

Abstract
Long noncoding RNA HOX transcript antisense RNA (HOTAIR) is involved in human tumorigenesis and is dysregulated in hepatocellular carcinoma (HCC). However, the molecular mechanisms underlying HOTAIR functions in HCC are largely unknown. Here, we employed an integrated transcriptomic and quantitative proteomic analysis to systematically explore the regulatory role of HOTAIR in HCC. A total of 673 transcripts and 293 proteins were found to be dysregulated after HOTAIR inhibition. Bioinformatics studies indicated that differentially expressed genes (DEGs) and differentially expressed proteins (DEPs) are involved in many biological processes, especially cancer-related signaling pathways. A set of DEGs and DEPs were validated by quantitative RT-PCR, Western blot and parallel reaction monitoring (PRM) analysis, respectively. Further functional studies of the opioid growth factor receptor (OGFr), a negative biological regulator of cell proliferation in HCC, revealed that HOTAIR exerts its effects on cell proliferation, at least in part, through the regulation of OGFr expression. By correlating the omics data with functional studies, the current results provide novel insights into the functional mechanisms of HOTAIR in HCC cells.
It has been shown that less than 2% of the human genome sequence encodes proteins (1), whereas more than 90% is transcribed into noncoding RNAs (ncRNAs). NcRNAs have been extensively studied and found to be involved in the regulation of many fundamental biological processes (2). Long noncoding RNAs (lncRNAs)1 constitute a group of mRNA-like nonprotein coding transcripts with lengths of at least 200 nucleotides (3–5). In recent years, lncRNAs have attracted increasing attention because of their critical regulatory functions in human diseases, especially in human cancers (6, 7). Hepatocellular carcinoma (HCC) is one of the most prevalent and deadly cancers among the human population, especially in many Asian and African countries (8, 9). Many lncRNAs have already been shown to be dysregulated in HCC, and their aberrant expression is related to tumorigenesis, metastasis, prognosis and diagnosis (10–15). HOX transcript antisense intergenic RNA (HOTAIR) is a 2158-nt lncRNA that is located within the Homeobox C (HOXC) gene cluster (between HoxC11 and HoxC12) on human chromosome 12q13.13 (16, 17). HOTAIR acts as an oncogenic lncRNA in different types of cancer, including HCC (10–12, 18–27). High expression of HOTAIR in HCC primary tumors was reported to be associated with a poor prognosis (10, 11, 28, 29). HOTAIR inhibition could markedly reduce HCC cell proliferation, migration, and invasion (10, 28–30).
The function of HOTAIR has been extensively studied (7, 31, 32). Studies pioneered by Chang and colleagues revealed that HOTAIR functions as a molecular scaffold to direct polycomb repressive complex 2 (PRC2, consists of EZH2, SUZ12 and EED) and lysine-specific demethylase 1A (LSD1) to the HOXD locus, trimethylate histone H3 at lysine 27 (H3K27me3), and epigenetically alter the expression of hundreds of genes (7, 33). Subsequent studies have uncovered more molecular regulatory mechanisms of HOTAIR (21, 23, 34, 35). The regulatory roles of HOTAIR in HCC have also been studied (12, 29, 30, 36, 37). HOTAIR may exert its function in HCC by regulating the Wnt/β-catenin signaling pathway (29). HOTAIR promotes cell migration and invasion by regulating RNA binding motif protein 38 (RBM38) in HCC cells (30). HOTAIR negatively regulates P16Ink4a and P14ARF signaling by enhancing the expression of miR-218 with subsequent inhibition of tumorigenesis in HCC (12). HOTAIR can be activated by FOXC1 and function through the repression of miR-1 (37). However, a global view of the actions of HOTAIR in HCC cells is lacking and can be explored with a systematic screen of HOTAIR-regulated genes and proteins.
High throughput omics strategies have already been applied to explore the function of ncRNAs. Transcriptomic studies have revealed extensive gene expression changes in response to HOTAIR dysregulation in cancer cells (7, 21, 32, 38–40), giving insight into the functional mechanisms of HOTAIR. However, mRNA expression levels do not necessarily correlate with cellular protein levels because of the existence of numerous post-transcriptional regulatory mechanisms (20, 41). In a previous study, we employed a quantitative proteomic approach to identify the downstream effectors of HOTAIR in HeLa cells (42). By comparing our proteomic data with previous transcriptomic results (21, 32, 38–40), we found that the correlation between global mRNA and protein expression was rather poor. We suppose that this lack of correlation is mainly because of the existence of post-transcriptional regulation. Moreover, we found that the overlap among different transcriptomic data sets is also limited (42). It is likely that the exact functional mechanisms of HOTAIR vary in different cancer cells. Thus, it is necessary to systematically screen the potential regulatory targets of HOTAIR in HCC at both the transcriptional and translational levels.
Here, an integrated analysis using a combined RNA sequencing (RNA-Seq)-based transcriptomic and quantitative proteomic analysis was employed to detect the potential regulatory targets of HOTAIR in HCC cells. We found that the expression of 673 transcripts and 293 proteins was dysregulated after HOTAIR suppression. Bioinformatics analysis revealed that many differentially expressed genes (DEGs) and differentially expressed proteins (DEPs) were implicated in pivotal signaling pathways. Further functional studies revealed that HOTAIR exerted its effects on cell proliferation, at least in part, through the regulation of opioid growth factor receptor (OGFr) expression. Our omics data provide new insights into the mechanism underlying the function of HOTAIR in HCC.
EXPERIMENTAL PROCEDURES
pX330-mCherry-gHOTAIR Plasmid Construction
The sgRNA design was based on the CRISPR design (http://crispr.mit.edu/) or CHOPCHOP (https://chopchop.rc.fas.harvard.edu/). The pX330-mCherry plasmid expressing hSpCas9 and sgRNA was modified based on the pX330: hSpCas9 + chimeric guide RNA vector (Addgene plasmid # 42230). The sgRNA oligos targeting the upstream and downstream regions of the HOTAIR gene were synthesized and cloned into the BbsI site of pX330-mCherry (pX330-mCherry-HOTAIR) as described (43). Then, the plasmids were transformed into Stbl3 competent cells and sequenced using the U6 sequencing primer.
Cell Culture and Transfection
Cell lines used in this study were purchased from the Cell Bank of Type Culture Collection (Chinese Academy of Sciences, Shanghai, China). Cells were cultured and maintained in Dulbecco's modified Eagle's high glucose medium (Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Gibco), 50 U/ml penicillin and 50 mg/ml streptomycin at 37 °C in a humidified incubator in the presence of 5% CO2.
For siRNA-mediated gene knockdown, cells were transfected with siRNA targeting HOTAIR (siHOTAIR), siRNA targeting OGFr (siOGFr) or a negative control siRNA (siNC) using Lipofectamine RNAiMAX (Invitrogen, Gaithersburg, MD), according to the manufacturer's instructions. All siRNAs were synthesized by GenePharma Co. Ltd. (Shanghai, China). The knockdown efficiency was confirmed by quantitative real-time reverse transcription-PCR (qRT-PCR). siRNA sequences are listed in supplemental Table S1.
For OGFr overexpression, the human OGFr cDNA was cloned into the mammalian expression vector pCMV-HA (Invitrogen). HepG2 and Huh-7 cells were transfected with the plasmid pCMV-HA + human OGFr (pOGFr) or pCMV-HA (empty vector, EV) using Lipofectamine 2000 (Invitrogen). Cells were harvested at 48 h post-transfection, and the overexpression of OGFr was confirmed by Western blot.
Cell Proliferation Assay
The cell proliferation assay was carried out using Cell Counting Kit-8 (Bossed, Beijing, China). Cells transfected with HOTAIR/OGFr siRNAs or the OGFr overexpression vector were seeded into 96-well plates at a density of 2 × 103 cells/well. After 0, 1, 2, 3, 4, and 6 days of incubation, CCK-8 reagents were added to each well, and cells were further incubated at 37 °C for 2 h. Each measurement was performed in quintuplicate, and the experiments were repeated three times. The relative numbers of viable cells were estimated using the absorbance OD at 450 nm.
Cell Cycle Progression Assay
Cells transfected with HOTAIR/OGFr siRNAs or the OGFr overexpression vector for 48 h were collected using trypsin and fixed in ice-cold 75% ethanol at −20 °C for 24 h. Then, the cells were washed with ice-cold phosphate buffered saline (PBS) twice and stained with propidium iodide (PI) containing RNase A (Beyotime, Haimen, China) for 30 min at 37 °C. Finally, the stained cells were analyzed using the BD FACSAria III Cell Sorting System (Becton Dickinson, Bedford, UK). A total of 10,000 events were acquired for each sample, and the percentage of cells in each cell cycle phase was determined using the ModFit LT software (Becton Dickinson). Experiments were performed in triplicate.
Wound Healing Assay
Wound healing assays were used to measure cell migration. Transfected cells were seeded into 6-well plates and incubated overnight to 80–90% confluence. The monolayers were scratched to form straight lines with a sterile 10 μl tip and washed with PBS to remove the cell debris. After the scratch, the cells were continuously grown in DMEM containing 1% FBS, and migrating cells were observed and imaged under an inverted microscope every 24 h until the wound healed.
