, the resolution of proximity measurements is defined by the length of the spacer group in the cross-linking reagent. In 2015, Heck and co-workers introduced a proteome-scale workflow for studying protein assemblies via cross-linked peptides, and they used a back-to-back combination of collisional activation and ETD MS/MS scans on the same precursor to identify 1622 intraprotein and 200 interprotein cross-links (134 protein–protein interactions) at a 1% false discovery rate (Figure 7).255 They recently followed up on this strategy by optimizing fragmentation conditions for cross-linked peptides, showing that MS2-MS3 collisional dissociation in concert with ETD MS2 scans provided the best characterization of cross-linked peptides.256 Rappsilber and colleagues extended studies of collision- and ETD-based methods to cross-linked peptides for development of decision tree methods including EThcD,257,258 and other groups have worked to develop cross-linking reagents that improve ETD performance.259,260
Figure 7.
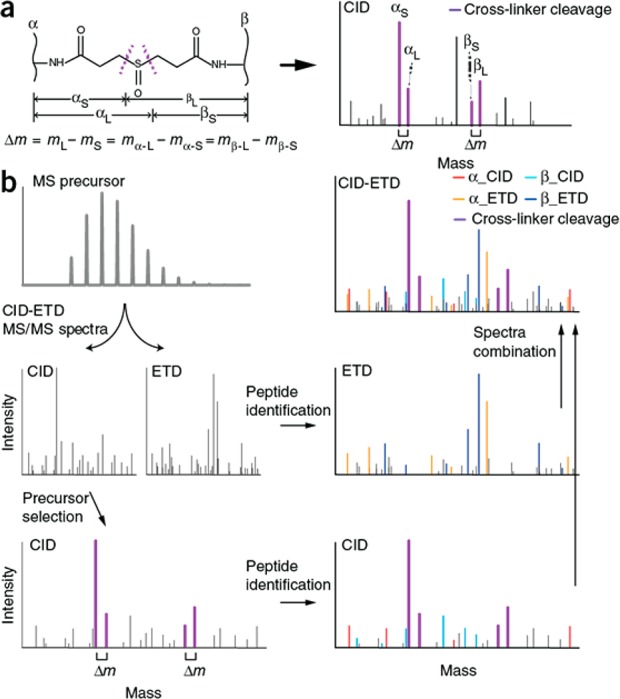
Strategy to identify cross-linked peptides. (a) Schematic structure of a DSSO interpeptide cross-link (left) and its specific fragmentation pattern under CID (right). The four signature MS/MS fragment ions are derived via the equation presented below the structure (i.e., the Δm principle). (b) The XlinkX workflow to identify interpeptide cross-links. The MS precursor ion is subjected to sequential CID-ETD fragmentation. Only CID spectra are used to obtain the precursor masses of both linked peptides by the Δm principle. The four signature fragment ions resulting from cross-linker cleavage are represented by purple peaks in the MS/MS fragmentation spectra. Subsequently, CID spectra are used to match b- and y-ions, and ETD spectra are used to search for c- and z-ion series. Reprinted by permission from Macmillan Publishers Ltd.: NATURE, Liu, F.; Rijkers, D. T. S.; Post, H.; Heck, A. J. R. Nat. Methods2015, 12, 1179–1184 (ref (255)). Copyright 2015.
Ion Mobility and Native Proteomics
As noted above, ion mobility separates ions based on their movement through a carrier gas, and complementing ion mobility with ETD provides access to detailed higher-order structures of peptides, proteins, and protein complexes. Lermyte and Sobott used a q-IM-TOF instrument to investigate native proteins and protein complexes with ETD.261 They demonstrated that post-ion-mobility collisional activation of ETD products could release noncovalently bound product ions to improve sequence coverage of exposed regions of proteins, while pre-ETD activation of tetramer complexes caused unfolding without monomer ejection that showed efficient fragmentation in some regions which are not sequenced under more gentle MS conditions (Figure 8). They also used incremental increases in pre-ETD collision-activation to trace initial steps of gas-phase protein unfolding. Sobott and co-workers also used ion mobility, ETD, and supplemental activation to study the topology of the native form of tetrameric alcohol dehydrogenase (∼150 kDa) by showing that regions of ETD fragmentation mapped to exposed regions on the complex.262 Furthermore, they used ion mobility separations to study the stability of ETnoD complexes, showing that more linear complexes are more likely to release c/z-type products (which has implications for understanding supplemental activation in ETD reactions at the intact protein level).263
Figure 8.
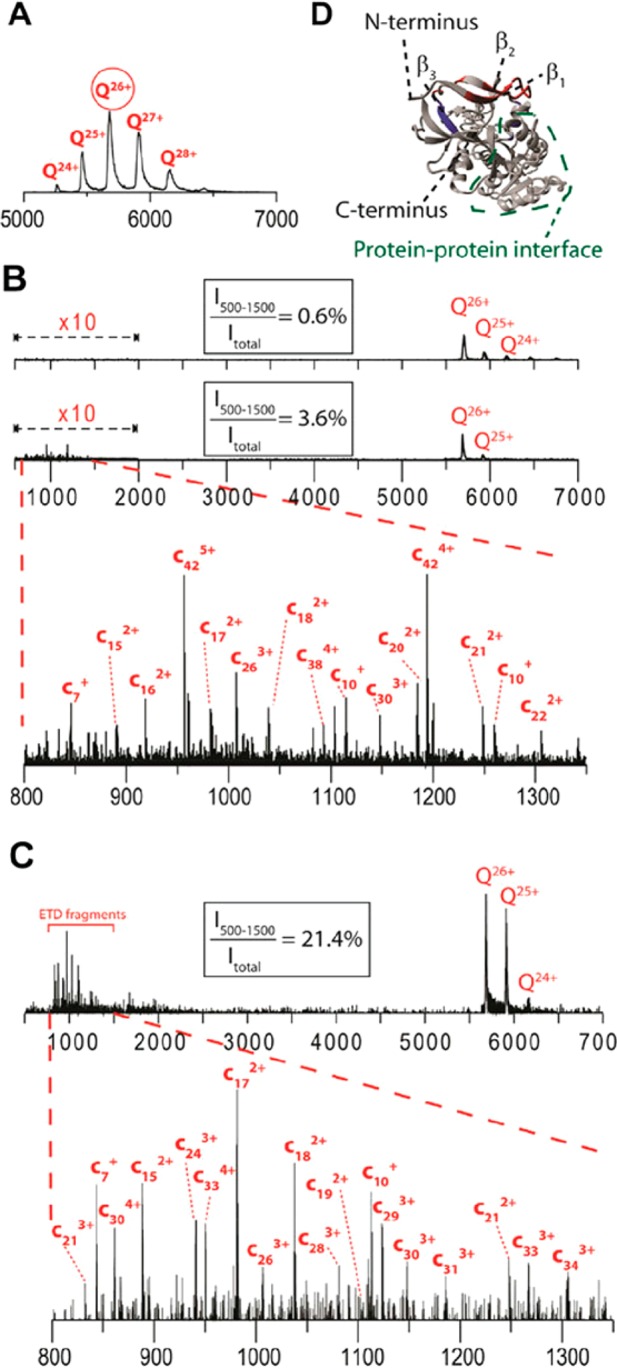
Using ETD and ion mobility to discern structure. (A) Charge state distribution observed in native ESI of the ADH tetramer, with the 26+ charge state isolated in the quadrupole for subsequent top-down dissociation. (B) ETD products at a sampling cone voltage of 40 V and a supplemental activation of (top) 10 V and (bottom) 70 V applied in the transfer cell and (C) at sampling cone voltage 120 V and supplemental activation 10 V. (D) Crystal structure of ADH tetramer (only one subunit shown for clarity). The fragmentation sites observed with a sampling cone of 80 V, without supplemental activation, are shown in red. The additional cleavage sites observed at a higher sampling cone voltage (causing partial unfolding) are shielded in the native structure and shown in blue. Reproduced from Electron transfer dissociation provides higher-order structural information on native and partially unfolded protein complexes Lermyte, F.; Sobott, F. Proteomics, Vol. 15, pp 2813–2822 (ref (261)). Copyright 2015 Wiley.
Combinations of ETD and collisional activation have been valuable in studying electrostatic interactions (i.e., salt bridges) in native protein structures too. In this approach, differences between pre- or postreaction collisionally activated ETD spectra and nonactivated ETD spectra indicate presence of electrostatic interactions in standard proteins where known salt bridge contacts in solution occur.264,265 ETD of native proteins complexes can also identify differences in fragmentation between subunits that arise from asymmetric charge portioning, in addition to characterizing domains of secondary-structure present in dimers, ejected monomers, and monomers obtained directly from electrospray ionization.266
Other approaches can be used in tandem with ETD to investigate protein structures. Kaltashov and co-workers coupled online ion exchange chromatographies with native electrospray ionization to characterize complex and heterogeneous therapeutic proteins and protein conjugates with intact conformational integrity, using ETD to provide online top-down structural analysis for identification PTMs that could not be identified via intact mass analysis alone.267 Cassou et al. showed that electrothermal supercharging can increase the precursor ion charge state distributions for proteins in buffered aqueous solutions used for native electrospray, and the higher charge state species facilitated collection of high-quality ETD spectra for structural analysis of intact proteins.268 Xie and Sharp used online size exclusion chromatography to ensure that peptide isomers eluted together and then quantified relative amounts of each isomer based on the presence of fragment ions in a single ETD MS/MS spectrum.269
Hydrogen–Deuterium Exchange
Hydrogen–deuterium exchange (HDX) is a noncovalent labeling approach for mapping protein structure based on the principle that hydrogens on amino acid residues at solvent-exposed regions of the protein backbone will exchange with deuterium in D2O, while amino acids that are buried in the folded protein core (or by an interacting protein) will not. The mass difference in exchanged deuterium atoms can then be measured by MS, with site-specific assignment enabled by tandem MS. Collision-based dissociation of HDX-labeled peptides and proteins can cause hydrogen scrambling that obscures assignment of heavy labeled residues, while ETD minimized hydrogen scrambling to enable measurement of deuterium incorporation with single-residue resolution.270−273
HDX strategies that leverage ETD are used at both the peptide and protein level. Masson et al. used HDX-ETD in a bottom-up strategy to screen inhibitors of the oncogene phosphoinositide 3-kinase (PI3K) catalytic p110α subunit, highlighting its potential use in pharmaceutical development for screening therapeutics.274 Seger et al. engineered new disulfide bonds in human growth hormone and used HDX-ETD to investigate conformational and functional consequences of new bond positions, showing how a different disulfide bond could stabilize the protein.275 HDX-ETD has also played a valuable role in determination of site-specific changes in enzyme activities (like coagulation factors) upon cofactor binding,276 structural changes upon binding of epidermal growth factor receptor inhibitors in cancer treatments,277 and oligomerization of apolipoprotein E, which can be a risk factor in Alzheimer’s disease.278 In an interesting study, Borchers and co-workers showed how specific phosphorylation events can affect protein structure using top-down HDX-ETD experiments, providing an avenue to study how PTMs affect protein activity and binding via structural changes.279
HDX can be combined with ion mobility (and subsequent beam type supplemental activation) with minimal hydrogen scrambling to expand the structural information obtained in an experiment.280,281 Interestingly, Rand et al. showed that this combination allows gas-phase HDX inside a mass spectrometer.282 The reaction occurs in milliseconds, offers complementary information to solution phase HDX and can be combined with ETD to provide orthogonal modes of structure characterization on the chromatographic time scale. Note, a combination of HDX and ETD also enabled analysis of the structure of antibodies in middle-down and top-down approaches discussed above.244,248,252 Beyond HDX, ETD has been used with other surface labeling techniques, as well,283−285 although HDX is the more common approach to use ETD because of the hydrogen scrambling concerns with collisional activation.
Other Uses of ETD and Related Ion–Ion Reactions
Ion–ion reactions like ETD can be used for proteome characterization in unique ways beyond those discussed above. Many of these approaches involve the ability to generate radical peptide cations and radical product ions that can be further probed to elucidate peptide structural information via radical ion chemistry. The body of work involving various manipulation of peptide radical cations for analytical means is extensive and technical, making it difficult to sufficiently review it in the space available here. Instead, we discuss specific applications peptide radical chemistry that we find particularly interesting and useful, and we point readers to recent reviews for further information.9,286,287 We also describe other uses of ETD and related ion–ion reactions for proteomics and briefly cover how ETD is coupled with quantitative strategies.
One valuable nontraditional application of ETD is for differentiating peptide isomers and isomeric residues.288 Lebedev et al. and Xiao et al. both recently described methods to perform HCD on z•-type generated from ETD to create diagnostic w-ions for distinguishing leucine and isoleucine residues in peptides.289,290 Lebedev and co-workers further extended this study to EThcD methods for more globally applicable leucine/isoleucine discrimination,291 although they also described limitations and considerations when using these approaches.292 Lyon et al. recently showed that ETD reactions can identify peptide isomers through a strategy that leverages hemolytic cleavage of carbon–iodine bonds that drives radical directed dissociation upon supplemental activation.293 Additionally, Turacek and colleagues have used radical product ions, created via ETD, to extensively study structure and ion chemistry of peptides with a variety of approaches for theoretical and analytical purposes.294−300
Proton Transfer and Other Ion–Ion Reactions
As mentioned in the Principles of ETD section, the choice of reagent anion used in ion–ion reactions governs the amount of electron transfer versus proton transfer between the anion and precursor cation populations. Proton transfer reactions (PTR), pioneered by McLuckey, Stephenson, and co-workers, have several analytical uses in proteomics, including signal concentration into a small distribution of charge states and simplification of spectra of highly charged precursors.301−304 Building on the work of Stephenson and McLuckey, Hunt and co-workers showed in 2005 that ETD and PTR are a powerful combination for sequencing intact proteins, using ETD to drive sequence informative product ion generation and then performing PTR on highly charged ETD products to decongest spectra for straightforward product ion assignments.17 The Hunt group has recently expanded on this sequential ion–ion reaction approach to Orbitrap systems with front end ETD sources, using ion–ion proton transfer reactions (PTR) to simplify ETD spectra and to disperse fragment ions over the entire mass range in a controlled manner (Figure 9).305 Additionally, multiple fills of ETD-PTR product ions can be collected prior to mass analysis in this approach, considerably enhancing observed ion current and product ion S/N without the need for time-consuming averaging of data from multiple mass measurements. The ETD/PTR technique proved extremely valuable in middle down analyses of antibodies,243 and they further extended its utility in a recent top-down characterization of histones by incorporating parallel ion parking35,306 during the PTR reactions to specifically control reaction of precursor ions to remain in a targeted product m/z range.237 The value of these strategies is clear for analysis of large peptides and proteins, and we expect this technology will be widely implemented in top-down experiments in coming years. Unsurprisingly, similar approaches have recently been extended to combinations of UVPD and PTR reactions for top-down analyses of denatured and native proteins.307,308 We note PTR has proven useful for gas-phase purification in bottom-up quantitative proteomic experiments as well.309,310
Figure 9.

