

Abstract
A big challenge in the clinical use of Cerenkov luminescence (CL) imaging is its low signal intensity, which is several orders of magnitude below ambient light. Consequently, highly sensitive cameras, sufficient shielding from background light, and long acquisition times are required. To alleviate this problem, we hypothesized a strategy to convert the weak CL signal into a stronger fluorescence signal by using CL-activated formation of nitrenes from azides to locally fix a fluorescent probe in tissue by the formation of a covalent bond. CL-activated drug delivery was also evaluated using the same azide chemistry. The specific delivery of the CL-activated drug to cancer cells could reduce systemic toxicity, which is a limitation in chemotherapy. Methods: A cyanine-class near-infrared fluorescent dye, Cy7, and doxorubicin were synthetically attached to polyfluorinated aryl azide to form Cy7 azide and DOX azide, respectively. Fibrosarcoma cells were incubated with 18F-FDG and exposed to Cy7 azide with subsequent fluorescence imaging. For CL-activated tagging in vivo, tumor-bearing mice were injected first with 90Y-DOTA-RGD, targeting αvβ3 integrins, and then with the Cy7 azide. Fluorescence signal was imaged over time. Breast cancer cells were incubated with DOX azide and 68Ga, after which cell viability was quantified using an assay. Results: CL photoactivation of Cy7 azide in vitro showed significantly higher fluorescence signal from 18F-FDG–treated than untreated cells. In vivo, CL photoactivation could be shown by using the tumor-specific, integrin-targeting 90Y-DOTA-RGD and the localized activation of Cy7 azide. Here, localized CL-induced fluorescence was detected in the tumors and remained significantly higher over several days than in tumors without CL. We also established as a next step CL-activated drug delivery of DOX azide by showing significantly decreasing cell viability of breast cancer cells in a CL dose–dependent manner in vitro using CL photoactivation of DOX azide. Conclusion: We were able to develop a CL-activated “sticky tag” that converts the low CL signal into a stable and long-lasting, highly intense fluorescence signal. This fluorescent footprint of the radioactive signal might be clinically used for intraoperative surgery. The CL-targeted drug delivery strategy may potentially be used for dual-step targeted therapy.
Keywords: Cerenkov luminescence, photoactivatable, azide, fluorescence, sticky tag, near-infrared optical imaging
Cerenkov luminescence (CL) has emerged over the last few years as a robust optical modality in preclinical and clinical applications (1–6). When a charged subatomic particle passes through a medium at speeds higher than the speed of light in that medium, the particle polarizes nearby molecules. CL arises on subsequent relaxation of these molecules from the polarized state. Most isotopes that are already being used for nuclear imaging and radiotherapy generate CL (7). The CL from positron- or electron-emitting radioisotopes can be used as an intrinsic source of light and suggests photoactivation of agents, overcoming the limited penetration and lack of accessibility of an external light source (8). However, the major limitation of CL is the low signal intensity and, depending on the isotope used, the ephemerality of the signal. The low signal intensity requires longer scanning times, which inherently could lower image quality because of subject movement during the imaging process. Also, for quickly decaying isotopes, the time restrictions for imaging are more constrained, especially if other imaging modalities, such as PET or SPECT, are being used concurrently, requiring time for their acquisition.
Radioisotopes are of course also used for targeted radiation therapy, which is often the first line of defense in the treatment of patients with thyroid cancer, metastatic prostate cancer, and lymphoma (9). Here, 90Y is one of the preferred therapeutic isotopes because it is a pure, high-energy β-emitter (maximum energy, 2.3 MeV) without γ-radiation and enables a localized cell death (10). RGD-based small molecules labeled with 90Y have been used for internal targeted radiotherapy, showing delayed growth of tumors (11,12). In addition, a combination of targeted internal radiation therapy with chemotherapy has been shown to be superior in progression-free survival and overall survival to radiotherapy or chemotherapy alone in the treatment of high-grade gliomas and a wide variety of different cancers (13–17). However, the unspecific distribution of the chemotherapeutic drugs is the major reason for the toxicity of this combination (18,19). To circumvent these side effects, this combination treatment modality needs to become more specific and selective to the tumor site.
To solve the abovementioned limitations in imaging and therapy, we hypothesized a mechanism by which CL could trigger the formation of a “sticky tag” to localize a CL-targeted fluorophore or a cytotoxic drug, as shown in Figure 1. The mechanism of formation of a sticky tag was adapted from polyfluorinated aryl azide chemistry, which has been shown to generate singlet nitrene species on irradiation with ultraviolet light (20). Photoaffinity ligands such as polyfluorinated aryl azides are preferable as they can be excited by the blue-weighted CL, leading to the generation of reactive singlet nitrene species, and the presence of fluorines on the aryl ring prevents intramolecular insertion (21). These nitrenes then immediately and irreversibly insert into C–H and N–H bonds of extracellular proteins or lipids by the formation of covalent bonds in an intermolecular fashion, acting like a sticky tag (22). Polyfluorinated aryl azide has been used previously for studying protein ligand binding interactions, identifying drug targets, and studying endogenous biologic molecules (21,23). However, to our knowledge they have never been applied to an in vivo or CL-activated dye or drug delivery system as in our study.
FIGURE 1.

Principle of CL-activatable azide probes for targeting of tumor tissue.