Cell Invasion Assay
Cell invasion assays were measured using Transwell chambers (Corning, 8.0 μm pore size) coated with Matrigel (BD Biosciences, Franklin Lakes, NJ). Transfected cell (3 × 105) suspensions in serum-free medium were added to the upper chambers, and media containing 10% FBS was added to the lower chambers. After 48 h of incubation, cells that migrated to the lower chamber insert were fixed in methanol and stained with crystal violet stain. The permeated cells were calculated and imaged in five random fields under an inverted microscope.
qRT-PCR
Cells were harvested, and total RNA was extracted using the Trizol reagent (Invitrogen) and reverse transcribed into first strand cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany). Complementary DNA was amplified and quantified using the LightCycler 480 Real-Time PCR system (Roche) with the SYBR Green PCR Master Mix (Roche). Each sample was analyzed in triplicate, and the relative mRNA expression fold changes, calculated with the 2 − ΔΔCT method, were normalized to a housekeeping gene, GAPDH. Primer sequences are listed in supplemental Table S1.
RNA Sequencing and Identification of DEGs
HepG2 cells were transfected with either siHOTAIR or siNC for 48 h. Then RNA were extracted with the Trizol reagent (Invitrogen) according to the manufacturer's instructions. rRNA was subsequently eliminated with the RiboMinusTM Human Transcriptome Isolation Kits (Thermo Fisher Scientific, Waltham, MA). Sequencing libraries were generated using the TruSeq Stranded total RNA sample preparation kit (Illumina, San Diego, CA). The libraries were sequenced on an Illumina HiSeq 2000 platform (Illumina), and 126-bp paired ends were generated. Clean reads were mapped to the human reference genome (hg19) using TOPHAT (44). In addition, HTSeq (version 0.6.1) (45) was used to calculate the number of aligned reads per gene. The read count values from each biological replicate were first normalized using DESeq2 package (46) (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) and the normalized read count values were used for subsequent differential analysis.
Protein Extraction
Cells were harvested by washing twice with room temperature (RT) PBS. Lysate cells directly with ice-cold lysis buffer (50 mm Tris-HCL, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) with 1% protease inhibitor mixture (Roche). Rotate lysis at 4 °C for 30 min. Then lysates were clarified by centrifugation at 20,000 g at 4 °C for 15 min. The supernatants were precipitated by addition of at least 5 volumes of ice-cold acetone at −20 °C overnight. Centrifugation at 20,000 × g at 4 °C for 15 min. Remove and discard supernatant and allow pellet to dry.
Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) Labeling
HepG2 cells were transfected with either siHOTAIR or siNC. At 48 h post-transfection, cells were harvested and protein was extracted as described above. A total of 100 μg protein from each sample was run on a 10% SDS-PAGE gel (Invitrogen) to 1 cm. Then, the samples were subjected to in-gel tryptic digestion after reduction with DTT (10 mm) at 60 °C for 30 min and alkylated with iodoacetamide (IAA) (40 mm) in the dark for 1 h. Trypsin was added to the proteins at 1:50 (w:w) and incubated at 37 °C overnight. The digested peptides were extracted from the gel with 60% ACN, 5% formic acid (FA) and SpeedVac-dried and washed with 50% ACN to a neutral pH. Peptides were labeled using the iTRAQ 4-plex reagent kit (Applied Biosystems, Foster City, CA), combined after labeling and dried by SpeedVac.
First Dimensional Separation of iTRAQ-labeled Peptides
The Alliance 2695 HPLC system (Waters, Milford, MA) was used for peptide fractionation. The iTRAQ-labeled and combined samples were solubilized in 200 μl ammonium formate (20 mm, pH 10) and injected onto an Xbridge C18 column (2.1 × 150 mm, 3.5 μm, Waters) using a linear gradient of 1% buffer B increase/min from 2–45% buffer B (buffer A: 20 mm ammonium formate, pH 10, B: 20 mm ammonium formate in 90% ACN, pH 10). One-minute fractions were collected and combined into 18 fractions and vacuum-dried and SpeedVac-dried.
LC - tandem Mass Spectrometry (MS/MS)
Peptides from each fraction were analyzed by nano-LC-MS/MS using an UltiMate 3000 RSLC System (Dionex, Sunnyvalue, CA) coupled with a Q Exactive™ Hybrid Quadrupole-Orbitrap MS (Thermo Fisher Scientific, Bremen, Germany). Samples were loaded onto a trap cartridge (100 μm × 2 cm) self-packed with 200 Å, 5 μm Magic C18AQ resin (Michrom Bioresources, Auburn, CA) and washed with 0.2% FA (buffer A) at a flow rate of 10 μl/min for 5 min. The trap was brought in-line with the homemade analytical column (Magic C18AQ, 200 Å, 3 μm, 75 μm × 50 cm), and peptides were fractionated at 300 nl/min with a multistepped gradient of buffer B (0.16% FA, 80% ACN; 4 to 15% B for 30 min, 15–25% B for 60 min and 25–50% B for 50 min). A data-dependent acquisition mode was used to acquire MS data with a cyclic series of a full scan with Orbitrap detection at a resolution of 120,000 followed by MS/MS scans (30% of the collision energy in the HCD cell) of the 20 most intense ions with a repeat count of 2 and a dynamic exclusion duration of 30 s.
MS/MS Data Analysis
The MS/MS data were searched using three search engines (MyriMatch v2.2.8634, X! Tandem v2015.04.01.1 and MS-GF+ v2016.06.29) against the UniProt human protein database (UP000005640, 70615 sequences, release 2016_06) with decoy sequences. Parameters for database searching were set as follows: Fixed modifications, Carbamidomethyl (C), iTRAQ4plex (N-term) and iTRAQ4plex (K); Variable modifications, Oxidation (M), iTRAQ4plex (Y) and Deamidated (NQ); Precursor ion mass tolerance, 10 ppm; MS/MS mass tolerance, 0.05 Da; Enzyme specificity, trypsin; maximum missed cleavages, 1. Then, the search results were integrated by IPeak (47, 48), a powerful software that combines multiple search engine results. The following criteria were set as a requirement for protein identification: peptide length ≥ 7, 2) FDR ≤ 1% at both the PSM and protein levels. The protein quantification was performed using IQuant (49) with variance stabilization normalization (VSN) normalization (50, 51). At least two unique peptides are required for protein quantitation. The protein ratios were calculated as weighted average ratios and only unique peptides were used. IQuant calculated a p value for each protein by permutation test and then the p value was corrected by Benjamini-Hochberg approach (52) to get q-value.
Parallel Reaction Monitoring (PRM) Analysis
HepG2 cells were transfected with either siHOTAIR or siNC. At 48 h post-transfection, cells were harvested and protein was extracted. Protein reduced, alkylated and digested as described above. The tryptic peptides were dissolved in 0.1% FA (solvent A), directly loaded onto a home-made reversed-phase analytical column (75 μm × 15 cm). The gradient was comprised of an increase from 7% to 25% solvent B (0.1% FA in 90% ACN) over 40 min, 25% to 40% solvent B in 14 min and climbing to 80% in 4 min, then holding at 80% for the last 4 min, all at a constant flow rate of 400 nL/min on an EASY-nLC 1000 UPLC system. The peptides were subjected to NSI source followed by MS/MS in Q Exactive™ Hybrid Quadrupole-Orbitrap Plus MS (Thermo Fisher Scientific) coupled online to the UPLC. The electrospray voltage applied was 2.0 kV. The m/z scan range was 350 to 1400 for full scan, and intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were then selected for MS/MS using NCE setting as 27 and the fragments were detected in the Orbitrap at a resolution of 35,000. A data-independent procedure that alternated among one MS scan followed by 20 MS/MS scans. Automatic gain control was set at 3 × 106 for full MS and 1 × 105 for MS/MS. The maximum IT was set at 50 ms for full MS and auto for MS/MS. The isolation window for MS/MS was set at 2.0 m/z.
The resulting MS data were processed using Skyline version.3.6. Peptide settings: enzyme was set as Trypsin [KR/P], max missed cleavage set as 0, the peptide length was set as 7–25, fixed modification was set to carbamidomethyl on Cys. Transition settings: precursor charges were set as 2, 3, ion charges were set as 1, ion types were set as b, y. The product ions were set as from ion 3 to last ion, the ion match tolerance was set as 0.02 Da. Peptides were quantified by extracting the peak areas of accurate fragment ions, and they were then integrated across the peptides' elution profiles. For each peptide, transition peak areas were normalized by the average of the sum of the transition peak areas for all the peptides across the runs.