ETD and PTR reactions (labeled here as ion–ion proton transfer, IIPT) combined for top-down intact protein analysis. (A) ETD spectrum recorded on (M + 26H)26+ ions from apomyoglobin using a reaction time of 5 ms. (B–E) Spectra obtained by performing PTR reactions on the ETD fragment ions in (A) for 20, 40, 80, and 160 ms, respectively. Reprinted from Int. J. Mass Spectrom., Vol. 377, Anderson, L. C.; English, A. M.; Wang, W.-H.; Bai, D. L.; Shabanowitz, J.; Hunt, D. F. A Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry, pp 617–624 (ref (305)). Copyright 2015, with permission from Elsevier.
PTR can also aid in structural proteomic experiments. Bush and co-workers have used PTR reactions to reduce the charge states of m/z-selected, native-like ions of proteins and protein complexes, which helps interpretation of complicated mass spectra that often represent contributions from multiple, coexisting species.311 Since its introduction for this purpose in 2015, PTR has enabled several detailed studies of gas-phase protein structure, folding, and dynamics.312−314 Jhingree et al. performed similar experiments but used ETnoD products instead of PTR products to study the effects of charge reduction on protein structure.315 Moreover, the Sobott group investigated how ETD and PTR can be balanced to generate sequencing ions and spectral decongestion.316
McLuckey and co-workers have recently used a variety of gas-phase ion–ion chemistry approaches for less traditional proteomic applications.317 Such methods include converting cations to anions in the gas-phase,318 performing 1,3-dipolar cycloadditions between azides and alkynes (click chemistry) through ion–ion reactions,319 creating dehydroalanine residues that provide specific backbone cleavages in peptide and protein cations320,321 and mapping of cysteine modification states and disulfide bonds through ion–ion oxidation reactions.322,323 Of particular note, they demonstrated the ability to form peptide bonds in the gas phase, providing a unique means for generating peptide linkages that is fast (<1 s), efficient (tens of percent), and flexible.324,325 Ion–ion reactions were also harnessed by Brodbelt and co-workers to derivative peptides with 4-formyl-1,3-benezenedisulfonic acid (FBDSA) anions to improve collisional dissociation of phosphopeptides and UVPD fragmentation efficiencies.326,327
Negative Electron Transfer Dissociation
Negative electron transfer dissociation (NETD) is the negative mode analogue of ETD, where precursor anions are sequenced through reactions with radical reagent cations. In NETD, radical reagent cations oxidize peptide precursor anions by abstracting an electron, which drives electron rearrangement steps that promote cleavage of the C–Cα backbone bond and produce odd-electron a•- and even-electron x-type product ions.328 Analysis of peptide anions offers several benefits, including the ability to sequence acidic peptides that do not readily ionize upon positive mode electrospray ionization329−332 and access to acid-labile PTMs that are difficult to characterize via positive mode approaches.
NETD is a notably valuable technique for peptide anion analysis because collisional activation often fails to reproducibly provide sequencing information in MS/MS spectra, making electron-driven fragmentation one of the main approaches to characterizing peptides in the negative mode. In fact, NETD enabled the first large-scale negative mode analysis of the proteome and demonstrated that systematic analysis of peptide anions at a proteome scale was a viable approach.333 NETD spectra often contain more side-chain neutral losses than their positive mode ETD counterparts, but the losses can have diagnostic value.334 Specific cleavages in NETD can also be used to create diagnostic fragments for peptide anion identification, such as enhanced c/z-type ion formation N-terminal to tyrosine residues.335
NETD suffers from nondissociative negative electron transfer (NETnoD) to a similar or even more severe degree than ETD for low charge-density precursors.336 We and others have demonstrated that supplemental activation for NETD using concurrent photoactivation (AI-NETD) is superior to collision-based supplemental activation (NETcaD) (Figure 10),337,338 and AI-NETD allowed characterization of nearly the entire yeast proteome exclusively in the negative mode338 and the largest study of the human proteome via peptide anion analysis.339 The radical cation of fluoranthene is a suitable NETD reagent for proteomic and phosphoproteomic analysis,340 but we recently demonstrated that the radical SF5•+ cation can be a preferred NETD reagent instead of fluoranthene as it decreases NETnoD, improves spectral quality through the generation of more a•- and x-type product ions, and increases peptide identifications in LC–MS/MS analyses.341 Although AI-NETD is generally a better strategy to improve NETD analyses, the use of SF5•+ reagent is a valuable approach when instrument modifications for AI-NETD are not available. Note, these studies also required modification of spectral interpretation tools, like the Open Mass Spectrometry Search Algorithm (OMSSA) and Byonic, to assign peptide spectral matches to NETD spectra.333,339
Figure 10.
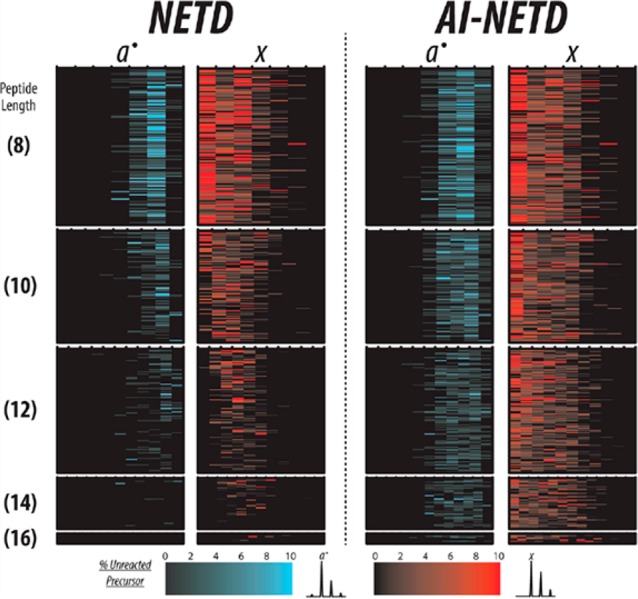
Fragment map of peptides identified with both NETD and AI-NETD. Here, each row is a unique peptide and each subcolumn corresponds a peptide backbone bond. The numbers in parentheses to the left show peptide length in number of residues, and all peptides shown here are z = −2. With NETD, a•- and x-type fragments decrease in number and intensity as precursor charge density decreases (i.e., as peptide length increases). AI-NETD maintains superior fragment ion generation even with decreasing precursor charge density, greatly increasing peptide dissociation and sequence coverage compared with NETD. This research was originally published in Molecular & Cellular Proteomics. Riley, N. M.; Rush, M. J. P.; Rose, C. M.; Richards, A. L.; Kwiecien, N. W.; Bailey, D. J.; Hebert, A. S.; Westphall, M. S.; Coon, J. J. The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation (AI-NETD). Mol. Cell. Proteomics2015, 14, 2644–266 (ref (330)). Copyright the American Society for Biochemistry and Molecular Biology.
NETD has currently been implemented on a handful of MS systems but has yet to be commercialized. Although it has admittedly niche applications compared to positive mode ETD analyses, the availability of NETD on commercially available platforms in the coming years, in addition to the NETD-compatible informatics tools now available, will boost its application to more biological problems (e.g., labile PTMs like tyrosine sulfation342 and histidine phosphorylation343) and, thus, its impact on proteomics. It is also worth noting that NETD has proven valuable beyond the realm of proteomics, as well, benefiting MS-based oligonucleotide and glycosaminoglycan analyses.344−351
Quantitative Proteomic Strategies
With the various implementations and applications of ETD, it is important to be able to leverage common quantitative proteomic approaches while using ETD for characterization. Perhaps the most straightforward quantitative approach is label-free quantitation, which has been a component of several ETD experiments discussed above.85,120,203,234,236 Stable isotope labeling is another widely used approach to enable multiplexed quantitation, and several strategies can be employed to incorporate stable isotopes into samples.352 ETD is readily coupled with MS1-based labeling approaches, including dimethyl labeling, stable isotope labeling in amino acid cell culture (SILAC), and neutron-encoded (NeuCode) SlLAC.233,353−357 In NeuCode SILAC, quantitative channels are spaced very closely together (several to tens of mDa) to allow controlled masking or revealing of quantitative peaks based on resolving power. This is typically employed at the MS1 level, but quantitative information can also be revealed in high-resolution ETD MS/MS spectra of both peptides356 and proteins.357
Isobaric labels, e.g., tandem mass tags (TMT) and isobaric tag for relative and absolute quantification (iTRAQ), are another approach to chemical labeling that enable mutliplexed quantitative comparisons at the MS/MS level. iTRAQ labels were first shown to be compatible with ETD, although the amount plexing was limited and required a second collisional activation step to generate the fully array of reporter ions for quantitative comparisons.358,359 TMT labels were modified to accommodate the fragmentation pathways of ETD, a process that required switching heavy carbon and nitrogen atoms to account for relocation of the proximal heavy carbon from the reporter ion to the balance region upon ETD fragmentation.360 This modification serendipitously led to the discovery that neutron-encoded mass differences could be exploited to increase the plexing of TMT reagents, and this phenomenon also led to the rise of the NeuCode SILAC approach.361 Just this year Li and co-workers investigated EThcD for TMT multiplexed quantification, looking for sequence information and reporter ion intensities for quantitative comparisons in the same MS/MS spectra in global proteomic and phosphoproteomic experiments.362 They showed through careful optimization and balancing of parameters that EThcD could generate extensive sequence-informative product ions while also preserving the presence of reporter ion signal and that this improved analyses over a combination of separate ETD and HCD scans.
New Informatic Tools for ETD and Related Technologies
In addition to improvements in ETD instrumentation and methodology, development of informatics tools to process ETD data is crucial to its utility in proteomics. Statistical analyses of ETD spectra have provided insight into how to design search algorithms to properly assess ETD spectra,363,364 and a number of widely available search engines exist to process ETD spectra.365 Incorporation of ETD search capabilities into flexible pipelines that offer many dimensions of data analysis (e.g., MaxQuant366 and OpenMS367) is important and enables straightforward integration of ETD methods with a variety of other tools. Moreover, resources like the Web-based tutorial recently presented by Hunt and colleagues for training in ETD spectral interpretation are valuable in making ETD methods more broadly accessible to a growing proteomics community.368 ProteomeTools, a project that analyzed >330 000 synthetic tryptic peptides representing essentially all canonical human gene products with ETD, ETcaD, and EThcD (in addition to collisional dissociation approaches), represents another fantastic resource for development of software tools ranging from intelligent decision-tree acquisition routines to search engines that will benefit ETD-based analyses and proteomic efforts in general.369
Even with the improvements made in ETD spectral interpretation, an important and often overlooked strategy to improve scoring of ETD spectra is the removal of interfering ions prior to searching.370−372 Charge-reduced precursor ions and neutral loss peaks resulting from intact peptide radical products in ETD are present373 but do not contribute sequence information, and they can be systematically cleaned from spectra while retaining sequence informative c/z•-type product ions. This can make a considerable difference in peptide identifications because presence of unexplained signal can penalize scoring in many search algorithms that only look for sequencing ions derived from expected peptide backbone cleavages.
Development of new algorithms to integrate ETD spectra into de novo sequencing strategies and PTM analyses are also emerging. The majority of de novo sequencing approaches, such as pNovo+, rely on pairing complementary dissociation from HCD and ETD spectra.374−376 NovoExD, recently described by Yan et al., enables de novo sequencing from ETD (or ECD) spectra alone,377 and the increasing prevalence of supplemental activation methods like EThcD and AI-ETD may drive development of similar approaches that do not require HCD complements.
Glycoproteomic applications are another active arena for development of ETD-enabled informatic tools. In 2013, Desaire and co-workers introduced GlycoPep Detector, a Web-based tool designed to identify intact N-glycopeptides from ETD spectra. Their glycoproteomic search engine applies filtering functions followed by correlation of glycopeptide compositions with the ETD spectra and intensity-weighted scoring based on independent assessment of multiple ion series (c-, z-, and y-type ions). They followed GlycoPep Detector with another tool called GlycoPep Evaluator to improve false discovery rate calculations in ETD-based intact glycopeptide analysis.378 Around the same time, Tang and co-workers described a search strategy for N-glycopeptides that combined scoring of collision-based dissociation spectra and ETD spectra to identify intact glycopeptides from complex samples (e.g., human serum).379 Most recently, Lee et al. described the benefits of the Byonic search algorithm for automated N-glycopeptide profiling,380 and although they did not use ETD in that particular study, Byonic has been widely used in recent glycoproteomic experiments, including several of those discussed above.
Investigations into ETD spectra of O-glycopeptides specifically have improved data interpretation. Darula et al. showed that altering scoring of O-glycopeptide ETD spectra with Protein Prospector can sizeably improve identifications by weighting product ion scores based on several spectral features.381 Zhu et al. conducted a similar study on ETD spectra from O-glycopeptides, finding that flexible scoring of c- or z-type ion series based on the precursor ion and inclusion of multiply charged c/z-type product ions can significantly benefit glycopeptide identifications.382 Beyond spectral scoring, the databases used for querying spectra can influence data quality and Chalkley and Baker recently employed a reference glycosite database based on known glycosites to improve identification of glycopeptide ETD spectra for both N- and O-glycopeptides.383
Informatics tools for analysis of ETD-based top-down proteomic data continue to improve as well. Kelleher et al. launched the freely available ProSight Lite in 2015, which is a simple and intuitive platform to characterize proteoforms based on MS/MS spectra.384 ProSight Lite is compatible with a range of dissociation methods, including ETD, EThcD, and AI-ETD, and it has provided a straightforward, flexible alternative to the commercially available ProSight software. MASH Suite Pro, created by Ge and co-workers, is a freely available comprehensive software package for top-down proteomics that is capable of processing high-resolution MS and MS/MS data (including ETD) using two deconvolution algorithms, enables PTM and sequence variant characterization, and provides relative quantitation of multiple proteoforms in different experimental conditions.385 Several other recently developed top-down proteomics software platforms, including Informed-Proteomics and Protein Goggle, can process ETD spectra as well.386−388
In addition to development of these data analysis pipelines, new strategies for spectral interpretation of MS/MS spectra of intact proteins are being developed. The C-score, for example, assesses proteoform identification and characterization to improve how both collisional dissociation and ETD spectra are interpreted in high-throughput top-down proteomic experiments.389 Sobott and co-workers recently introduced a method to examine the prevalence of ETD and PTR products ion MS/MS spectra of intact proteins, providing both an avenue to compare reaction conditions on different instrument platforms and a strategy for understanding how different reaction conditions and supplemental activation schemes effect product ions.390 They have also used relative quantities of fragmentation products in top-down ETD spectra to estimate ETD reaction rates on highly charged intact protein cations.391 Finally, Pevzner and co-workers described de novo sequencing approaches for intact proteins based on generating high-quality sequence tags, both with combinations of bottom-up and top-down spectra392 and with only intact protein MS/MS spectra.393
Looking Forward
The application of ETD to a diverse array of biological questions highlights its impact on proteome characterization. Its ease of implementation on a variety of instrument platforms and a concerted effort among many researchers to advance ETD methodology for widespread use laid a foundation for this success. As more complex questions about the proteome come to the forefront, ETD is poised to play an important role in research ranging from epigenetic and post-translational regulation of health and disease to the roles of protein–protein interactions and higher order protein structure in molecular biology.