To evaluate CL-activatable fluorescence imaging, polyfluorinated aryl azide is functionalized with a cyanine-class near-infrared fluorescent dye, Cy7, to make Cy7 azide. CL-mediated localized activation and thus accumulation provide a durable fluorescence signal for standard fluorescence imaging. A Cy7 fluoride is used as a nonactivatable control dye in which the polyfluorinated aryl azides moiety of a Cy7 azide is synthetically replaced with fluorine. To induce the activation, 18F-FDG or 90Y-labeled DOTA-RGD peptide is used. Since 90Y-DOTA-RGD is targeted to αvβ3-integrins expressed on the cancer cells as well as the neovasculature (24), the CL is generated at the tumor site, enabling a selective activation of the dye-labeled azide injected after free activity has cleared. Of course, by choosing other targeting moieties for the CL generator, one could target other structures (tumor cells, vascular plaques, inflammation). Here, a multitude of clinical radiotracers could be harvested for image-guided surgery but circumventing the shortcomings of CL by translating the CL signal with a single CL-activated fluorescent sticky tag into a fluorescent signal that allows for facile intraoperative imaging.
To evaluate a CL-activatable drug delivery system, a chemotherapeutic drug, doxorubicin, is functionalized with polyfluorinated aryl azide to make DOX azide. This strategy could essentially allow for a dual-step targeting strategy, separating the delivery of radioactivity from the delivery of a CL-activatable DOX azide and potentially reducing nonspecific cytotoxicity.
MATERIALS AND METHODS
Reagents
All chemicals, unless otherwise noted, were acquired from Sigma-Aldrich. The near-infrared dye sulfo-Cy7 N-hydroxysuccinimide (NHS) ester was purchased from Lumiprobe. Boc-1-amino-3,6-dioxa-8-octanediamine was purchased from Chem-Impex International. DOTA-Glu-[cyclo(Arg-Gly-Asp-D-Phe-Lys)]2 was purchased from Peptides International and is referred to as DOTA-RGD. Doxorubicin was purchased as an HCl salt from Selleckchem. 18F-FDG was obtained as a saline solution from IBA Molecular through the Nuclear Pharmacy at Memorial Sloan Kettering Cancer Center. 90YCl3 was obtained as a 0.05 M HCl solution from Perkin Elmer. 64CuCl2 was produced on the cyclotron at Washington University Medical School (model CS-15; Cyclotron Corp.) by the 64Ni(p,n)64Cu reaction and purified to yield a specific activity of 7.4–14.8 GBq/mg in a 0.1 M HCl solution. 68Ga was eluted from a 68Ge/68Ga generator using 0.5 M KOH, neutralized using 30% HCl, and mixed with phosphate-buffered saline (PBS) powder concentrate. (2,5-dioxopyrrolidin-1-yl)4-azido-2,3,5,6-tetrafluorobenzoate (Fig. 2A, compound 1) was synthesized from 4-azido-2,3,5,6-tetrafluorobenzoic acid according to a previously published protocol (25). N-(2-aminoethyl)-4-azido-2,3,5,6-tetrafluorobenzamide (Supplemental Fig. 1, compound 2; supplemental materials are available at http://jnm.snmjournals.org) was synthesized from (2,5-dioxopyrrolidin-1-yl)4-azido-2,3,5,6-tetrafluorobenzoate according to a previously published protocol (26). All azide-containing syntheses were protected from exposure to direct light, as was the final product. High-performance liquid chromatography (HPLC) for qualitative analysis and quantitative purification was performed on a Shimadzu HPLC system equipped with LC20AD Prominence pumps, an SPD-M10AVP photodiode array detector, an FRC-10A fraction collector, and a DGU-20A degasser. Reverse-phase HPLC was performed using an Atlantis T3 analytic column (100 Å, 3 μm, 4.6 × 150 mm; Waters) and an Atlantis T3 OBD semiprep column (100 Å, 5 μm, 19 × 150 mm; Waters), along with Milli-Q water (0.1% formic acid; MilliporeSigma) as solvent A and acetonitrile (0.1% formic acid) as solvent B.
FIGURE 2.

Synthesis of Cy7 azide and Cy7 fluoride (A) and of DOX azide (B). RT = room temperature.
Radiochemistry reactions were performed in Chelex (Bio-Rad) purified Milli-Q water. The radiolabeling was monitored using instant thin-layer chromatography (iTLC) by spotting on silicic acid–coated chromatography paper (Agilent Technologies), eluting with 50 mM ethylenediaminetetraacetic acid (pH 5.5), and analyzing the developed strip on a System 200 imaging scanner coupled to an Autochanger 1000 using Win-Scan radio-TLC software, version 2.2 (these last 3 all from Bioscan Inc.). Radiochemical purity was also determined using a Flow-RAM radio-HPLC detector (LabLogic Systems) with the HPLC described above, and signal was recorded using Shimadzu software. The radio-HPLC column and solvent setup was the same as that used in the nonradiosynthesis application. Activity was measured using a CRC-15R dose calibrator (Capintec).
Cells and Animals
Human fibrosarcoma cell line HT1080 and human breast cancer cell line MDA-MB-231 were obtained from the American Type Culture Collection. Human breast cancer cell line 4T1-luc2-GFP was obtained from Perkin Elmer. All cell lines were cultured using standard conditions.
All animal experiments were conducted in accordance with institutional guidelines and the protocol approved by the Institutional Animal Care and Use Committee. Female Hsd:athymic nude-Foxn1nu mice (7–9 wk old; weight, 18–20 g) were obtained from Envigo, and all procedures were performed using 2% isoflurane (Baxter Healthcare) as inhalation anesthesia. Xenografts of HT1080 cells were implanted by subcutaneous injection of 1.5 × 106 cells suspended in 100 μL of Matrigel (BD Biosciences) in the left and right flanks of the mice. Experiments were performed when the tumors reached a volume of approximately 150 mm3. The mice were fed an irradiated nonfluorescent diet before fluorescence imaging to lower autofluorescence.