Western Blotting
Western blotting were carried out as previously described (42). In brief, cells were lysed with ice-cold Western and IP lysis buffer (Beyotime) containing 1% PMSF and protease inhibitor mixture (Thermo Fisher Scientific). Protein concentration in the lysates was quantitated using a BCA Protein Assay Kit (Thermo Fisher Scientific). All primary antibodies and peroxidase-conjugated anti-mouse or anti-rabbit IgG used in this study were purchased from Abclonal Technology (Wuhan, China). Immunoreactive bands were developed with a SuperSignal West Pico kit and visualized with an Image Scanner (GE Healthcare, Waukesha, WI). Protein loading was normalized using the GAPDH antibody, and blot densitometry analysis was performed using ImageJ (National Institutes of Health, Bethesda, MD).
Immunofluorescence (IF) Microscopy
Immunofluorescence staining was performed as previously described (42). Briefly, the transiently transfected HCC cells were grown in 35-mm glass-bottom dishes at 37 °C for 48 h, and washed and fixed with ice-cold 100% ethanol. Subsequently, the cells were permeabilized in 0.5% Triton-X100 and blocked with 10% BSA for 1 h at RT. Then, the cells were washed with PBS and incubated with rabbit anti-OGFr polyclonal antibody at a dilution of 1:200 at 4 °C overnight. After washing with PBS, the cells were incubated with Dylight488-conjugated Goat anti-Rabbit IgG (H+L) (Abbkine, Los Angeles, CA) at a dilution of 1:1000 for 1 h at RT in darkness. For nucleic acid labeling, cells were embedded in ProLong Gold antifade reagent with 4′6-diamidino-2-phenylindole (DAPI). Cells were washed thoroughly with PBS and examined using an LSM 710 laser scanning confocal microscope (Carl Zeiss, Thornwood, NY).
Tissue Microarray and Immunohistochemistry (IHC) Analysis
Human HCC tissue microarray slides (Cat: OD-CT-DgLiv04–001) were purchased from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). A total of 41 matched pairs of HCC samples and their corresponding adjacent liver tissues were obtained with detailed patient information, including age, gender, metastasis status, pathological grades, tumor size, TNM stage, and AJCC stage. The human tissue samples were used on the basis of the guidelines of the Nanfang Hospital. Written informed consent forms were signed by each participant before his or her inclusion in the study. For immunohistochemical assays, the tissue microarrays were deparaffinized in xylene, rehydrated through decreasing concentrations of ethanol, washed with PBS for 5 min, submerged into prewarmed antigenic retrieval buffer (0.5 mg/ml trypsin in Tris-HCl pH 8.0) and kept in retrieval buffer at 37 °C for 20 min for antigenic retrieval. Sections were then incubated in 2% BSA for 30 min to block nonspecific binding, followed by incubation with rabbit anti-OGFr polyclonal antibody at a dilution of 1:150 at 4 °C overnight. Immunostaining continued with Dylight488-conjugated secondary antibodies (Abbkine) at a dilution of 1:200 for 1 h at RT in darkness. After washing with PBS, sections were mounted in ProLong Gold antifade reagent with DAPI for nucleic acid labeling. The tissue microarrays were washed three times with PBS and scanned using a digital microscopy scanner Pannoramic MIDI (3DHISTECH Ltd., Budapest, Hungary) with a 20 × microscope objective. The Pannoramic Viewer 1.15.2 software (3DHISTECH) was used for image viewing.
Experimental Design and Statistical Rationale
For mRNA expression profiling, an RNA-Seq-based transcriptomic analysis was conducted for siHOTAIR group and siNC group. Three biological replicates are included for each group. Sample siNC-2 was not used for quantification because less than 70% of its reads were mapped to the reference genome. We use “q-value ≤ 0.001 and the absolute value of log2 ratio ≥ 1” as the threshold to determine the significance of the gene expression difference. For protein expression profiling, an iTRAQ-based quantitative proteomic approach was employed to screen DEPs in siHOTAIR group and siNC group. Two biological replicates are included in the proteomics experiments. siHOTAIR/siNC ratio of each quantitated protein was normalized using z-score analysis, as described previously (53–55). In brief, siHOTAIR/siNC ratios from two biological replicates were averaged, and converted into log2 space to determine geometric means and facilitate normalization. Then the average and standard deviations (S.D.) of log2 ratios for the data set were calculated. The log2 siHOTAIR/siNC ratio of each protein were converted into a z-score, using the following formula:
| (Eq. 1) |
in which b represents an individual protein in a data set population (a….n), and the z-score is the measure of how many standard deviation units (expressed as “σ”) that protein's log2 L/H ratio is away from its population mean. Thus, a protein with a z-score of 1.960 indicates that it is outside the 95% confidence level. “q-value ≤ 0.05 for both biological replicates, coefficient of variation (CV) < 20% and the absolute value of z-score ≥ 1.96” were used as criteria to define the DEPs in the HOTAIR knockdown cells. Three biological replicates were performed for PRM analysis. Peptide ratios from replicate experiments were averaged, and the Grubbs' method was used for detecting and removing outliers. For all other experiments carried out in this study, including cell proliferation assay, cell cycle progression assay, wound healing assay, cell invasion assay, Western blot, IF microscopy and IHC assay, at least three independent biological replicates were performed. All data are presented as the means ± S.D., and a p value < 0.05 was considered to indicate statistical significance. Statistical analyses were performed using SPSS version 17.0 (Released 2008. Chicago: SPSS Inc.) and GraphPad Prism version 6.01. Student's t test (two-tailed) was used to calculate the significance of differences among different groups.
RESULTS
Comparison of CRISPRi and siRNA-mediated Knockdown of HOTAIR
The CRISPR/Cas9 technique has emerged as a simple and powerful genome editing tool for DNA modification in recent years, and it has already been applied to lncRNA deletion (56, 57). At the beginning of this study, we tested the feasibility of HOTAIR knockdown with the CRISPR system. Modified plasmid px330:hspcas9+chimeric (Addgene plasmid # 42230) was used to establish HOTAIR deletion cell lines. pX330-mCherry-gHOTAIR was successfully transfected into HeLa cells (supplemental Fig. S1A). After single cell screening using flow cytometry and single cell culturing, we obtained three CRISPRi cell lines, named gRNA-1, gRNA-2 and gRNA-3 (Fig. 1A). We found that HOTAIR was still detectable in these cell lines, but the expression levels were significantly reduced (Fig. 1B). The sequencing results are shown in supplemental Fig. S1B. Meanwhile, we also examined the expression level of the HOXC11 gene in these CRISPRi cell lines, which is adjacent to the HOTAIR gene. The results showed that CRISPRi-mediated targeting using three independent gRNAs resulted in simultaneous knockdown of HOXC11, but siRNA-mediated knockdown of HOTAIR had no significant effect on HOXC11 expression (Fig. 1B). A recent study evaluated whether the CRISRP approach was applicable to target lncRNAs, and the results showed that over 60% of the lncRNA loci were at risk for inadvertent deregulation of neighboring genes, including HOTAIR (58). Taken together, the CRISPR approach is not suitable for HOTAIR deletion. Thus, siRNA-mediated HOTAIR knockdown was employed in this study.
Fig. 1.
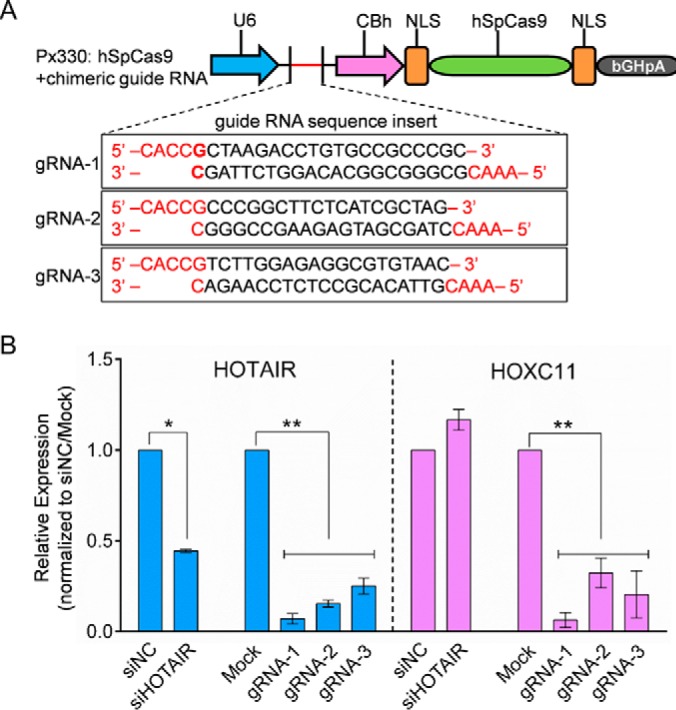
Comparison of CRISPRi and siRNA-mediated knockdown of HOTAIR. A, The map of pX330 vector and deletion site of HOTAIR. B, The relative expression of HOTAIR and HOXC11 in siRNA transfected or CRISPRi-knockdown cells. Hela cells were transfected with either siNC or siHOTAIR. Also, Hela cells were transfected with px330-mCherry containing one of the three indicated sgRNAs targeting HOTAIR. qPCR results for HOXC11 and HOTAIR normalized to siNC and Mock. Data are presented as means ± S.D. and represent results from three independent experiments. Statistically significant differences are indicated: *p < 0.05; **p < 0.01.