ETD technology is fundamentally dependent on continued progress in MS instrumentation, and its utility will expand as mass spectrometers generally improve in sensitivity and resolution. ETD will specifically benefit from advances in robust reagent ion sources, data acquisitions schemes to improve product ion S/N (e.g., the multiple fill approach and spectral decongestion via PTR), and supplemental activation implementations, all of which have been active areas of development in recent years. ETD is particularly valuable when coupled with collisional dissociation, especially in real-time decisions tree strategies for characterizing modified peptides and intact proteins in complex samples, and the implementation of ETD on the newest generations of hybrid instruments will only extend this further. From our perspective, supplemental activation methods, especially EThcD and AI-ETD, are the future of ETD as well, as they offer substantial benefits with few drawbacks. We expect that the majority of ETD research in the coming years will use supplemental activation, especially as the various methods become default options on commercially available instruments.
One intriguing area of growth for ETD is in reaction cell design for both speed and S/N considerations. Collision-based fragmentation has more markedly benefitted from the speed and parallelized acquisition schemes on new instruments thus far, but little work has focused on how to optimize ETD reaction cell design to capitalize on these faster instruments. Improved reaction cells will not only minimize ion–ion reaction times, but they will also allow efficient storage of large precursor and reagent ion populations and provide easy access to supplemental activation methods, ideally infrared photoactivation for concurrent rather than postreaction activation. Most of the reaction cells in use today were described in the early years after the introduction of ETD, and it is now high time for a concentrated effort in improving the speed at which ETD spectra can be acquired.
In all, ETD is arguably the most valuable and widely used alternative dissociation method in peptide and protein characterization, and its utility is expanding to other biomolecules as well. The ETD community is active and continues to grow, especially as more ETD-enabled instruments make it into laboratories around the world. Success will depend on a sustained push to improve hardware and data acquisition strategies, and these technologies must reach beyond the expert laboratories and developers of ETD methodologies. We foresee ETD contributing to new insights in a wide swath of proteomic experiments in the coming years as it further expands to users across the realm of proteome research.
Acknowledgments
The authors gratefully acknowledge support from National Institutes of Health NIH Grant R35 GM118110 awarded to J.J.C. N.M.R. is funded through an NIH Predoctoral to Postdoctoral Transition Award (Grant F99 CA212454). The authors acknowledge the researchers whose original research work, both before and during the period covered by this review, could not be discussed in great detail due to space limitations.
Biographies
Nicholas M. Riley is a chemistry Ph.D. candidate and NIH F99 Graduate Fellow in the research group of Professor Joshua J. Coon at the University of Wisconsin-Madison. He completed his ACS Certified B.S. degree in Chemistry from the University of South Carolina in 2012 with Honors from the South Carolina Honors College, and Nick anticipates his Ph.D. in Analytical Chemistry in 2018. His research focuses on development of new ETD-based methodology and instrumentation for characterizing peptides, proteins, and post-translational modifications, with a concentration on protein phosphorylation and glycosylation.
Joshua J. Coon is a Professor of Chemistry and Biomolecular Chemistry at the University of Wisconsin-Madison and the Thomas and Margaret Pyle Chair at the Morgridge Institute for Research. Coon earned his B.S. degree at Central Michigan University in 1998 and took his Ph.D. at the University of Florida in 2002. At Florida Coon studied ambient ionization processes under the guidance of Professor Willard Harrison. From 2003 to 2005 he was an NIH postdoctoral fellow with Professor Donald Hunt at the University of Virginia. During his time at Virginia he, with Hunt and John Syka, coinvented electron transfer dissociation (ETD). Coon’s research program at Wisconsin is focused on all aspects of biomolecular mass spectrometry.
The authors declare no competing financial interest.
References
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Nature 2016, 537, 347–355. 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M.-C.; Yates J. R. Chem. Rev. 2013, 113, 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Wu S.; Stenoien D. L.; Paša-Tolić L. Annu. Rev. Anal. Chem. 2014, 7, 427–454. 10.1146/annurev-anchem-071213-020216. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Coon J. J. Cell Syst. 2016, 2, 142–143. 10.1016/j.cels.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Brodbelt J. S. Anal. Chem. 2016, 88, 30–51. 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. J. Am. Chem. Soc. 1998, 120, 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Coon J. J. Anal. Chem. 2009, 81, 3208–3215. 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tureček F.; Julian R. R. Chem. Rev. 2013, 113, 6691–6733. 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- Zhurov K. O.; Fornelli L.; Wodrich M. D.; Laskay Ü. A.; Tsybin Y. O. Chem. Soc. Rev. 2013, 42, 5014. 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Volmer D. A. Mass Spectrom. Rev. 2017, 36, 4–15. 10.1002/mas.21482. [DOI] [PubMed] [Google Scholar]
- Wiesner J.; Premsler T.; Sickmann A. Proteomics 2008, 8, 4466–4483. 10.1002/pmic.200800329. [DOI] [PubMed] [Google Scholar]
- Kim M.-S.; Pandey A. Proteomics 2012, 12, 530–542. 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon J. J.; Syka J. E. P.; Schwartz J. C.; Shabanowitz J.; Hunt D. F. Int. J. Mass Spectrom. 2004, 236, 33–42. 10.1016/j.ijms.2004.05.005. [DOI] [Google Scholar]
- Gunawardena H. P.; He M.; Chrisman P. A.; Pitteri S. J.; Hogan J. M.; Hodges B. D. M.; McLuckey S. A. J. Am. Chem. Soc. 2005, 127, 12627–12639. 10.1021/ja0526057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton P. D.; Strukl J. V.; Bai D. L.; Shabanowitz J.; Hunt D. F. Anal. Chem. 2012, 84, 1781–1785. 10.1021/ac202807h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon J. J.; Ueberheide B.; Syka J. E. P.; Dryhurst D. D.; Ausio J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9463–9468. 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D. F.; Coon J. J.; Syka J. E. P.; Marto J. A.. Electron transfer dissociation for biopolymer sequence analysis. European Patent ApplicationEP 2835642 A1, February 11, 2015.
- McAlister G. C.; Berggren W. T.; Griep-Raming J.; Horning S.; Makarov A.; Phanstiel D.; Stafford G.; Swaney D. L.; Syka J. E. P.; Zabrouskov V.; Coon J. J. J. Proteome Res. 2008, 7, 3127–3136. 10.1021/pr800264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good D. M.; Wirtala M.; McAlister G. C.; Coon J. J. Mol. Cell. Proteomics 2007, 6, 1942–1951. 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- Liu J.; McLuckey S. A. Int. J. Mass Spectrom. 2012, 330–332, 174–181. 10.1016/j.ijms.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteri S. J.; Chrisman P. A.; Hogan J. M.; McLuckey S. A. Anal. Chem. 2005, 77, 1831–1839. 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteri S. J.; Chrisman P. A.; McLuckey S. A. Anal. Chem. 2005, 77, 5662–5669. 10.1021/ac050666h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko B. J.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2012, 23, 1991–2000. 10.1007/s13361-012-0458-z. [DOI] [PubMed] [Google Scholar]
- Frey B. L.; Ladror D. T.; Sondalle S. B.; Krusemark C. J.; Jue A. L.; Coon J. J.; Smith L. M. J. Am. Soc. Mass Spectrom. 2013, 24, 1710–1721. 10.1007/s13361-013-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney D. L.; McAlister G. C.; Coon J. J. Nat. Methods 2008, 5, 959–964. 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; Hennrich M. L.; Nolting D.; Zeller M.; Griep-Raming J.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2011, 10, 2377–2388. 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Zubarev A. R.; Savitski M. M. J. Am. Soc. Mass Spectrom. 2008, 19, 753–761. 10.1016/j.jasms.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Horn D. M.; Ge Y.; McLafferty F. W. Anal. Chem. 2000, 72, 4778–4784. 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- Horn D. M.; Breuker K.; Frank A. J.; McLafferty F. W. J. Am. Chem. Soc. 2001, 123, 9792–9799. 10.1021/ja003143u. [DOI] [PubMed] [Google Scholar]
- Sze S. K.; Ge Y.; Oh H.; McLafferty F. W. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 1774–1779. 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsybin Y. O.; Witt M.; Baykut G.; Kjeldsen F.; Håkansson P. Rapid Commun. Mass Spectrom. 2003, 17, 1759–1768. 10.1002/rcm.1118. [DOI] [PubMed] [Google Scholar]
- Cooper H. J.; Håkansson K.; Marshall A. G. Mass Spectrom. Rev. 2005, 24, 201–222. 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- Mikhailov V. A.; Cooper H. J. J. Am. Soc. Mass Spectrom. 2009, 20, 763–771. 10.1016/j.jasms.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey S. A.; Reid G. E.; Wells J. M. Anal. Chem. 2002, 74, 336–346. 10.1021/ac0109671. [DOI] [PubMed] [Google Scholar]
- Han H.; Xia Y.; McLuckey S. A. Rapid Commun. Mass Spectrom. 2007, 21, 1567–1573. 10.1002/rcm.2994. [DOI] [PubMed] [Google Scholar]
- Wu S.-L.; Hühmer A. F. R.; Hao Z.; Karger B. L. J. Proteome Res. 2007, 6, 4230–4244. 10.1021/pr070313u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Han H.; McLuckey S. A. Anal. Chem. 2008, 80, 1111–1117. 10.1021/ac702188q. [DOI] [PubMed] [Google Scholar]
- Liu J.; Gunawardena H. P.; Huang T. Y.; McLuckey S. A. Int. J. Mass Spectrom. 2008, 276, 160–170. 10.1016/j.ijms.2008.07.028. [DOI] [Google Scholar]
- Campbell J. L.; Hager J. W.; Le Blanc J. C. Y. J. Am. Soc. Mass Spectrom. 2009, 20, 1672–1683. 10.1016/j.jasms.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Rathore D.; Aboufazeli F.; Dodds E. D. Analyst 2015, 140, 7175–7183. 10.1039/C5AN01225B. [DOI] [PubMed] [Google Scholar]
- Swaney D. L.; McAlister G. C.; Wirtala M.; Schwartz J. C.; Syka J. E. P.; Coon J. J. Anal. Chem. 2007, 79, 477–485. 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Anal. Chem. 2012, 84, 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- O’Connor P. B.; Lin C.; Cournoyer J. J.; Pittman J. L.; Belyayev M.; Budnik B. A. J. Am. Soc. Mass Spectrom. 2006, 17, 576–585. 10.1016/j.jasms.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Tsybin Y. O.; He H.; Emmett M. R.; Hendrickson C. L.; Marshall A. G. Anal. Chem. 2007, 79, 7596–7602. 10.1021/ac071165u. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Kjeldsen F.; Nielsen M. L.; Zubarev R. A. J. Am. Soc. Mass Spectrom. 2007, 18, 113–120. 10.1016/j.jasms.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Ledvina A. R.; McAlister G. C.; Gardner M. W.; Smith S. I.; Madsen J. A.; Schwartz J. C.; Stafford G. C.; Syka J. E. P.; Brodbelt J. S.; Coon J. J. Angew. Chem., Int. Ed. 2009, 48, 8526–8528. 10.1002/anie.200903557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Hebert A. S.; Coon J. J. Anal. Chem. 2017, 89, 6358–6366. 10.1021/acs.analchem.7b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledvina A. R.; Beauchene N. A.; McAlister G. C.; Syka J. E. P.; Schwartz J. C.; Griep-Raming J.; Westphall M. S.; Coon J. J. Anal. Chem. 2010, 82, 10068–10074. 10.1021/ac1020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley R. J.; Medzihradszky K. F.; Lynn A. J.; Baker P. R.; Burlingame A. L. Anal. Chem. 2010, 82, 579–584. 10.1021/ac9018582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Holden D. D.; Brodbelt J. S. Anal. Chem. 2014, 86, 10970–10977. 10.1021/ac5036082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A.; Damoc E.; Lange O.; Denisov E.; Nolting D.; Müller M.; Viner R.; Schwartz J.; Remes P.; Belford M.; Dunyach J.-J.; Cox J.; Horning S.; Mann M.; Makarov A. Mol. Cell. Proteomics 2012, 11, O111.013698. 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C. M.; Russell J. D.; Ledvina A. R.; McAlister G. C.; Westphall M. S.; Griep-Raming J.; Schwartz J. C.; Coon J. J.; Syka J. E. P. J. Am. Soc. Mass Spectrom. 2013, 24, 816–827. 10.1007/s13361-013-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledvina A. R.; Rose C. M.; McAlister G. C.; Syka J. E. P.; Westphall M. S.; Griep-Raming J.; Schwartz J. C.; Coon J. J. J. Am. Soc. Mass Spectrom. 2013, 24, 1623–1633. 10.1007/s13361-013-0621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Nolting D.; Altelaar A. F. M.; Griep-Raming J.; Mohammed S.; Heck A. J. R. J. Am. Soc. Mass Spectrom. 2013, 24, 1663–1670. 10.1007/s13361-013-0618-9. [DOI] [PubMed] [Google Scholar]