Synthesis of Calcein Azide
Calcein azide was synthesized by conjugating the free amine of the N-(2-aminoethyl)-4-azido-2,3,5,6-tetrafluorobenzamide (Supplemental Fig. 1, compound 2) to the activated carboxylic acid of calcein (21 mg, 0.034 mmol). Calcein was dissolved in 1 mL of dimethyl formamide (DMF) and cooled to 0°C, followed by the addition of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (56 mg, 0.034 mmol) and NHS (9 mg, 0.08 mmol). After 30 min, N-(2-aminoethyl)-4-azido-2,3,5,6-tetrafluorobenzamide (14 mg, 0.051 mmol) was added and stirred overnight at room temperature. The reaction mixture was diluted with water and washed with ethyl acetate, and the final product was isolated after lyophilization.
Synthesis of Cy7 Azide
Boc-1-amino-3,6-dioxa-8-octanediamine (6.5 mg, 0.026 mmol) and 2,5-dioxopyrrolidin-1-yl 4-azido-2,3,5,6-tetrafluorobenzoate (10.3 mg, 0.031 mmol) were dissolved in 0.5 mL of DMF. N,N-diisopropylethylamine (DIEA) (54 μL, 0.31 mmol) was added and stirred for 1 h at room temperature. After the addition of 100 μL of water, the reaction mixture was lyophilized. Neat TFA was added to the residue and stirred for 2 h, after which the free amine was purified by preparative HPLC. Purified free amine (2.48 mg, 0.006 mmol) and sulfo-Cy7 NHS ester (5 mg, 0.006 mmol) were dissolved in 2 mL of DMF. DIEA (10.5 μL, 0.06 mmol) was added, and the reaction mixture was stirred for 2 h, followed by purification using HPLC and lyophilization.
Synthesis of Cy7 Fluoride (Nonactivatable Control)
Boc-1-amino-3,6-dioxa-8-octanediamine (14 mg, 0.056 mmol) and DIEA (22.2 μL, 0.127 mmol) were dissolved in 0.2 mL of dichloromethane and added to an ice-cold solution of 2,3,4,5,6-pentafluorobenzoyl chloride (19.5 mg, 0.085 mmol) in 0.1 mL of dichloromethane. Afterward, the reaction mixture was gradually warmed to room temperature and stirred for 2 h. The reaction was then quenched with 0.1 mL of water and concentrated using rotary evaporation. Neat TFA was mixed with the crude product and stirred for 4 h. The reaction mixture was concentrated using rotary evaporation and purified using HPLC to get free amine. Purified free amine (2.4 mg, 0.007 mmol) was dissolved in 21.4 μL of dimethyl sulphoxide and mixed with sulfo-Cy7 NHS ester (5 mg, 0.006 mmol), DIEA (10.5 μL, 0.06 mmol), and DMF (2 mL). The reaction mixture was stirred for 3 h, rotary evaporated, and purified using preparative HPLC, and the desired fractions were lyophilized.
Synthesis of DOX Azide
Doxorubicin (5 mg, 8.6 μM) was dissolved in 86.2 μL of DMF and added to (2,5-dioxopyrrolidin-1-yl)4-azido-2,3,5,6-tetrafluorobenzoate (10.3 mg, 0.031 mmol) and DIEA (15 μL, 86 μM) and diluted by adding 250 μL of DMF. The reaction mixture was stirred for 24 h and purified using preparative HPLC, and the desired fractions were lyophilized.
Synthesis of 90Y-DOTA-RGD
A 200-MBq activity of 90YCl3 in 9 μL of 0.05 M HCl was added to a mixture of 27 μL of sodium ascorbate (20 mg/mL), 135 μL of 200 mM ammonium acetate (pH 5.5), and 7.325 μL of 1 mM DOTA-RGD (12.5 μg, 7.3 nmol). The resulting reaction mixture was incubated in a ThermoMixer (Eppendorf) at 37°C with moderate mixing for 1 h. Afterward, the radiochemical purity of 90Y-DOTA-RGD was verified using iTLC and radio-HPLC. A serum stability analysis was performed by adding 5 μL (0.37 MBq) of 90Y-DOTA-RGD to 850 μL of fetal bovine serum and determining the radiochemical purity at different time points using iTLC.
Synthesis of 64Cu-DOTA-RGD
A 53-MBq activity of 64CuCl2 in 72 μL of 0.1 M HCl was added to a mixture of 11.1 μL of sodium ascorbate (20 mg/mL), 55.5 μL of 200 mM ammonium acetate (pH 5.5), and 7.325 μL of 1 mM DOTA-RGD (5.1 μg, 3 nmol). The resulting reaction mixture was incubated in a ThermoMixer at 37°C with moderate mixing for 40 min, after which the radiochemical purity of 64Cu-DOTA-RGD was verified using iTLC.
Monitoring of Ultraviolet-Catalyzed Aryl Azide Photocleavage Using Absorption Spectra
A 12-μg quantity of 4-azido-2,3,5,6-tetrafluorobenzoic acid was dissolved in 1 mL of CHCl3 and irradiated using an ultraviolet-C–radiating germicidal Puritec HNS 30-watt G13 mercury lamp (Osram) at a distance of about 70 cm. Ultraviolet–visible spectra were recorded after 2, 5, 10, 15, and 20 min of irradiation (Supplemental Fig. 2).