Functional Effects of HOTAIR Inhibition on HCC Cells
To study the physiological function of HOTAIR in HCC progression, siRNAs were used to knockdown HOTAIR expression in HCC cells, and the knockdown efficiency was over 50% at 48 h post-transfection (Fig. 2A). CCK-8 assay results showed that HOTAIR inhibition significantly suppressed HepG2 cell proliferation (Fig. 2B). Meanwhile, the percentage of G1-phase cells was significantly increased after HOTAIR inhibition, whereas cells in the G2/M phase decreased (Fig. 2C), indicating a G1-phase arrest. Then, we examined the effect of HOTAIR suppression on cell migration and cell invasion using wound-healing assays and Boyden chamber assays, respectively. The migration and invasion ability of HepG2 cells were both significantly decreased at 48 h after HOTAIR knockdown (Fig. 2D–2G). These results indicated that HOTAIR played a role in the regulation of cell growth, cell cycle, cell migration and cell invasion of HCC cells. Then, an integrated transcriptomics and proteomics analysis was employed to explore the functional mechanisms of HOTAIR (Fig. 3A).
Fig. 2.
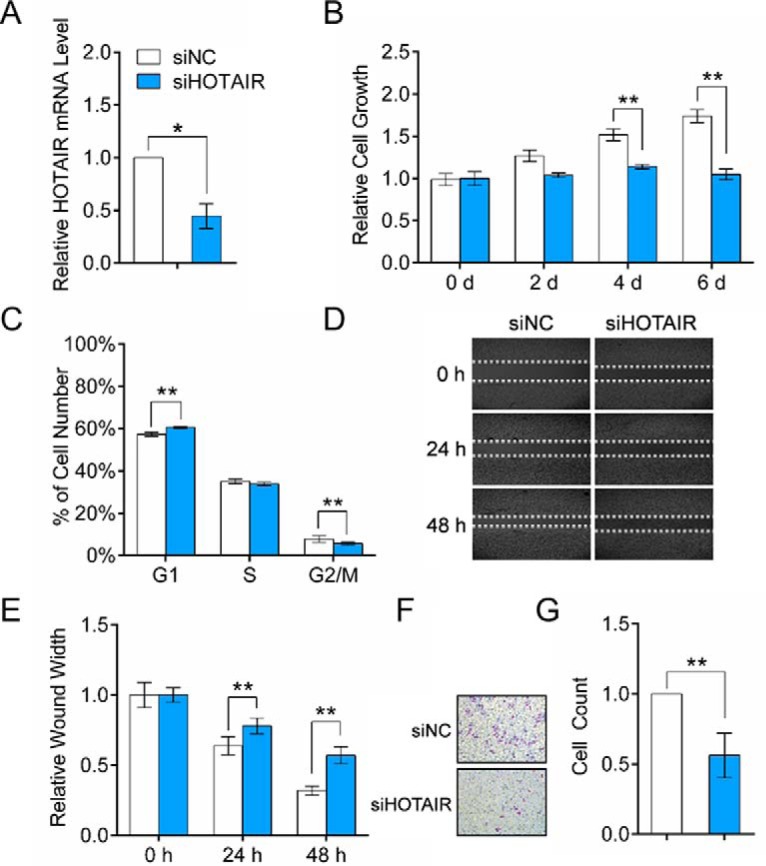
Functional effects of HOTAIR inhibition on HepG2 cells. A, HepG2 cells were transfected with siHOTAIR or siNC for 48 h. HOTAIR knockdown efficiency was determined by qRT-PCR. The expression level of HOTAIR was normalized to GAPDH. B, HOTAIR knockdown in HepG2 cells significantly inhibits cell growth. C, Effect of HOTAIR knockdown on cell cycle progress. D, HOTAIR inhibition led to a significant reduction of cell migration as determined by a wound-healing assay. E, Quantification of the wound healing assay. F, Effect of HOTAIR knockdown on cell invasion, as determined in a Boyden chamber assay. G, Numbers of cells on the underside of the filter. Significantly enhanced invasion (p < 0.05) is indicated.
Fig. 3.

Workflow and overview of transcriptomic and proteomic data. A, The overall workflow of this study. B, The Venn diagram shows the number of transcripts and proteins quantified. C, Scatterplot of the correlation between genes quantified in both transcriptomic and proteomic data sets. The red plot indicates DEPs and black plot indicates non-DEPs. D, Heatmap showing all the quantified proteins and their corresponding mRNAs. E Heatmap showing all the DEPs and their corresponding mRNAs.
Transcriptomic Analysis
A total of 19,589 genes were quantitated after calculating the expression level of each mapped gene. Detailed information of the transcriptomic results such as the base mean, fold change and q value are shown in supplemental Table S2. Based on the criteria described in the Experimental Design and Statistical Rationale subsection, 673 genes were regarded to be DEGs after HOTAIR inhibition, of which 268 were upregulated and 405 were downregulated (supplemental Table S3). Correlation analysis of the biological replicates showed good reproducibility (supplemental Fig. S2A).
Quantitative Proteomic Analysis
The iTRAQ-based quantitative proteomics strategy was applied to investigate the HOTAIR-regulated proteins. A total of 6547 proteins were quantified (supplemental Table S4). Based on the criteria described in the Experimental Design and Statistical Rationale subsection, 293 proteins were defined as DEPs after HOTAIR inhibition, among which 147 were upregulated and 146 were downregulated (supplemental Table S5). Correlation analysis of the biological replicates showed good reproducibility (supplemental Fig. S2B).
Comparison of Transcriptome and Proteome Data
A correlation analysis between the RNA-Seq transcriptomic and quantitative proteomic data was conducted. Among the 6547 quantitated proteins, 6042 were also detected in the transcriptomic data sets (Fig. 3B). The expression levels of all quantified proteins and their corresponding mRNAs showed a moderate correlation (r = 0.5859 (Fig. 3C and 3D), whereas a high correlation between the DEPs and their corresponding mRNAs was indicated (r = 0.8584) (Fig. 3C and 3E). The transcriptomic analysis identified 673 DEGs, and 245 (36.4%) of them were also quantitated in the quantitative proteomics (supplemental Table S3). Eighty-six genes were detected to be dysregulated at both the transcription and translation levels. It is intriguing that these 86 genes show the same direction of change at the two levels (supplemental Table S3), implying that the expression changes of these proteins are mainly driven by transcriptional changes. However, the mRNA expression of 203 DEPs was unchanged, suggesting the existence of post-transcriptional regulation.
Bioinformatics Analyses of the DEGs and DEPs
To understand the biological function of the DEGs and DEPs, we first classified them using the PANTHER Classification Systems (59, 60). The 673 DEGs and 293 DEPs were mainly assigned to 24 and 21 PANTHER Protein Class categories, respectively (Fig. 4A and supplemental Table S6). Most of the DEGs and DEPs were assigned to the same protein classes, the top six of which were “nucleic acid binding,” “enzyme modulator,” “hydrolase,” “transferase,” “cytoskeletal protein,” and “transcription factor.” It is worth noting that numerous transcription factors (TFs) were dysregulated after HOTAIR inhibition. Sixteen TFs were dysregulated at the protein level, and 48 were dysregulated at the transcription level, among which 6 TFs were modulated at both levels (supplemental Table S6).
Fig. 4.

Bioinformatics analysis of the DEGs and DEPs. A, PANTHER Protein Class ontology classification of DEGs and DEPs after HOTAIR knockdown. B, KEGG pathway enrichment analysis of the DEPs (p < 0 .05). KEGG pathways which were also significantly enriched in DEGs are shown. C, DEGs and DEPs involved in pathways in cancer. DEGs and DEPs after HOTAIR suppression were mapped to the “pathways in cancer,” according to the “pathways in cancer” map in KEGG with some modifications. DEPs are colored in red; DEGs are colored in blue, genes which are regulated at both mRNA and protein levels are colored green.
We next performed GO and KEGG pathway enrichment analysis of the DEGs and DEPs using DAVID Bioinformatics Resources 6.8 (61, 62). Functional enrichment of DEPs showed that the top-ranked biological processes GO terms (ranked by protein count) were “intracellular protein transport,” “apoptotic process,” “protein folding,” “cell proliferation,”and “negative regulation of cell proliferation” (supplemental Table S7–1). Besides, “positive regulation of apoptotic process,” “cell migration,” “regulation of cell migration,” and “cell motility” was also enriched (supplemental Table S7–1). These results are consistent with the functional effects caused by HOTAIR knockdown in HCC cells. Moreover, some biological processes were significantly enriched only in the upregulated proteins, such as “negative regulation of cell proliferation,” “cell migration,” which is correlated with the inhibited cell growth and cell migration after HOTAIR knockdown (supplemental Table S7–3). And some were only enriched in the downregulated proteins, such as “mRNA splicing, via spliceosome,” “response to virus,” and “autophagy,” suggesting that proteins involved in these cellular processes may positively regulated by HOTAIR (supplemental Table S7–4). KEGG pathway enrichment analysis revealed that both DEPs and DEGs are significantly enriched in “Pathways in cancer (hsa05200)” with the most protein count (Fig. 4B and supplemental Table S8). Fourteen DEPs and 22 DEGs (4 of which are both DEGs and DEPs) were mapped to the “Pathways in cancer,” and most of these DEGs/DEPs are involved in cell proliferation, cell cycle, and cell apoptosis (Fig. 4C). The other two pathways that are enriched in both DEPs and DEGs are “Focal adhesion” and “Proteoglycans in cancer.” These results indicate the important role of HOTAIR in regulating cancer cell physiology.