- Earley L.; Anderson L. C.; Bai D. L.; Mullen C.; Syka J. E. P.; English A. M.; Dunyach J.-J.; Stafford G. C.; Shabanowitz J.; Hunt D. F.; Compton P. D.; Compton P. D. Anal. Chem. 2013, 85, 8385–8390. 10.1021/ac401783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senko M. W.; Remes P. M.; Canterbury J. D.; Mathur R.; Song Q.; Eliuk S. M.; Mullen C.; Earley L.; Hardman M.; Blethrow J. D.; Bui H.; Specht A.; Lange O.; Denisov E.; Makarov A.; Horning S.; Zabrouskov V. Anal. Chem. 2013, 85, 11710–11714. 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Mullen C.; Weisbrod C. R.; Sharma S.; Senko M. W.; Zabrouskov V.; Westphall M. S.; Syka J. E. P.; Coon J. J. J. Am. Soc. Mass Spectrom. 2016, 27, 520–531. 10.1007/s13361-015-1306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C. M.; Rush M. J. P.; Riley N. M.; Merrill A. E.; Kwiecien N. W.; Holden D. D.; Mullen C.; Westphall M. S.; Coon J. J. J. Am. Soc. Mass Spectrom. 2015, 26, 1848–1857. 10.1007/s13361-015-1183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan D. A.; Hartmer R.; Speir J. P.; Stoermer C.; Gumerov D.; Easterling M. L.; Brekenfeld A.; Kim T.; Laukien F.; Park M. A. Rapid Commun. Mass Spectrom. 2008, 22, 271–278. 10.1002/rcm.3356. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Chrisman P. A.; Erickson D. E.; Liu J.; Liang X.; Londry F. A.; Yang M. J.; McLuckey S. A. Anal. Chem. 2006, 78, 4146–4154. 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson C. L.; Quinn J. P.; Kaiser N. K.; Smith D. F.; Blakney G. T.; Chen T.; Marshall A. G.; Weisbrod C. R.; Beu S. C. J. Am. Soc. Mass Spectrom. 2015, 26, 1626–1632. 10.1007/s13361-015-1182-2. [DOI] [PubMed] [Google Scholar]
- Weisbrod C. R.; Kaiser N. K.; Syka J. E. P.; Early L.; Mullen C.; Dunyach J. J.; English A. M.; Anderson L. C.; Blakney G. T.; Shabanowitz J.; Hendrickson C. L.; Marshall A. G.; Hunt D. F. J. Am. Soc. Mass Spectrom. 2017, 28, 1787–1795. 10.1007/s13361-017-1702-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Wojcik R.; Zhang X.; Ibrahim Y. M.; Burnum-Johnson K. E.; Orton D. J.; Monroe M. E.; Moore R. J.; Smith R. D.; Baker E. S. Annu. Rev. Anal. Chem. 2017, 10, 71–92. 10.1146/annurev-anchem-061516-045212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. P.; Brown J. M.; Campuzano I.; Sadler P. J. Chem. Commun. 2010, 46, 5458–5460. 10.1039/c0cc00358a. [DOI] [PubMed] [Google Scholar]
- Lermyte F.; Verschueren T.; Brown J. M.; Williams J. P.; Valkenborg D.; Sobott F. Methods 2015, 89, 22–29. 10.1016/j.ymeth.2015.05.019. [DOI] [PubMed] [Google Scholar]
- Cramer C. N.; Brown J. M.; Tomczyk N.; Nielsen P. K.; Haselmann K. F. J. Am. Soc. Mass Spectrom. 2017, 28, 384–388. 10.1007/s13361-016-1538-2. [DOI] [PubMed] [Google Scholar]
- Baird M. A.; Shvartsburg A. A. J. Am. Soc. Mass Spectrom. 2016, 27, 2064–2070. 10.1007/s13361-016-1498-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe G. C.; Maleki H.; Arndt J. R.; Khakinejad M.; Yi J.; McBride C.; Nurkiewicz T. R.; Valentine S. J. Anal. Chem. 2014, 86, 8121–8128. 10.1021/ac501527y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Lietz C. B.; Li L. Anal. Bioanal. Chem. 2017, 1–11. 10.1007/s00216-017-0611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall D. D.; Inutan E. D.; Wang B.; Liu C.-W.; Thawoos S.; Wager-Miller J.; Mackie K.; Trimpin S. Proteomics 2016, 16, 1695–1706. 10.1002/pmic.201500530. [DOI] [PubMed] [Google Scholar]
- He M.; Jiang Y.; Guo D.; Xiong X.; Fang X.; Xu W. J. Am. Soc. Mass Spectrom. 2017, 28, 1262–1270. 10.1007/s13361-016-1504-z. [DOI] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Martens J.; Grzetic J.; Berden G.; Oomens J. Nat. Commun. 2016, 7, 11754. 10.1038/ncomms11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Oomens J. Anal. Chem. 2016, 88, 6126–6129. 10.1021/acs.analchem.6b01483. [DOI] [PubMed] [Google Scholar]
- Munshi M. U.; Craig S. M.; Berden G.; Martens J.; DeBlase A. F.; Foreman D. J.; McLuckey S. A.; Oomens J.; Johnson M. A. J. Phys. Chem. Lett. 2017, 8, 5047–5052. 10.1021/acs.jpclett.7b02223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. M.; Kelleher N. L. Nat. Methods 2013, 10, 186–187. 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toby T. K.; Fornelli L.; Kelleher N. L. Annu. Rev. Anal. Chem. 2016, 9, 499–519. 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner O. S.; Catherman A. D.; Early B. P.; Thomas P. M.; Compton P. D.; Kelleher N. L. Anal. Chem. 2014, 86, 4627–4634. 10.1021/ac500864w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlf D. R.; Compton P. D.; Tran J. C.; Early B. P.; Thomas P. M.; Kelleher N. L. J. Proteome Res. 2012, 11, 4308–4314. 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rožman M.; Gaskell S. J. Rapid Commun. Mass Spectrom. 2012, 26, 282–286. 10.1002/rcm.5330. [DOI] [PubMed] [Google Scholar]
- Tran J. C.; Zamdborg L.; Ahlf D. R.; Lee J. E.; Catherman A. D.; Durbin K. R.; Tipton J. D.; Vellaichamy A.; Kellie J. F.; Li M.; Wu C.; Sweet S. M. M.; Early B. P.; Siuti N.; LeDuc R. D.; Compton P. D.; Thomas P. M.; Kelleher N. L. Nature 2011, 480, 254–258. 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman A. D.; Durbin K. R.; Ahlf D. R.; Early B. P.; Fellers R. T.; Tran J. C.; Thomas P. M.; Kelleher N. L. Mol. Cell. Proteomics 2013, 12, 3465–3473. 10.1074/mcp.M113.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin K. R.; Fellers R. T.; Ntai I.; Kelleher N. L.; Compton P. D. Anal. Chem. 2014, 86, 1485–1492. 10.1021/ac402904h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin K. R.; Fornelli L.; Fellers R. T.; Doubleday P. F.; Narita M.; Kelleher N. L. J. Proteome Res. 2016, 15, 976–982. 10.1021/acs.jproteome.5b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner A. M.; Lössl P.; Liu F.; Huguet R.; Mullen C.; Yamashita M.; Zabrouskov V.; Makarov A.; Altelaar A. F. M.; Heck A. J. R. Anal. Chem. 2015, 87, 4152–4158. 10.1021/acs.analchem.5b00162. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. Anal. Chem. 2015, 87, 7109–7116. 10.1021/acs.analchem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. J. Proteome Res. 2017, 16, 2653–2659. 10.1021/acs.jproteome.7b00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Westphall M. S.; Coon J. J. J. Am. Soc. Mass Spectrom. 2017, 10.1007/s13361-017-1808-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Dürnberger G.; Stanek F.; Mechtler K.; Westphall M. S.; Coon J. J. Anal. Chem. 2017, 89, 6367–6376. 10.1021/acs.analchem.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Li W.; Holden D. D.; Zhang Y.; Griep-Raming J.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. J. Am. Chem. Soc. 2013, 135, 12646–12651. 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Riley N. M.; Sun L.; Hebert A. S.; Yan X.; Westphall M. S.; Rush M. J. P.; Zhu G.; Champion M. M.; Medie F. M.; Champion P. A. D.; Coon J. J.; Dovichi N. J. Anal. Chem. 2015, 87, 5422–5429. 10.1021/acs.analchem.5b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornelli L.; Parra J.; Hartmer R.; Stoermer C.; Lubeck M.; Tsybin Y. O. Anal. Bioanal. Chem. 2013, 405, 8505–8514. 10.1007/s00216-013-7267-5. [DOI] [PubMed] [Google Scholar]
- Anderson L. C.; DeHart C. J.; Kaiser N. K.; Fellers R. T.; Smith D. F.; Greer J. B.; LeDuc R. D.; Blakney G. T.; Thomas P. M.; Kelleher N. L.; Hendrickson C. L. J. Proteome Res. 2017, 16, 1087–1096. 10.1021/acs.jproteome.6b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho Graça D.; Lescuyer P.; Clerici L.; Tsybin Y. O.; Hartmer R.; Meyer M.; Samii K.; Hochstrasser D. F.; Scherl A. J. Am. Soc. Mass Spectrom. 2012, 23, 1750–1756. 10.1007/s13361-012-0446-3. [DOI] [PubMed] [Google Scholar]
- Coelho Graça D.; Hartmer R.; Jabs W.; Beris P.; Clerici L.; Stoermer C.; Samii K.; Hochstrasser D.; Tsybin Y. O.; Scherl A.; Lescuyer P. Anal. Bioanal. Chem. 2015, 407, 2837–2845. 10.1007/s00216-015-8525-5. [DOI] [PubMed] [Google Scholar]
- Sarsby J.; Martin N. J.; Lalor P. F.; Bunch J.; Cooper H. J. J. Am. Soc. Mass Spectrom. 2014, 25, 1953–1961. 10.1007/s13361-014-0967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarsby J.; Griffiths R. L.; Race A. M.; Bunch J.; Randall E. C.; Creese A. J.; Cooper H. J. Anal. Chem. 2015, 87, 6794–6800. 10.1021/acs.analchem.5b01151. [DOI] [PubMed] [Google Scholar]
- Schey K. L.; Anderson D. M.; Rose K. L. Anal. Chem. 2013, 85, 6767–6774. 10.1021/ac400832w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S.; Burlingame A. L. ACS Chem. Biol. 2015, 10, 63–71. 10.1021/cb500904b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Mann M. Mol. Cell. Proteomics 2013, 12, 3444–3452. 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Coon J. J. Anal. Chem. 2016, 88, 74–94. 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey S. J.; James D. E.; Mann M. Trends Endocrinol. Metab. 2015, 26, 676. 10.1016/j.tem.2015.09.013. [DOI] [PubMed] [Google Scholar]
- von Stechow L.; Francavilla C.; Olsen J. V. Expert Rev. Proteomics 2015, 12, 469–487. 10.1586/14789450.2015.1078730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaysen-Andersen M.; Packer N. H.; Schulz B. L. Mol. Cell. Proteomics 2016, 15, 1773–1790. 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J. Glycoconjugate J. 2016, 33, 261–272. 10.1007/s10719-016-9649-3. [DOI] [PubMed] [Google Scholar]
- Leymarie N.; Zaia J. Anal. Chem. 2012, 84, 3040–3048. 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras-Yakushi T. R.; Sweredoski M. J.; Hess S. J. Am. Soc. Mass Spectrom. 2015, 26, 1580–1587. 10.1007/s13361-015-1168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe S. O.; Rana A. S. J. B.; Deol K. K.; Ge Y.; Strieter E. R. Anal. Chem. 2017, 89, 4428–4434. 10.1021/acs.analchem.6b03675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A. S. J. B.; Ge Y.; Strieter E. R. J. Proteome Res. 2017, 16, 3363–3369. 10.1021/acs.jproteome.7b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels C. M.; Ong S.-E.; Leung A. K. L. Methods in Molecular Biology 2017, 1608, 79–93. 10.1007/978-1-4939-6993-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal F.; Nanni P.; Barkow-Oesterreicher S.; Hottiger M. O. J. Proteome Res. 2015, 14, 4072–4079. 10.1021/acs.jproteome.5b00432. [DOI] [PubMed] [Google Scholar]
- Bilan V.; Leutert M.; Nanni P.; Panse C.; Hottiger M. O. Anal. Chem. 2017, 89, 1523–1530. 10.1021/acs.analchem.6b03365. [DOI] [PubMed] [Google Scholar]
- Erce M. A.; Abeygunawardena D.; Low J. K. K.; Hart-Smith G.; Wilkins M. R. Mol. Cell. Proteomics 2013, 12, 3184–3198. 10.1074/mcp.M113.031500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui S.; Gathiaka S.; Li J.; Qu J.; Acevedo O.; Hevel J. M. J. Biol. Chem. 2014, 289, 9320–9327. 10.1074/jbc.M113.535278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J. K. K.; Im H.; Erce M. A.; Hart-Smith G.; Snyder M. P.; Wilkins M. R. Proteomics 2016, 16, 465–476. 10.1002/pmic.201400564. [DOI] [PubMed] [Google Scholar]
- Snijders A. P.; Hautbergue G. M.; Bloom A.; Williamson J. C.; Minshull T. C.; Phillips H. L.; Mihaylov S. R.; Gjerde D. T.; Hornby D. P.; Wilson S. A.; Hurd P. J.; Dickman M. J. RNA 2015, 21, 347–359. 10.1261/rna.045138.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk J. C.; Li J.; Wang H.; Aletta J. M.; Qu J.; Read L. K. Mol. Cell. Proteomics 2013, 12, 302–311. 10.1074/mcp.M112.022533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheron L.; van den Toorn H.; Heck A. J. R.; Mohammed S. Anal. Chem. 2014, 86, 8312–8320. 10.1021/ac501803z. [DOI] [PubMed] [Google Scholar]
- de Graaf E. L.; Giansanti P.; Altelaar A. F. M.; Heck A. J. R. Mol. Cell. Proteomics 2014, 13, 2426–2434. 10.1074/mcp.O113.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayapuram N.; Bonhomme L.; Bigeard J.; Haddadou K.; Przybylski C.; Hirt H.; Pflieger D. J. Proteome Res. 2014, 13, 2137–2151. 10.1021/pr401268v. [DOI] [PubMed] [Google Scholar]
- Giansanti P.; Aye T. T.; van den Toorn H.; Peng M.; van Breukelen B.; Heck A. J. R. Cell Rep. 2015, 11, 1834–1843. 10.1016/j.celrep.2015.05.029. [DOI] [PubMed] [Google Scholar]