In Vitro Activation of Calcein Azide Using Ultraviolet Light
Four thousand MDA-MB-231 cells were seeded into each well of a 4-well chamber slide and incubated overnight to form a monolayer. On the following day, the cells were fixed with 4% paraformaldehyde for 15 min and washed once with PBS. Afterward, 2 wells were incubated with either PBS only, a 67 μg/mL solution of pure calcein, or a 67 μg/mL solution of calcein azide. Calcein- and calcein azide–treated cells were further divided into 2 subgroups. One well of each group was kept in the dark, and the other was ultraviolet-C–irradiated for 10 min using a germicidal Puritec HNS 30-watt G13 mercury lamp at a distance of about 70 cm. Afterward, all slides were washed with PBS, and the cells were imaged using an Eclipse fluorescence microscope (exposure time 36 ms; Nikon) to detect fluorescence of the activated calcein azide (Supplemental Fig. 3A). Quantitative analysis was performed using the signal-to-noise ratio calculated by NIS-Elements AR, version 4.0 (Nikon), for each condition (n = 3) (Supplemental Fig. 3B).
In Vitro Activation of Cy7 Azide Using CL
Trypsinized HT1080 cells (2 × 106) were washed with PBS and incubated with either 9.25 MBq of 18F-FDG or PBS (n = 3). After 30 min, the cells were centrifuged at 1,000 rpm, the supernatant decanted, and the cells washed with PBS. A 10-μL volume of 10 μM Cy7 azide was added and incubated for 2 h. After incubation, the cells were washed with PBS and the fluorescence of the cells was measured using an IVIS Spectrum system (Perkin Elmer) with a 745-nm excitation filter and an 800-nm emission filter with binning of 4, field of view of 24.4, and f2 of 0.75 s. Quantitative analysis was performed by drawing regions of interest over the cell pellets using Living Image software, version 4.2.
Tumor Accumulation and Biodistribution of 64Cu-DOTA-RGD
To investigate the tumor accumulation of 64Cu-DOTA-RGD over time, 3 HT1080 tumor–bearing mice were administered 11.1 MBq of 64Cu-DOTA-RGD in sterile saline via retroorbital injection. Ten-minute static PET scans were acquired, followed by 2-min CT scans on an Inveon small-animal PET/CT scanner (Siemens) at 1, 3, 7, 10, 19, and 25 h after injection. Quantitative analysis of the tumor uptake was done by drawing a region of interest over the tumor using AsiPro VM software (Concorde Microsystems) and Inveon Research Workplace software (version 4.1; Siemens). For biodistribution analysis, 20 HT1080 tumor–bearing mice were administered 0.56 MBq of 64Cu-DOTA-RGD in sterile saline via retroorbital injection. Mice were sacrificed at 1, 3, 7, 10, 19, and 25 h after injection (3 animals per time point) and dissected. Tumors, blood, heart, lung, liver, kidneys, stomach, intestine, muscle, and spleen were placed into tubes and weighed, and the activity was measured in a γ-counter (Wizard2; Perkin Elmer). The percentage of injected dose per gram of tissue (%ID/g) was calculated for every organ (n = 3).
In Vivo Activation of Cy7 Azide Using CL from 90Y-DOTA-RGD
A 29.6-MBq activity of 90Y-DOTA-RGD in 100 μL of PBS was administered to 5 tumor-bearing animals by retroorbital injection. Saline was injected into 4 more mice as a control. CL images were acquired using the IVIS Spectrum system from 5 min to 6 h after injection at different time points with an open emission filter, a blocked excitation filter, binning of 8, and an f1 and exposure time of 30 s. After 7 h, 100 μL of 1 mM Cy7 azide were retroorbitally administered in the alternate eye of the previously injected 9 mice. In a separate control experiment, 5 mice were injected with 100 μL of 1 mM Cy7 fluoride at 7 h after a 29.6-MBq injection of 90Y-DOTA-RGD, and 4 mice were injected with saline and 100 μL of 1 mM Cy7 fluoride to determine nonspecific uptake of the control agent. Fluorescence images of all mice were acquired over time until 158 h after injection using the IVIS Spectrum system as described before. Quantitative analysis was done by drawing regions of interest over the tumors and the muscle, and the tumor-to-muscle ratio was calculated.
In Vitro Activation of DOX Azide Using CL
Five thousand 4T1 cells per well were seeded in a black 96-well plate with a clear bottom (Costar) and incubated overnight. On the next day, the medium was removed and the cells were covered with 50 μL of fresh medium with or without 1 nmol of DOX azide. Wells with and without the DOX azide were further treated with or without 3.7 MBq and 11.1 MBq of 68Ga in PBS buffer (3 wells per condition). The cells were incubated for 48 h. Afterward, a viability assay was performed by adding 10 μL of AlamarBlue reagent (Invitrogen) and 90 μL of medium into each well and incubating for 4 h. The fluorescence of the plate was recorded using a plate reader (SpectraMax M5; Molecular Devices) with an excitation at 570 nm and emission at 585 nm and analyzed quantitatively.
Statistical Analysis
Data and statistical analyses were performed using Prism (version 5.01; GraphPad Software, Inc.). Data were analyzed using the unpaired, 2-tailed Student t test. Differences at the 95% confidence level (P < 0.05) were considered to be statistically significant. For time-series data, statistical significance was evaluated for each time slice, and a significant difference over the entire time series was represented using square brackets at the tail of the plot.
RESULTS
Synthesis and Characterization of Cy7 Azide, Cy7 Fluoride, and DOX Azide
The Cy7 azide was synthesized by coupling sulfo-Cy7 NHS ester to the free amine (Fig. 2A, compound 1, X = N3), and the desired product was obtained after preparative HPLC with greater than 90% yield. LCMS showed that the product was more than 98% pure, with a retention time of 5.38 min. Mass spectroscopy showed a peak at 1,056.40 m/z [M+H]+. The Cy7 fluoride was synthesized by coupling the Cy7 NHS ester to the free amine (Fig. 2A, compound 1, X = F), and the desired product was obtained after preparative HPLC with greater than 90% yield. LCMS showed that the product was more than 98% pure, with a retention time of 5.45 min. Mass spectroscopy showed a peak at 1,033.30 m/z [M+H]+. DOX azide was prepared by coupling free amine of doxorubicin with 4-azido-2,3,5,6-tetrafluorobenzoyl chloride in greater than 99% yield, as shown in Figure 2B. LCMS revealed that the product was more than 98% pure, with a retention time of 7.25 min. Mass spectroscopy showed a peak at 783.0 m/z [M+Na]+.