To explore the interaction relationship among these DEPs, a protein-protein interaction (PPI) analysis was carried out. Because only 245 of the DEGs have corresponding protein expression information, it was unclear whether the rest of the DEGs were dysregulated at the protein level. To gain more potential PPI information, all DEPs and DEGs without protein expression information were combined and searched against the STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) database version 10.0 (63) and visualized by Cytoscape (64). An interaction network of the HOTAIR-regulated genes and proteins was generated, involving 137/293 (48.7%) DEPs and 115 DEGs (interaction sources were set to “experiments,” 38 genes were regulated at both levels) (supplemental Fig. S3 and supplemental Table S9).
Validation of the Transcriptomic and Quantitative Proteomic Data
Twenty-one DEPs (CDK6, OGFr, PAFAH1B2, GRPEL2, CASP7, TGFBR1, AP2A1, RTFDC1, POFUT1, MLH1, EFEMP1, CCND1, RBM25, MAP2K1, FAF2, BCL2L1, VAMP8, PODXL, CHKA, STAT5A, and UBAC2) that are involved in cancer-related pathways were chosen to validate the proteomic data by Western blot. As shown in Fig. 5A, the Western blot results for all selected proteins were consistent with the quantitative proteomic results. To further validate the proteomic data, we select 10 proteins for PRM analysis, in which 8 were successfully quantitated. The results also showed that the PRM results have good correlation with proteomics results (Fig. 5B and supplemental Table S10). qRT-PCR was also performed to examine the mRNA expression levels of these selected proteins. The mRNA expression levels of most selected proteins were in agreement with the transcriptomic data (Fig. 5C). These results demonstrated that our omics results are reliable. We also examined the expression of these DEPs in another HCC cancer cell line, Huh-7 cells. Interestingly, the trends in the protein expression changes were the same as those in HCC cells (supplemental Fig. S4).
Fig. 5.

Validation of the proteome data and OGFr expression in HCC cells and tissues. A, A set of DEPs were validated by Western blot in HepG2 cells. GAPDH was used as an internal control. B, A set of DEPs were selected and validated by PRM in HepG2 cells. The dashed line was drawn at scale 1. Greater or less than scale 1 represent up- or downregulation after HOTAIR inhibition, respectively. Each data point is calculated from averages of biological triplicates. Results of the PRM analysis were consistent with ITRAQ data. C, Comparison of relative protein expression of 11 selected differentially regulated proteins measured by MS and qPCR. The dashed line was drawn at scale 1. Greater or less than scale 1 represent up- or downregulation after HOTAIR inhibition, respectively. Each data point is calculated from averages of biological triplicates. D, Representative confocal microscopy images showing the expression level of OGFr in HepG2 cells after HOTAIR knockdown. The nucleus was stained with DAPI. The images were acquired by fluorescence microscopy under a 60 × objective lens (scale bar, 11 μm). E, A representative image from the HCC tissue microarray showing HE staining of the HCC cancer tissues and their corresponding adjacent liver tissues. F, Digital microscopy scanner images showing the expression level of OGFr in HCC tissues was downregulated compared with their corresponding adjacent liver tissues. The images were acquired by Pannoramic MIDI with a 20 × microscope objective (scale bar, 500 μm).
HCC Cell Growth Was Negatively Regulated by OGFr after HOTAIR Knockdown
The opioid growth factor (OGF)-opioid growth factor receptor (OGFr) axis is an important physiological regulator of cell growth in diverse human cancers, and OGFr overexpression can inhibit cell growth (65–69). We have shown that HepG2 cell growth is significantly suppressed after HOTAIR knockdown (Fig. 2B), whereas OGFr is significantly induced at both mRNA and protein levels (Fig. 5A and 5C). The upregulation of OGFr in HCC cells was also validated by IF staining (Fig. 5C and supplemental Fig. S5). We also examined OGFr expression in human HCC tissues using an HCC tissue microarray, and the results of the immunohistochemical assays showed that the expression of OGFr in HCC tissues was significantly lower than that in adjacent liver tissues (Fig. 5E, 5F, and supplemental Fig. S6). Then, we studied the functions of OGFr in HCC through either inhibition or overexpression of OGFr. The knockdown and overexpression efficiency was validated by both qRT-PCR and Western blot (Fig. 6A, 6B and Fig. 7A, 7B). The CCK-8 assay results showed that the OGFr knockdown significantly promoted HCC cell proliferation (Fig. 6C), whereas OGFr overexpression resulted in significantly reduced cell proliferation (Fig. 7C). We further demonstrated that knockdown the expression of OGFr in HCC cells resulted in promotion of the G1/S phase transition (Fig. 6D), whereas OGFr overexpression induced a G1-phase arrest in HCC cells (Fig. 7D).
Fig. 6.

Functional effects of OGFr inhibition on HCC cells. A, HepG2 and Huh-7 cells were transfected with siOGFr or siNC for 48 h. OGFr knockdown efficiency was determined by qRT-PCR. The expression level of OGFr was normalized to GAPDH. B, Western blot analysis of OGFr protein expression 48 h after transfection with siOGFr or siNC. GAPDH was used as an internal control. C, OGFr inhibition in HCC cells significantly promotes cell growth. D, Effect of OGFr knockdown on HCC cell cycle. E, Effect of OGFr knockdown on cell invasion, as determined in a Boyden chamber assay. F, Relative cell invasion on the underside of the filter. Significantly enhanced invasion (p < 0.05) is indicated. G, OGFr inhibition had no effect on HCC cell migration as determined by a wound-healing assay. H, Quantification of the wound healing assay. Data are presented as means ± S.D. and represent results from three independent experiments. Statistically significant differences are indicated: *p < 0.05; **p < 0.01. (I) Western blot showing the expression levels of p16INK4a and p21WAF1/CIP1 after OGFr inhibition.
Fig. 7.

Functional effects of OGFr overexpression on HCC cells. A, HepG2 and Huh-7 cells were transfected with empty vector (EV) or OGFr overexpression vector (pOGFr) for 48 h. OGFr overexpression efficiency was determined by qRT-PCR. The expression level of OGFr was normalized to GAPDH. B, Western blot analysis of OGFr protein expression 48 h after transfection with EV (pCMV-HA) or pOGFr (pCMV-HA-OGFr). GAPDH was used as an internal control. C, OGFr overexpression in HCC cells significantly inhibits cell growth. D, Effect of OGFr overexpression on HCC cell cycle. E, Effect of OGFr overexpression on cell invasion, as determined in a Boyden chamber assay. F, Relative cell invasion on the underside of the filter. Significantly enhanced invasion (p < 0.05) is indicated. G, OGFr overexpression had no effect on HCC cell migration as determined by a wound-healing assay. H, Quantification of the wound healing assay. I, Western blot showing the expression levels of p16INK4a and p21WAF1/CIP1 after OGFr overexpression or HOTAIR inhibition. Data are presented as means ± S.D. and represent results from three independent experiments. Statistically significant differences are indicated: *p < 0.05; **p < 0.01.
Furthermore, the invasion capability of HCC cells was significantly increased after the OGFr knockdown (Fig. 6E and 6F), whereas the number of invaded cells was significantly reduced after OGFr overexpression (Fig. 7E and 7F). However, the migration ability of HCC cells was not affected by OGFr expression (Fig. 6G and 6H, Fig. 7G and 7H). In summary, these results suggested that HOTAIR functions as a positive regulator of HCC cell proliferation, whereas OGFr functions as a negative regulator of HCC cell proliferation. HOTAIR may exert its effects on HCC cell proliferation and invasion by regulating the OGFr expression level.
DISCUSSION
HOTAIR has emerged as a prognostic biomarker and potential therapeutic target in HCC (10, 11, 28, 29). In the present study, we found that HOTAIR expression could significantly influence the physical function of HCC cells (Fig. 2), which is consistent with previous reports (10, 28–30). However, the mechanisms underlying its function remain largely unknown. Therefore, we conducted a combined transcriptomics and quantitative proteomics study to systematically screen the potential regulatory targets of HOTAIR in HCC cells. A total of 673 transcripts and 293 proteins were found to be dysregulated in HCC cells after HOTAIR inhibition, which provides a global view of the downstream effectors of HOTAIR.