- Joshi R. N.; Binai N. A.; Marabita F.; Sui Z.; Altman A.; Heck A. J. R.; Tegnér J.; Schmidt A. Front. Immunol. 2017, 8, 1163. 10.3389/fimmu.2017.01163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D. J.; Rose C. M.; McAlister G. C.; Brumbaugh J.; Yu P.; Wenger C. D.; Westphall M. S.; Thomson J. A.; Coon J. J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8411–8416. 10.1073/pnas.1205292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M. O.; Wright J. C.; Jones M.; Rayner J. C.; Choudhary J. S. J. Proteomics 2014, 103, 1–14. 10.1016/j.jprot.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese H.; Kuhlmann K.; Wiese S.; Stoepel N. S.; Pawlas M.; Meyer H. E.; Stephan C.; Eisenacher M.; Drepper F.; Warscheid B. J. Proteome Res. 2014, 13, 1128–1137. 10.1021/pr400402s. [DOI] [PubMed] [Google Scholar]
- Marx H.; Lemeer S.; Schliep J. E.; Matheron L.; Mohammed S.; Cox J.; Mann M.; Heck A. J. R.; Kuster B. Nat. Biotechnol. 2013, 31, 557–564. 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
- Penkert M.; Yates L. M.; Schümann M.; Perlman D.; Fiedler D.; Krause E. Anal. Chem. 2017, 89, 3672–3680. 10.1021/acs.analchem.6b05095. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Zhou H.; Taus T.; Altelaar A. F. M.; Mechtler K.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2013, 12, 1520–1525. 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus T.; Köcher T.; Pichler P.; Paschke C.; Schmidt A.; Henrich C.; Mechtler K. J. Proteome Res. 2011, 10, 5354–5362. 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- Lössl P.; Brunner A. M.; Liu F.; Leney A. C.; Yamashita M.; Scheltema R. A.; Heck A. J. R. ACS Cent. Sci. 2016, 2, 445–455. 10.1021/acscentsci.6b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamara S.; Scheltema R. A.; Heck A. J. R.; Leney A. C. Angew. Chem. 2017, 129, 13829–13832. 10.1002/ange.201706749. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Ammerer G.; Mechtler K. Proteomics 2013, 13, 945–954. 10.1002/pmic.201200240. [DOI] [PubMed] [Google Scholar]
- Bertran-Vicente J.; Serwa R. A.; Schümann M.; Schmieder P.; Krause E.; Hackenberger C. P. R. J. Am. Chem. Soc. 2014, 136, 13622–13628. 10.1021/ja507886s. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Trentini D. B.; Spiess S.; Fuhrmann J.; Ammerer G.; Mechtler K.; Clausen T. Mol. Cell. Proteomics 2014, 13, 537–550. 10.1074/mcp.M113.032292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Vicente J.; Schümann M.; Hackenberger C. P. R.; Krause E. Anal. Chem. 2015, 87, 6990–6994. 10.1021/acs.analchem.5b01389. [DOI] [PubMed] [Google Scholar]
- Trentini D. B.; Suskiewicz M. J.; Heuck A.; Kurzbauer R.; Deszcz L.; Mechtler K.; Clausen T. Nature 2016, 539, 48–53. 10.1038/nature20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott N. E.; Parker B. L.; Connolly A. M.; Paulech J.; Edwards A. V. G.; Crossett B.; Falconer L.; Kolarich D.; Djordjevic S. P.; Højrup P.; Packer N. H.; Larsen M. R.; Cordwell S. J. Mol. Cell. Proteomics 2011, 10, M000031-MCP201. 10.1074/mcp.M000031-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford K. L.; Zeng W.; Heazlewood J. L.; Bacic A. Front. Plant Sci. 2015, 6, 674. 10.3389/fpls.2015.00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. P.; Pringle S.; Richardson K.; Gethings L.; Vissers J. P. C.; De Cecco M.; Houel S.; Chakraborty A. B.; Yu Y. Q.; Chen W.; Brown J. M. Rapid Commun. Mass Spectrom. 2013, 27, 2383–2390. 10.1002/rcm.6684. [DOI] [PubMed] [Google Scholar]
- Barallobre-Barreiro J.; Baig F.; Fava M.; Yin X.; Mayr M. J. Visualized Exp. 2017, e55674–e55674. 10.3791/55674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker B. L.; Thaysen-Andersen M.; Solis N.; Scott N. E.; Larsen M. R.; Graham M. E.; Packer N. H.; Cordwell S. J. J. Proteome Res. 2013, 12, 5791–5800. 10.1021/pr400783j. [DOI] [PubMed] [Google Scholar]
- Trinidad J. C.; Schoepfer R.; Burlingame A. L.; Medzihradszky K. F. Mol. Cell. Proteomics 2013, 12, 3474–3488. 10.1074/mcp.M113.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzihradszky K. F.; Kaasik K.; Chalkley R. J. Mol. Cell. Proteomics 2015, 14, 2103–2110. 10.1074/mcp.M115.050393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.-L.; Medzihradszky K. F.; Wang Z.-Y.; Burlingame A. L.; Chalkley R. J. Mol. Cell. Proteomics 2016, 15, 2048–2054. 10.1074/mcp.M115.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darula Z.; Medzihradszky K. F. J. Am. Soc. Mass Spectrom. 2014, 25, 977–987. 10.1007/s13361-014-0852-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba J.; Dutta S.; Hemenway E.; Viner R. Int. J. Proteomics 2012, 2012, 560391. 10.1155/2012/560391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh C.; Zampronio C. G.; Creese A. J.; Cooper H. J. J. Proteome Res. 2012, 11, 4517–4525. 10.1021/pr300257c. [DOI] [PubMed] [Google Scholar]
- Wu S. W.; Pu T. H.; Viner R.; Khoo K. H. Anal. Chem. 2014, 86, 5478–5486. 10.1021/ac500945m. [DOI] [PubMed] [Google Scholar]
- Levery S. B.; Steentoft C.; Halim A.; Narimatsu Y.; Clausen H.; Vakhrushev S. Y. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 33–42. 10.1016/j.bbagen.2014.09.026. [DOI] [PubMed] [Google Scholar]
- Thaysen-Andersen M.; Wilkinson B. L.; Payne R. J.; Packer R. H. Electrophoresis 2011, 32, 3536–3545. 10.1002/elps.201100294. [DOI] [PubMed] [Google Scholar]
- Darula Z.; Sherman J.; Medzihradszky K. F. Mol. Cell. Proteomics 2012, 11, 1–10. 10.1074/mcp.O111.016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darula Z.; Sarnyai F.; Medzihradszky K. F. Glycoconjugate J. 2016, 33, 435–445. 10.1007/s10719-015-9630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Marx K.; Reichl U.; Wuhrer M.; Rapp E. Mol. Cell. Proteomics 2016, 15, 624–641. 10.1074/mcp.M115.053546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houel S.; Hilliard M.; Yu Y. Q.; McLoughlin N.; Martin S. M.; Rudd P. M.; Williams J. P.; Chen W. Anal. Chem. 2014, 86, 576–584. 10.1021/ac402726h. [DOI] [PubMed] [Google Scholar]
- Steentoft C.; Vakhrushev S. Y.; Vester-Christensen M. B.; Schjoldager K. T. B. G.; Kong Y.; Bennett E. P.; Mandel U.; Wandall H.; Levery S. B.; Clausen H. Nat. Methods 2011, 8, 977–982. 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- Steentoft C.; Vakhrushev S. Y.; Joshi H. J.; Kong Y.; Vester-Christensen M. B.; Schjoldager K. T. B. G.; Lavrsen K.; Dabelsteen S.; Pedersen N. B.; Marcos-Silva L.; Gupta R.; Paul Bennett E.; Mandel U.; Brunak S.; Wandall H. H.; Levery S. B.; Clausen H. EMBO J. 2013, 32, 1478–1488. 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Halim A.; Narimatsu Y.; Joshi H. J.; Steentoft C.; Schjoldager K. T. B. G.; Schulz M. A.; Sealover N. R.; Kayser K. J.; Bennett E. P.; Levery S. B.; Vakhrushev S. Y.; Clausen H. Mol. Cell. Proteomics 2014, 13, 3224–3235. 10.1074/mcp.M114.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert P.; Halim A.; Zauser M.; Essig A.; Joshi H. J.; Zatorska E.; Larsen I. S. B.; Loibl M.; Castells-Ballester J.; Aebi M.; Clausen H.; Strahl S. Mol. Cell. Proteomics 2016, 15, 1323–1337. 10.1074/mcp.M115.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windwarder M.; Altmann F. J. Proteomics 2014, 108, 258–268. 10.1016/j.jprot.2014.05.022. [DOI] [PubMed] [Google Scholar]
- Ma J.; Hart G. W. Analysis of Protein O-GlcNAcylation by Mass Spectrometry. Current Protocols in Protein Science 2017, 2017 (John Wiley & Sons, Inc.: Hoboken, NJ), 24.10.1–24.10.16. 10.1002/cpps.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. A.; Daou S.; Affar E. B.; Burlingame A. Proteomics 2013, 13, 982–991. 10.1002/pmic.201200332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leney A. C.; El Atmioui D.; Wu W.; Ovaa H.; Heck A. J. R. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E7255–E7261. 10.1073/pnas.1620529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad J. C.; Barkan D. T.; Gulledge B. F.; Thalhammer A.; Sali A.; Schoepfer R.; Burlingame A. L. Mol. Cell. Proteomics 2012, 11, 215–229. 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M.; Knudsen G. M.; Maeda S.; Trinidad J. C.; Ioanoviciu A.; Burlingame A. L.; Mucke L. Nat. Neurosci. 2015, 18, 1183–1189. 10.1038/nn.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro J. F.; Gong C.-X.; Monroe M. E.; Aldrich J. T.; Clauss T. R. W.; Purvine S. O.; Wang Z.; Camp D. G.; Shabanowitz J.; Stanley P.; Hart G. W.; Hunt D. F.; Yang F.; Smith R. D. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 7280–7285. 10.1073/pnas.1200425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik K.; Kivimäe S.; Allen J. J.; Chalkley R. J.; Huang Y.; Baer K.; Kissel H.; Burlingame A. L.; Shokat K. M.; Ptáček L. J.; Fu Y.-H. Cell Metab. 2013, 17, 291–302. 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen J. W.; Balsbaugh J. L.; Chanda D.; Shabanowitz J.; Hunt D. F.; Neumann D.; Hart G. W. J. Biol. Chem. 2014, 289, 10592–10606. 10.1074/jbc.M113.523068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q.; Wang B.; Chen Z.; Urabe G.; Glover M. S.; Shi X.; Guo L. W.; Kent K. C.; Li L. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764. 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q.; Canales A.; Glover M. S.; Das R.; Shi X.; Liu Y.; Keller M. P.; Attie A. D.; Li L. Anal. Chem. 2017, 89, 9184–9191. 10.1021/acs.analchem.7b01926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover M. S.; Yu Q.; Chen Z.; Shi X.; Kent K. C.; Li L. Int. J. Mass Spectrom. 2017, 10.1016/j.ijms.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker B. L.; Thaysen-Andersen M.; Fazakerley D. J.; Holliday M.; Packer N. H.; James D. E. Mol. Cell. Proteomics 2016, 15, 141–153. 10.1074/mcp.M115.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totten S. M.; Feasley C. L.; Bermudez A.; Pitteri S. J. J. Proteome Res. 2017, 16, 1249–1260. 10.1021/acs.jproteome.6b00849. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Westphall M. S.; Coon J. J.. Thousands of Glycosites Characterized via Intact Glycopeptide Analysis using Activated Ion Electron Transfer Dissociation. In Proceedings of the 65th ASMS Conference on Mass Spectrometry and Allied Topics, Indianapolis, IN, June 4–8, 2017.
- Bourgoin-Voillard S.; Leymarie N.; Costello C. E. Proteomics 2014, 14, 1174–1184. 10.1002/pmic.201300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Iavarone A. T.; Spiciarich D. R.; Palaniappan K. K.; Bertozzi C. R. Nat. Methods 2015, 12, 561–567. 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Felix A.; Byrd W. E.; Zuegel D. K.; Ishihara M.; Azadi P.; Iavarone A. T.; Pitteri S. J.; Bertozzi C. R. J. Proteome Res. 2017, 16, 1706–1718. 10.1021/acs.jproteome.6b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. M.; Felix A.; Zhang L.; Elias J. E.; Bertozzi C. R. Anal. Bioanal. Chem. 2017, 409, 579–588. 10.1007/s00216-016-9934-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Liu F.; Franc V.; Halim L. A.; Schellekens H.; Heck A. J. R. Nat. Commun. 2016, 7, 13397. 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc V.; Yang Y.; Heck A. J. R. Anal. Chem. 2017, 89, 3483–3491. 10.1021/acs.analchem.6b04527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronker M. F.; Lemstra S.; Snijder J.; Heck A. J. R.; Thies-Weesie D. M. E.; Pasterkamp R. J.; Janssen B. J. C. Nat. Commun. 2016, 7, 13584. 10.1038/ncomms13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard J. C.; Burlingame A. L.; Medzihradszky K. F. Mol. Cell. Proteomics 2016, 15, 3405–3411. 10.1074/mcp.M116.061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani N.; Ashour A.; Thornalley P. J. Glycoconjugate J. 2016, 33, 553–568. 10.1007/s10719-016-9709-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkovska-Stamenova S.; Hoffmann R. Food Chem. 2017, 221, 489–495. 10.1016/j.foodchem.2016.10.092. [DOI] [PubMed] [Google Scholar]
- Milkovska-Stamenova S.; Hoffmann R. J. Proteomics 2016, 134, 102–111. 10.1016/j.jprot.2015.12.022. [DOI] [PubMed] [Google Scholar]
- Bern M.; Beniston R.; Mesnage S. Anal. Bioanal. Chem. 2017, 409, 551–560. 10.1007/s00216-016-9857-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-L.; Jiang H.; Lu Q.; Dai S.; Hancock W. S.; Karger B. L. Anal. Chem. 2009, 81, 112–122. 10.1021/ac801560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-L.; Jiang H.; Hancock W. S.; Karger B. L. Anal. Chem. 2010, 82, 5296–5303. 10.1021/ac100766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massonnet P.; Upert G.; Smargiasso N.; Gilles N.; Quinton L.; De Pauw E. Anal. Chem. 2015, 87, 5240–5246. 10.1021/acs.analchem.5b00245. [DOI] [PubMed] [Google Scholar]