Synthesis of 90Y-DOTA-RGD and 64Cu-DOTA-RGD
The radiolabeling of DOTA-RGD with 90YCl3 was successful, and a high specific activity of 27.4 MBq/nmol was achieved. 90Y-DOTA-RGD was prepared with a radiochemical purity of more than 99%, as shown by the Rf 0 peak on iTLC (Supplemental Fig. 4A) and a retention time of 7.7 min on radio-HPLC (Supplemental Fig. 4B). As a control, free 90Y was shown to have an Rf of 0.86 on iTLC (Supplemental Fig. 4C). The serum stability study with 90Y-DOTA-RGD showed that the radiochemical purity was retained over 6 d (Supplemental Fig. 4D).
Monitoring Photoactivation of 4-Azido-2,3,5,6-Tetrafluorobenzoic Acid
Cumulative ultraviolet-C doses to 4-azido-2,3,5,6-tetrafluorobenzoic acid showed a gradual decrease in the absorbance at 265 nm and more tailing above 310 nm (Supplemental Fig. 2). The isosbestic point for the photochemical transformation was found to be 290 nm.
In Vitro Photoactivation of Calcein Azide
Incubation of MDA-MB-231 cells with calcein azide led to a detectable fluorescence in those cells additionally treated with ultraviolet light (Supplemental Fig. 3A). In comparison, cells without ultraviolet treatment or the pure calcein control dye did not show a fluorescence signal. After quantification, the fluorescence signal-to-noise ratio of the cells treated with calcein azide and ultraviolet light was 3.5 times higher (P < 0.0001) than that of the cells treated with calcein azide only, at 15.56 ± 0.03 and 3.55 ± 0.06, respectively (Supplemental Fig. 3B). There was no autofluorescence of the untreated cells themselves, of the cells treated with pure calcein with or without ultraviolet light, or of the cells treated with calcein azide only.
In Vitro Activation of Cy7 Azide Using CL
Two million HT1080 cells incubated with 18F-FDG had an average uptake of 4.33 ± 0.07 MBq of 18F-FDG per cell pellet (2.22 Bq/cell). Subsequent incubation with Cy7 azide resulted in a strong fluorescence from the 18F-FDG–incorporated cell pellets, compared with the pellets incubated with Cy7 azide only (Fig. 3A). The quantitative analysis confirmed the observation by revealing a significantly higher fluorescence signal for the cells incubated with 18F-FDG and Cy7 azide than for the cells incubated with Cy7 azide alone, with average radiant efficiencies of (7.770 ± 0.348) × 107 and (3.993 ± 0.071) × 107, respectively (P = 0.0004) (Fig. 3B).
FIGURE 3.

CL-activated cell labeling using Cy7 azide. (A) Fluorescence images of HT1080 cell pellets incubated with 18F-FDG and Cy7 azide or Cy7 azide alone. (B) Quantitative analysis of HT1080 cell pellet fluorescence signal after incubation with Cy7 azide alone or together with 18F-FDG (P = 0.0004). IVIS Spectrum system was used for fluorescence imaging (emission filter, 800 nm; excitation filter, 745 nm; epiillumination; bins, 4; field of view, 24.4; f2, 0.75 s). *P < 0.05.
Tumor Accumulation and Biodistribution of 64Cu-DOTA-RGD
The PET/CT images at different time points after intravenous injection of 64Cu-DOTA-RGD showed a rapid accumulation of 64Cu-DOTA-RGD at the tumor site, as was also revealed by the quantitative analysis of these images (Fig. 4A). Furthermore, the biodistribution studies quantified the %ID/g for each tumor to be 8.1 ± 1.8 at 1 h after injection, 12.5 ± 4.1 at 3 h, 9.3 ± 1.5 at 7 h, 7.8 ± 2.2 at 10 h, 8.5 ± 2.7 at 19 h, and 11.3 ± 1.5 at 25 h, a pattern that shows a rapid and steady uptake in the tumors (Fig. 4B). The conjugate was excreted mostly through the liver, with 42.4 ± 9.7 %ID/g at 1 h dropping to 12.0 ± 2.4 %ID/g at 25 h.
FIGURE 4.

In vivo PET/CT imaging and biodistribution studies of 64Cu-DOTA-RGD. (A) Representative PET/CT image with left tumor (LT) and right tumor (RT) at 7 h after injection of 64Cu-DOTA-RGD. Graph shows quantitative analysis of activity in tumor as function of time. (B) Biodistribution of 64Cu-DOTA-RGD at different time points after injection as %ID/g of organ.
CL Imaging of 90Y-DOTA-RGD and In Vivo CL-Activated Accumulation of Cy7 Azide
Mice injected with 90Y-DOTA-RGD showed a rapid and specific accumulation of activity in the tumor at 6 h after injection (Fig. 5A). The corresponding quantitative analysis showed an increase in the tumor CL signal within the first 2 h after injection (Fig. 5B). The CL signal stayed at this level up to 6 h after injection. Control mice received just saline injection before the Cy7 azide injection and thus lacked the CL-emitting radioisotope required to activate the azide. Mice that were injected with the Cy7 azide and 90Y-DOTA-RGD demonstrated a significantly higher fluorescence within the tumors (site of CL generation) than did the control mice (average radiant efficiency at 36 h, 5.0 × 109 ± 1.1 × 109 vs. 2.4 × 109 ± 0.4 × 109; P < 0.0002) (Figs. 5C and 5D). Even 6.5 d (153 h) after Cy7 azide injection, fluorescence within the tumors remained significantly higher and more stable than in the tumors of control mice (1.6 × 109 ± 0.5 × 109 vs. 0.8 × 109 ± 0.2 × 109; P < 0.002). Plotting the tumor-to-muscle ratios of the Cy7 fluorescence for 90Y-DOTA-RGD–treated and –untreated mice also helped visualize the contrast enhancement within the tumor and showed maximum contrast to be at 72 h (Fig. 5E).