HOTAIR can modulate gene expression at different levels. It may serve as a molecular scaffold mediating the PRC2 and LSD1/CoREST/REST protein complexes to modulate the transcription of hundreds of genes (33). It can also function as a competitive endogenous RNA to regulate gene expression post-transcriptionally (23, 34). MicroRNAs have already been reported to be negatively regulated by HOTAIR in HCC, such as miR-218 and miR-1 (12, 37). Thus, a systematic comparison of the protein and RNA expression changes after HOTAIR inhibition could provide information about which protein changes are because of post-transcriptional regulatory events, and which are mainly driven by transcriptional changes. Combining data on RNA and protein changes could also provide insights about the protein changes caused directly by HOTAIR dysregulation (primary targets) and changes that are because of more downstream effects (secondary and tertiary targets). In the present study, eighty-six genes were dysregulated at both the mRNA and protein levels with changes in the same direction, suggesting that the expression changes of these proteins are mainly caused by transcriptional changes. However, the transcriptional level of 203 DEPs remains unchanged, suggesting that these proteins may be modulated by some post-transcriptional mechanisms. We also compared our transcriptomic data with previously published transcriptomic data after HOTAIR dysregulation (21, 38, 39, 70) and found that the overlap among these transcriptomic data was rather limited. Therefore, we supposed that HOTAIR may have different regulatory mechanisms in different cancers.
The bioinformatics analysis revealed that a total of 115 DEGs (without protein expression information) and 137 DEPs (in which 36 genes were both DEPs and DEGs) are implicated in PPI networks, suggesting that these downstream effectors of HOTAIR tend to interact with each other and are functionally related (supplemental Fig. S3). The best way to fully understand the functional roles of these HOTAIR-regulated proteins in cancer physiology was to map them to well-defined biological pathways. The KEGG pathway enrichment analysis revealed that the DEGs and DEPs were enriched in some fundamental biological regulatory pathways (supplemental Table S8), providing a global view of the downstream pathways of HOTAIR. Importantly, 16 DEPs and 18 DEGs (4 of which were both DEGs and DEPs) were enriched in “pathways in cancer” (supplemental Table S8). These DEGs/DEPs participate in several important signaling pathways, including the PI3K-Akt, JAK-STAT and MAPK signaling pathways, which are key regulators of cell proliferation, cell cycle, cell apoptosis and angiogenesis (Fig. 4C). Identification of these cancer related genes/proteins will be very informative for deciphering the modulating mechanisms of HOTAIR in cancer cell biology. It is intriguing that several DEGs and DEPs were identified to be TFs (supplemental Table S6), among which 24 were involved in the PPI network (supplemental Fig. S3). These TFs may play important roles in regulating the transcription of several genes after HOTAIR knockdown, and elucidating the function of these TFs may be crucial toward revealing the pleiotropic functions of HOTAIR. For example, YAP1, which is known to play a role in the development and progression of multiple cancers as a transcriptional regulator of Hippo signaling pathway (71–75), can interact with 11 other DEGs/DEPs, suggestion that it may be responsible for the expression changes of some DEGs/DEPs.
It is notable that the expression of OGFr was clearly upregulated at both the mRNA and protein levels after HOTAIR knockdown. OGFr is a receptor for the opioid growth factor (OGF). OGF, also known as [Met5]-enkephalin, is a constitutively expressed native opioid peptide (67, 68, 76). The OGF-OGFr axis was demonstrated to be a negative regulator of cell growth in diverse human cancers (65–69), including HCC (76), and the mechanism was related to inhibition of DNA synthesis rather than the apoptotic or necrotic pathways (76). The unbound OGFr is localized to the outer nuclear envelope, where it binds to OGF and is translocated into the nucleus with the aid of nuclear localization signals in OGFr (76, 77). It has been reported that the OGF-OGFr axis uses the p16INK4a and p21WAF1/CIP1 pathway to restrict proliferation in head and neck cancers and pancreatic cancer cells, as well as in normal cells (67, 68, 78). The OGF-OGFr axis can induce the expression of two cyclin-dependent kinase inhibitors, p16INK4a and p21WAF1/CIP1, leading to a retardation of cells in the G1 phase of the cell cycle and a decrease in cell proliferation (67, 68, 78). In the present study, we found that after HOTAIR knockdown, the HCC cells were arrested in the G1 phase, cell growth was inhibited (Fig. 2C), and the expression levels of p16INK4a and p21WAF1/CIP1 were significantly increased (Fig. 7I). Interestingly, the effects of HOTAIR suppression on cell proliferation and the cell cycle in HCC cells can be mimicked by OGFr overexpression (Fig. 7C and 7D). And the expression levels of p16INK4a and p21WAF1/CIP1 were also significantly increased after OGFr overexpression (Fig. 7I), whereas significantly decreased after OGFr knockdown (Fig. 6I). These results proved that the inhibited HCC cell proliferation caused by HOTAIR knockdown was, at least in part, mediated by OGF-OGFr axis and the downstream p16INK4a and p21WAF1/CIP1pathways. In our previous study, we have revealed that HOTAIR may not directly interact with OGFr through RNA pull-down experiment (unpublished data). We deduce that OGFr may be regulated by HOTAIR through two mechanisms: (1) HOTAIR may negatively regulate a transcription factor, which binds to the promoter region of OGFR, and enhance its transcription level (2). HOTAIR may modulate the mRNA expression of OGFr by regulating the expression of one or more miRNAs which targeting the 3′UTR of OGFR mRNA. We will validate our hypothesis in our further studies.
HOTAIR can promote cell proliferation in a variety of cancers, which seems to be a general action of HOTAIR (19, 25, 29, 79). To investigate whether HOTAIR suppression can induce OGFr expression in other cancer cells or noncancer cells, we examined the expression of OGFr in HeLa, MCF-7, and 293T after HOTAIR inhibition. Upregulation of OGFr was also detected in these cell lines (supplemental Fig. S7). Therefore, it is supposed that HOTAIR suppression- induced OGFr expression may be a general mechanism for regulating cell proliferation. The GO enrichment analysis of the DEPs revealed that “cell proliferation” and “negative regulation of cell proliferation” were significantly enriched (supplemental Table S7). Intriguingly, we found that besides OGFr, some other negative regulators of cell proliferation were also upregulated, including YAP1, CDK6, PTPRJ, FABP3, DLG5, and FABP3. These proteins may also contribute to the decreased cell growth after HOTAIR inhibition. In other words, HOTAIR knockdown induced suppression of HCC cell proliferation may be a combination effect of several proteins.
In conclusion, we have performed an integrated omic analysis to screen the downstream effectors of HOTAIR in HCC cells. Hundreds of genes and proteins were found to be dysregulated after HOTAIR inhibition, providing a global view of the mechanisms of action of HOTAIR in HCC and revealing the complexity of HOTAIR regulation. These DEGs and DEPs are implicated in many critical signaling pathways in cancer progression. Further functional studies revealed that HOTAIR exerts its effects on cell proliferation, at least in part, through the regulation of opioid growth factor receptor (OGFr) expression. We expect that our omics data may shed light on the molecular basis of the regulatory role of HOTAIR in the progression of human HCC.
DATA AVAILABILITY
The MS raw data for iTRAQ-LC-MS/MS have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (80) with data set identifier PXD006554. The MS raw data for PRM have been deposited in the PeptideAtlas database (http://www.PeptideAtlas.org) with the identifier PASS01010. The RNA-Seq data have been deposited in NCBI's Gene Expression Omnibus (81) and are accessible through GEO Series accession number GSE98091 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98091).
Supplementary Material
Acknowledgments
We thank the Center for Instrumental Analysis and Metrology, Institute of Hydrobiology, Chinese Academy of Science for our Flow Cytometry analysis (Ms Yan Wang). We also thank The Core Facility and Technique Support, Wuhan Institute of Virology for technique support in digital microscopy scanner Pannoramic MIDI (Ms Juan Min).
Footnotes
* This work was supported by the National Natural Science Foundation of China (Grant No. 31370746 and No. 91540102), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB14030202).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- lncRNA
- long noncoding RNA
- HOTAIR
- HOX Transcript Antisense Intergenic RNA
- OGFr
- Opioid Growth Factor Receptor
- HCC
- Hepatocellular carcinoma
- DEG
- differentially expressed gene
- DEP
- differentially expressed protein
- PRM
- parallel reaction monitoring
- ncRNA
- noncoding RNA
- HOXC
- Homeobox C
- PRC2
- polycomb repressive complex 2
- LSD1
- lysine-specific demethylase 1A
- H3K27me3
- trimethylate histone H3 at lysine 27
- RBM38
- RNA binding motif protein 38
- RNA-Seq
- RNA sequencing
- qRT-PCR
- quantitative real-time reverse transcription-PCR
- RT
- room temperature
- iTRAQ
- Isobaric Tags for Relative and Absolute Quantitation
- IAA
- iodoacetamide
- FA
- formic acid
- MS/MS
- tandem Mass Spectrometry
- VSN
- variance stabilization normalization
- IF
- Immunofluorescence
- DAPI
- 4′6-diamidino-2-phenylindole
- IHC
- Immunohistochemistry
- TF
- transcription factor
- PPI
- protein-protein interaction.