- Wen D.; Xiao Y.; Vecchi M. M.; Gong B. J.; Dolnikova J.; Pepinsky R. B. Anal. Chem. 2017, 89, 4021–4030. 10.1021/acs.analchem.6b04600. [DOI] [PubMed] [Google Scholar]
- Tan L.; Durand K. L.; Ma X.; Xia Y. Analyst 2013, 138, 6759. 10.1039/c3an01333b. [DOI] [PubMed] [Google Scholar]
- Liu F.; van Breukelen B.; Heck A. J. R. Mol. Cell. Proteomics 2014, 13, 2776–2786. 10.1074/mcp.O114.039057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Khani A.; Ye X.; Petruzziello F.; Gao H.; Zhang X.; Rainer G. Anal. Chem. 2015, 87, 11646–11651. 10.1021/ac504872z. [DOI] [PubMed] [Google Scholar]
- Clark D. F.; Go E. P.; Desaire H. Anal. Chem. 2013, 85, 1192–1199. 10.1021/ac303124w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombouts I.; Lagrain B.; Scherf K. A.; Lambrecht M. A.; Koehler P.; Delcour J. A. Sci. Rep. 2015, 5, 12210. 10.1038/srep12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisiol S.; Ayoub D.; Lesur A.; Ancheva L.; Gallien S.; Domon B. Proteomics 2016, 16, 715–728. 10.1002/pmic.201500379. [DOI] [PubMed] [Google Scholar]
- Tsiatsiani L.; Heck A. J. R. FEBS J. 2015, 282, 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- Swaney D. L.; Wenger C. D.; Coon J. J. J. Proteome Res. 2010, 9, 1323–1329. 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardiello D.; Palermo C.; Natale A.; Quinto M.; Centonze D. Anal. Chim. Acta 2015, 854, 106–117. 10.1016/j.aca.2014.10.053. [DOI] [PubMed] [Google Scholar]
- Somasundaram P.; Koudelka T.; Linke D.; Tholey A. J. Proteome Res. 2016, 15, 1369–1378. 10.1021/acs.jproteome.6b00146. [DOI] [PubMed] [Google Scholar]
- van der Post S.; Thomsson K. A.; Hansson G. C. J. Proteome Res. 2014, 13, 6013–6023. 10.1021/pr500874f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthals A.; Clauser K. R.; Frank A. M.; Bandeira N. J. Proteome Res. 2013, 12, 2846–2857. 10.1021/pr400173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatsiani L.; Giansanti P.; Scheltema R. A.; van den Toorn H.; Overall C. M.; Altelaar A. F. M.; Heck A. J. R. J. Proteome Res. 2017, 16, 852–861. 10.1021/acs.jproteome.6b00825. [DOI] [PubMed] [Google Scholar]
- Ebhardt H. A.; Nan J.; Chaulk S. G.; Fahlman R. P.; Aebersold R. Rapid Commun. Mass Spectrom. 2014, 28, 2735–2743. 10.1002/rcm.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J. D.; Greer S. M.; Brodbelt J. S. Anal. Chem. 2017, 89, 11772–11778. 10.1021/acs.analchem.7b03396. [DOI] [PubMed] [Google Scholar]
- Wu C.; Tran J. C.; Zamdborg L.; Durbin K. R.; Li M.; Ahlf D. R.; Early B. P.; Thomas P. M.; Sweedler J. V.; Kelleher N. L. Nat. Methods 2012, 9, 822–824. 10.1038/nmeth.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristobal A.; Marino F.; Post H.; van den Toorn H. W. P.; Mohammed S.; Heck A. J. R. Anal. Chem. 2017, 89, 3318–3325. 10.1021/acs.analchem.6b03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas D. C.; Guerrero A.; Parker E. A.; Robinson R. C.; Gan J.; German J. B.; Barile D.; Lebrilla C. B. Proteomics 2015, 15, 1026–1038. 10.1002/pmic.201400310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K.; Osaki T.; Minamino N. Mol. Cell. Proteomics 2013, 12, 700–709. 10.1074/mcp.M112.017400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Petruzziello F.; Zhao N.; Zhao H.; Ye X.; Zhang X.; Rainer G. Proteomics 2017, 17, 1600419. 10.1002/pmic.201600419. [DOI] [PubMed] [Google Scholar]
- Frese C. K.; Boender A. J.; Mohammed S.; Heck A. J. R.; Adan R. A. H.; Altelaar A. F. M. Anal. Chem. 2013, 85, 4594–4604. 10.1021/ac400232y. [DOI] [PubMed] [Google Scholar]
- Hart S. R.; Kenny L. C.; Myers J. E.; Baker P. N. Int. J. Mass Spectrom. 2015, 391, 41–46. 10.1016/j.ijms.2015.08.025. [DOI] [Google Scholar]
- Gucinski A. C.; Boyne M. T. Rapid Commun. Mass Spectrom. 2014, 28, 1757–1763. 10.1002/rcm.6957. [DOI] [PubMed] [Google Scholar]
- Juba M. L.; Russo P. S.; Devine M.; Barksdale S.; Rodriguez C.; Vliet K. A.; Schnur J. M.; van Hoek M. L.; Bishop B. M. J. Proteome Res. 2015, 14, 4282–4295. 10.1021/acs.jproteome.5b00447. [DOI] [PubMed] [Google Scholar]
- Bishop B. M.; Juba M. L.; Russo P. S.; Devine M.; Barksdale S. M.; Scott S.; Settlage R.; Michalak P.; Gupta K.; Vliet K.; Schnur J. M.; van Hoek M. L. J. Proteome Res. 2017, 16, 1470–1482. 10.1021/acs.jproteome.6b00857. [DOI] [PubMed] [Google Scholar]
- Trevisan-Silva D.; Bednaski A. V.; Fischer J. S. G.; Veiga S. S.; Bandeira N.; Guthals A.; Marchini F. K.; Leprevost F. V.; Barbosa V. C.; Senff-Ribeiro A.; Carvalho P. C. Sci. Data 2017, 4, 170090. 10.1038/sdata.2017.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt D. F.; Henderson R. A.; Shabanowitz J.; Sakaguchi K.; Michel H.; Sevilir N.; Cox A. L.; Appella E.; Engelhard V. H. Science 1992, 255, 1261–1263. 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- Abelin J. G.; Trantham P. D.; Penny S. A.; Patterson A. M.; Ward S. T.; Hildebrand W. H.; Cobbold M.; Bai D. L.; Shabanowitz J.; Hunt D. F. Nat. Protoc. 2015, 10, 1308–1318. 10.1038/nprot.2015.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold M.; De La Peña H.; Norris A.; Polefrone J. M.; Qian J.; English A. M.; Cummings K. L.; Penny S.; Turner J. E.; Cottine J.; Abelin J. G.; Malaker S. A.; Zarling A. L.; Huang H.-W.; Goodyear O.; Freeman S. D.; Shabanowitz J.; Pratt G.; Craddock C.; Williams M. E.; Hunt D. F.; Engelhard V. H. Sci. Transl. Med. 2013, 5, 203ra125. 10.1126/scitranslmed.3006061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mommen G. P. M.; Marino F.; Meiring H. D.; Poelen M. C. M.; van Gaans-van den Brink J. A. M.; Mohammed S.; Heck A. J. R.; van Els C. A. C. M. Mol. Cell. Proteomics 2016, 15, 1412–1423. 10.1074/mcp.M115.055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaker S. A.; Penny S. A.; Steadman L. G.; Myers P. T.; Loke J. C.; Raghavan M.; Bai D. L.; Shabanowitz J.; Hunt D. F.; Cobbold M. Cancer Immunol. Res. 2017, 5, 376–384. 10.1158/2326-6066.CIR-16-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino F.; Bern M.; Mommen G. P. M.; Leney A. C.; van Gaans-van den Brink J. A. M.; Bonvin A. M. J. J.; Becker C.; van Els C. A. C. M.; Heck A. J. R. J. Am. Chem. Soc. 2015, 137, 10922–10925. 10.1021/jacs.5b06586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino F.; Mommen G. P. M.; Jeko A.; Meiring H. D.; van Gaans-van den Brink J. A. M.; Scheltema R. A.; van Els C. A. C. M.; Heck A. J. R. J. Proteome Res. 2017, 16, 34–44. 10.1021/acs.jproteome.6b00528. [DOI] [PubMed] [Google Scholar]
- Liepe J.; Marino F.; Sidney J.; Jeko A.; Bunting D. E.; Sette A.; Kloetzel P. M.; Stumpf M. P. H.; Heck A. J. R.; Mishto M. Science 2016, 354, 354–358. 10.1126/science.aaf4384. [DOI] [PubMed] [Google Scholar]
- Pavlos R.; McKinnon E. J.; Ostrov D. A.; Peters B.; Buus S.; Koelle D.; Chopra A.; Schutte R.; Rive C.; Redwood A.; Restrepo S.; Bracey A.; Kaever T.; Myers P.; Speers E.; Malaker S. A.; Shabanowitz J.; Jing Y.; Gaudieri S.; Hunt D. F.; Carrington M.; Haas D. W.; Mallal S.; Phillips E. J. Sci. Rep. 2017, 7, 8653. 10.1038/s41598-017-08876-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaker S. A.; Ferracane M. J.; Depontieu F. R.; Zarling A. L.; Shabanowitz J.; Bai D. L.; Topalian S. L.; Engelhard V. H.; Hunt D. F. J. Proteome Res. 2017, 16, 228–237. 10.1021/acs.jproteome.6b00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradian A.; Kalli A.; Sweredoski M. J.; Hess S. Proteomics 2014, 14, 489–497. 10.1002/pmic.201300256. [DOI] [PubMed] [Google Scholar]
- Önder Ö.; Sidoli S.; Carroll M.; Garcia B. A. Expert Rev. Proteomics 2015, 12, 499–517. 10.1586/14789450.2015.1084231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S.; Garcia B. A. Middle-down proteomics: a still unexploited resource for chromatin biology. Expert Rev. Proteomics 2017, 14, 617–626. 10.1080/14789450.2017.1345632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benevento M.; Tonge P. D.; Puri M. C.; Nagy A.; Heck A. J. R.; Munoz J. Proteomics 2015, 15, 3219–3231. 10.1002/pmic.201500031. [DOI] [PubMed] [Google Scholar]
- Wang W.-L.; Anderson L. C.; Nicklay J. J.; Chen H.; Gamble M. J.; Shabanowitz J.; Hunt D. F.; Shechter D. Epigenet. Chromatin 2014, 7, 22. 10.1186/1756-8935-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch K. R.; Langelier M.-F.; Pascal J. M.; Garcia B. A. Mol. BioSyst. 2017, 13, 2660. 10.1039/C7MB00498B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszowy P.; Donnelly M. R.; Lee C.; Ciborowski P. Proteome Sci. 2015, 13, 24. 10.1186/s12953-015-0080-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schräder C. U.; Lee L.; Rey M.; Sarpe V.; Man P.; Sharma S.; Zabrouskov V.; Larsen B.; Schriemer D. C. Mol. Cell. Proteomics 2017, 16, 1162–1171. 10.1074/mcp.M116.066803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S.; Schwämmle V.; Ruminowicz C.; Hansen T. A.; Wu X.; Helin K.; Jensen O. N. Proteomics 2014, 14, 2200–2211. 10.1002/pmic.201400084. [DOI] [PubMed] [Google Scholar]
- Tian Z.; Tolić N.; Zhao R.; Moore R. J.; Hengel S. M.; Robinson E. W.; Stenoien D. L.; Wu S.; Smith R. D.; Paša-Tolić L. Genome Biol. 2012, 13, r86. 10.1186/gb-2012-13-10-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faserl K.; Sarg B.; Maurer V.; Lindner H. H. J. Chromatogr. A 2017, 1498, 215–223. 10.1016/j.chroma.2017.01.086. [DOI] [PubMed] [Google Scholar]
- Shliaha P. V.; Baird M. A.; Nielsen M. M.; Gorshkov V.; Bowman A. P.; Kaszycki J. L.; Jensen O. N.; Shvartsburg A. A. Anal. Chem. 2017, 89, 5461–5466. 10.1021/acs.analchem.7b00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweredoski M. J.; Pekar Second T.; Broeker J.; Moradian A.; Hess S. Int. J. Mass Spectrom. 2015, 390, 155–162. 10.1016/j.ijms.2015.06.018. [DOI] [Google Scholar]
- Sidoli S.; Lin S.; Karch K. R.; Garcia B. A. Anal. Chem. 2015, 87, 3129–3133. 10.1021/acs.analchem.5b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S.; Lu C.; Coradin M.; Wang X.; Karch K. R.; Ruminowicz C.; Garcia B. A. Epigenet. Chromatin 2017, 10, 34. 10.1186/s13072-017-0139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweredoski M. J.; Moradian A.; Raedle M.; Franco C.; Hess S. Anal. Chem. 2015, 87, 8360–8366. 10.1021/acs.analchem.5b01542. [DOI] [PubMed] [Google Scholar]
- Dang X.; Scotcher J.; Wu S.; Chu R. K.; Tolić N.; Ntai I.; Thomas P. M.; Fellers R. T.; Early B. P.; Zheng Y.; Durbin K. R.; LeDuc R. D.; Wolff J. J.; Thompson C. J.; Pan J.; Han J.; Shaw J. B.; Salisbury J. P.; Easterling M.; Borchers C. H.; Brodbelt J. S.; Agar J. N.; Paša-Tolić L.; Kelleher N. L.; Young N. L. Proteomics 2014, 14, 1130–1140. 10.1002/pmic.201300438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang X.; Singh A.; Spetman B. D.; Nolan K. D.; Isaacs J. S.; Dennis J. H.; Dalton S.; Marshall A. G.; Young N. L. J. Proteome Res. 2016, 15, 3196–3203. 10.1021/acs.jproteome.6b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L. C.; Karch K. R.; Ugrin S. A.; Coradin M.; English A. M.; Sidoli S.; Shabanowitz J.; Garcia B. A.; Hunt D. F. Mol. Cell. Proteomics 2016, 15, 975–988. 10.1074/mcp.O115.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Fornelli L.; Compton P. D.; Sharma S.; Canterbury J.; Mullen C.; Zabrouskov V.; Fellers R. T.; Thomas P. M.; Licht J. D.; Senko M. W.; Kelleher N. L. Mol. Cell. Proteomics 2016, 15, 776–790. 10.1074/mcp.M115.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molden R. C.; Bhanu N. V.; LeRoy G.; Arnaudo A. M.; Garcia B. A. Epigenet. Chromatin 2015, 8, 15. 10.1186/s13072-015-0006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Gross M. L. FEBS Lett. 2014, 588, 308–317. 10.1016/j.febslet.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthals A.; Gan Y.; Murray L.; Chen Y.; Stinson J.; Nakamura G.; Lill J. R.; Sandoval W.; Bandeira N. J. Proteome Res. 2017, 16, 45–54. 10.1021/acs.jproteome.6b00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y.; Wang W.-H.; Reid G. E.; Hunt D. F.; Bruening M. L. Anal. Chem. 2015, 87, 10942–10949. 10.1021/acs.analchem.5b02739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; English A. M.; Bai D. L.; Ugrin S. A.; Shabanowitz J.; Ross M. M.; Hunt D. F.; Wang W.-H. Mol. Cell. Proteomics 2016, 15, 1479–1488. 10.1074/mcp.O115.056721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Chou A.; Borchers C. H. Chem. Sci. 2016, 7, 1480–1486. 10.1039/C5SC03420E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srzentić K.; Fornelli L.; Laskay Ü. A.; Monod M.; Beck A.; Ayoub D.; Tsybin Y. O. Anal. Chem. 2014, 86, 9945–9953. 10.1021/ac502766n. [DOI] [PubMed] [Google Scholar]