FIGURE 5.

CL-activated tumor labeling using Cy7 azide. (A) CL images of mice injected with 90Y-DOTA-RGD (6-h time point). (B) Quantitative analysis of CL radiance of tumors 6 h after injection of 90Y-DOTA-RGD. (C) Fluorescence images of mice injected with Cy7 azide 7 h after 90Y-DOTA-RGD or saline injection, or 90Y-DOTA-RGD alone. (D) Quantitative analysis of fluorescence of tumors with Cy7 azide with and without 90Y-DOTA-RGD over time (P < 0.05). (E) Quantitative plot of tumor-to-muscle ratio in mice with 90Y-DOTA-RGD–activated Cy7 azide compared with saline control as function of time (P < 0.05). (F) Quantitative analysis of fluorescence of tumors from mice injected with Cy7 fluoride control 7 h after 90Y-DOTA-RGD or saline injection over time. IVIS Spectrum system was used for optical imaging (CLI: emission filter, open; excitation filter, blocked; bins [medium; HR], 8; field of view, 24.4; f1, 30 s) (fluorescence imaging: emission filter, 800 nm; excitation filter, 745 nm; epiillumination; bins [high resolution; HR], 4; field of view, 24.4; f2, 0.75 s).
As a second control, we used a nonactivatable Cy7 fluoride control dye. Animals injected with 90Y-DOTA-RGD and Cy7 fluoride had no significant fluorescence enhancement in their tumors, in comparison to mice injected with Cy7 fluoride alone (average radiance fluorescence, 6.6 × 1010 ± 1.4 × 1010 vs. 6.1 × 1010 ± 0.982 × 1010, respectively, at 36 h) (Fig. 5F).
In Vitro Activation of DOX Azide Using CL
To demonstrate the possibility of developing a therapy based on this application, we developed an activatable doxorubicin analog. Treatment of MDA-MB-231 cells with DOX azide alone reduced the cell viability by only 3.8%, compared with untreated cells (P = 0.0034) (Fig. 6). In comparison, treatment of the cells with DOX azide in combination with 3.7 or 11.1 MBq of 68Ga decreased the cell viability significantly by 9% (P < 0.0001) and 20% (P = 0.0001), respectively, compared with the untreated control. A treatment with 3.7 or 11.1 MBq of 68Ga alone did not affect the viability of the cells. This pilot demonstration suggests the possible further application of our approach.
FIGURE 6.
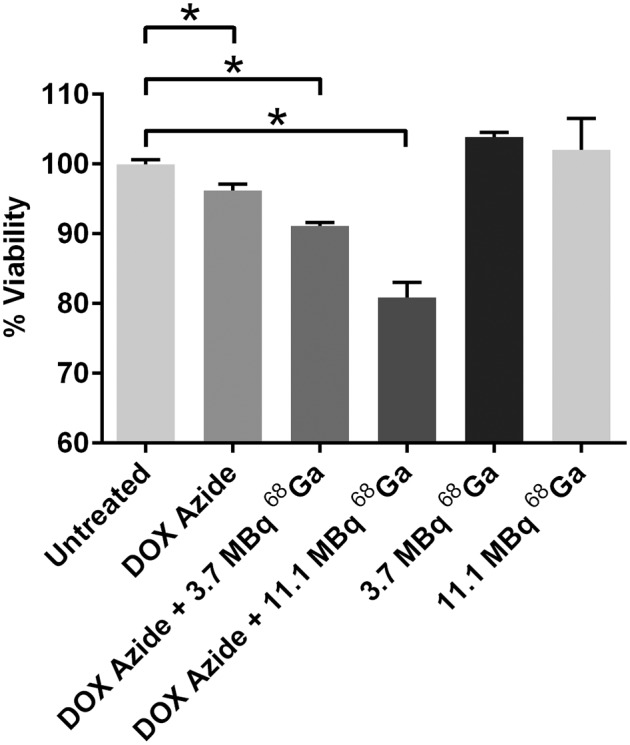
Dose-dependent CL-activated in vitro therapy using DOX azide. Viability is shown for 4T1 cells treated with 1 nmol of DOX azide and different doses of 68Ga. *P < 0.05.
DISCUSSION
Our study demonstrated that attaching an azide group to various molecules leads to a specific and localized targeted delivery of those molecules to CL-emitting tumors. This strategy was successful with both CL-activatable azide conjugated to fluorescent dyes and chemotherapeutics. With this method, we converted a low CL signal into a stronger, long-lasting fluorescence signal that will facilitate the use of radiotracers for intraoperative imaging. We also showed enhanced cytotoxicity after the combination of CL with an activatable drug.
In our first experiments, we showed that the azide could be cleaved using ultraviolet light and monitored using ultraviolet–visible spectra. Also using ultraviolet light, we showed in vitro activation of calcein azide. We showed an enhanced delivery of calcein using ultraviolet light to activate calcein azide on tumor cells. Hence, in the presence of ultraviolet light, our calcein azide was also found to be bound to the tumor cells. Pure calcein was used as a control to show that the dye alone did not result in any accumulation with or without ultraviolet light. This finding points out that the azide moiety is essential for the attachment to the cells.