REFERENCES
- 1. Esteller M. (2011) Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874 [DOI] [PubMed] [Google Scholar]
- 2. Kugel J. F., and Goodrich J. A. (2012) Non-coding RNAs: key regulators of mammalian transcription. Trends Biochem. Sci. 37, 144–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ponting C. P., Oliver P. L., and Reik W. (2009) Evolution and functions of long noncoding RNAs. Cell 136, 629–641 [DOI] [PubMed] [Google Scholar]
- 4. Rinn J. L., and Chang H. Y. (2012) Genome regulation by long noncoding RNAs. Ann. Rev. Biochem. 81, 145–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ernst C., and Morton C. C. (2013) Identification and function of long non-coding RNA. Front. Cell. Neurosci. 7, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cech T. R., and Steitz J. A. (2014) The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94 [DOI] [PubMed] [Google Scholar]
- 7. Gupta R. A., Shah N., Wang K. C., Kim J., Horlings H. M., Wong D. J., Tsai M. C., Hung T., Argani P., Rinn J. L., Wang Y. L., Brzoska P., Kong B., Li R., West R. B., van de Vijver M. J., Sukumar S., and Chang H. Y. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, U1071–U1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. El-Serag H. B., and Rudolph K. L. (2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576 [DOI] [PubMed] [Google Scholar]
- 9. Venook A. P., Papandreou C., Furuse J., and de Guevara L. L. (2010) The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist 15, 5–13 [DOI] [PubMed] [Google Scholar]
- 10. Yang Z., Zhou L., Wu L. M., Lai M. C., Xie H. Y., Zhang F., and Zheng S. S. (2011) Overexpression of Long Non-coding RNA HOTAIR Predicts Tumor Recurrence in Hepatocellular Carcinoma Patients Following Liver Transplantation. Ann. Surg. Oncol. 18, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 11. Ishibashi M., Kogo R., Shibata K., Sawada G., Takahashi Y., Kurashige J., Akiyoshi S., Sasaki S., Iwaya T., Sudo T., Sugimachi K., Mimori K., Wakabayashi G., and Mori M. (2013) Clinical significance of the expression of long non-coding RNA HOTAIR in primary hepatocellular carcinoma. Oncol. Reports 29, 946–950 [DOI] [PubMed] [Google Scholar]
- 12. Fu W. M., Zhu X., Wang W. M., Lu Y. F., Hu B. G., Wang H., Liang W. C., Wang S. S., Ko C. H., Waye M. M. Y., Kung H. F., Li G., and Zhang J. F. (2015) Hotair mediates hepatocarcinogenesis through suppressing miRNA-218 expression and activating P14 and P16 signaling. J. Hepatol. 63, 886–895 [DOI] [PubMed] [Google Scholar]
- 13. Wang J. Y., Liu X. F., Wu H. C., Ni P. H., Gu Z. D., Qiao Y. X., Chen N., Sun F. Y., and Fan Q. S. (2010) CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 38, 5366–5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cui M., Xiao Z. L., Wang Y., Zheng M. Y., Song T. Q., Cai X. L., Sun B. D., Ye L. H., and Zhang X. D. (2015) Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res. 75, 846–857 [DOI] [PubMed] [Google Scholar]
- 15. Lai M. C., Yang Z., Zhou L., Zhu Q. Q., Xie H. Y., Zhang F., Wu L. M., Chen L. M., and Zheng S. S. (2012) Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med. Oncol. 29, 1810–1816 [DOI] [PubMed] [Google Scholar]
- 16. Rinn J. L., Kertesz M., Wang J. K., Squazzo S. L., Xu X., Brugmann S. A., Goodnough L. H., Helms J. A., Farnham P. J., Segal E., and Chang H. Y. (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shah N., and Sukumar S. (2010) The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 10, 361–371 [DOI] [PubMed] [Google Scholar]
- 18. Kim H. J., Lee D. W., Yim G. W., Nam E. J., Kim S., Kim S. W., and Kim Y. T. (2015) Long non-coding RNA HOTAIR is associated with human cervical cancer progression. Int. J. Oncol. 46, 521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee M., Kim H. J., Kim S. W., Park S. A., Chun K. H., Cho N. H., Song Y. S., and Kim Y. T. (2016) The long non-coding RNA HOTAIR increases tumour growth and invasion in cervical cancer by targeting the Notch pathway. Oncotarget 7, 44558–44571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen F. J., Sun M., Li S. Q., Wu Q. Q., Ji L., Liu Z. L., Zhou G. Z., Cao G., Jin L., Xie H. W., Wang C. M., Lv J., De W., Wu M., and Cao X. F. (2013) Upregulation of the Long Non-Coding RNA HOTAIR Promotes Esophageal Squamous Cell Carcinoma Metastasis and Poor Prognosis. Mol. Carcinogen 52, 908–915 [DOI] [PubMed] [Google Scholar]
- 21. Ge X. S., Ma H. J., Zheng X. H., Ruan H. L., Liao X. Y., Xue W. Q., Chen Y. B., Zhang Y., and Jia W. H. (2013) HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci. 104, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu Z. Y., Yu Q. M., Du Y. A., Yang L. T., Dong R. Z., Huang L., Yu P. F., and Cheng X. D. (2013) Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 9, 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu X. H., Sun M., Nie F. Q., Ge Y. B., Zhang E. B., Yin D. D., Kong R., Xia R., Lu K. H., Li J. H., De W., Wang K. M., and Wang Z. X. (2014) Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331–3p in gastric cancer. Mol. Cancer 13, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Du M., Wang W., Jin H., Wang Q., Ge Y., Lu J., Ma G., Chu H., Tong N., Zhu H., Wang M., Qiang F., and Zhang Z. (2015) The association analysis of lncRNA HOTAIR genetic variants and gastric cancer risk in a Chinese population. Oncotarget 6, 31255–31262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yiwei T., Hua H., Hui G., Mao M., and Xiang L. (2015) HOTAIR Interacting with MAPK1 regulates ovarian cancer skov3 cell proliferation, migration, and invasion. Med. Sci. Monitor 21, 1856–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ozes A. R., Miller D. F., Ozes O. N., Fang F., Liu Y., Matei D., Huang T., and Nephew K. P. (2016) NF-kappaB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene 35, 5350–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dong L. J., and Hu L. N. (2016) HOTAIR Promotes Proliferation, Migration, and Invasion of Ovarian Cancer SKOV3 Cells Through Regulating PIK3R3. Med. Sci. Monitor 22, 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geng Y. J., Xie S. L., Li Q., Ma J., and Wang G. Y. (2011) Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int.Med. Res. 39, 2119–2128 [DOI] [PubMed] [Google Scholar]
- 29. Gao J. Z., Li J., Du J. L., and Li X. L. (2016) Long non-coding RNA HOTAIR is a marker for hepatocellular carcinoma progression and tumor recurrence. Oncol. Lett. 11, 1791–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding C., Cheng S., Yang Z., Lv Z., Xiao H., Du C., Peng C., Xie H., Zhou L., Wu J., and Zheng S. (2014) Long non-coding RNA HOTAIR promotes cell migration and invasion via down-regulation of RNA binding motif protein 38 in hepatocellular carcinoma cells. Int. J. Mol. Sci. 15, 4060–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hajjari M., and Salavaty A. (2015) HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol. Med. 12, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li L., Liu B., Wapinski O. L., Tsai M.-C., Qu K., Zhang J., Carlson J. C., Lin M., Fang F., and Gupta R. A. (2013) Targeted Disruption of Hotair Leads to Homeotic Transformation and Gene Derepression. Cell Reports 5, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsai M. C., Manor O., Wan Y., Mosammaparast N., Wang J. K., Lan F., Shi Y., Segal E., and Chang H. Y. (2010) Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 329, 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma M. Z., Li C. X., Zhang Y., Weng M. Z., Zhang M. D., Qin Y. Y., Gong W., and Quan Z. W. (2014) Long non-coding RNA HOTAIR, a c-Myc activated driver of malignancy, negatively regulates miRNA-130a in gallbladder cancer. Mol. Cancer 13, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoon J. H., Abdelmohsen K., Kim J., Yang X., Martindale J. L., Tominaga-Yamanaka K., White E. J., Orjalo A. V., Rinn J. L., Kreft S. G., Wilson G. M., and Gorospe M. (2013) Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nature Commun. 4, 2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou J. J., Cheng D., He X. Y., Meng Z., Li W. Z., and Chen R. F. (2017) Knockdown of Hotair suppresses proliferation and cell cycle progression in hepatocellular carcinoma cell by downregulating CCND1 expression. Mol. Med. Reports 16, 4980–4986 [DOI] [PubMed] [Google Scholar]
- 37. Su D. N., Wu S. P., Chen H. T., and He J. H. (2016) HOTAIR, a long non-coding RNA driver of malignancy whose expression is activated by FOXC1, negatively regulates miRNA-1 in hepatocellular carcinoma. Oncol. Lett. 12, 4061–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Niinuma T., Suzuki H., Nojima M., Nosho K., Yamamoto H., Takamaru H., Yamamoto E., Maruyama R., Nobuoka T., Miyazaki Y., Nishida T., Bamba T., Kanda T., Ajioka Y., Taguchi T., Okahara S., Takahashi H., Nishida Y., Hosokawa M., Hasegawa T., Tokino T., Hirata K., Imai K., Toyota M., and Shinomura Y. (2012) Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 72, 1126–1136 [DOI] [PubMed] [Google Scholar]