- Fornelli L.; Ayoub D.; Aizikov K.; Beck A.; Tsybin Y. O. Anal. Chem. 2014, 86, 3005–3012. 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- Tran B. Q.; Barton C.; Feng J.; Sandjong A.; Yoon S. H.; Awasthi S.; Liang T.; Khan M. M.; Kilgour D. P. A.; Goodlett D. R.; Goo Y. A. J. Proteomics 2016, 134, 93–101. 10.1016/j.jprot.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Jensen P. F.; Larraillet V.; Schlothauer T.; Kettenberger H.; Hilger M.; Rand K. D. Mol. Cell. Proteomics 2015, 14, 148–161. 10.1074/mcp.M114.042044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Anderson L. C.; Barnidge D. R.; Murray D. L.; Hendrickson C. L.; Marshall A. G. J. Am. Soc. Mass Spectrom. 2017, 28, 827–838. 10.1007/s13361-017-1602-6. [DOI] [PubMed] [Google Scholar]
- Tsybin Y. O.; Fornelli L.; Stoermer C.; Luebeck M.; Parra J.; Nallet S.; Wurm F. M.; Hartmer R. Anal. Chem. 2011, 83, 8919–8927. 10.1021/ac201293m. [DOI] [PubMed] [Google Scholar]
- Fornelli L.; Damoc E.; Thomas P. M.; Kelleher N. L.; Aizikov K.; Denisov E.; Makarov A.; Tsybin Y. O. Mol. Cell. Proteomics 2012, 11, 1758–1767. 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Chou A.; Hardie D. B.; Borchers C. H. Anal. Chem. 2015, 87, 5884–5890. 10.1021/ac504809r. [DOI] [PubMed] [Google Scholar]
- Fornelli L.; Ayoub D.; Aizikov K.; Liu X.; Damoc E.; Pevzner P. A.; Makarov A.; Beck A.; Tsybin Y. O. J. Proteomics 2017, 159, 67–76. 10.1016/j.jprot.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lössl P.; van de Waterbeemd M.; Heck A. J. EMBO J. 2016, 35, 2634–2657. 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Rijkers D. T. S.; Post H.; Heck A. J. R. Nat. Methods 2015, 12, 1179–1184. 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Liu F.; Lössl P.; Scheltema R.; Viner R.; Heck A. J. R. Nat. Commun. 2017, 8, 15473. 10.1038/ncomms15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese S. H.; Belsom A.; Rappsilber J. Anal. Chem. 2016, 88, 8239–8247. 10.1021/acs.analchem.6b02082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbowski L.; Mendes M. L.; Rappsilber J. Anal. Chem. 2017, 89, 5311–5318. 10.1021/acs.analchem.6b04935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn C. H.; Agnew H. D.; Lee J. E.; Sweredoski M. J.; Graham R. L. J.; Smith G. T.; Hess S.; Czerwieniec G.; Loo J. A.; Heath J. R.; Deshaies R. J.; Beauchamp J. L. Anal. Chem. 2012, 84, 2662–2669. 10.1021/ac202637n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen H. H. F.; Gomes A. F.; Schwab N. V.; Eberlin M. N.; Gozzo F. C. J. Am. Soc. Mass Spectrom. 2014, 25, 1181–1191. 10.1007/s13361-014-0900-5. [DOI] [PubMed] [Google Scholar]
- Lermyte F.; Sobott F. Proteomics 2015, 15, 2813–2822. 10.1002/pmic.201400516. [DOI] [PubMed] [Google Scholar]
- Lermyte F.; Konijnenberg A.; Williams J. P.; Brown J. M.; Valkenborg D.; Sobott F. J. Am. Soc. Mass Spectrom. 2014, 25, 343–350. 10.1007/s13361-013-0798-3. [DOI] [PubMed] [Google Scholar]
- Lermyte F.; Łącki M. K.; Valkenborg D.; Gambin A.; Sobott F. J. Am. Soc. Mass Spectrom. 2017, 28, 69–76. 10.1007/s13361-016-1444-7. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Browne S. J.; Vachet R. W. J. Am. Soc. Mass Spectrom. 2014, 25, 604–613. 10.1007/s13361-013-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Vachet R. W. Gas-phase protein salt bridge stabilities from collisional activation and electron transfer dissociation. Int. J. Mass Spectrom. 2017, 420, 51–56. 10.1016/j.ijms.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton P. D.; Fornelli L.; Kelleher N. L.; Skinner O. S. Int. J. Mass Spectrom. 2015, 390, 132–136. 10.1016/j.ijms.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muneeruddin K.; Nazzaro M.; Kaltashov I. A. Anal. Chem. 2015, 87, 10138–10145. 10.1021/acs.analchem.5b02982. [DOI] [PubMed] [Google Scholar]
- Cassou C. A.; Sterling H. J.; Susa A. C.; Williams E. R. Anal. Chem. 2013, 85, 138–146. 10.1021/ac302256d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B.; Sharp J. S. J. Am. Soc. Mass Spectrom. 2016, 27, 1322–1327. 10.1007/s13361-016-1403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehl M.; Rand K. D.; Jensen O. N.; Jørgensen T. J. D. J. Am. Chem. Soc. 2008, 130, 17453–17459. 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
- Abzalimov R. R.; Kaplan D. A.; Easterling M. L.; Kaltashov I. A. J. Am. Soc. Mass Spectrom. 2009, 20, 1514–1517. 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf R. R.; Chalmers M. J.; Griffin P. R. J. Am. Soc. Mass Spectrom. 2012, 23, 301–309. 10.1007/s13361-011-0298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y. J. Am. Soc. Mass Spectrom. 2017, 28, 971–977. 10.1007/s13361-017-1612-4. [DOI] [PubMed] [Google Scholar]
- Masson G. R.; Maslen S. L.; Williams R. L. Biochem. J. 2017, 474, 1867–1877. 10.1042/BCJ20170127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger S. T.; Breinholt J.; Faber J. H.; Andersen M. D.; Wiberg C.; Schjødt C. B.; Rand K. D. Anal. Chem. 2015, 87, 5973–5980. 10.1021/ac504782v. [DOI] [PubMed] [Google Scholar]
- Song H.; Olsen O. H.; Persson E.; Rand K. D. J. Biol. Chem. 2014, 289, 35388–35396. 10.1074/jbc.M114.614297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob R. E.; Chen G.; Ahn J.; Houel S.; Wei H.; Mo J.; Tao L.; Cohen D.; Xie D.; Lin Z.; Morin P. E.; Doyle M. L.; Tymiak A. A.; Engen J. R. J. Am. Soc. Mass Spectrom. 2014, 25, 2093–2102. 10.1007/s13361-014-0973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.-C.; Garai K.; Frieden C.; Gross M. L. Biochemistry 2011, 50, 9273–9282. 10.1021/bi2010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Borchers C. H. J. Proteomics 2016, 134, 138–143. 10.1016/j.jprot.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Rand K. D.; Pringle S. D.; Morris M.; Engen J. R.; Brown J. M. J. Am. Soc. Mass Spectrom. 2011, 22, 1784–1793. 10.1007/s13361-011-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakinejad M.; Ghassabi Kondalaji S.; Tafreshian A.; Valentine S. J. J. Am. Soc. Mass Spectrom. 2017, 28, 960–970. 10.1007/s13361-017-1641-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistarz U. H.; Brown J. M.; Haselmann K. F.; Rand K. D. Anal. Chem. 2014, 86, 11868–11876. 10.1021/ac5035456. [DOI] [PubMed] [Google Scholar]
- Xie B.; Sood A.; Woods R. J.; Sharp J. S. Sci. Rep. 2017, 7, 4552. 10.1038/s41598-017-04689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasicek L.; O’Brien J. P.; Browning K. S.; Tao Z.; Liu H.-W.; Brodbelt J. S. Mol. Cell. Proteomics 2012, 11, O111.015826. 10.1074/mcp.O111.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budelier M. M.; Cheng W. W. L.; Bergdoll L.; Chen Z.-W.; Abramson J.; Krishnan K.; Qian M.; Covey D. F.; Janetka J. W.; Evers A. S. Anal. Chem. 2017, 89, 2636–2644. 10.1021/acs.analchem.6b05003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice B. M.; McLuckey S. A. Chem. Commun. 2013, 49, 947–965. 10.1039/C2CC36577D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J. D.; Fisher C. M.; Bu J.; Prentice B. M.; Redwine J. G.; McLuckey S. A. J. Mass Spectrom. 2015, 50, 418–426. 10.1002/jms.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado P. P.; O’Connor P. B. Mass Spectrom. Rev. 2012, 31, 609–625. 10.1002/mas.20357. [DOI] [PubMed] [Google Scholar]
- Lebedev A. T.; Damoc E.; Makarov A. A.; Samgina T. Y. Anal. Chem. 2014, 86, 7017–7022. 10.1021/ac501200h. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Vecchi M. M.; Wen D. Anal. Chem. 2016, 88, 10757–10766. 10.1021/acs.analchem.6b03409. [DOI] [PubMed] [Google Scholar]
- Zhokhov S. S.; Kovalyov S. V.; Samgina T. Y.; Lebedev A. T. J. Am. Soc. Mass Spectrom. 2017, 28, 1600–1611. 10.1007/s13361-017-1674-3. [DOI] [PubMed] [Google Scholar]
- Kovalyov S. V.; Zhokhov S. S.; Onoprienko L. V.; Vaskovsky B. V.; Lebedev A. T. Eur. J. Mass Spectrom. 2017, 23, 376–384. 10.1177/1469066717730705. [DOI] [PubMed] [Google Scholar]
- Lyon Y. A.; Beran G.; Julian R. R. J. Am. Soc. Mass Spectrom. 2017, 28, 1365–1373. 10.1007/s13361-017-1627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledvina A. R.; Coon J. J.; Tureček F. Int. J. Mass Spectrom. 2015, 377, 44–53. 10.1016/j.ijms.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. T. H.; Shaffer C. J.; Ledvina A. R.; Coon J. J.; Tureček F. Int. J. Mass Spectrom. 2015, 378, 20–30. 10.1016/j.ijms.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. T. H.; Shaffer C. J.; Tureček F. J. Phys. Chem. B 2015, 119, 3948–3961. 10.1021/jp511717c. [DOI] [PubMed] [Google Scholar]
- Nguyen H. T. H.; Tureček F. J. Am. Soc. Mass Spectrom. 2017, 28, 1333–1344. 10.1007/s13361-016-1586-7. [DOI] [PubMed] [Google Scholar]
- Shaffer C. J.; Marek A.; Pepin R.; Slovakova K.; Turecek F. J. Mass Spectrom. 2015, 50, 470–475. 10.1002/jms.3551. [DOI] [PubMed] [Google Scholar]
- Shaffer C. J.; Pepin R.; Tureček F. J. Mass Spectrom. 2015, 50, 1438–1442. 10.1002/jms.3717. [DOI] [PubMed] [Google Scholar]
- Pepin R.; Layton E. D.; Liu Y.; Afonso C.; Tureček F. J. Am. Soc. Mass Spectrom. 2017, 28, 164–181. 10.1007/s13361-016-1512-z. [DOI] [PubMed] [Google Scholar]
- McLuckey S. A.; Stephenson J. L. Mass Spectrom. Rev. 1998, 17, 369–407. . [DOI] [PubMed] [Google Scholar]
- Reid G. E.; McLuckey S. A. J. Mass Spectrom. 2002, 37, 663–675. 10.1002/jms.346. [DOI] [PubMed] [Google Scholar]
- McLuckey S. A.; Huang T.-Y. Anal. Chem. 2009, 81, 8669–8676. 10.1021/ac9014935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabik A.; Bodzon-Kulakowska A.; Suder P. J. Mass Spectrom. 2012, 47, 1347–1352. 10.1002/jms.3086. [DOI] [PubMed] [Google Scholar]
- Anderson L. C.; English A. M.; Wang W.-H.; Bai D. L.; Shabanowitz J.; Hunt D. F. Int. J. Mass Spectrom. 2015, 377, 617–624. 10.1016/j.ijms.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisman P. A.; Pitteri S. J.; McLuckey S. A. Anal. Chem. 2006, 78, 310–316. 10.1021/ac0515778. [DOI] [PubMed] [Google Scholar]
- Holden D. D.; McGee W. M.; Brodbelt J. S. Anal. Chem. 2016, 88, 1008–1016. 10.1021/acs.analchem.5b03911. [DOI] [PubMed] [Google Scholar]
- Holden D. D.; Brodbelt J. S. Anal. Chem. 2016, 88, 12354–12362. 10.1021/acs.analchem.6b03565. [DOI] [PubMed] [Google Scholar]
- Wenger C. D.; Lee M. V.; Hebert A. S.; McAlister G. C.; Phanstiel D. H.; Westphall M. S.; Coon J. J. Nat. Methods 2011, 8, 933–935. 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent C. E.; Rensvold J. W.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Anal. Chem. 2013, 85, 2079–2086. 10.1021/ac302156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo K. J.; Bush M. F. J. Am. Soc. Mass Spectrom. 2015, 26, 2152–2161. 10.1007/s13361-015-1245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo K. J.; Munger E. B.; Bush M. F. J. Am. Chem. Soc. 2016, 138, 9581–9588. 10.1021/jacs.6b04282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo K. J.; Bush M. F. Anal. Chem. 2017, 89, 7607–7614. 10.1021/acs.analchem.7b01474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo K. J.; Buckner J. H.; Munger E. B.; Bush M. F. J. Am. Soc. Mass Spectrom. 2017, 28, 1382–1391. 10.1007/s13361-017-1620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhingree J. R.; Beveridge R.; Dickinson E. R.; Williams J. P.; Brown J. M.; Bellina B.; Barran P. E. Int. J. Mass Spectrom. 2017, 413, 43–51. 10.1016/j.ijms.2016.08.006. [DOI] [Google Scholar]
- Lermyte F.; Williams J. P.; Brown J. M.; Martin E. M.; Sobott F. J. Am. Soc. Mass Spectrom. 2015, 26, 1068–1076. 10.1007/s13361-015-1124-z. [DOI] [PubMed] [Google Scholar]
- Pilo A. L.; Zhao F.; McLuckey S. A. J. Am. Soc. Mass Spectrom. 2017, 28, 991–1004. 10.1007/s13361-016-1554-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J. D.; Prentice B. M.; McLuckey S. A. J. Am. Soc. Mass Spectrom. 2015, 26, 818–825. 10.1007/s13361-015-1077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu J.; Pilo A. L.; McLuckey S. A. Int. J. Mass Spectrom. 2015, 390, 118–123. 10.1016/j.ijms.2015.05.010. [DOI] [Google Scholar]