After showing proof of concept using ultraviolet light, we used CL to activate Cy7 azide. The accumulation of 18F-FDG in HT1080 cells could be seen by virtue of the Warburg effect (27). On incubation of the 18F-FDG–enriched cells with Cy7 azide, the CL activated the azide moiety, generating nitrenes and resulting in binding of the Cy7 construct to the cell surface. Cells without 18F-FDG had a significantly lower Cy7 signal, confirming our proposed mechanism.
In vivo experiments were performed with 90Y as the source of CL, as this isotope produces more CL than other isotopes because of the emission of energetic electrons (28). The attachment of DOTA-RGD made this CL source highly specific through targeting of αVβ3 integrins on the tumor (29). The tumor-targeting efficiency of the peptide was quantified by biodistribution studies using 64Cu-labeled DOTA-RGD. Instead of the 90Y that was used for CL activation of the azide, we switched to 64Cu because of its usability in PET imaging and γ-counting. This quantification lets us gauge the tumor uptake of 90Y-DOTA-RGD for CL activation of Cy7 azide activation. After accumulation of 90Y-DOTA-RGD at the tumor site, an enhanced labeling with Cy7 azide over time was detected in only these tumors, not in control tumors, which did not receive 90Y-DOTA-RGD. The retained fluorescence contrast even after several days indicates a stable covalent binding of the dye to the CL-enhanced tissue. The use of the inactive Cy7 fluoride instead of the active azide did not lead to a CL-dependent accumulation of fluorescence within the tumor, demonstrating that the presence of the azide is necessary for the accumulation of the dye. This indeed clarifies that the phenomenon we observed is truly a CL-triggered activation.
In future experiments, the fluorescence output gained from the Cy7 azide in the CL-enhanced tissue could be used for fluorescence-guided intraoperative resection. The near-infrared fluorescence probes are particularly important in clinical application because of their greater penetration depth and low autofluorescence. Translating CL into long-lasting fluorescence circumvents the disadvantage of the low signal intensity of the CL and the need for radioactivity. Surgeons could inject first the radiotracer and then the fluorescent CL-activated sticky tag. Once the activity has decreased or fully decayed, the surgeon can perform surgery, using the fluorescent tag for image-guided surgery. This technique can be used with the multitude of existing radiotracers, requiring only one azide label (one-stop shop), thus preserving the advantage of having many clinical tracers to mark tumors. The detection of cancer using such targeted fluorophores could possibly improve survival rates and outcomes in patients (30).
Using this system, we also investigated its possible application for a therapeutic approach. We introduced a CL-triggered, targeted drug delivery system by demonstrating a significant CL dose-dependent increase in toxicity of DOX azide to breast cancer cells. This finding can be particularly useful in therapy settings that combine radiotherapy (as additional CL source) and chemotherapy. A multistep drug delivery strategy has previously been shown to be better for patients receiving combination therapy rather than chemotherapy or radiotherapy alone (17). Because DOX azide alone had a minor toxicity effect on the cells, the compound needs to be improved. However, a significantly higher toxicity was attained in combination with CL. Thus, the delivered drug is shown to benefit from the advantage that the azide version of the drug effectively is a pro-drug, activated only in the presence of CL excitation. This characteristic could increase the therapeutic efficacy because of the CL-activated targeting. The fact that the used 68Ga doses showed no toxicity is promising for the potential use of even higher doses and the generation of more CL. The use of 68Ga is not directly translatable but could easily be conjugated to DOTA-RGD, similar to the 90Y conjugation techniques that we have shown and used to create targeted agents. The modest drop in cell viability calls for further research into more effective photochemistry and drug–azide conjugates. Nonetheless, the indications for this new modality of small-molecule drug delivery are very promising.
CONCLUSION
We were able to photoactivate probes for tumor imaging and therapy by using CL. We could specifically activate Cy7 dye–labeled azides in vitro and in vivo to convert a low-intensity CL signal into a higher and long-lasting fluorescence signal. This conversion is suitable for circumventing the known disadvantages of CL imaging. Furthermore, we could introduce the first CL-photoactivated drug delivery system. Our targeting strategy can be used to enable a more specific fluorescent intraoperative resection or to assess tissue radiation dose. With our approach, we could successfully establish a new CL application in the field of tumor imaging and therapy.
DISCLOSURE
This work was supported by the National Institute of Health (R01EB014944 and R01 CA183953 to Jan Grimm and the MSKCC NIH Core Grant P30-CA008748). No other potential conflict of interest relevant to this article was reported.
Supplementary Material
Acknowledgments
We thank Drs. Jason Lewis, Nagavarakishore Pillarsetty, and Thomas Reiner for help with radiochemistry. Also, we thank the MSKCC Radiopharmacy, Small-Animal Imaging Core Facility, Nuclear Magnetic Resonance Analytical Core Facility, and Design and Creative Services.