- 39. Ono H., Motoi N., Nagano H., Miyauchi E., Ushijima M., Matsuura M., Okumura S., Nishio M., Hirose T., and Inase N. (2014) Long noncoding RNA HOTAIR is relevant to cellular proliferation, invasiveness, and clinical relapse in small-cell lung cancer. Cancer Med. 3, 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim K., Jutooru I., Chadalapaka G., Johnson G., Frank J., Burghardt R., Kim S., and Safe S. (2013) HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 42. Zheng P., Xiong Q., Wu Y., Chen Y., Chen Z., Fleming J., Gao D., Bi L. J., and Ge F. (2015) Quantitative proteomics analysis reveals novel insights into mechanisms of action of long noncoding RNA Hox transcript antisense intergenic RNA (HOTAIR) in HeLa cells. Mol. Cell. Proteomics 14, 1447–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protocols 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trapnell C., Pachter L., and Salzberg S. L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anders S., Pyl P. T., and Huber W. (2015) HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wen B., Du C., Li G., Ghali F., Jones A. R., Kall L., Xu S., Zhou R., Ren Z., Feng Q., Xu X., and Wang J. (2015) IPeak: An open source tool to combine results from multiple MS/MS search engines. Proteomics 15, 2916–2920 [DOI] [PubMed] [Google Scholar]
- 48. Wen B., Li G., Wright J. C., Du C., Feng Q., Xu X., Choudhary J. S., and Wang J. (2014) The OMSSAPercolator: an automated tool to validate OMSSA results. Proteomics 14, 1011–1014 [DOI] [PubMed] [Google Scholar]
- 49. Wen B., Zhou R., Feng Q., Wang Q., Wang J., and Liu S. (2014) IQuant: an automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 14, 2280–2285 [DOI] [PubMed] [Google Scholar]
- 50. Huber W., von Heydebreck A., Sultmann H., Poustka A., and Vingron M. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18, S96–104 [DOI] [PubMed] [Google Scholar]
- 51. Karp N. A., Huber W., Sadowski P. G., Charles P. D., Hester S. V., and Lilley K. S. (2010) Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 9, 1885–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Benjamini Y., and Hochberg Y. (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Roy. Statistical Soc. 57, 289–300 [Google Scholar]
- 53. Berard A. R., Cortens J. P., Krokhin O., Wilkins J. A., Severini A., and Coombs K. M. (2012) Quantification of the host response proteome after mammalian reovirus T1L infection. PLoS ONE 7, e51939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Armenta J. M., Hoeschele I., and Lazar I. M. (2009) Differential Protein Expression Analysis Using Stable Isotope Labeling and PQD Linear Ion Trap MS Technology. J. Am. Soc. Mass Spectrom. 20, 1287–1302 [DOI] [PubMed] [Google Scholar]
- 55. Coombs K. M., Berard A., Xu W., Krokhin O., Meng X., Cortens J. P., Kobasa D., Wilkins J., and Brown E. G. (2010) Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J. Virol 84, 10888–10906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Han J., Zhang J., Chen L., Shen B., Zhou J., Hu B., Du Y., Tate P. H., Huang X., and Zhang W. (2014) Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 11, 829–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ho T. T., Zhou N., Huang J., Koirala P., Xu M., Fung R., Wu F., and Mo Y. Y. (2015) Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 43, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goyal A., Myacheva K., Gross M., Klingenberg M., Duran Arque B., and Diederichs S. (2016) Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 45, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mi H., Muruganujan A., Casagrande J. T., and Thomas P. D. (2013) Large-scale gene function analysis with the PANTHER classification system. Nat. Protocols 8, 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., and Thomas P. D. (2017) PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 63. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zagon I. S., Donahue R. N., and McLaughlin P. J. (2009) Opioid growth factor-opioid growth factor receptor axis is a physiological determinant of cell proliferation in diverse human cancers. Am. J. Physiol.-Reg I 297, R1154–R1161 [DOI] [PubMed] [Google Scholar]
- 66. Donahue R. N., McLaughlin P. J., and Zagon I. S. (2009) Cell proliferation of human ovarian cancer is regulated by the opioid growth factor-opioid growth factor receptor axis. Am. J. Physiol.-Reg I 296, R1716–R1725 [DOI] [PubMed] [Google Scholar]
- 67. Cheng F., McLaughlin P. J., Verderame M. F., and Zagon I. S. (2008) The OGF-OGFr axis uses the p21 pathway to restrict progression of human pancreatic cancer. Molecular cancer 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cheng F., Zagon I. S., Verderame M. F., and McLaughlin P. J. (2007) The opioid growth factor (OGF)-OGF receptor axis uses the p16 pathway to inhibit head and neck cancer. Cancer Res. 67, 10511–10518 [DOI] [PubMed] [Google Scholar]
- 69. Zagon I. S., Kreiner S., Heslop J. J., Conway A. B., Morgan C. R., and McLaughlin P. J. (2008) Prevention and delay in progression of human pancreatic cancer by stable overexpression of the opioid growth factor receptor. Int. J. Oncol. 33, 317–323 [PubMed] [Google Scholar]
- 70. Kim K., Jutooru I., Chadalapaka G., Johnson G., Frank J., Burghardt R., Kim S., and Safe S. (2012) HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee K., Lee K. B., Jung H. Y., Yi N. J., Lee K. W., Suh K. S., and Jang J. J. (2017) The correlation between poor prognosis and increased yes-associated protein 1 expression in keratin 19 expressing hepatocellular carcinomas and cholangiocarcinomas. BMC Cancer 17, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ma X., Zhang H., Xue X., and Shah Y. M. (2017) Hypoxia-inducible factor 2alpha (HIF-2alpha) promotes colon cancer growth by potentiating Yes-associated protein 1 (YAP1) activity. J. Biol. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ou C., Sun Z., Li X., Li X., Ren W., Qin Z., Zhang X., Yuan W., Wang J., Yu W., Zhang S., Peng Q., Yan Q., Xiong W., Li G., and Ma J. (2017) MiR-590–5p, a density-sensitive microRNA, inhibits tumorigenesis by targeting YAP1 in colorectal cancer. Cancer Lett. 399, 53–63 [DOI] [PubMed] [Google Scholar]
- 74. Salcedo Allende M. T., Zeron-Medina J., Hernandez J., Macarulla T., Balsells J., Merino X., Allende H., Tabernero J., Ramon Y. C. A. S. (2017) Overexpression of Yes associated protein 1, an independent prognostic marker in patients with pancreatic ductal adenocarcinoma, correlated with liver metastasis and poor prognosis. Pancreas 46, 913–920 [DOI] [PubMed] [Google Scholar]
- 75. Xia J., Zeng M., Zhu H., Chen X., Weng Z., and Li S. (2017) Emerging role of Hippo signalling pathway in bladder cancer. J. Cell. Mol. Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Avella D. M., Kimchi E. T., Donahue R. N., Tagaram H. R. S., McLaughlin P. J., Zagon I. S., and Staveley-O'Carroll K. F. (2010) The opioid growth factor-opioid growth factor receptor axis regulates cell proliferation of human hepatocellular cancer. Am. J. Physiol.-Reg I 298, R459–R466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zagon I. S., Ruth T. B., Leure-duPree A. E., Sassani J. W., and McLaughlin P. J. (2003) Immunoelectron microscopic localization of the opioid growth factor receptor (OGFr) and OGF in the cornea. Brain Res. 967, 37–47 [DOI] [PubMed] [Google Scholar]
- 78. Cheng F., McLaughlin P. J., Verderame M. F., and Zagon I. S. (2009) The OGF-OGFr axis utilizes the p16INK4a and p21WAF1/CIP1 pathways to restrict normal cell proliferation. Mol. Biol. Cell 20, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu Y., Liu J., Zheng Y., You L., Kuang D., and Liu T. (2014) Suppressed expression of long non-coding RNA HOTAIR inhibits proliferation and tumourigenicity of renal carcinoma cells. Tumour Biol. 35, 11887–11894 [DOI] [PubMed] [Google Scholar]
- 80. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Edgar R., Domrachev M., and Lash A. E. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS raw data for iTRAQ-LC-MS/MS have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (80) with data set identifier PXD006554. The MS raw data for PRM have been deposited in the PeptideAtlas database (http://www.PeptideAtlas.org) with the identifier PASS01010. The RNA-Seq data have been deposited in NCBI's Gene Expression Omnibus (81) and are accessible through GEO Series accession number GSE98091 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98091).