- Pilo A. L.; Peng Z.; McLuckey S. A. J. Mass Spectrom. 2016, 51, 857–866. 10.1002/jms.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z.; Bu J.; McLuckey S. A. J. Am. Soc. Mass Spectrom. 2017, 28, 1765–1774. 10.1007/s13361-017-1672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilo A. L.; Zhao F.; McLuckey S. A. J. Proteome Res. 2016, 15, 3139–3146. 10.1021/acs.jproteome.6b00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilo A. L.; McLuckey S. A. Anal. Chem. 2016, 88, 8972–8979. 10.1021/acs.analchem.6b01043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee W. M.; McLuckey S. A. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 1288–1292. 10.1073/pnas.1317914111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z.; Mcluckey S. A. Int. J. Mass Spectrom. 2015, 391, 17–23. 10.1016/j.ijms.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotham V. C.; McGee W. M.; Brodbelt J. S. Anal. Chem. 2016, 88, 8158–8165. 10.1021/acs.analchem.6b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotham V. C.; Shaw J. B.; Brodbelt J. S. Anal. Chem. 2015, 87, 9396–9402. 10.1021/acs.analchem.5b02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon J. J.; Shabanowitz J.; Hunt D. F.; Syka J. E. P. J. Am. Soc. Mass Spectrom. 2005, 16, 880–882. 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Commodore J. J.; Cassady C. J. J. Am. Soc. Mass Spectrom. 2016, 27, 1499–1509. 10.1007/s13361-016-1428-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commodore J. J.; Cassady C. J. J. Mass Spectrom. 2017, 52, 218–229. 10.1002/jms.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer C. E.; Stover M. L.; Bokatzian S. S.; Davis J. T. M.; Dixon D. A.; Cassady C. J. J. Phys. Chem. B 2015, 119, 9661–9669. 10.1021/acs.jpcb.5b04486. [DOI] [PubMed] [Google Scholar]
- McMillen C. L.; Wright P. M.; Cassady C. J. J. Am. Soc. Mass Spectrom. 2016, 27, 847–855. 10.1007/s13361-016-1345-9. [DOI] [PubMed] [Google Scholar]
- McAlister G. C.; Russell J. D.; Rumachik N. G.; Hebert A. S.; Syka J. E. P.; Geer L. Y.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Anal. Chem. 2012, 84, 2875–2882. 10.1021/ac203430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumachik N. G.; McAlister G. C.; Russell J. D.; Bailey D. J.; Wenger C. D.; Coon J. J. J. Am. Soc. Mass Spectrom. 2012, 23, 718–727. 10.1007/s13361-011-0331-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Ledvina A. R.; Zhang X.; Julian R. R.; Brodbelt J. S. J. Am. Chem. Soc. 2012, 134, 15624–15627. 10.1021/ja3032086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Madsen J. A.; Xu H.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2012, 23, 1707–1715. 10.1007/s13361-012-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Kaplan D. A.; Brodbelt J. S. Anal. Chem. 2013, 85, 4721–4728. 10.1021/ac4005315. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Rush M. J. P.; Rose C. M.; Richards A. L.; Kwiecien N. W.; Bailey D. J.; Hebert A. S.; Westphall M. S.; Coon J. J. Mol. Cell. Proteomics 2015, 14, 2644–2660. 10.1074/mcp.M115.049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Bern M.; Westphall M. S.; Coon J. J. J. Proteome Res. 2016, 15, 2768–2776. 10.1021/acs.jproteome.6b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huzarska M.; Ugalde I.; Kaplan D. A.; Hartmer R.; Easterling M. L.; Polfer N. C. Anal. Chem. 2010, 82, 2873–2878. 10.1021/ac9028592. [DOI] [PubMed] [Google Scholar]
- Rush M. J. P.; Riley N. M.; Westphall M. S.; Syka J. E. P.; Coon J. J. J. Am. Soc. Mass Spectrom. 2017, 28, 1324–1332. 10.1007/s13361-017-1600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. S.; Wang C. C.; Chen B. H.; Hou Y. H.; Hung K. S.; Mao Y. C. Molecules 2015, 20, 2138–2164. 10.3390/molecules20022138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhs S. R.; Hunter T. pHisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Curr. Opin. Cell Biol. 2017, 45, 8–16. 10.1016/j.ceb.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; McLuckey S. A. Rapid Commun. Mass Spectrom. 2013, 27, 249–257. 10.1002/rcm.6428. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Yang J.; Cancilla M. T.; Meng F.; McLuckey S. A. Anal. Chem. 2013, 85, 4713–4720. 10.1021/ac400448t. [DOI] [PubMed] [Google Scholar]
- Gao Y.; McLuckey S. A. Fragmentation Reactions of Nucleic Acid Ions in the Gas Phase. In Nucleic Acids in the Gas Phase. Physical Chemistry in Action; Gabelica V., Ed.; Springer: Berlin, Heidelberg, Germany, 2014; pp 131–182, DOI: 10.1007/978-3-642-54842-0_6. [DOI] [Google Scholar]
- Wolff J. J.; Leach F. E.; Laremore T. N.; Kaplan D. A.; Easterling M. L.; Linhardt R. J.; Amster I. J.; Amster I. J. Anal. Chem. 2010, 82, 3460–3466. 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach F. E.; Wolff J. J.; Xiao Z.; Ly M.; Laremore T. N.; Arungundram S.; Al-Mafraji K.; Venot A.; Boons G.-J.; Linhardt R. J.; Amster I. J.; Amster I. J. Eur. Mass Spectrom. 2011, 17, 167–176. 10.1255/ejms.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Yu X.; Mao Y.; Costello C. E.; Zaia J.; Lin C. Anal. Chem. 2013, 85, 11979–11986. 10.1021/ac402931j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Huang Y.; Mao Y.; Yu X.; Xu Y.; Liu J.; Zong C.; Boons G.-J.; Lin C.; Xia Y.; Zaia J. Mol. Cell. Proteomics 2014, 13, 2490–2502. 10.1074/mcp.M114.039560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach F. E.; Riley N. M.; Westphall M. S.; Coon J. J.; Amster I. J. J. Am. Soc. Mass Spectrom. 2017, 28, 1844–1854. 10.1007/s13361-017-1709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. E.; Coon J. J. Curr. Opin. Chem. Biol. 2013, 17, 779–786. 10.1016/j.cbpa.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennrich M. L.; Marino F.; Groenewold V.; Kops G. J. P. L.; Mohammed S.; Heck A. J. R. J. Proteome Res. 2013, 12, 2214–2224. 10.1021/pr400074f. [DOI] [PubMed] [Google Scholar]
- Munoz J.; Low T. Y.; Kok Y. J.; Chin A.; Frese C. K.; Ding V.; Choo A.; Heck A. J. R. Mol. Syst. Biol. 2011, 7, 550. 10.1038/msb.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholten A.; Mohammed S.; Low T. Y.; Zanivan S.; van Veen T. A. B.; Delanghe B.; Heck A. J. R. Mol. Cell. Proteomics 2011, 10, O111.008474. 10.1074/mcp.O111.008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A. L.; Vincent C. E.; Guthals A.; Rose C. M.; Westphall M. S.; Bandeira N.; Coon J. J. Mol. Cell. Proteomics 2013, 12, 3812–3823. 10.1074/mcp.M113.028951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads T. W.; Rose C. M.; Bailey D. J.; Riley N. M.; Molden R. C.; Nestler A. J.; Merrill A. E.; Smith L. M.; Hebert A. S.; Westphall M. S.; Pagliarini D. J.; Garcia B. A.; Coon J. J. Anal. Chem. 2014, 86, 2314–2319. 10.1021/ac403579s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H.; Pappin D. J.; Ross P. L.; McLuckey S. A. J. Proteome Res. 2008, 7, 3643–3648. 10.1021/pr8001113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel D.; Unwin R.; McAlister G. C.; Coon J. J. Anal. Chem. 2009, 81, 1693–1698. 10.1021/ac8019202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Huttlin E. L.; Haas W.; Ting L.; Jedrychowski M. P.; Rogers J. C.; Kuhn K.; Pike I.; Grothe R. A.; Blethrow J. D.; Gygi S. P. Anal. Chem. 2012, 84, 7469–7478. 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Merrill A. E.; Bailey D. J.; Still A. J.; Westphall M. S.; Strieter E. R.; Pagliarini D. J.; Coon J. J. Nat. Methods 2013, 10, 332–334. 10.1038/nmeth.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q.; Shi X.; Feng Y.; Kent K. C.; Li L. Anal. Chim. Acta 2017, 968, 40–49. 10.1016/j.aca.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley R. J.; Medzihradszky K. F.; Lynn A. J.; Baker P. R.; Burlingame A. L. Anal. Chem. 2010, 82, 579–584. 10.1021/ac9018582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Song C.; Bailey D. J.; Tseng G. C.; Coon J. J.; Wysocki V. H. Anal. Chem. 2011, 83, 9540–9545. 10.1021/ac202327r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askenazi M.; Bandeira N.; Chalkley R. J.; Deutsch E.; Lam H. H. N.; McDonald W. H.; Neubert T.; Rudnick P. A.; Martens L. J. Biomol. Technol. 2011, 22, S20. [Google Scholar]
- Tyanova S.; Temu T.; Cox J. Nat. Protoc. 2016, 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Röst H. L.; Sachsenberg T.; Aiche S.; Bielow C.; Weisser H.; Aicheler F.; Andreotti S.; Ehrlich H.-C.; Gutenbrunner P.; Kenar E.; Liang X.; Nahnsen S.; Nilse L.; Pfeuffer J.; Rosenberger G.; Rurik M.; Schmitt U.; Veit J.; Walzer M.; Wojnar D.; Wolski W. E.; Schilling O.; Choudhary J. S.; Malmström L.; Aebersold R.; Reinert K.; Kohlbacher O. Nat. Methods 2016, 13, 741–748. 10.1038/nmeth.3959. [DOI] [PubMed] [Google Scholar]
- Hunt D. F.; Shabanowitz J.; Bai D. L. J. Am. Soc. Mass Spectrom. 2015, 26, 1256–1258. 10.1007/s13361-015-1105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolg D. P.; Wilhelm M.; Schnatbaum K.; Zerweck J.; Knaute T.; Delanghe B.; Bailey D. J.; Gessulat S.; Ehrlich H.-C.; Weininger M.; Yu P.; Schlegl J.; Kramer K.; Schmidt T.; Kusebauch U.; Deutsch E. W.; Aebersold R.; Moritz R. L.; Wenschuh H.; Moehring T.; Aiche S.; Huhmer A.; Reimer U.; Kuster B. Nat. Methods 2017, 14, 259–262. 10.1038/nmeth.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good D. M.; Wenger C. D.; McAlister G. C.; Bai D. L.; Hunt D. F.; Coon J. J. J. Am. Soc. Mass Spectrom. 2009, 20, 1435–1440. 10.1016/j.jasms.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good D. M.; Wenger C. D.; Coon J. J. Proteomics 2010, 10, 164–167. 10.1002/pmic.200900570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhara V.; Bai D. L.; Chi A.; Shabanowitz J.; Hunt D. F.; Bryant S. H.; Geer L. Y. Proteome Sci. 2012, 10, 8. 10.1186/1477-5956-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q.; Lee M. V.; Rose C. M.; Marsh A. J.; Hubler S. L.; Wenger C. D.; Coon J. J. J. Am. Soc. Mass Spectrom. 2011, 22, 255–264. 10.1007/s13361-010-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H.; Chen H.; He K.; Wu L.; Yang B.; Sun R.-X.; Liu J.; Zeng W.-F.; Song C.-Q.; He S.-M.; Dong M.-Q. J. Proteome Res. 2013, 12, 615–625. 10.1021/pr3006843. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Kusalik A. J.; Wu F.-X. IEEE Trans. Nanobioscience 2015, 14, 478–484. 10.1109/TNB.2015.2419194. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Zhang K. BMC Bioinf. 2016, 17, 538. 10.1186/s12859-016-1330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Kusalik A. J.; Wu F.-X. IEEE/ACM Trans. Comput. Biol. Bioinf. 2017, 14, 337–344. 10.1109/TCBB.2015.2389813. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Su X.; Go E. P.; Desaire H. Anal. Chem. 2014, 86, 9212–9219. 10.1021/ac502176n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayampurath A.; Yu C.-Y.; Song E.; Balan J.; Mechref Y.; Tang H. Anal. Chem. 2014, 86, 453–463. 10.1021/ac402338u. [DOI] [PubMed] [Google Scholar]
- Lee L. Y.; Moh E. S. X.; Parker B. L.; Bern M.; Packer N. H.; Thaysen-Andersen M. J. Proteome Res. 2016, 15, 3904–3915. 10.1021/acs.jproteome.6b00438. [DOI] [PubMed] [Google Scholar]
- Darula Z.; Chalkley R. J.; Lynn A.; Baker P. R.; Medzihradszky K. F. Amino Acids 2011, 41, 321–328. 10.1007/s00726-010-0692-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Su X.; Clark D. F.; Go E. P.; Desaire H. Anal. Chem. 2013, 85, 8403–8411. 10.1021/ac401814h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley R. J.; Baker P. R. Anal. Bioanal. Chem. 2017, 409, 571–577. 10.1007/s00216-016-9981-2. [DOI] [PubMed] [Google Scholar]
- Fellers R. T.; Greer J. B.; Early B. P.; Yu X.; LeDuc R. D.; Kelleher N. L.; Thomas P. M. Proteomics 2015, 15, 1235–1238. 10.1002/pmic.201400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W.; Guner H.; Gregorich Z. R.; Chen A. J.; Ayaz-Guner S.; Peng Y.; Valeja S. G.; Liu X.; Ge Y. Mol. Cell. Proteomics 2016, 15, 703–714. 10.1074/mcp.O115.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Paša-Tolić L.; Stenoien D. L. J. Proteome Res. 2017, 16, 599–608. 10.1021/acs.jproteome.6b00694. [DOI] [PubMed] [Google Scholar]
- Xiao K.; Yu F.; Tian Z. J. Proteomics 2017, 152, 41–47. 10.1016/j.jprot.2016.10.010. [DOI] [PubMed] [Google Scholar]
- Li L.; Tian Z. Rapid Commun. Mass Spectrom. 2013, 27, 1267–1277. 10.1002/rcm.6565. [DOI] [PubMed] [Google Scholar]
- LeDuc R. D.; Fellers R. T.; Early B. P.; Greer J. B.; Thomas P. M.; Kelleher N. L. J. Proteome Res. 2014, 13, 3231–3240. 10.1021/pr401277r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lermyte F.; Łacki M. K.; Valkenborg D.; Baggerman G.; Gambin A.; Sobott F. Int. J. Mass Spectrom. 2015, 390, 146–154. 10.1016/j.ijms.2015.08.008. [DOI] [Google Scholar]
- Ciach M. A.; Łącki M. K.; Miasojedow B.; Lermyte F.; Valkenborg D.; Sobott F.; Gambin A. J. Comput. Biol. 2017, 96–107. 10.1089/cmb.2017.0156. [DOI] [PubMed] [Google Scholar]
- Liu X.; Dekker L. J. M.; Wu S.; Vanduijn M. M.; Luider T. M.; Tolić N.; Kou Q.; Dvorkin M.; Alexandrova S.; Vyatkina K.; Paša-Tolić L.; Pevzner P. A. J. Proteome Res. 2014, 13, 3241–3248. 10.1021/pr401300m. [DOI] [PubMed] [Google Scholar]
- Vyatkina K.; Wu S.; Dekker L. J. M.; VanDuijn M. M.; Liu X.; Tolić N.; Dvorkin M.; Alexandrova S.; Luider T. M.; Paša-Tolić L.; Pevzner P. A. J. Proteome Res. 2015, 14, 4450–4462. 10.1021/pr501244v. [DOI] [PubMed] [Google Scholar]