REFERENCES
- 1.Robertson R, Germanos MS, Li C, Mitchell GS, Cherry SR, Silva MD. Optical imaging of Cerenkov light generation from positron-emitting radiotracers. Phys Med Biol. 2009;54:N355–N365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thorek DL, Robertson R, Bacchus WA, et al. Cerenkov imaging: a new modality for molecular imaging. Am J Nucl Med Mol Imaging. 2012;2:163–173. [PMC free article] [PubMed] [Google Scholar]
- 3.Thorek DLJ, Riedl CC, Grimm J. Clinical Cerenkov luminescence imaging of 18F-FDG. J Nucl Med. 2014;55:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorek DLJ, Ogirala A, Beattie BJ, Grimm J. Quantitative imaging of disease signatures through radioactive decay signal conversion. Nat Med. 2013;19:1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorek DL, Das S, Grimm J. Molecular imaging using nanoparticle quenchers of Cerenkov luminescence. Small. 2014;10:3729–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czupryna J, Kachur AV, Blankemeyer E, et al. Cerenkov-specific contrast agents for detection of pH in vivo. J Nucl Med. 2015;56:483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das S, Thorek DL, Grimm J. Cerenkov imaging. Adv Cancer Res. 2014;124:213–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ran C, Zhang Z, Hooker J, Moore A. In vivo photoactivation without “light”: use of Cherenkov radiation to overcome the penetration limit of light. Mol Imaging Biol. 2012;14:156–162. [DOI] [PubMed] [Google Scholar]
- 9.Zukotynski K, Jadvar H, Capala J, Fahey F. Targeted radionuclide therapy: practical applications and future prospects. Biomark Cancer. 2016;8:35–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chinol M, Paganelli G, Sudati F, Meares C, Fazio F. Biodistribution in tumour-bearing mice of two 90Y-labelled biotins using three-step tumour targeting. Nucl Med Commun. 1997;18:176–182. [DOI] [PubMed] [Google Scholar]
- 11.Hnatowich DJ, Virzi F, Doherty PW. DTPA-coupled antibodies labeled with yttrium-90. J Nucl Med. 1985;26:503–509. [PubMed] [Google Scholar]
- 12.Janssen ML, Oyen WJ, Dijkgraaf I, et al. Tumor targeting with radiolabeled αvβ3 integrin binding peptides in a nude mouse model. Cancer Res. 2002;62:6146–6151. [PubMed] [Google Scholar]
- 13.Sofou S. Radionuclide carriers for targeting of cancer. Int J Nanomedicine. 2008;3:181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong G, Johnston V, Ramdave S, Lau E, Rischin D, Hicks RJ. High-administered activity In-111 octreotide therapy with concomitant radiosensitizing 5FU chemotherapy for treatment of neuroendocrine tumors: preliminary experience. Cancer Biother Radiopharm. 2009;24:527–533. [DOI] [PubMed] [Google Scholar]
- 15.DeNardo SJ. Combined molecular targeting for cancer therapy: a new paradigm in need of molecular imaging. J Nucl Med. 2006;47:4–5. [PubMed] [Google Scholar]
- 16.Rusckowski M, Wang Y, Blankenberg FG, Levashova Z, Backer MV, Backer JM. Targeted scVEGF/177Lu radiopharmaceutical inhibits growth of metastases and can be effectively combined with chemotherapy. EJNMMI Res. 2016;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence TS, Haffty BG, Harris JR. Milestones in the use of combined-modality radiation therapy and chemotherapy. J Clin Oncol. 2014;32:1173–1179. [DOI] [PubMed] [Google Scholar]
- 18.Al-Sarraf M, LeBlanc M, Giri PG, et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: phase III randomized intergroup study 0099. J Clin Oncol. 1998;16:1310–1317. [DOI] [PubMed] [Google Scholar]
- 19.Penack O, Becker C, Buchheidt D, et al. Management of sepsis in neutropenic patients: 2014 updated guidelines from the Infectious Diseases Working Party of the German Society of Hematology and Medical Oncology (AGIHO). Ann Hematol. 2014;93:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gritsan NP, Gudmundsdóttir AD, Tigelaar D, et al. A laser flash photolysis and quantum chemical study of the fluorinated derivatives of singlet phenylnitrene. J Am Chem Soc. 2001;123:1951–1962. [DOI] [PubMed] [Google Scholar]
- 21.Lapinsky DJ. Tandem photoaffinity labeling-bioorthogonal conjugation in medicinal chemistry. Bioorg Med Chem. 2012;20:6237–6247. [DOI] [PubMed] [Google Scholar]
- 22.Schnapp KA, Poe R, Leyva E, Soundararajan N, Platz MS. Exploratory photochemistry of fluorinated aryl azides: implications for the design of photoaffinity labeling reagents. Bioconjug Chem. 1993;4:172–177. [DOI] [PubMed] [Google Scholar]
- 23.Xia Y, Peng L. Photoactivatable lipid probes for studying biomembranes by photoaffinity labeling. Chem Rev. 2013;113:7880–7929. [DOI] [PubMed] [Google Scholar]
- 24.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. [DOI] [PubMed] [Google Scholar]
- 25.Marchand-Brynaert J, Welle A, Billard F. Tri- and tetravalent photoactivable cross-linking agents. Synthesis. 2012;44:2249–2254. [Google Scholar]
- 26.Rajagopalan R, Kuntz RR, Sharma U, Volkert WA, Pandurangi RS. Chemistry of bifunctional photoprobes. 6. Synthesis and characterization of high specific activity metalated photochemical probes: development of novel rhenium photoconjugates of human serum albumin and Fab fragments. J Org Chem. 2002;67:6748–6757. [DOI] [PubMed] [Google Scholar]
- 27.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. [DOI] [PubMed] [Google Scholar]
- 28.Carpenter CM, Ma X, Liu H, et al. Cerenkov luminescence endoscopy: improved molecular sensitivity with beta–emitting radiotracers. J Nucl Med. 2014;55:1905–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Conti PS, Moats RA. In vivo near-infrared fluorescence imaging of integrin αvβ3 in brain tumor xenografts. Cancer Res. 2004;64:8009–8014. [DOI] [PubMed] [Google Scholar]
- 30.Zhang RR, Schroeder AB, Grudzinski JJ, et al. Beyond the margins: real-time detection of cancer using targeted fluorophores. Nat Rev Clin Oncol. 2017;14:347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.